Label: CARAFATE- sucralfate suspension

-
Contains inactivated NDC Code(s)
NDC Code(s): 17856-0170-1, 17856-0170-3 - Packager: ATLANTIC BIOLOGICALS CORP.
- This is a repackaged label.
- Source NDC Code(s): 58914-170
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated January 26, 2021
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
DESCRIPTION
CARAFATE Suspension contains sucralfate and sucralfate is an α-D-glucopyranoside, β-D-fructofuranosyl-, octakis-(hydrogen sulfate), aluminum complex.
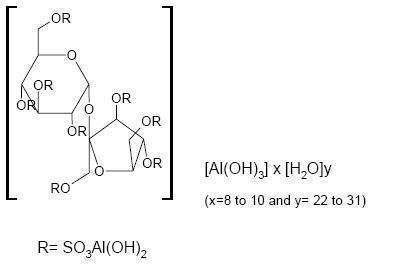
CARAFATE Suspension for oral administration contains 1 g of sucralfate per 10 mL.
CARAFATE Suspension also contains: colloidal silicon dioxide NF, FD&C Red #40, flavor, glycerin USP, methylcellulose USP, methylparaben NF, microcrystalline cellulose NF, purified water USP, simethicone USP, and sorbitol solution USP. Therapeutic category: antiulcer.
-
CLINICAL PHARMACOLOGY
Sucralfate is only minimally absorbed from the gastrointestinal tract. The small amounts of the sulfated disaccharide that are absorbed are excreted primarily in the urine.
Although the mechanism of sucralfate’s ability to accelerate healing of duodenal ulcers remains to be fully defined, it is known that it exerts its effect through a local, rather than systemic, action. The following observations also appear pertinent:
- Studies in human subjects and with animal models of ulcer disease have shown that sucralfate forms an ulcer-adherent complex with proteinaceous exudate at the ulcer site.
- In vitro, a sucralfate-albumin film provides a barrier to diffusion of hydrogen ions.
- In human subjects, sucralfate given in doses recommended for ulcer therapy inhibits pepsin activity in gastric juice by 32%.
- In vitro, sucralfate adsorbs bile salts.
These observations suggest that sucralfate’s antiulcer activity is the result of formation of an ulcer-adherent complex that covers the ulcer site and protects it against further attack by acid, pepsin, and bile salts. There are approximately 14 to 16 mEq of acid-neutralizing capacity per 1 g dose of sucralfate.
-
CLINICAL TRIALS
In a multicenter, double-blind, placebo-controlled study of CARAFATE Suspension, a dosage regimen of 1 g (10 mL) four times daily was demonstrated to be superior to placebo in ulcer healing.
Results From Clinical Trials Healing Rates for Acute Duodenal Ulcer*P=0.016
†P=0.001
‡P=0.0001
Treatment n Week 2 Healing Rates Week 4 Healing Rates Week 8 Healing Rates
CARAFATE Suspension 145 23(16%) * 66(46%) † 95(66%) ‡
Placebo 147 10(7%) 39(27%) 58(39%)Equivalence of sucralfate suspension to sucralfate tablets has not been demonstrated.
- INDICATIONS AND USAGE
- CONTRAINDICATIONS
-
PRECAUTIONS
The physician should read the "PRECAUTIONS" section when considering the use of CARAFATE in pregnant or pediatric patients, or patients of childbearing potential.
Duodenal ulcer is a chronic, recurrent disease. While short-term treatment with sucralfate can result in complete healing of the ulcer, a successful course of treatment with sucralfate should not be expected to alter the post healing frequency or severity of duodenal ulceration.
Episodes of hyperglycemia have been reported in diabetic patients. Close monitoring of glycemia in diabetic patients treated with sucralfate suspension is recommended. Adjustment of the anti-diabetic treatment dose during the use of sucralfate suspension might be necessary.
Special Populations: Chronic Renal Failure and Dialysis Patients
When sucralfate is administered orally, small amounts of aluminum are absorbed from the gastrointestinal tract. Concomitant use of sucralfate with other products that contain aluminum, such as aluminum-containing antacids, may increase the total body burden of aluminum. Patients with normal renal function receiving the recommended doses of sucralfate and aluminum-containing products adequately excrete aluminum in the urine. Patients with chronic renal failure or those receiving dialysis have impaired excretion of absorbed aluminum. In addition, aluminum does not cross dialysis membranes because it is bound to albumin and transferrin plasma proteins. Aluminum accumulation and toxicity (aluminum osteodystrophy, osteomalacia, encephalopathy) have been described in patients with renal impairment. Sucralfate should be used with caution in patients with chronic renal failure.
Drug Interactions
Some studies have shown that simultaneous sucralfate administration in healthy volunteers reduced the extent of absorption (bioavailability) of single doses of the following: cimetidine, digoxin, fluoroquinolone antibiotics, ketoconazole, l-thyroxine, phenytoin, quinidine, ranitidine, tetracycline, and theophylline. Subtherapeutic prothrombin times with concomitant warfarin and sucralfate therapy have been reported in spontaneous and published case reports. However, two clinical studies have demonstrated no change in either serum warfarin concentration or prothrombin time with the addition of sucralfate to chronic warfarin therapy.
The mechanism of these interactions appears to be nonsystemic in nature, presumably resulting from sucralfate binding to the concomitant agent in the gastrointestinal tract. In all cases studied to date (cimetidine, ciprofloxacin, digoxin, norfloxacin, ofloxacin, and ranitidine), dosing the concomitant medication 2 hours before sucralfate eliminated the interaction. Due to CARAFATE's potential to alter the absorption of some drugs, CARAFATE should be administered separately from other drugs when alterations in bioavailability are felt to be critical. In these cases, patients should be monitored appropriately.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Chronic oral toxicity studies of 24 months’ duration were conducted in mice and rats at doses up to 1 g/kg (12 times the human dose).
There was no evidence of drug-related tumorigenicity. A reproduction study in rats at doses up to 38 times the human dose did not reveal any indication of fertility impairment. Mutagenicity studies were not conducted.
Pregnancy
Teratogenic effects. Pregnancy Category B.
Teratogenicity studies have been performed in mice, rats, and rabbits at doses up to 50 times the human dose and have revealed no evidence of harm to the fetus due to sucralfate. There are, however, no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed.
Nursing Mothers
It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when sucralfate is administered to a nursing woman.
Geriatric Use
Clinical studies of CARAFATE Suspension did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy. (See ) DOSAGE AND ADMINISTRATION
This drug is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function (See ). Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function. PRECAUTIONS Special Populations: Chronic Renal Failure and Dialysis Patients
-
ADVERSE REACTIONS
Adverse reactions to sucralfate tablets in clinical trials were minor and only rarely led to discontinuation of the drug. In studies involving over 2700 patients treated with sucralfate, adverse effects were reported in 129 (4.7%).
Constipation was the most frequent complaint (2%). Other adverse effects reported in less than 0.5% of the patients are listed below by body system:
diarrhea, dry mouth, flatulence, gastric discomfort, indigestion, nausea, vomiting Gastrointestinal:
pruritus, rash Dermatological:
dizziness, insomnia, sleepiness, vertigo Nervous System:
back pain, headache Other:
cases of hypersensitivity have been reported with the use of sucralfate suspension, including anaphylactic reactions, dyspnea, lip swelling, edema of the mouth, pharyngeal edema, pruritus, rash, swelling of the face and urticaria. Post-marketing
Cases of bronchospasm, laryngeal edema and respiratory tract edema have been reported with an unknown oral formulation of sucralfate.
Cases of hyperglycemia have been reported with sucralfate
Bezoars have been reported in patients treated with sucralfate. The majority of patients had underlying medical conditions that may predispose to bezoar formation (such as delayed gastric emptying) or were receiving concomitant enteral tube feedings.
Inadvertent injection of insoluble sucralfate and its insoluble excipients has led to fatal complications, including pulmonary and cerebral emboli. Sucralfate is intended for intravenous administration. not
-
OVERDOSAGE
Due to limited experience in humans with overdosage of sucralfate, no specific treatment recommendations can be given. Acute oral studies in animals, however, using doses up to 12 g/kg body weight, could not find a lethal dose. Sucralfate is only minimally absorbed from the gastrointestinal tract. Risks associated with acute overdosage should, therefore, be minimal. In rare reports describing sucralfate overdose, most patients remained asymptomatic. Those few reports where adverse events were described included symptoms of dyspepsia, abdominal pain, nausea, and vomiting.
-
DOSAGE AND ADMINISTRATION
The recommended adult oral dosage for duodenal ulcer is 1 g (10 mL/2 teaspoons) four times per day. CARAFATE should be administered on an empty stomach. Active Duodenal Ulcer.
Antacids may be prescribed as needed for relief of pain but should not be taken within one-half hour before or after sucralfate.
While healing with sucralfate may occur during the first week or two, treatment should be continued for 4 to 8 weeks unless healing has been demonstrated by x-ray or endoscopic examination.
In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy (See ). Elderly:PRECAUTIONS Geriatric Use
Call your doctor for medical advice about side effects. You may report side effects to Aptalis Pharma US, Inc. at 1-800-472-2634 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch
- HOW SUPPLIED
- CARAFATE (SUCRALFATE) SUSPENSION
-
INGREDIENTS AND APPEARANCE
CARAFATE
sucralfate suspensionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:17856-0170(NDC:58914-170) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Sucralfate (UNII: XX73205DH5) (Sucralfate - UNII:XX73205DH5) Sucralfate 1 g in 10 mL Inactive Ingredients Ingredient Name Strength FD&C RED NO. 40 (UNII: WZB9127XOA) 0.004 mg in 10 mL Glycerin (UNII: PDC6A3C0OX) 87.92 mg in 10 mL Methylparaben (UNII: A2I8C7HI9T) 1.78 mg in 10 mL Sorbitol (UNII: 506T60A25R) 177.62 mg in 10 mL Water (UNII: 059QF0KO0R) 618.006 mg in 10 mL Product Characteristics Color pink Score Shape Size Flavor CHERRY (Maraschino Cherry Artificial Flavor #2359) Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:17856-0170-1 10 mL in 1 CUP; Type 0: Not a Combination Product 01/26/2021 2 NDC:17856-0170-3 10 mL in 1 CUP, UNIT-DOSE; Type 0: Not a Combination Product 01/26/2021 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA019183 12/16/1993 Labeler - ATLANTIC BIOLOGICALS CORP. (047437707) Registrant - ATLANTIC BIOLOGICALS CORP. (047437707) Establishment Name Address ID/FEI Business Operations ATLANTIC BIOLOGICALS CORP. 047437707 RELABEL(17856-0170) , REPACK(17856-0170)
