ROFERON-A- interferon alfa-2a injection, solution
Genentech, Inc.
----------
ROFERON®-A
(Interferon alfa-2a, recombinant)
Alpha-interferons, including Interferon alfa-2a, cause or aggravate fatal or life-threatening neuropsychiatric, autoimmune, ischemic, and infectious disorders. Patients should be monitored closely with periodic clinical and laboratory evaluations. Patients with persistently severe or worsening signs or symptoms of these conditions should be withdrawn from therapy. In many, but not all cases, these disorders resolve after stopping Interferon alfa-2a therapy (see WARNINGS and ADVERSE REACTIONS).
DESCRIPTION
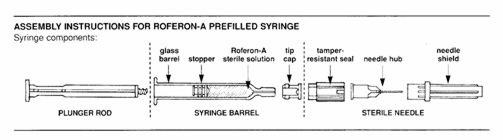
Roferon-A (Interferon alfa-2a, recombinant) is a sterile protein product for use by injection. Roferon-A is manufactured by recombinant DNA technology that employs a genetically engineered Escherichia coli bacterium containing DNA that codes for the human protein. Interferon alfa-2a, recombinant is a highly purified protein containing 165 amino acids, and it has an approximate molecular weight of 19,000 daltons. Fermentation is carried out in a defined nutrient medium containing the antibiotic tetracycline hydrochloride, 5 mg/L. However, the presence of the antibiotic is not detectable in the final product. Roferon-A is supplied in prefilled syringes. Each glass syringe barrel contains 0.5 mL of product. In addition, there is a needle, which is ½ inch in length.
Single Use Prefilled Syringes
3 million IU (11.1 mcg/0.5 mL) Roferon-A per syringe — The solution is colorless and each 0.5 mL contains 3 MIU of Interferon alfa-2a, recombinant, 3.605 mg sodium chloride, 0.1 mg polysorbate 80, 5 mg benzyl alcohol as a preservative and 0.385 mg ammonium acetate.
6 million IU (22.2 mcg/0.5 mL) Roferon-A per syringe — The solution is colorless and each 0.5 mL contains 6 MIU of Interferon alfa-2a, recombinant, 3.605 mg sodium chloride, 0.1 mg polysorbate 80, 5 mg benzyl alcohol as a preservative and 0.385 mg ammonium acetate.
9 million IU (33.3 mcg/0.5 mL) Roferon-A per syringe — The solution is colorless and each 0.5 mL contains 9 MIU of Interferon alfa-2a, recombinant, 3.605 mg sodium chloride, 0.1 mg polysorbate 80, 5 mg benzyl alcohol as a preservative and 0.385 mg ammonium acetate.
The route of administration is by subcutaneous injection.
CLINICAL PHARMACOLOGY
The mechanism by which Interferon alfa-2a, recombinant, or any other interferon, exerts antitumor or antiviral activity is not clearly understood. However, it is believed that direct antiproliferative action against tumor cells, inhibition of virus replication and modulation of the host immune response play important roles in antitumor and antiviral activity.
The biological activities of Interferon alfa-2a, recombinant are species-restricted, i.e., they are expressed in a very limited number of species other than humans. As a consequence, preclinical evaluation of Interferon alfa-2a, recombinant has involved in vitro experiments with human cells and some in vivo experiments.1 Using human cells in culture, Interferon alfa-2a, recombinant has been shown to have antiproliferative and immunomodulatory activities that are very similar to those of the mixture of interferon alfa subtypes produced by human leukocytes. In vivo, Interferon alfa-2a, recombinant has been shown to inhibit the growth of several human tumors growing in immunocompromised (nude) mice. Because of its species-restricted activity, it has not been possible to demonstrate antitumor activity in immunologically intact syngeneic tumor model systems, where effects on the host immune system would be observable. However, such antitumor activity has been repeatedly demonstrated with, for example, mouse interferon-alfa in transplantable mouse tumor systems. The clinical significance of these findings is unknown.
The metabolism of Interferon alfa-2a, recombinant is consistent with that of alpha-interferons in general. Alpha-interferons are totally filtered through the glomeruli and undergo rapid proteolytic degradation during tubular reabsorption, rendering a negligible reappearance of intact alfa interferon in the systemic circulation. Small amounts of radiolabeled Interferon alfa-2a, recombinant appear in the urine of isolated rat kidneys, suggesting near complete reabsorption of Interferon alfa-2a, recombinant catabolites. Liver metabolism and subsequent biliary excretion are considered minor pathways of elimination for alfa interferons.
The serum concentrations of Interferon alfa-2a, recombinant reflected a large intersubject variation in both healthy volunteers and patients with disseminated cancer.
In healthy people, Interferon alfa-2a, recombinant exhibited an elimination half-life of 3.7 to 8.5 hours (mean 5.1 hours), volume of distribution at steady-state of 0.223 to 0.748 L/kg (mean 0.400 L/kg) and a total body clearance of 2.14 to 3.62 mL/min/kg (mean 2.79 mL/min/kg) after a 36 MIU (2.2×108pg) intravenous infusion. After intramuscular and subcutaneous administrations of 36 MIU, peak serum concentrations ranged from 1500 to 2580 pg/mL (mean 2020 pg/mL) at a mean time to peak of 3.8 hours and from 1250 to 2320 pg/mL (mean 1730 pg/mL) at a mean time to peak of 7.3 hours, respectively. The apparent fraction of the dose absorbed after intramuscular injection was greater than 80%.
The pharmacokinetics of Interferon alfa-2a, recombinant after single intramuscular doses to patients with disseminated cancer were similar to those found in healthy volunteers. Dose proportional increases in serum concentrations were observed after single doses up to 198 MIU. There were no changes in the distribution or elimination of Interferon alfa-2a, recombinant during twice daily (0.5 to 36 MIU), once daily (1 to 54 MIU), or three times weekly (1 to 136 MIU) dosing regimens up to 28 days of dosing. Multiple intramuscular doses of Interferon alfa-2a, recombinant resulted in an accumulation of two to four times the single dose serum concentrations. There is no pharmacokinetic information in patients with chronic hepatitis C, hairy cell leukemia, and chronic myelogenous leukemia.
Serum neutralizing activity, determined by a highly sensitive enzyme immunoassay, and a neutralization bioassay, was detected in approximately 25% of all patients who received Roferon-A.2 Antibodies to human leukocyte interferon may occur spontaneously in certain clinical conditions (cancer, systemic lupus erythematosus, herpes zoster) in patients who have never received exogenous interferon.3 The significance of the appearance of serum neutralizing activity is not known.
Clinical Studies
Studies have shown that Roferon-A can normalize serum ALT, improve liver histology and reduce viral load in patients with chronic hepatitis C. Other studies have shown that Roferon-A can produce clinically meaningful tumor regression or disease stabilization in patients with hairy cell leukemia.4,5 In Ph-positive Chronic Myelogenous Leukemia, Roferon-A supplemented with intermittent chemotherapy has been shown to prolong overall survival and to delay disease progression compared to patients treated with chemotherapy alone.6 In addition, Roferon-A has been shown to produce sustained complete cytogenetic responses in a small subset of patients with CML in chronic phase. The activity of Roferon-A in Ph-negative CML has not been determined.
Effects On Chronic Hepatitis C
The safety and efficacy of Roferon-A was evaluated in multiple clinical trials involving over 2000 patients 18 years of age or older with hepatitis, with or without cirrhosis, who had elevated serum alanine aminotransferase (ALT) levels and tested positive for antibody to hepatitis C. Roferon-A was given three times a week (tiw) by subcutaneous (SC) or intramuscular (IM) injection in a variety of dosing regimens, including dose escalation and de-escalation regimens. Normalization of serum ALT was defined in all studies as two consecutive normal serum ALT values at least 21 days apart. A sustained response (SR) was defined as normalization of ALT both at the end of treatment and at the end of at least 6 months of treatment-free follow-up.
In trials in which Roferon-A was administered for 6 months, 6 MIU, 3 MIU, and 1 MIU were directly compared. Six MIU was associated with higher SR rates but greater toxicity (see ADVERSE REACTIONS). In studies in which the same dose of Roferon-A was administered for 6 or 12 months, the longer duration was associated with higher SR rates and adverse events were no more severe or frequent in the second 6 months than in the first 6 months. Based on these data, the recommended regimens are 3 MIU for 12 months or 6 MIU for the first 3 months followed by 3 MIU for the next 9 months (see Table 1 and DOSAGE AND ADMINISTRATION). There are no direct comparisons of these two regimens.
Younger patients (e.g., less than 35 years of age) and patients without cirrhosis on liver biopsy were more likely to respond completely to Roferon-A than those patients greater than 35 years of age or patients with cirrhosis on liver biopsy.
In the two studies in which Roferon-A was administered subcutaneously three times weekly for 12 months, 20/173 (12%) patients experienced a sustained response to therapy (see Table 1). Of these patients, 15/173 (9%) maintained this sustained response during continuous follow-up for up to four years. Patients who have ALT normalization but who fail to have a sustained response following an initial course of therapy may benefit from retreatment with higher doses of Roferon-A (see DOSAGE AND ADMINISTRATION).
A subset of patients had liver biopsies performed both before and after treatment with Roferon-A. An improvement in liver histology as assessed by Knodell Histology Activity Index was generally observed.
A retrospective subgroup analysis of 317 patients from two studies suggested a correlation between improvement in liver histology, durable serum ALT response rates, and decreased viral load as measured by the polymerase chain reaction (PCR).
| Study No. | Dose (MIU) | N | End of Treatment [% (95% CI)] | End of Observation (Sustained Response SR) [% (95% CI)]* |
|---|---|---|---|---|
| 1† | 3 | 56 | 23 | 11 |
| 2 | 3 | 117 | 23 | 12 |
| 1 and 2 Combined | 3 | 173 | 23 (17-30) | 12 (7-17) |
| 3 | 6-3 | 210 | 25 (19-31) | 19 (14-25) |
Effects on Ph-Positive Chronic Myelogenous Leukemia (CML)
Roferon-A was evaluated in two trials of patients with chronic phase CML. Study DM84-38 was a single center phase II study conducted at the MD Anderson Cancer Center, which enrolled 91 patients, 81% were previously treated, 82% were Ph positive, and 63% received Roferon-A within 1 year of diagnosis. Study MI400 was a multicenter randomized phase III study conducted in Italy by the Italian Cooperative Study Group on CML in 335 patients; 226 Roferon-A and 109 chemotherapy. Patients with Ph-positive, newly diagnosed or minimally treated CML were randomized (ratio 2:1) to either Roferon-A or conventional chemotherapy with either hydroxyurea or busulfan. In study DM84-38, patients started Roferon-A at 9 MIU/day, whereas in study MI400, it was progressively escalated from 3 to 9 MIU/day over the first month. In both trials, dose escalation for insufficient hematologic response, and dose attenuation or interruption for toxicity was permitted. No formal guidelines for dose attenuation were given in the chemotherapy arm of study MI400. In addition, in the Roferon-A arm, the MI400 protocol allowed the addition of intermittent single agent chemotherapy for insufficient hematologic response to Roferon-A alone. In this trial, 44% of the Roferon-A treated patients also received intermittent single agent chemotherapy at some time during the study.
The two studies were analyzed according to uniform response criteria. For hematologic response: complete response (WBC <9×109/L, normalization of the differential with no immature forms in the peripheral blood, disappearance of splenomegaly), partial response (>50% decrease from baseline of WBC to <20%×109/L). For cytogenetic response: complete response (0% Ph-positive metaphases), partial response (1% to 34% Ph-positive metaphases).
In study DM84-38, the median survival from initiation of Roferon-A was 47 months. In study MI400, the median survival for the patients on the interferon arm was 69 months, which was significantly better than the 55 months seen in the chemotherapy control group (48 patients in study MI400 proceeded to BMT and in study DM84-38, 15 patients proceeded to BMT). Roferon-A treatment significantly delayed disease progression to blastic phase as evidenced by a median time to disease progression of 69 months to 46 months with chemotherapy.
By multivariate analysis of prognostic factors associated with all 335 patients entered into the randomized study, treatment with Roferon-A (with or without intermittent additional chemotherapy; p=0.006), Sokal index7 (p=0.006) and WBC (p=0.023) were the three variables associated with an improved survival, independent of other baseline characteristics (Karnofsky performance status and hemoglobin being the other factors entered into the model).
In study MI400, overall hematologic responses, [complete responses (CR) and partial responses (PR)], were observed in approximately 60% of patients treated with Roferon-A (40% CR, 20% PR), compared to 70% with chemotherapy (30% CR, 40% PR). The median time to reach a complete hematologic response was 5 months in the Roferon-A arm and 4 months in the chemotherapy arm. The overall cytogenetic response rate (CR+PR), in patients receiving Roferon-A, was 10% and 12% in studies MI400 and DM84-38, respectively, according to the intent-to-treat principle. In contrast, only 2% of the patients in the chemotherapy arm of study MI400 achieved a cytogenetic response (with no complete responses). Cytogenetic responses were observed only in patients who had complete hematologic responses. In study DM84-38, hematologic and cytogenetic response rates were higher in the subset of patients treated with Roferon-A within 1 year of diagnosis (76% and 17%, respectively) compared to the subset initiating Roferon-A therapy more than 1 year from diagnosis (29% and 4%, respectively). In an exploratory analysis, patients who achieved a cytogenetic response lived longer than those who did not.
Severe adverse events were observed in 66% and 31% of patients on study DM84-38 and MI400, respectively. Dose reduction and temporary cessation of therapy was required frequently. Permanent cessation of Roferon-A, due to intolerable side effects, was required in 15% and 23% of patients on studies DM84-38 and MI400, respectively (see ADVERSE REACTIONS).
Limited data are available on the use of Roferon-A in children with Ph-positive, adult-type CML. A published report on 15 children with CML suggests a safety profile similar to that seen in adult CML; clinical responses were also observed8 (see DOSAGE AND ADMINISTRATION).
Effects on Hairy Cell Leukemia
A multicenter US phase II study (N2752) enrolled 218 patients; 75 were evaluable for efficacy in a preliminary analysis; 218 patients were evaluable for safety. Patients were to receive a starting dose of Roferon-A up to 6 MIU/m2/day, for an induction period of 4 to 6 months. Responding patients were to receive 12 months maintenance therapy.
During the first 1 to 2 months of treatment of patients with hairy cell leukemia, significant depression of hematopoiesis was likely to occur. Subsequently, there was improvement in circulating blood cell counts. Of the 75 patients who were evaluable for efficacy following at least 16 weeks of therapy, 46 (61%) achieved complete or partial response. Twenty-one patients (28%) had a minor remission, 8 (11%) remained stable, and none had worsening of disease. All patients who achieved either a complete or partial response had complete or partial normalization of all peripheral blood elements including hemoglobin level, white blood cell, neutrophil, monocyte and platelet counts with a concomitant decrease in peripheral blood and bone marrow hairy cells. Responding patients also exhibited a marked reduction in red blood cell and platelet transfusion requirements, a decrease in infectious episodes and improvement in performance status. The probability of survival for 2 years in patients receiving Roferon-A (94%) was statistically increased compared to a historical control group (75%).
INDICATIONS AND USAGE
Roferon-A is indicated for the treatment of chronic hepatitis C and hairy cell leukemia in patients 18 years of age or older. In addition, it is indicated for chronic phase, Philadelphia chromosome (Ph) positive chronic myelogenous leukemia (CML) patients who are minimally pretreated (within 1 year of diagnosis).
For Patients With Chronic Hepatitis C
Roferon-A is indicated for use in patients with chronic hepatitis C diagnosed by HCV antibody and/or a history of exposure to hepatitis C who have compensated liver disease and are 18 years of age or older. A liver biopsy and a serum test for the presence of antibody to HCV should be performed to establish the diagnosis of chronic hepatitis C. Other causes of hepatitis, including hepatitis B, should be excluded prior to therapy with Roferon-A.
CONTRAINDICATIONS
Roferon-A is contraindicated in patients with:
- Hypersensitivity to Roferon-A or any of its components
- Autoimmune hepatitis
- Hepatic decompensation (Child-Pugh class B and C) before or during treatment
Roferon-A is contraindicated in neonates and infants because it contains benzyl alcohol. Benzyl alcohol is associated with an increased incidence of neurologic and other complications in neonates and infants, which are sometimes fatal.
WARNINGS
Roferon-A should be administered under the guidance of a qualified physician (see DOSAGE AND ADMINISTRATION). Appropriate management of the therapy and its complications is possible only when adequate facilities are readily available.
Neuropsychiatric Disorders
DEPRESSION AND SUICIDAL BEHAVIOR INCLUDING SUICIDAL IDEATION, SUICIDAL ATTEMPTS AND SUICIDES HAVE BEEN REPORTED IN ASSOCIATION WITH TREATMENT WITH ALFA INTERFERONS, INCLUDING ROFERON-A, IN PATIENTS WITH AND WITHOUT PREVIOUS PSYCHIATRIC ILLNESS. Roferon-A should be used with extreme caution in patients who report a history of depression. Patients should be informed that depression and suicidal ideation may be side effects of treatment and should be advised to report these side effects immediately to the prescribing physician. Patients receiving Roferon-A therapy should receive close monitoring for the occurrence of depressive symptomatology. Psychiatric intervention and/or cessation of treatment should be considered for patients experiencing depression. Although dose reduction or treatment cessation may lead to resolution of the depressive symptomatology, depression may persist and suicides have occurred after withdrawing therapy (see PRECAUTIONS and ADVERSE REACTIONS).
Central nervous system adverse reactions have been reported in a number of patients. These reactions included decreased mental status, dizziness, impaired memory, agitation, manic behavior and psychotic reactions. More severe obtundation and coma have been rarely observed. Most of these abnormalities were mild and reversible within a few days to 3 weeks upon dose reduction or discontinuation of Roferon-A therapy. Careful periodic neuropsychiatric monitoring of all patients is recommended. Roferon-A should be used with caution in patients with seizure disorders and/or compromised central nervous system function.
Cardiovascular Disorders
Roferon-A should be administered with caution to patients with cardiac disease or with any history of cardiac illness. Acute, self-limited toxicities (i.e., fever, chills) frequently associated with Roferon-A administration may exacerbate preexisting cardiac conditions. Rarely, myocardial infarction has occurred in patients receiving Roferon-A. Cases of cardiomyopathy have been observed on rare occasions in patients treated with alpha interferons.
Cerebrovascular Disorders
Ischemic and hemorrhagic cerebrovascular events have been observed in patients treated with interferon alfa-based therapies, including Roferon-A. Events occurred in patients with few or no reported risk factors for stroke, including patients less than 45 years of age. Because these are spontaneous reports, estimates of frequency cannot be made and a causal relationship between interferon alfa-based therapies and these events is difficult to establish.
Hypersensitivity
Serious, acute hypersensitivity reactions (e.g., urticaria, angioedema, bronchoconstriction and anaphylaxis), as well as skin rashes have been rarely observed during alpha-interferon therapy, including interferon alfa-2a. If a serious reaction develops during treatment with Roferon-A, discontinue treatment and institute appropriate medical therapy immediately. Transient rashes do not necessitate interruption of treatment.
Hepatic Disorders
In chronic hepatitis C, initiation of alfa-interferon therapy, including Roferon-A, has been reported to cause transient liver abnormalities, which in patients with poorly compensated liver disease can result in increased ascites, hepatic failure or death.
Gastrointestinal Disorders
Infrequently, severe or fatal gastrointestinal hemorrhage has been reported in association with alpha-interferon therapy.
Ulcerative, and hemorrhagic/ischemic colitis, sometimes fatal, have been observed within 12 weeks of starting alpha interferon treatment. Abdominal pain, bloody diarrhea, and fever are the typical manifestations of colitis. Roferon-A should be discontinued immediately if these symptoms develop. The colitis usually resolves within 1 to 3 weeks of discontinuation of alpha interferon.
Infections
While fever may be associated with the flu-like syndrome reported commonly during interferon therapy, other causes of high or persistent fever must be ruled out, particularly in patients with neutropenia. Serious and severe infections (bacterial, viral, fungal), some fatal, have been reported during treatment with alpha interferons including Roferon-A. Appropriate anti-infective therapy should be started immediately and discontinuation of therapy should be considered.
Bone Marrow Toxicity
Alpha-interferons suppress bone marrow function and may result in severe cytopenias and anemia including very rare events of aplastic anemia. Cytopenias (e.g., leukopenia, thrombocytopenia) can lead to an increased risk of infections or hemorrhage. It is advised that complete blood counts (CBC) be obtained pretreatment and monitored routinely during therapy. Alpha interferon therapy should be discontinued in patients who develop severe decreases in neutrophil (<0.5 × 109/L) or platelet counts (<25 × 109/L).
Caution should be exercised when administering Roferon-A to patients with myelosuppression or when Roferon-A is used in combination with other agents that are known to cause myelosuppression. Synergistic toxicity has been observed when Roferon-A is administered in combination with zidovudine (AZT).9
Endocrine Disorders
Roferon-A causes or aggravates hypothyroidism and hyperthyroidism. Hyperglycemia has been observed in patients treated with Roferon-A. Symptomatic patients should have their blood glucose measured and followed-up accordingly. Patients with diabetes mellitus may require adjustment of their anti-diabetic regimen.
Pulmonary Disorders
Dyspnea, pulmonary infiltrates, pneumonia, bronchiolitis obliterans, interstitial pneumonitis and sarcoidosis, some resulting in respiratory failure and/or patient deaths, may be induced or aggravated by alpha interferon therapy. Patients who develop persistent or unexplained pulmonary infiltrates or pulmonary function impairment should discontinue treatment with Roferon-A.
Ophthalmologic Disorders
Decrease or loss of vision, retinopathy including macular edema, retinal artery or vein thrombosis, retinal hemorrhages and cotton wool spots, optic neuritis, and papilledema are induced or aggravated by treatment with Interferon alfa-2a or other alpha interferons. All patients should receive an eye examination at baseline. Patients with preexisting ophthalmologic disorders (e.g., diabetic or hypertensive retinopathy) should receive periodic ophthalmologic exams during interferon alpha treatment. Any patient who develops ocular symptoms should receive a prompt and complete eye examination. Interferon alfa-2a treatment should be discontinued in patients who develop new or worsening ophthalmologic disorders.
Pancreatitis
Pancreatitis has been observed in patients receiving alpha interferon treatment, including those who developed marked triglyceride elevations. In some cases, fatalities have been observed. Although a causal relationship to Roferon-A has not been established, marked triglyceride elevation is a risk factor for development of pancreatitis. Roferon-A should be suspended if symptoms or signs suggestive of pancreatitis are observed. In patients diagnosed with pancreatitis, discontinuation of therapy with Roferon-A should be considered.
PRECAUTIONS
General
In all instances where the use of Roferon-A is considered for chemotherapy, the physician must evaluate the need and usefulness of the drug against the risk of adverse reactions. Most adverse reactions are reversible if detected early. If severe reactions occur, the drug should be reduced in dosage or discontinued and appropriate corrective measures should be taken according to the clinical judgment of the physician. Reinstitution of Roferon-A therapy should be carried out with caution and with adequate consideration of the further need for the drug and, alertness to possible recurrence of toxicity. The minimum effective doses of Roferon-A for treatment of hairy cell leukemia and chronic myelogenous leukemia have not been established.
Variations in dosage and adverse reactions exist among different brands of Interferon. Therefore, do not use different brands of Interferon in a single treatment regimen.
The safety and efficacy of Roferon-A have not been established in organ transplant recipients.
Renal Impairment
Dose-limiting renal toxicities were unusual. Infrequently, severe renal toxicities, sometimes requiring renal dialysis, have been reported with alpha-interferon therapy alone or in combination with IL-2. In patients with impaired renal function, signs and symptomsof interferon toxicity should be closely monitored. Roferon-A should be used with caution in patients with creatinine clearance <50 mL/min.
Autoimmune Disease
Development or exacerbation of autoimmune diseases including idiopathic thrombocytopenic purpura, vasculitis, Raynaud's phenomenon, rheumatoid arthritis, psoriasis, interstitial nephritis, thyroiditis, lupus erythematosus, hepatitis, myositis and rhabdomyolysis have been observed in patients treated with alpha-interferons. Any patient developing an autoimmune disorder during treatment should be closely monitored and, if appropriate, treatment should be discontinued.
Information for Patients
Patients should be cautioned not to change brands of Interferon without medical consultation, as a change in dosage may result. Patients should be informed regarding the potential benefits and risks attendant to the use of Roferon-A. If home use is determined to be desirable by the physician, instructions on appropriate use should be given, including review of the contents of the enclosed Medication Guide. Patients should be well hydrated, especially during the initial stages of treatment.
Patients should be thoroughly instructed in the importance of proper disposal procedures and cautioned against reusing syringes and needles. If home use is prescribed, a puncture-resistant container for the disposal of used syringes and needles should be supplied to the patient. The full container should be disposed of according to directions provided by the physician (see Medication Guide).
Patients should be advised that laboratory evaluations are required before starting therapy and periodically thereafter (see Laboratory Tests).
Patients receiving high-dose alpha-interferon should be cautioned against performing tasks that require complete mental alertness such as operating machinery or driving a motor vehicle. Patients to be treated with Roferon-A should be informed that depression and suicidal ideation may be side effects of treatment and should be advised to report these side effects immediately to the prescribing physician.
Laboratory Tests
Leukopenia and elevation of hepatic enzymes occurred frequently but were rarely dose-limiting. Thrombocytopenia occurred less frequently. Proteinuria and increased cells in urinary sediment were also seen infrequently.
Complete blood counts with differential platelet counts and clinical chemistry tests should be performed before initiation of Roferon-A therapy and at appropriate periods during therapy. Patients with neutrophil count<1500/mm3, platelet count <75,000/mm3, hemoglobin <10 g/dL and creatinine >1.5 mg/dL were excluded from several major chronic hepatitis C studies; patients with these laboratory abnormalities should be carefully monitored if treated with Roferon-A. Since responses of hairy cell leukemia, chronic hepatitis C and chronic myelogenous leukemia are not generally observed for 1 to 3 months after initiation of treatment, very careful monitoring for severe depression of blood cell counts is warranted during the initial phase of treatment.
Those patients who have preexisting cardiac abnormalities and/or are in advanced stages of cancer should have electrocardiograms taken before and during the course of treatment.
Liver Function
For patients being treated for chronic hepatitis C, serum ALT should be evaluated before therapy to establish baselines and repeated at week 2 and monthly thereafter following initiation of therapy for monitoring clinical response. Patients developing liver function abnormalities during Roferon-A treatment should be closely monitored and if necessary treatment should be discontinued. Use of alpha-interferons has been rarely associated with severe hepatic dysfunction and liver failure.
Thyroid Function
Patients with preexisting thyroid abnormalities may be treated if normal thyroid stimulating hormone (TSH) levels can be maintained by medication. Testing of TSH levels in these patients is recommended at baseline and every 3 months following initiation of therapy.
Triglycerides
Elevated triglyceride levels have been observed in patients treated with interferons including Roferon-A therapy. Triglyceride levels should be monitored periodically during treatment and elevated levels should be managed as clinically appropriate. Hypertriglyceridemia may result in pancreatitis. Discontinuation of Roferon-A therapy should be considered for patients with persistently elevated triglycerides (e.g., triglycerides >1000 mg/dL) associated with symptoms of potential pancreatitis, such as abdominal pain, nausea, or vomiting.
Drug Interactions
Roferon-A has been reported to reduce the clearance of theophylline.10,11 The clinical relevance of this interaction is presently unknown. Caution should be exercised when administering Roferon-A in combination with other potentially myelosuppressive agents. Synergistic toxicity has been observed when Roferon-A is administered in combination with zidovudine (AZT) (see WARNINGS: Bone Marrow Toxicity).
In transplant recipients, therapeutic immunosuppression may be weakened because interferons also exert an immunostimulatory action.
Alpha-interferons may affect the oxidative metabolic process by reducing the activity of hepatic microsomal cytochrome enzymes in the P450 group. Although the clinical relevance is still unclear, this should be taken into account when prescribing concomitant therapy with drugs metabolized by this route.
The neurotoxic, hematotoxic or cardiotoxic effects of previously or concurrently administered drugs may be increased by interferons. Interactions could occur following concurrent administration of centrally acting drugs. Use of Roferon-A in conjunction with interleukin-2 may potentiate risks of renal failure.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Mutagenesis
A. Internal Studies — Ames tests using six different tester strains, with and without metabolic activation, were performed with Roferon-A up to a concentration of 1920 µg/plate. There was no evidence of mutagenicity.
Human lymphocyte cultures were treated in vitro with Roferon-A at noncytotoxic concentrations. No increase in the incidence of chromosomal damage was noted.
B. Published Studies — There are no published studies on the mutagenic potential of Roferon-A. However, a number of studies on the genotoxicity of human leukocyte interferon have been reported.
A chromosomal defect following the addition of human leukocyte interferon to lymphocyte cultures from a patient suffering from a lymphoproliferative disorder has been reported.
In contrast, other studies have failed to detect chromosomal abnormalities following treatment of lymphocyte cultures from healthy volunteers with human leukocyte interferon.
It has also been shown that human leukocyte interferon protects primary chick embryo fibroblasts from chromosomal aberrations produced by gamma rays.
Impairment of Fertility
Roferon-A has been studied for its effect on fertility in Macaca mulatta (rhesus monkeys). Nonpregnant rhesus females treated with Roferon-A at doses of 5 and 25 MIU/kg/day have shown menstrual cycle irregularities, including prolonged or shortened menstrual periods and erratic bleeding; these cycles were considered to be anovulatory on the basis that reduced progesterone levels were noted and that expected increases in preovulatory estrogen and luteinizing hormones were not observed. These monkeys returned to a normal menstrual rhythm following discontinuation of treatment.
Pregnancy
Pregnancy Category C
Roferon-A has been associated with statistically significant, dose-related increases in abortions in pregnant rhesus monkeys treated with 1, 5, or 25 MIU/kg/day (approximately 20 to 500 times the human weekly dose, when scaled by body surface area) during the early to midfetal period of organogenesis (gestation day 22 to 70). Abortifacient activity was also observed in 2/6 pregnant rhesus monkeys treated with 25 MIU/kg/day Roferon-A (500 times the human dose) during the period of late fetal development (days 79 to 100 of gestation). No teratogenic effects were seen in either study. However, the validity of extrapolating doses used in animal studies to human doses is not established. Therefore, no direct comparison of the doses that induced fetal death in monkeys to dose levels of Roferon-A used clinically can be made. There are no adequate and well-controlled studies of Roferon-A in pregnant women. Roferon-A is to be used during pregnancy only if the potential benefit to the woman justifies the potential risk to the fetus. Roferon-A is recommended for use in women of childbearing potential and in men only when they are using effectivecontraception during therapy.
The injectable solution contains benzyl alcohol. The excipient benzyl alcohol can be transmitted via the placenta. The possibility of toxicity should be taken into account in premature infants after the administration of Roferon-A solution for injection immediately prior to birth or Cesarean section.
Male fertility and teratologic evaluations have yielded no significant adverse effects to date.
Nursing Mothers
It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from Roferon-A, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Pediatric Use
Use of Roferon-A in children with Ph-positive adult-type CML is supported by evidence from adequate and well-controlled studies of Roferon-A in adults with additional data from the literature on the use of alfa interferon in children with CML. A published report on 15 children with Ph-positive adult-type CML suggests a safety profile similar to that seen in adult CML; clinical responses were also observed8 (see DOSAGE AND ADMINISTRATION).
For all other indications, safety and effectiveness have not been established in patients below the age of 18 years.
The injectable solutions are not indicated for use in neonates or infants and should not be used by patients in that age group. There have been rare reports of death in neonates and infants associated with excessive exposure to benzyl alcohol (see CONTRAINDICATIONS).
Geriatric Use
In clinical studies of Roferon-A in chronic hepatitis C, 101 patients were 65 years old or older. The numbers were insufficient to determine if antiviral responses differ from younger subjects. There were greater proportions of geriatric patients with serious adverse reactions (9% vs. 6%), withdrawals due to adverse reactions (11% vs. 6%), and WHO grade III neutropenia and thrombocytopenia.
Clinical studies of Roferon-A in chronic myelogenous leukemia or hairy cell leukemia did not include sufficient numbers of subjects aged 65 or older to determine whether they respond differently from younger subjects.
This drug is known to be excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, these patients should receive careful monitoring, including renal function.
ADVERSE REACTIONS
Depressive illness and suicidal behavior, including suicidal ideation, suicide attempt, and suicides, have been reported in association with the use of alfa-interferon products. The incidence of reported depression has varied substantially among trials, possibly related to the underlying disease, dose, duration of therapy and degree of monitoring, but has been reported to be 15% or higher (see WARNINGS).
For Patients With Chronic Hepatitis C
The most frequent adverse experiences were reported to be possibly or probably related to therapy with 3 MIU tiw Roferon-A, were mostly mild to moderate in severity and manageable without the need for discontinuation of therapy. A relative increase in the incidence, severity and seriousness of adverse events was observed in patients receiving doses above 3 MIU tiw.
Adverse reactions associated with the 3 MIU dose include:
Flu-like Symptoms: Fatigue (58%), myalgia/arthralgia (51%), flu-like symptoms (33%), fever (28%), chills (23%), asthenia (6%), sweating (5%), leg cramps (3%) and malaise (1%).
Central and Peripheral Nervous System: Headache (52%), dizziness (13%), paresthesia (7%), confusion (7%), concentration impaired (4%) and change in taste or smell (3%).
Gastrointestinal: Nausea/vomiting (33%), diarrhea (20%), anorexia (14%), abdominal pain (12%), flatulence (3%), liver pain (3%), digestion impaired (2%) and gingival bleeding (2%).
Psychiatric: Depression (16%), irritability (15%), insomnia (14%), anxiety (5%) and behavior disturbances (3%).
Pulmonary and Cardiovascular: Dryness or inflammation of oropharynx (6%), epistaxis (4%), rhinitis (3%), arrhythmia (1%) and sinusitis (<1%).
Skin: Injection site reaction (29%), partial alopecia (19%), rash (8%), dry skin or pruritus (7%), hematoma (1%), psoriasis (<1%), cutaneous eruptions (<1%), eczema (<1%) and seborrhea (<1%).
Other: Conjunctivitis (4%), menstrual irregularity (2%) and visual acuity decreased (<1%).
Patients receiving 6 MIU tiw experienced a higher incidence of severe psychiatric events (9%) than those receiving 3 MIU tiw (6%) in two large US studies. In addition, more patients withdrew from these studies when receiving 6 MIU tiw (11%) than when receiving 3 MIU tiw (7%). Up to half of patients receiving 3 MIU or 6 MIU tiw withdrawing from the study experienced depression or other psychiatric adverse events. At higher doses anxiety, sleep disorders, and irritability were observed more frequently. An increased incidence of fatigue, myalgia/arthralgia, headache, fever, chills, alopecia, sleep disturbances and dry skin or pruritus was also generally observed during treatment with higher doses of Roferon-A.
Generally there were fewer adverse events reported in the second 6 months of treatment than in the first 6 months for patients treated with 3 MIU tiw. Patients tolerant of initial therapy with Roferon-A generally tolerate re-treatment at the same dose, but tend to experience more adverse reactions at higher doses.
Infrequent adverse events (>1% but <3% incidence) included: cold feeling, cough, muscle cramps, diaphoresis, dyspnea, eye pain, reactivation of herpes simplex, lethargy, edema, sexual dysfunction, shaking, skin lesions, stomatitis, tooth disorder, urinary tract infection, weakness in extremities.
Triglyceride levels were not evaluated in the clinical trials. However, hypertriglyceridemia has been reported postmarketing in patients receiving Roferon-A therapy for chronic hepatitis C.
For Patients With Chronic Myelogenous Leukemia
For patients with chronic myelogenous leukemia, the percentage of adverse events, whether related to drug therapy or not, experienced by patients treated with rIFNα-2a is given below. Severe adverse events were observed in 66% and 31% of patients on study DM84-38 and MI400, respectively. Dose reduction and temporary cessation of therapy were required frequently. Permanent cessation of Roferon-A, due to intolerable side effects, was required in 15% and 23% of patients on studies DM84-38 and MI400, respectively.
Flu-like Symptoms: Fever (92%), asthenia or fatigue (88%), myalgia (68%), chills (63%), arthralgia/bone pain (47%) and headache (44%).
Gastrointestinal: Anorexia (48%), nausea/vomiting (37%) and diarrhea (37%).
Central and Peripheral Nervous System: Headache (44%), depression (28%), decreased mental status (16%), dizziness (11%), sleep disturbances (11%), paresthesia (8%), involuntary movements (7%) and visual disturbance (6%).
Pulmonary and Cardiovascular: Coughing (19%), dyspnea (8%) and dysrhythmia (7%).
Skin: Hair changes (including alopecia) (18%), skin rash (18%), sweating (15%), dry skin (7%) and pruritus (7%).
Uncommon adverse events (<4%) reported in clinical studies included chest pain, syncope, hypotension, impotence, alterations in taste or hearing, confusion, seizures, memory loss, disturbances of libido, bruising and coagulopathy. Miscellaneous adverse events that were rarely observed included Coombs' positive hemolytic anemia, aplastic anemia, hypothyroidism, cardiomyopathy, hypertriglyceridemia and bronchospasm.
For Patients With Hairy Cell Leukemia
Constitutional (100%): Fever (92%), fatigue (86%), headache (64%), chills (64%), weight loss (33%), dizziness (21%) and flu-like symptoms (16%).
Integumentary (79%): Skin rash (44%), diaphoresis (22%), partial alopecia (17%), dry skin (17%) and pruritus (13%).
Musculoskeletal (73%): Myalgia (71%), joint or bone pain (25%) and arthritis or polyarthritis (5%).
Gastrointestinal (69%): Anorexia (43%), nausea/vomiting (39%) and diarrhea (34%).
Head and Neck (45%): Throat irritation (21%), rhinorrhea (12%) and sinusitis (11%).
Pulmonary (40%): Coughing (16%), dyspnea (12%) and pneumonia (11%).
Central Nervous System (39%): Dizziness (21%), depression (16%), sleep disturbance (10%), decreased mental status (10%), anxiety (6%), lethargy (6%), visual disturbance (6%) and confusion (5%).
Cardiovascular (39%): Chest pain (11%), edema (11%) and hypertension (11%).
Pain (34%): Pain (24%) and pain in back (16%).
Peripheral Nervous System (23%): Paresthesia (12%) and numbness (12%).
Rarely (<5%), central nervous system effects including gait disturbance, nervousness, syncope and vertigo, as well as cardiac adverse events including murmur, thrombophlebitis and hypotension were reported. Adverse experiences that occurred rarely, and may have been related to underlying disease, included ecchymosis, epistaxis, bleeding gums and petechiae. Urticaria and inflammation at the site of injection were also rarely observed.
In Other Investigational Studies of Roferon-A
The following infrequent adverse events have been reported with the investigational use of Roferon-A.
Gastrointestinal: Pancreatitis, colitis, gastrointestinal hemorrhage, stomatitis (<5%); constipation (<3%); hepatitis, abdominal fullness, hypermotility, excessive salivation, gastric distress (<1%).
Cardiovascular: Palpitations (<3%); myocardial infarction, congestive heart failure, ischemic retinopathy, Raynaud's phenomenon, hot flashes (<1%).
Pulmonary: Pneumonitis, some cases responded to interferon cessation and corticosteroid therapy (<5%); chest congestion (<3%); tachypnea (<1%).
Central Nervous System and Psychiatric: Stroke, coma, encephalopathy, transient ischemic attacks, dysphasia, hallucinations, gait disturbance, psychomotor retardation, apathy, sedation, irritability, hyperactivity, claustrophobia, loss of libido, ataxia, neuropathy, poor coordination, dysarthria, aphasia, aphonia, amnesia (<1%).
Autoimmune Disease: Vasculitis, arthritis, hemolytic anemia and lupus erythematosus syndrome (<3%).
Other: Thyroid dysfunction including hypothyroidism and hyperthyroidism, diabetes requiring insulin therapy in some patients (<5%); anaphylactic reactions, eye irritation, earache, cyanosis, flushing of skin (<1%).
Abnormal Laboratory Test Values
The percentage of patients with chronic hepatitis C, hairy cell leukemia, and with chronic myelogenous leukemia who experienced a significant abnormal laboratory test value (NCI or WHO grades III or IV) at least once during their treatment with Roferon-A is shown in Table 2:
| Chronic Hepatitis C | Chronic Myelogenous Leukemia* | Hairy Cell Leukemia | ||
|---|---|---|---|---|
| (n=203) 3 MIU tiw | US Study (n=91) | Non-US Study (n=219) | (n=218) | |
| NAP = Not applicable. | ||||
| NA = Not assessed. | ||||
| Leukopenia | 1.5% | 20% | 3% | 45%† |
| Neutropenia | 10% | 22% | 0% | 68%† |
| Thrombocytopenia | 4.5% | 27% | 5% | 62%† |
| Anemia (Hb) | 0% | 15% | 4% | 31%† |
| SGOT | NAP | 5% | 1% | 9% |
| Alk. Phosphatase | 0% | 3% | 1% | 3% |
| LDH | NAP | NA | NA | <1% |
| Proteinuria | 0% | NA | NA | 10%‡ |
Elevated triglyceride levels have been observed in patients receiving interferon therapy, including Roferon-A.
Chronic Hepatitis C
The incidence of neutropenia (WHO grades III or IV) was over twice as high in those treated with 6 MIU tiw (21%) as those treated with 3 MIU tiw (10%).
Chronic Myelogenous Leukemia
In the two clinical studies, a severe or life-threatening anemia was seen in up to 15% of patients. A severe or life-threatening leukopenia and thrombocytopenia were observed in up to 20% and 27% of patients, respectively. Changes were usually reversible when therapy was discontinued. One case of aplastic anemia and one case of Coombs' positive hemolytic anemia were seen in 310 patients treated with rIFNα-2a in clinical studies. Severe cytopenias led to discontinuation of therapy in 4% of all Roferon-A treated patients.
Transient increases in liver transaminases or alkaline phosphatase of any intensity were seen in up to 50% of patients during treatment with Roferon-A. Only 5% of patients had a severe or life-threatening increase in SGOT. In the clinical studies, such abnormalities required termination of therapy in less than 1% of patients.
Hairy Cell Leukemia
Increases in serum phosphorus (≥1.6 mmol/L) and serum uric acid (≥9.1 mg/dL) were observed in 9% and 10% of patients, respectively. The increase in serum uric acid is likely to be related to the underlying disease. Decreases in serum calcium (≤1.9 mmol/L) and serum phosphorus (≤0.9 mmol/L) were seen in 28% and 22% of patients, respectively.
Postmarketing
Central and Peripheral Nervous System: Somnolence, hearing impairment, hearing loss.
Vision: Retinopathy including retinal hemorrhages and cotton-wool spots, papilledema, retinal artery and vein thrombosis and optic neuropathy.
Skin: Injection site necrosis.
Blood: Idiopathic thrombocytopenic purpura, cyanosis.
Renal and Urinary System: Increased blood urea and serum creatinine, decreased renal function and acute renal failure.
Endocrine: Hyperglycemia.
Immune System Disorder: Sarcoidosis.
Respiratory: Pulmonary edema.
Metabolic and Nutritional: Cases of hypertriglyceridemia/hyperlipidemia have been reported including some occurring in association with pancreatitis.
OVERDOSAGE
There are no reports of overdosage, but repeated large doses of interferon can be associated with profound lethargy, fatigue, prostration, and coma. Such patients should be hospitalized for observation and appropriate supportive treatment given.
DOSAGE AND ADMINISTRATION
Roferon-A recommended dosing regimens are different for each of the following indications as described below.
Note: Parenteral drug products should be inspected visually for particulate matter and discoloration before administration, whenever solution and container permit.
Roferon-A is administered subcutaneously.
Chronic Hepatitis C
The recommended dosage of Roferon-A for the treatment of chronic hepatitis C is 3 MIU three times a week (tiw) administered subcutaneously for 12 months (48 to 52 weeks). As an alternative, patients may be treated with an induction dose of 6 MIU tiw for the first 3 months (12 weeks) followed by 3 MIU tiw for 9 months (36 weeks). Normalization of serum ALT generally occurs within a few weeks after initiation of treatment in responders. Approximately 90% of patients who respond to Roferon-A do so within the first 3 months of treatment; however, patients responding to Roferon-A with a reduction in ALT should complete 12 months of treatment. Patients who have no response to Roferon-A within the first 3 months of therapy are not likely to respond with continued treatment; treatment discontinuation should be considered in these patients.
Patients who tolerate and partially or completely respond to therapy with Roferon-A but relapse following its discontinuation may be re-treated. Re-treatment with either 3 MIU tiw or with 6 MIU tiw for 6 to 12 months may be considered. Please see ADVERSE REACTIONS regarding the increased frequency of adverse reactions associated with treatment with higher doses.
Temporary dose reduction by 50% is recommended in patients who do not tolerate the prescribed dose. If adverse events resolve, treatment with the original prescribed dose can be re-initiated. In patients who cannot tolerate the reduced dose, cessation of therapy, at least temporarily, is recommended.
Chronic Myelogenous Leukemia
For patients with Ph-positive CML in chronic phase: Prior to initiation of therapy, a diagnosis of Philadelphia chromosome positive CML in chronic phase by the appropriate peripheral blood, bone marrow and other diagnostic testing should be made. Monitoring of hematologic parameters should be done regularly (e.g., monthly). Since significant cytogenetic changes are not readily apparent until after hematologic response has occurred, and usually not until several months of therapy have elapsed, cytogenetic monitoring may be performed at less frequent intervals. Achievement of complete cytogenetic response has been observed up to 2 years following the start of Roferon-A treatment.
The recommended initial dose of Roferon-A is 9 MIU daily administered as a subcutaneous injection. Based on clinical experience,3 short-term tolerance may be improved by gradually increasing the dose of Roferon-A over the first week of administration from 3 MIU daily for 3 days to 6 MIU daily for 3 days to the target dose of 9 MIU daily for the duration of the treatment period.
The optimal dose and duration of therapy have not yet been determined. Even though the median time to achieve a complete hematologic response was 5 months in study MI400, hematologic responses have been observed up to 18 months after treatment start. Treatment should be continued until disease progression. If severe side effects occur, a treatment interruption or a reduction in either the dose or the frequency of injections may be necessary to achieve the individual maximally tolerated dose (see PRECAUTIONS).
Limited data are available on the use of Roferon-A in children with CML. In one report of 15 children with Ph-positive, adult-type CML doses between 2.5 to 5 MIU/m2/day given intramuscularly were tolerated.8 In another study, severe adverse effects including deaths were noted in children with previously untreated, Ph-negative, juvenile CML, who received interferon doses of 30 MIU/m2/day. 12
Hairy Cell Leukemia
Prior to initiation of therapy, tests should be performed to quantitate peripheral blood hemoglobin, platelets, granulocytes and hairy cells and bone marrow hairy cells. These parameters should be monitored periodically (e.g., monthly) during treatment to determine whether response to treatment has occurred. If a patient does not respond within 6 months, treatment should be discontinued. If a response to treatment does occur, treatment should be continued until no further improvement is observed and these laboratory parameters have been stable for about 3 months. Patients with hairy cell leukemia have been treated for up to 24 consecutive months. The optimal duration of treatment for this disease has not been determined.
The induction dose of Roferon-A is 3 MIU daily for 16 to 24 weeks, administered as a subcutaneous injection. The recommended maintenance dose is 3 MIU, tiw. Dose reduction by one-half or withholding of individual doses may be needed when severe adverse reactions occur. The use of doses higher than 3 MIU is not recommended in hairy cell leukemia.
HOW SUPPLIED
Single Use Prefilled Syringes
(for subcutaneous administration)
3 million IU Roferon-A per syringe — Each 0.5 mL contains 3 MIU of Interferon alfa-2a, recombinant, 3.605 mg sodium chloride, 0.1 mg polysorbate 80, 5 mg benzyl alcohol as a preservative and 0.385 mg ammonium acetate. Boxes of 1 (NDC 0004-2015-09); Boxes of 6 (NDC 0004-2015-07).
6 million IU Roferon-A per syringe — Each 0.5 mL contains 6 MIU of Interferon alfa-2a, recombinant, 3.605 mg sodium chloride, 0.1 mg polysorbate 80, 5 mg benzyl alcohol as a preservative and 0.385 mg ammonium acetate. Boxes of 1 (NDC 0004-2016-09); Boxes of 6 (NDC 0004-2016-07).
9 million IU Roferon-A per syringe — Each 0.5 mL contains 9 MIU of Interferon alfa-2a, recombinant, 3.605 mg sodium chloride, 0.1 mg polysorbate 80, 5 mg benzyl alcohol as a preservative and 0.385 mg ammonium acetate. Boxes of 1 (NDC 0004-2017-09); Boxes of 6 (NDC 0004-2017-07).
REFERENCES
- Trown PW, et al. Cancer. 1986; 57(suppl):1648-1656.
- Itri LM, et al. Cancer. 1987; 59:668-674.
- Jones GJ, Itri LM. Cancer. 1986; 57(suppl):1709-1715.
- Foon KA, et al. Blood. 1984; 64(suppl 1):164a.
- Quesada Jr, et al. Cancer. 1986; 57(suppl):1678-1680.
- The Italian Cooperative Study Group on CML. N Engl J Med. 1994; 330:820-825.
- Sokal JE, et al. Blood. 1984; 63(4):789-799.
- Dow LW, et al. Cancer. 1991; 68:1678-1684.
- Krown SE, et al. Proc Am Soc Clin Oncol. 1988; 7:1.
- Williams SJ, et al. Lancet. 1987; 2:939-941.
- Jonkman JHG, et al. Br J Clin Pharmacol. 1989; 2(27):795-802.
- Maybee D, et al. Proc Annu Meet Am Soc Clin Oncol. 1992; 11:A950.
MEDICATION GUIDE
Roferon®-A
(Interferon alfa-2a, recombinant)
Solution for Injection – Prefilled Syringes
Before you start taking Roferon-A (ro-FER-on), please read this Medication Guide carefully. Read this Medication Guide each time you refill your prescription in case new information has been added. This information does not take the place of talking with your healthcare provider.
What is the most important information I should know about Roferon-A?
Roferon-A is used to treat people with hepatitis C, hairy cell leukemia and Philadelphia chromosome positive chronic myelogenous leukemia (CML). However, Roferon-A can cause some serious side effects that may cause death in rare cases. Before starting Roferon-A, you should talk with your healthcare provider about the possible benefits and the possible side effects of treatment, to decide if Roferon-A is right for you. While taking Roferon-A, you will need to see your healthcare provider regularly for medical examinations and blood tests to make sure your treatment is working and to check for side effects.
The most serious possible side effects of Roferon-A treatment include:
- Mental health problems: Roferon-A may cause some patients to develop mood or behavioral problems. Signs of these problems include irritability (getting easily upset), depression (feeling low, feeling bad about yourself or feeling hopeless), and anxiety. Some patients may have aggressive behavior and think about hurting others. Some patients may develop thoughts about ending their lives (suicidal thoughts) and may attempt to do so. A few patients have even ended their lives. Former drug addicts may fall back into drug addiction or overdose. You must tell your healthcare provider if you are being treated for a mental illness or have a history of mental illness or if you are or have ever been addicted to drugs or alcohol. Call your healthcare provider immediately if you develop any of these problems while on Roferon-A treatment.
- Heart problems: Roferon-A may cause some patients to experience high blood pressure, a fast heartbeat, chest pain, and very rarely a heart attack. Tell your healthcare provider if you have or have had any heart problems in the past.
- Blood problems: Many patients taking Roferon-A have had a drop in the number of their white blood cells and their platelets. If the numbers of these blood cells are too low, you could be at risk for infections or bleeding.
Stop taking Roferon-A and call your healthcare provider immediately if you develop any of these symptoms:
- You become very depressed or think about suicide
- You have severe chest pain
- You have trouble breathing
- You have a change in your vision
- You notice unusual bleeding or bruising
- High fever
- Severe stomach pain. If the pain is in the lower part of your stomach area it could mean that your bowels are inflamed (colitis)
For more information on possible side effects with Roferon-A therapy, please read the section on "What are the possible side effects of Roferon-A?" in this Medication Guide.
What is Roferon-A?
Roferon-A is a treatment that is used for some people who are infected with the hepatitis C virus, hairy cell leukemia, and Philadelphia chromosome positive chronic myelogenous leukemia (CML). Patients with hepatitis C have the virus that causes hepatitis in their blood and liver. Patients with hairy cell leukemia produce abnormal white blood cells that travel to the spleen where they trap and destroy normal blood cells. In CML, your body produces too many of certain blood cells. Roferon-A works in these conditions by reducing the amount of virus in the body, destroying cells that may be harmful to your body and keeping the body from producing too many cells.
Who should not take Roferon-A?
Do not use Roferon-A if:
- You are pregnant or breast-feeding or are planning to become pregnant.
- You are allergic to alpha interferons, Escherichia coli-derived products or any component of Roferon-A.
- You have autoimmune hepatitis (hepatitis caused by your immune system attacking your liver).
Roferon-A should not be given to newborn or premature infants.
If you have or have had any of the following conditions or serious medical problems, discuss them with your doctor before taking Roferon-A:
- History of or current severe mental illness (such as depression or anxiety)
- Previous heart attack or heart problems
- Sleep problems
- High blood pressure
- Autoimmune disease (where the body's immune system attacks the body's own cells), such as vasculitis, psoriasis, systemic lupus erythematosus, rheumatoid arthritis
- Kidney problems
- Blood disorders-Low blood counts or bleeding problems
- You take a medicine called theophylline
- Diabetes (high blood sugar)
- Thyroid problems
- Liver problems, other than hepatitis C
- Hepatitis B infection
- HIV infection (the virus that causes AIDS)
- Problems with your vision
- Colitis
- Body organ transplant and are taking medicine that keeps your body from rejecting your transplant (suppresses your immune system)
- Alcoholism
- Drug abuse or addiction
If you have any doubts about your health condition or about taking Roferon-A, talk to your healthcare provider.
What should I avoid while taking Roferon-A?
- Female patients as well as female partners of male patients must avoid becoming pregnant while taking Roferon-A. Roferon-A may harm your unborn child or cause you to lose your baby (miscarry).
- You should not breast-feed your baby while taking Roferon-A.
How should I take Roferon-A?
To get the most benefit from this medicine, it is important to take Roferon-A exactly as your healthcare provider tells you.
Your healthcare provider will tell you how much medicine to take and how often to take it. Once you start treatment with Roferon-A, do not switch to another brand of interferon without talking to your doctor. Other interferons may not have the same effect on the treatment of your disease. Switching brands will also require a change in your dose. Your healthcare provider will tell you how long you need to use Roferon-A.
Over time, your healthcare provider may change your dose of Roferon-A. Do not change your dose unless your doctor tells you to change it.
Roferon-A is supplied in prefilled syringes. Whether you give yourself the injection or another person gives the injection to you, it is important to follow the instructions in this Medication Guide (see the appendix "Instructions for Preparing and Giving a Dose with a Roferon-A Prefilled Syringe").
If you miss a dose of Roferon-A, take the missed dose as soon as possible during the same day or the next day, then continue on your regular dosing schedule. If several days go by after you miss a dose, check with your doctor about what to do. Do not double the next dose or take more than one dose a day unless your doctor tells you to. Call your doctor right away if you take more than your prescribed Roferon-A dose. Your doctor may wish to examine you more closely and take blood for testing.
You must get regular blood tests to help your healthcare provider check how the treatment is working and to check for side effects.
Tell your doctor if you are taking or planning to take other prescription or non-prescription medicines, including vitamins and mineral supplements and herbal medicines.
What are the possible side effects of Roferon-A?
Possible, serious side effects include:
- Mental health problems including suicide, suicidal thoughts, heart problems, and blood problems: See the section "What is the most important information I should know about Roferon-A?".
- Other body organ problems: Some patients may experience lung problems (such as difficulty breathing or pneumonia) and vision problems.
- New or worsening autoimmune disease: Some patients may develop an autoimmune disease (a disease where the body's own immune system begins to attack itself) while on Roferon-A therapy. These diseases can include vasculitis (an inflammation of your blood vessels), rheumatoid arthritis or lupus erythematosus, psoriasis or thyroid problems. In some patients who already have an autoimmune disease, the disease may worsen while on Roferon-A therapy.
Common, but less serious, side effects include:
- Flu-like symptoms: Most patients who take Roferon-A have flu-like symptoms that usually lessen after the first few weeks of treatment. Flu-like symptoms may include unusual tiredness, fever, chills, muscle aches, and joint pain. Taking acetaminophen or ibuprofen before you take Roferon-A can help with these symptoms. You can also try taking Roferon-A at night. You may be able to sleep through the symptoms.
- Extreme fatigue (tiredness): Many patients may become extremely tired while on Roferon-A therapy.
- Upset stomach: Nausea, taste changes, diarrhea, and loss of appetite occur commonly.
- Blood sugar problems: Some patients may develop a problem with the way their body controls their blood sugar and may develop diabetes.
- Thyroid problems: Some patients may develop changes in their thyroid function. Symptoms of these changes may include feeling hot or cold all the time, trouble concentrating, changes in your skin (your skin may become very dry), and changes in your weight.
- Skin reactions: Some patients may develop a rash, dry or itchy skin, and redness and swelling at the site of injection.
- Sleep disturbances and headache: Trouble sleeping and headaches may also occur during Roferon-A therapy.
- Hair thinning: Hair loss is not uncommon while using Roferon-A. This hair loss is temporary and hair growth should return after you stop taking Roferon-A.
These are not all of the side effects of Roferon-A. Your doctor or pharmacist can give you a more complete list.
Talk to your healthcare provider if you are worried about side effects or find them very bothersome.
General advice about prescription medicines
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. If you have any concerns or questions about Roferon-A, contact your healthcare provider. Do not use Roferon-A for a condition or person other than that for which it is prescribed. If you want to know more about Roferon-A, your healthcare provider or pharmacist will be able to provide you with detailed information that is written for healthcare providers.
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Keep this and all other medications out of the reach of children.
Revised: October 2004
Medication Guide Appendix: Instructions for Preparing and Giving a Dose with a Roferon-A Prefilled Syringe
How should I store Roferon-A?
Roferon-A must be stored in the refrigerator at a temperature of 36°F to 46°F (2°C to 8°C). Do not leave Roferon-A outside of the refrigerator for more than 24 hours. Do not freeze Roferon-A. Keeping Roferon-A at temperatures outside the recommended range can destroy the medicine. Do not shake Roferon-A. Shaking can destroy Roferon-A so that it will not work. Protect Roferon-A from light during storage.
How do I inject Roferon-A?
The instructions that follow will help you learn how to use Roferon-A prefilled syringes. Please read all of these directions before trying to take your medicine. It is important to follow these directions carefully. Talk to your healthcare provider if you have any concerns about how to use Roferon-A. Whether you are giving yourself an injection or if you are giving this injection to someone else, a healthcare provider must teach you how to inject.
The prefilled syringes are used for injecting Roferon-A under the surface of the skin (subcutaneous).
- Collect all the materials you will need before you start to give the injection:
- •
- one sterile Roferon-A prefilled syringe with needle
- •
- alcohol swabs
- •
- puncture-resistant disposable container
- Check the expiration date on the package to make sure that it has not passed and check the solution in the syringe. The solution in the syringe should be clear or colorless to light yellow in color.
- •
- Do not use Roferon-A if:
- –
- the medicine is cloudy
- –
- the medicine has particles floating in it
- –
- the medicine is any color besides clear or colorless to light yellow
- –
- it has passed the expiration date
- Warm the refrigerated medicine by gently rolling the syringe in the palms of your hands for about one minute.
- Wash your hands with soap and warm water. This step is very important to help prevent infection.
- Roferon-A prefilled syringe:
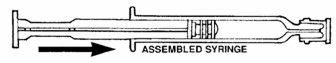
- Assemble syringe:
- •
- Place the plunger rod into the open end of the syringe barrel.
- •
- Gently screw the rod into the plunger stopper until snug.
DO NOT USE FORCE.
- Prepare the needle:
- •
- Turn and pull off the bright yellow tamper-resistant seal from needle. A "click" sound means that the needle is OK to use.
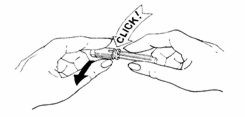
IF YOU DO NOT HEAR A "CLICK", DO NOT USE THE NEEDLE AND DO NOT REMOVE THE CLEAR NEEDLE SHIELD. DISCARD THE NEEDLE IN THE PUNCTURE-PROOF CONTAINER.
If you have another needle, proceed again with Step 7. If no alternate needle is available, contact your healthcare provider to make arrangements for a replacement needle.
- To attach the needle to the prefilled syringe:
-
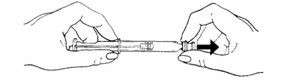
- •
- Remove the grey tip cap from syringe barrel.
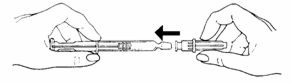
- •
- Place the needle onto the end of the syringe barrel so it fits snugly. Do not remove the clear needle shield.
- Choose an injection site:
- •
- You should choose a different spot each time you give or receive an injection. The common sites to use are:
- •
- abdomen, avoiding the navel and waistline area
- •
- thigh
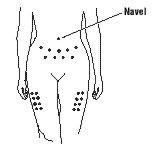
- •
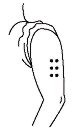
- If someone else is giving you the injection, then the upper, outer arm can be used as an injection site.
- Preparing the injection site:
- •
- Clean the skin where the injection will be given with an alcohol swab and allow the site to dry for 10 seconds.
- Injecting Roferon-A:
- •
- Hold the pale yellow hub between your thumb and forefinger and carefully (to avoid a needle-stick) remove the clear needle shield with your other hand. The syringe is ready for injection.
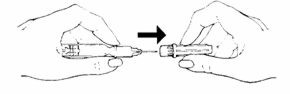
- •
- Keep the syringe in a horizontal position until ready for use.
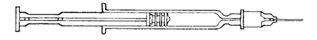
- •
- Holding the syringe with the needle facing up, tap the syringe barrel to bring air bubbles to the top.
- •
- Press the plunger slightly to push the air bubbles out through the needle.
- •
- Hold the syringe horizontally, and position the bevel of the needle so the point of the needle is facing up.
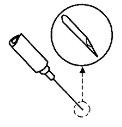
- •
- Pinch an area of skin firmly between your thumb and forefinger.
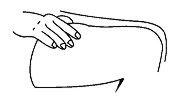
- •
- Hold the needle like a pencil at a 45° to 90° angle to skin and using a quick dart-like motion, insert the needle as far as it will go.
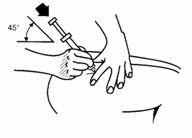
- •
- Once inserted, draw back slowly on the syringe. If blood appears in the syringe, the needle has entered a blood vessel.
Do not inject Roferon-A at that site and discard the syringe. Use a new syringe for the injection and use at a different injection site.
-
- •
- If blood does not appear in the syringe then slowly push the plunger all the way down so that you get all of your medicine.
- •
- Withdraw the needle at same angle it was inserted. See instructions for disposal of the needle and syringe in the section "How should I dispose of materials used to inject Roferon-A?".
- •
- When you are finished, place an alcohol swab over the injection site and press slightly.
- •
- Do not reuse syringes and needles. Use a new prefilled syringe and needle for each injection.
How should I dispose of materials used to inject Roferon-A?
- Do not recap the needle.
- Place the entire syringe and needle in a puncture-resistant container. A home "Sharps Container" may be purchased at your pharmacy or you can use a hard plastic container with a screw top or a coffee can with a plastic lid. You should talk to your healthcare provider about how to properly dispose of a full container of used syringes. There may be special state or local laws about disposing used syringes and needles, so please check with your physician, nurse or pharmacist for instructions. DO NOT throw the filled container in the household trash and DO NOT recycle.
- The needle cover and alcohol swabs can be thrown in the regular trash. You should always keep your syringes and disposal container out of the reach of children.
Appendix revision date: September 2003
Hoffmann-La Roche Inc.
340 Kingsland Street
Nutley, New Jersey 07110-1199
U.S. Govt. Lic. No. 0136
Copyright © 1999–2008 by Hoffmann-La Roche Inc. All rights reserved.
Representative sample of labeling (see HOW SUPPLIED section for complete listing):
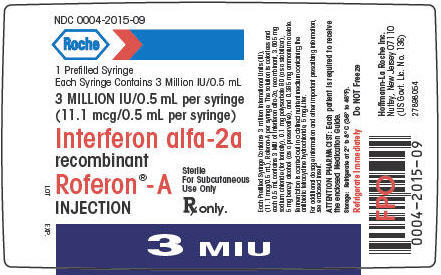
PRINCIPAL DISPLAY PANEL - 11.1 mcg/0.5 mL Syringe Label
NDC 0004-2015-09
Roche
1 Prefilled Syringe
Each Syringe Contains 3 Million IU/0.5 mL
3 MILLION IU/0.5 mL per syringe
(11.1 mcg/0.5 mL per syringe)
Interferon alfa-2a
recombinant
Roferon®-A
INJECTION
Sterile
For Subcutaneous
Use Only
Rx only.
3 MIU


| ROFERON-A
interferon alfa-2a injection, solution |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| ROFERON-A
interferon alfa-2a injection, solution |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| ROFERON-A
interferon alfa-2a injection, solution |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| Labeler - Genentech, Inc. (080129000) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| F. Hoffmann-La Roche | 482242971 | API MANUFACTURE(0004-2015, 0004-2016, 0004-2017), MANUFACTURE(0004-2015, 0004-2016, 0004-2017) | |