ELOXATIN- oxaliplatin injection, solution, concentrate
sanofi-aventis U.S. LLC
----------
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use ELOXATIN safely and effectively. See full prescribing information for ELOXATIN.
ELOXATIN (oxaliplatin) injection, for intravenous use Initial U.S. Approval: 2002 WARNING: HYPERSENSITIVITY REACTIONS, INCLUDING ANAPHYLAXISSee full prescribing information for complete boxed warning.Serious and fatal hypersensitivity adverse reactions, including anaphylaxis, can occur with ELOXATIN within minutes of administration and during any cycle. ELOXATIN is contraindicated in patients with hypersensitivity reactions to oxaliplatin and other platinum-based drugs. Immediately and permanently discontinue ELOXATIN for hypersensitivity reactions and administer appropriate treatment. (4, 5.1) INDICATIONS AND USAGEDOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHSInjection: 50 mg (5 mg/mL) or 100 mg (5 mg/mL) in a single-dose vial (3) CONTRAINDICATIONSWARNINGS AND PRECAUTIONS
ADVERSE REACTIONSMost common adverse reactions (incidence greater than or equal to 40%) were peripheral sensory neuropathy, neutropenia, thrombocytopenia, anemia, nausea, increase in transaminases and alkaline phosphatase, diarrhea, emesis, fatigue, and stomatitis. (6.1) To report SUSPECTED ADVERSE REACTIONS, contact sanofi-aventis U.S. LLC at 1-800-633-1610 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling. Revised: 6/2023 |
FULL PRESCRIBING INFORMATION
WARNING: HYPERSENSITIVITY REACTIONS, INCLUDING ANAPHYLAXIS
Serious and fatal hypersensitivity adverse reactions, including anaphylaxis, can occur with ELOXATIN within minutes of administration and during any cycle. ELOXATIN is contraindicated in patients with hypersensitivity reactions to oxaliplatin and other platinum-based drugs [see Contraindications (4)]. Immediately and permanently discontinue ELOXATIN for hypersensitivity reactions and administer appropriate treatment for management of the hypersensitivity reaction [see Warnings and Precautions (5.1)].
1 INDICATIONS AND USAGE
ELOXATIN, in combination with infusional fluorouracil and leucovorin, is indicated for:
- adjuvant treatment of stage III colon cancer in patients who have undergone complete resection of the primary tumor.
- treatment of advanced colorectal cancer.
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
Administer ELOXATIN in combination with fluorouracil and leucovorin every 2 weeks.
- For adjuvant treatment, continue treatment for up to 12 cycles or unacceptable toxicity.
- For advanced colorectal cancer, continue treatment until disease progression or unacceptable toxicity.
Day 1
Administer ELOXATIN 85 mg/m2 as an intravenous infusion over 120 minutes and leucovorin 200 mg/m2 as an intravenous infusion over 120 minutes at the same time in separate bags, followed by fluorouracil 400 mg/m2 as intravenous bolus over 2–4 minutes, followed by fluorouracil 600 mg/m2 as a 22-hour continuous infusion.
Day 2
Administer leucovorin 200 mg/m2 as an intravenous infusion over 120 minutes, followed by fluorouracil 400 mg/m2 as intravenous bolus over 2–4 minutes, followed by fluorouracil 600 mg/m2 as a 22-hour continuous infusion.
Refer to the prescribing information for fluorouracil and leucovorin for additional information.
2.2 Dose Modifications for Adverse Reactions
Prolongation of infusion time for ELOXATIN from 2 hours to 6 hours may mitigate acute toxicities, such as non–life-threatening infusion-related reactions.
Permanently discontinue ELOXATIN for any of the following:
- Hypersensitivity Reactions [see Warnings and Precautions (5.1)]
- Posterior reversible encephalopathy syndrome (PRES) [see Warnings and Precautions (5.4)]
- Confirmed interstitial lung disease or pulmonary fibrosis [see Warnings and Precautions (5.5)]
- Rhabdomyolysis [see Warnings and Precautions (5.8)]
Refer to the fluorouracil and leucovorin prescribing information for dosage modifications for adverse reactions.
Dosage Modifications for Adjuvant Treatment
Dosage modifications for adverse reactions for adjuvant treatment are presented in Table 1.
| Adverse Reactions | Severity | ELOXATIN Dosage Modifications |
|---|---|---|
| Peripheral Sensory Neuropathy [see Warnings and Precautions (5.2)] | Persistent Grade 2 | Consider reducing ELOXATIN dose to 75 mg/m2. |
| Persistent Grade 3 | Consider discontinuing ELOXATIN. | |
| Grade 4 | Discontinue ELOXATIN. | |
| Myelosuppression [see Warnings and Precautions (5.3), Adverse Reactions (6.1)]. | Grade 4 neutropenia or febrile neutropenia | Delay the next dose until neutrophils greater than or equal to 1.5 × 109/L and platelets greater than or equal to 75 × 109/L. Reduce ELOXATIN dose to 75 mg/m2. |
| Grade 3–4 thrombocytopenia | ||
| Gastrointestinal Adverse Reactions [see Adverse Reactions (6.1)] | Grade 3–4 | After recovery, reduce ELOXATIN dose to 75 mg/m2 along with a dose reduction of fluorouracil to 300 mg/m2 as an intravenous bolus and 500 mg/m2 as a 22-hour continuous infusion. |
Dosage Modifications for Advanced Colorectal Cancer
Dosage modifications for adverse reactions for advanced colorectal cancer are presented in Table 2.
| Adverse Reactions | Severity | ELOXATIN Dosage Modifications |
|---|---|---|
| Neuropathy [see Warnings and Precautions (5.2)] | Persistent Grade 2 | Consider reducing ELOXATIN dose to 65 mg/m2. |
| Persistent Grade 3 | Consider discontinuing ELOXATIN. | |
| Grade 4 | Discontinue ELOXATIN. | |
| Myelosuppression [see Warnings and Precautions (5.3), Adverse Reactions (6.1)] | Grade 4 neutropenia or febrile neutropenia | Delay the next dose until neutrophils greater than or equal to 1.5 × 109/L and platelets greater than or equal to 75 × 109/L. Reduce ELOXATIN dose to 65 mg/m2. |
| Grade 3–4 thrombocytopenia | ||
| Gastrointestinal Adverse Reactions [see Adverse Reactions (6.1)] | Grade 3–4 | After recovery, reduce ELOXATIN dose to 65 mg/ m2 along with a dose reduction of fluorouracil to 300 mg/m2 as an intravenous bolus and 500 mg/m2 as a 22-hour continuous infusion. |
2.3 Dose Modifications for Patients with Renal Impairment
In patients with severe renal impairment (creatinine clearance [CLcr] less than 30 mL/min, calculated by the Cockcroft-Gault equation), reduce the ELOXATIN dose to 65 mg/m2 [see Use in Specific Populations (8.6), Clinical Pharmacology (12.3)].
2.4 Preparation and Administration
- ELOXATIN is a cytotoxic drug. Follow applicable special handling and disposal procedures.1
- Do not freeze.
- Protect the concentrated solution from light.
- Dilute concentrated solution with 250 to 500 mL of 5% Dextrose Injection, USP. Do not dilute with sodium chloride solution or other chloride-containing solutions.
- Store diluted solution for no more than 6 hours at room temperature (20°C to 25°C [68°F to 77°F]) or 24 hours under refrigeration (2°C to 8°C [36°F to 46°F]). Protection from light is not required.
- Visually inspect for particulate matter and discoloration prior to administration and discard if present.
- Do not mix ELOXATIN or administer ELOXATIN through the same infusion line concurrently with alkaline medications or media (such as basic solutions of fluorouracil).
- Flush the infusion line with 5% Dextrose Injection, USP prior to administration of any concomitant medication.
- Do not use needles or intravenous administration sets containing aluminum parts for the preparation or mixing of ELOXATIN. Aluminum has been reported to cause degradation of platinum compounds.
- Administer ELOXATIN as an intravenous infusion over 120 minutes concurrently with leucovorin over 120 minutes in separate bags.
3 DOSAGE FORMS AND STRENGTHS
Injection: 50 mg (5 mg/mL) or 100 mg (5 mg/mL) clear, colorless solution in a single-dose vial.
4 CONTRAINDICATIONS
ELOXATIN is contraindicated in patients with a history of a hypersensitivity reaction to oxaliplatin or other platinum-based drugs. Reactions have included anaphylaxis [see Warnings and Precautions (5.1)].
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions
Serious and fatal hypersensitivity reactions, including anaphylaxis, can occur with ELOXATIN within minutes of administration and during any cycle. Grade 3–4 hypersensitivity reactions, including anaphylaxis, occurred in 2% to 3% of patients with colon cancer who received ELOXATIN. Hypersensitivity reactions, including rash, urticaria, erythema, pruritus, and rarely, bronchospasm and hypotension, were similar in nature and severity to those reported with other platinum-based drugs.
ELOXATIN is contraindicated in patients with hypersensitivity reactions to platinum-based drugs [see Contraindications (4)]. Immediately and permanently discontinue ELOXATIN for hypersensitivity reactions and administer appropriate treatment for management of hypersensitivity reactions.
5.2 Peripheral Sensory Neuropathy
ELOXATIN can cause acute and delayed neuropathy. Reduce the dose or permanently discontinue ELOXATIN for persistent neurosensory reactions based on the severity of the adverse reaction [see Dosage and Administration (2.2)].
Acute Neuropathy
Acute neuropathy typically presents as a reversible, primarily peripheral sensory neuropathy that occurs within hours or 2 days following a dose, resolves within 14 days, and frequently recurs with further dosing. The symptoms can be precipitated or exacerbated by exposure to cold temperature or cold objects and they usually present as transient paresthesia, dysesthesia and hypoesthesia in the hands, feet, perioral area, or throat. Jaw spasm, abnormal tongue sensation, dysarthria, eye pain, and a feeling of chest pressure have also been observed. The acute, reversible pattern of sensory neuropathy was observed in about 56% of patients who received ELOXATIN with fluorouracil/leucovorin. In any individual cycle, acute neuropathy occurred in approximately 30% of patients. For grade 3 peripheral sensory neuropathy, the median time to onset was 9 cycles for adjuvant treatment and 6 cycles for previously treated advanced colorectal cancer.
An acute syndrome of pharyngolaryngeal dysesthesia occurred in 1% to 2% (grade 3–4) of patients previously untreated for advanced colorectal cancer. Subjective sensations of dysphagia or dyspnea, without any laryngospasm or bronchospasm (no stridor or wheezing) occurred in patients previously treated for advanced colorectal cancer.
Avoid topical application of ice for mucositis prophylaxis or other conditions, because cold temperature can exacerbate acute neurological symptoms.
Delayed Neuropathy
Delayed neuropathy typically presents as a persistent (greater than 14 days), primarily peripheral sensory neuropathy that is usually characterized by paresthesias, dysesthesias, and hypoesthesias, but may also include deficits in proprioception that can interfere with daily activities (e.g., writing, buttoning, swallowing, and difficulty walking from impaired proprioception). These forms of neuropathy occurred in 48% of patients receiving ELOXATIN. Delayed neuropathy can occur without any prior acute neuropathy. Most patients (80%) who developed grade 3 persistent neuropathy progressed from prior grade 1 or 2 reactions. These symptoms may improve in some patients upon discontinuation of ELOXATIN.
Adjuvant treatment
In the adjuvant treatment trial, neuropathy was graded using NCI CTC, version 1 as summarized in Table 3.
| Grade | Definition |
|---|---|
| 0 | No change or none |
| 1 | Mild paresthesias, loss of deep tendon reflexes |
| 2 | Mild or moderate objective sensory loss, moderate paresthesias |
| 3 | Severe objective sensory loss or paresthesias that interfere with function |
| 4 | Not applicable |
Peripheral sensory neuropathy occurred in 92% of patients (all grades), including 13% of patients (grade 3) who received ELOXATIN with fluorouracil/leucovorin. At the 28-day follow-up after the last treatment cycle, 60% of patients had any grade (grade 1=40%, grade 2=16%, grade 3=5%) peripheral sensory neuropathy, decreasing to 39% at 6 months of follow-up (grade 1=31%, grade 2=7%, grade 3=1%) and 21% at 18 months of follow-up (grade 1=17%, grade 2=3%, grade 3=1%).
Advanced colorectal cancer
In the advanced colorectal cancer trials, neuropathy was graded using the neurotoxicity scale summarized in Table 4.
| Grade | Definition |
|---|---|
| 1 | Resolved and did not interfere with functioning |
| 2 | Interfered with function but not daily activities |
| 3 | Pain or functional impairment that interfered with daily activities |
| 4 | Persistent impairment that is disabling or life-threatening |
Neuropathy occurred in 82% (all grades) of patients previously untreated for advanced colorectal cancer, including 19% grade 3–4; and in 74% (all grades) of patients previously treated for advanced colorectal cancer, including 7% grade 3–4.
5.3 Severe Myelosuppression
Grade 3 or 4 neutropenia occurred in 41% to 44% of patients with colorectal cancer who received ELOXATIN with fluorouracil/leucovorin. Sepsis, neutropenic sepsis and septic shock, including fatal outcomes, occurred in patients who received ELOXATIN [see Adverse Reactions (6.1, 6.2)].
Grade 3 or 4 thrombocytopenia occurred in 2% to 5% of patients with colorectal cancer who received ELOXATIN with fluorouracil/leucovorin.
Monitor complete blood cell count at baseline, before each subsequent cycle and as clinically indicated. Delay ELOXATIN until neutrophils are greater than or equal to 1.5 × 109/L and platelets are greater than or equal to 75 × 109/L. Withhold ELOXATIN for sepsis or septic shock. Dose reduce ELOXATIN after recovery from grade 4 neutropenia, febrile neutropenia or grade 3–4 thrombocytopenia as recommended [see Dosage and Administration (2.2)].
5.4 Posterior Reversible Encephalopathy Syndrome
PRES occurred in less than 0.1% of patients across clinical trials [see Adverse Reactions (6.1)]. Signs and symptoms of PRES can include headache, altered mental functioning, seizures, abnormal vision from blurriness to blindness, associated or not with hypertension. Confirm the diagnosis of PRES with magnetic resonance imaging. Permanently discontinue ELOXATIN in patients who develop PRES.
5.5 Pulmonary Toxicity
ELOXATIN has been associated with pulmonary fibrosis (less than 1% of patients), which may be fatal [see Adverse Reactions (6.1)].
In the adjuvant treatment trial, the combined incidence of cough and dyspnea was 7.4% (any grade), including less than 1% (grade 3) in the ELOXATIN arm. One patient died from eosinophilic pneumonia in the ELOXATIN arm.
In the previously untreated advanced colorectal cancer trial, the combined incidence of cough, dyspnea, and hypoxia was 43% (any grade), including 7% (grade 3–4) in the ELOXATIN with fluorouracil/leucovorin arm.
In case of unexplained respiratory symptoms, such as non-productive cough, dyspnea, crackles, or radiological pulmonary infiltrates, withhold ELOXATIN until further pulmonary investigation excludes interstitial lung disease or pulmonary fibrosis. Permanently discontinue ELOXATIN for confirmed interstitial lung disease or pulmonary fibrosis.
5.6 Hepatotoxicity
In the adjuvant treatment trial, increased transaminases (57% vs 34%) and alkaline phosphatase (42% vs 20%) occurred more commonly in the ELOXATIN arm than in the fluorouracil/leucovorin arm [see Adverse Reactions (6.1)]. The incidence of increased bilirubin was similar on both arms. Changes noted on liver biopsies include: peliosis, nodular regenerative hyperplasia or sinusoidal alterations, perisinusoidal fibrosis, and veno-occlusive lesions.
Consider evaluating patients who develop abnormal liver tests or portal hypertension, which cannot be explained by liver metastases, for hepatic vascular disorders. Monitor liver function tests at baseline, before each subsequent cycle, and as clinically indicated.
5.7 QT Interval Prolongation and Ventricular Arrhythmias
QT prolongation and ventricular arrhythmias, including fatal torsade de pointes, have been reported with ELOXATIN [see Adverse Reactions (6.2)].
Avoid ELOXATIN in patients with congenital long QT syndrome. Monitor electrocardiograms (ECG) in patients with congestive heart failure, bradyarrhythmias, and electrolyte abnormalities and in patients taking drugs known to prolong the QT interval, including Class Ia and III antiarrhythmics [see Drug Interactions (7.1)]. Monitor and correct electrolyte abnormalities prior to initiating ELOXATIN and periodically during treatment.
5.8 Rhabdomyolysis
Rhabdomyolysis, including fatal cases, has been reported with ELOXATIN [see Adverse Reactions (6.2)]. Permanently discontinue ELOXATIN for any signs or symptoms of rhabdomyolysis.
5.9 Hemorrhage
The incidence of hemorrhage in clinical trials was higher on the ELOXATIN combination arm compared to the fluorouracil/leucovorin arm. These reactions included gastrointestinal bleeding, hematuria, and epistaxis. In the adjuvant treatment trial, 2 patients died from intracerebral hemorrhage [see Adverse Reactions (6.1)].
Prolonged prothrombin time and INR occasionally associated with hemorrhage have been reported in patients who received ELOXATIN with fluorouracil/leucovorin while on anticoagulants [see Adverse Reactions (6.2)]. Increase frequency of monitoring in patients who are receiving ELOXATIN with fluorouracil/leucovorin and oral anticoagulants [see Drug Interactions (7.3)].
Thrombocytopenia and immune-mediated thrombocytopenia have been observed with ELOXATIN. Rapid onset of thrombocytopenia and greater risk of bleeding have been observed in immune-mediated thrombocytopenia. In this case, consider discontinuing ELOXATIN.
5.10 Embryo-Fetal Toxicity
Based on findings from animal studies and its mechanism of action, ELOXATIN can cause fetal harm when administered to a pregnant woman. The available human data do not establish the presence or absence of major birth defects or miscarriage related to the use of ELOXATIN. Reproductive toxicity studies demonstrated adverse effects on embryo-fetal development in rats at maternal doses that were below the recommended human dose based on body surface area. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with ELOXATIN and for 9 months after the final dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with ELOXATIN and for 6 months after the final dose [see Use in Specific Populations (8.1, 8.3)].
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in labeling:
- Hypersensitivity Reactions [see Warnings and Precautions (5.1)]
- Peripheral Sensory Neuropathy [see Warnings and Precautions (5.2)]
- Severe Myelosuppression [see Warnings and Precautions (5.3)]
- Reversible Posterior Leukoencephalopathy Syndrome [see Warnings and Precautions (5.4)]
- Pulmonary Toxicity [see Warnings and Precautions (5.5)]
- Hepatotoxicity [see Warnings and Precautions (5.6)]
- QT Interval Prolongation and Ventricular Arrhythmias [see Warnings and Precautions (5.7)]
- Rhabdomyolysis [see Warnings and Precautions (5.8)]
- Hemorrhage [see Warnings and Precautions (5.9)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
More than 1100 patients with stage II or III colon cancer and more than 4,000 patients with advanced colorectal cancer were treated in trials with ELOXATIN. The most common adverse reactions in patients with stage II or III colon cancer receiving adjuvant treatment were peripheral sensory neuropathy, neutropenia, thrombocytopenia, anemia, nausea, increase in transaminases and alkaline phosphatase, diarrhea, emesis, fatigue and stomatitis. The most common adverse reactions in previously untreated and treated patients with advanced colorectal cancer were peripheral sensory neuropathies, fatigue, neutropenia, nausea, emesis, and diarrhea.
Adjuvant Treatment
The safety of ELOXATIN in combination with fluorouracil (FU)/leucovorin (LV) was evaluated in patients with stage II or III colon cancer, who had undergone complete resection of the primary tumor in the adjuvant treatment trial [see Clinical Studies (14.1)].
Fatal adverse reactions in patients who received ELOXATIN in the combination arm included sepsis/neutropenic sepsis (n=3), intracerebral hemorrhage (n=2), and eosinophilic pneumonia (n=1).
Thromboembolic events occurred in 6% (grade 3–4, 1.2%) of patients in the ELOXATIN arm.
Grade 3 or 4 adverse reactions occurred in 70% of patients in the ELOXATIN arm. Grade 3–4 gastrointestinal bleeding occurred in 0.2% of patients. Febrile neutropenia occurred in 0.7% and documented infection with concomitant grade 3–4 neutropenia occurred in 1.1%.
Discontinuation due to an adverse reaction occurred in 15% of the patients in the ELOXATIN arm.
Tables 5, 6, and 7 summarize the adverse reactions reported in patients with colon cancer receiving adjuvant treatment.
| Adverse Reaction* | ELOXATIN + FU/LV N=1108 | FU/LV N=1111 |
||
|---|---|---|---|---|
| All Grades (%) | Grade 3–4 (%) | All Grades (%) | Grade 3–4 (%) |
|
| Neurology | ||||
| Peripheral Sensory Neuropathy | 92 | 12 | 16 | <1 |
| Gastrointestinal | ||||
| Nausea | 74 | 5 | 61 | 2 |
| Diarrhea | 56 | 11 | 48 | 7 |
| Vomiting | 47 | 6 | 24 | 1 |
| Stomatitis | 42 | 3 | 40 | 2 |
| Anorexia | 13 | 1 | 8 | <1 |
| Constitutional Symptoms/Pain | ||||
| Fatigue | 44 | 4 | 38 | 1 |
| Abdominal Pain | 18 | 1 | 17 | 2 |
| Dermatology/Skin | ||||
| Skin Disorder | 32 | 2 | 36 | 2 |
| Injection Site Reaction† | 11 | 3 | 10 | 3 |
| Fever/Infection | ||||
| Fever | 27 | 1 | 12 | 1 |
| Infection | 25 | 4 | 25 | 3 |
| Allergy/Immunology | ||||
| Allergic Reaction | 10 | 3 | 2 | <1 |
| Adverse Reaction* | ELOXATIN + FU/LV N=1108 | FU/LV N=1111 |
|---|---|---|
| All Grades (%) | All Grades (%) |
|
| Dermatology/Skin | ||
| Alopecia† | 30 | 28 |
| Gastrointestinal | ||
| Constipation | 22 | 19 |
| Taste Perversion | 12 | 8 |
| Dyspepsia | 8 | 5 |
| Constitutional Symptoms/Pain/Ocular/Visual | ||
| Epistaxis | 16 | 12 |
| Weight Increase | 10 | 10 |
| Conjunctivitis | 9 | 15 |
| Headache | 7 | 5 |
| Dyspnea | 5 | 3 |
| Pain | 5 | 5 |
| Abnormal Lacrimation | 4 | 12 |
| Neurology | ||
| Sensory Disturbance | 8 | 1 |
| Allergy/Immunology | ||
| Rhinitis | 6 | 8 |
In females, the following grade 3–4 adverse reactions were more frequent: diarrhea, fatigue, neutropenia, nausea, and vomiting.
In patients greater than or equal to 65 years old, the incidence of grade 3–4 diarrhea and neutropenia was higher than in younger adults.
Clinically relevant adverse reactions were reported in greater than or equal to 2% and less than 5% of the patients in the ELOXATIN arm (listed in decreasing order of frequency) were pain, leukopenia, weight loss, and cough.
| Laboratory-Related Adverse Reaction | ELOXATIN with FU/LV N=1108 | FU/LV N=1111 |
||
|---|---|---|---|---|
| All Grades (%) | Grades 3–4 (%) | All Grades (%) | Grades 3–4 (%) |
|
| Hematology | ||||
| Neutropenia | 79 | 41 | 40 | 5 |
| Thrombocytopenia | 77 | 2 | 19 | <1 |
| Anemia | 76 | 1 | 67 | <1 |
| Hepatic | ||||
| Increased Transaminases | 57 | 2 | 34 | 1 |
| Increased Alkaline Phosphatase | 42 | <1 | 20 | <1 |
| Hyperbilirubinemia | 20 | 4 | 20 | 5 |
Previously Untreated Advanced Colorectal Cancer
The safety of ELOXATIN in combination with fluorouracil (FU)/leucovorin (LV) was evaluated in a randomized trial of patients with previously untreated advanced colorectal cancer [see Clinical Studies (14.2)]. The adverse reaction profile in this trial was similar to that seen in other trials.
Tables 8, 9, and 10 summarize the adverse reactions reported in the previously untreated advanced colorectal cancer trial.
| Adverse Reaction* | ELOXATIN + FU/LV N=259 | Irinotecan + FU/LV N=256 | ELOXATIN + Irinotecan N=258 |
|||
|---|---|---|---|---|---|---|
| All Grades (%) | Grades 3–4 (%) | All Grades (%) | Grades 3–4 (%) | All Grades (%) | Grades 3–4 (%) |
|
| Neurology | ||||||
| Neuropathy | 82 | 19 | 18 | 2 | 69 | 7 |
| Paresthesias | 77 | 18 | 16 | 2 | 62 | 6 |
| Pharyngo-laryngeal Dysesthesias | 38 | 2 | 1 | 0 | 28 | 1 |
| Neuro-sensory | 12 | 1 | 2 | 0 | 9 | 1 |
| Neuro NOS† | 1 | 0 | 1 | 0 | 1 | 0 |
| Gastrointestinal | ||||||
| Nausea | 71 | 6 | 67 | 15 | 83 | 19 |
| Diarrhea | 56 | 12 | 65 | 29 | 76 | 25 |
| Vomiting | 41 | 4 | 43 | 13 | 64 | 23 |
| Stomatitis | 38 | 0 | 25 | 1 | 19 | 1 |
| Anorexia | 35 | 2 | 25 | 4 | 27 | 5 |
| Constipation | 32 | 4 | 27 | 2 | 21 | 2 |
| Diarrhea-colostomy | 13 | 2 | 16 | 7 | 16 | 3 |
| Gastrointestinal NOS† | 5 | 2 | 4 | 2 | 3 | 2 |
| Constitutional Symptoms/Pain/Ocular/Visual | ||||||
| Fatigue | 70 | 7 | 58 | 11 | 66 | 16 |
| Abdominal Pain | 29 | 8 | 31 | 7 | 39 | 10 |
| Myalgia | 14 | 2 | 6 | 0 | 9 | 2 |
| Pain | 7 | 1 | 5 | 1 | 6 | 1 |
| Abnormal Vision | 5 | 0 | 2 | 1 | 6 | 1 |
| Neuralgia | 5 | 0 | 0 | 0 | 2 | 1 |
| Pulmonary | ||||||
| Cough | 35 | 1 | 25 | 2 | 17 | 1 |
| Dyspnea | 18 | 7 | 14 | 3 | 11 | 2 |
| Hiccups | 5 | 1 | 2 | 0 | 3 | 2 |
| Hepatic/Metabolic/Laboratory/Renal | ||||||
| Hyperglycemia | 14 | 2 | 11 | 3 | 12 | 3 |
| Hypokalemia | 11 | 3 | 7 | 4 | 6 | 2 |
| Dehydration | 9 | 5 | 16 | 11 | 14 | 7 |
| Hypoalbuminemia | 8 | 0 | 5 | 2 | 9 | 1 |
| Hyponatremia | 8 | 2 | 7 | 4 | 4 | 1 |
| Urinary Frequency | 5 | 1 | 2 | 1 | 3 | 1 |
| Hematology/Infection | ||||||
| Infection Normal ANC‡ | 10 | 4 | 5 | 1 | 7 | 2 |
| Infection Low ANC‡ | 8 | 8 | 12 | 11 | 9 | 8 |
| Lymphopenia | 6 | 2 | 4 | 1 | 5 | 2 |
| Febrile Neutropenia | 4 | 4 | 15 | 14 | 12 | 11 |
| Dermatology/Skin | ||||||
| Hand/Foot Syndrome | 7 | 1 | 2 | 1 | 1 | 0 |
| Injection Site Reaction | 6 | 0 | 1 | 0 | 4 | 1 |
| Cardiovascular | ||||||
| Thrombosis | 6 | 5 | 6 | 6 | 3 | 3 |
| Hypotension | 5 | 3 | 6 | 3 | 4 | 3 |
| Adverse Reaction* | ELOXATIN + FU/LV N=259 | Irinotecan + FU/LV N=256 | ELOXATIN + Irinotecan N=258 |
|---|---|---|---|
| All Grades (%) | All Grades (%) | All Grades (%) |
|
| Dermatology/Skin | |||
| Alopecia† | 38 | 44 | 67 |
| Flushing | 7 | 2 | 5 |
| Pruritus | 6 | 4 | 2 |
| Dry Skin | 6 | 2 | 5 |
| Hematology/Infection | |||
| Fever Normal ANC‡ | 16 | 9 | 9 |
| Cardiovascular | |||
| Edema | 15 | 13 | 10 |
| Gastrointestinal | |||
| Taste Perversion | 14 | 6 | 8 |
| Dyspepsia | 12 | 7 | 5 |
| Flatulence | 9 | 6 | 5 |
| Mouth Dryness | 5 | 2 | 3 |
| Constitutional Symptoms/Pain/Ocular/Visual | |||
| Headache | 13 | 6 | 9 |
| Weight Loss | 11 | 9 | 11 |
| Epistaxis | 10 | 2 | 2 |
| Tearing | 9 | 1 | 2 |
| Rigors | 8 | 2 | 7 |
| Dysphasia | 5 | 3 | 3 |
| Sweating | 5 | 6 | 12 |
| Arthralgia | 5 | 5 | 8 |
| Neurology | |||
| Insomnia | 13 | 9 | 11 |
| Depression | 9 | 5 | 7 |
| Dizziness | 8 | 6 | 10 |
| Anxiety | 5 | 2 | 6 |
| Allergy/Immunology | |||
| Rash | 11 | 4 | 7 |
| Rhinitis Allergic | 10 | 6 | 6 |
| Hepatic/Metabolic/Laboratory/Renal | |||
| Hypocalcemia | 7 | 5 | 4 |
| Elevated Creatinine | 4 | 4 | 5 |
Clinically relevant adverse reactions that occurred in greater than or equal to 2% and less than 5% of the patients in the ELOXATIN and fluorouracil/leucovorin combination arm (listed in decreasing order of frequency) were: metabolic, pneumonitis, catheter infection, vertigo, prothrombin time, pulmonary, rectal bleeding, dysuria, nail changes, chest pain, rectal pain, syncope, hypertension, hypoxia, unknown infection, bone pain, pigmentation changes, and urticaria.
| Laboratory-Related Adverse Reaction | ELOXATIN and FU/LV N=259 | Irinotecan and FU/LV N=256 | ELOXATIN and Irinotecan N=258 |
|||
|---|---|---|---|---|---|---|
| All Grades (%) | Grades 3–4 (%) | All Grades (%) | Grades 3–4 (%) | All Grades (%) | Grades 3–4 (%) |
|
| Hematology | ||||||
| Leukopenia | 85 | 20 | 84 | 23 | 76 | 24 |
| Neutropenia | 81 | 53 | 77 | 44 | 71 | 36 |
| Thrombocytopenia | 71 | 5 | 26 | 2 | 44 | 4 |
| Anemia | 27 | 3 | 28 | 4 | 25 | 3 |
| Hepatic | ||||||
| Increased AST* | 17 | 1 | 2 | 1 | 11 | 1 |
| Increased Alkaline Phosphatase | 16 | 0 | 8 | 0 | 14 | 2 |
| Hyperbilirubinemia | 6 | 1 | 3 | 1 | 3 | 2 |
| Increased ALT† | 6 | 1 | 2 | 0 | 5 | 2 |
Previously Treated Advanced Colorectal Cancer
The safety of ELOXATIN in combination with fluorouracil (FU)/leucovorin (LV) was evaluated in a randomized trial in patients with refractory and relapsed colorectal cancer [see Clinical Studies (14.3)]. The adverse reaction profile in this trial was similar to that seen in other trials.
Three patients who received ELOXATIN in the combination arm experienced fatal adverse reactions: gastrointestinal bleeding and dehydration.
Grade 3 and 4 neutropenia were reported in 27% and 17% of patients, respectively, in the ELOXATIN with fluorouracil/leucovorin combination arm. Grade 3–4 increased serum creatinine occurred in 1% of patients in the ELOXATIN with combination fluorouracil/leucovorin arm.
Thirteen percent of patients in the ELOXATIN with fluorouracil/leucovorin combination arm discontinued treatment; the most frequent reasons were gastrointestinal adverse reactions, hematologic adverse reactions and neuropathies.
Tables 11, 12, and 13 summarize the adverse reactions reported in the previously treated advanced colorectal cancer trial.
| Adverse Reaction* | ELOXATIN + FU/LV N=150 | ELOXATIN N=153 | FU/LV N=142 |
|||
|---|---|---|---|---|---|---|
| All Grades (%) | Grades 3–4 (%) | All Grades (%) | Grades 3–4 (%) | All Grades (%) | Grades 3–4 (%) |
|
|
||||||
| Neurology | ||||||
| Neuropathy | 74 | 7 | 76 | 7 | 17 | 0 |
| Acute | 56 | 2 | 65 | 5 | 10 | 0 |
| Persistent | 48 | 6 | 43 | 3 | 9 | 0 |
| Constitutional Symptoms/Pain | ||||||
| Fatigue | 68 | 7 | 61 | 9 | 52 | 6 |
| Back Pain | 19 | 3 | 11 | 0 | 16 | 4 |
| Pain | 15 | 2 | 14 | 3 | 9 | 3 |
| Gastrointestinal | ||||||
| Diarrhea | 67 | 11 | 46 | 4 | 44 | 3 |
| Nausea | 65 | 11 | 64 | 4 | 59 | 4 |
| Vomiting | 40 | 9 | 37 | 4 | 27 | 4 |
| Stomatitis | 37 | 3 | 14 | 0 | 32 | 3 |
| Abdominal Pain | 33 | 4 | 31 | 7 | 31 | 5 |
| Anorexia | 29 | 3 | 20 | 2 | 20 | 1 |
| Gastroesophageal Reflux | 5 | 2 | 1 | 0 | 3 | 0 |
| Hematology/Infection | ||||||
| Fever | 29 | 1 | 25 | 1 | 23 | 1 |
| Febrile Neutropenia | 6 | 6 | 0 | 0 | 1 | 1 |
| Cardiovascular | ||||||
| Dyspnea | 20 | 4 | 13 | 7 | 11 | 2 |
| Coughing | 19 | 1 | 11 | 0 | 9 | 0 |
| Edema | 15 | 1 | 10 | 1 | 13 | 1 |
| Thromboembolism | 9 | 8 | 2 | 1 | 4 | 2 |
| Chest Pain | 8 | 1 | 5 | 1 | 4 | 1 |
| Dermatology/Skin | ||||||
| Injection Site Reaction | 10 | 3 | 9 | 0 | 5 | 1 |
| Hepatic/Metabolic/Laboratory/Renal | ||||||
| Hypokalemia | 9 | 4 | 3 | 2 | 3 | 1 |
| Dehydration | 8 | 3 | 5 | 3 | 6 | 4 |
| Adverse Reaction* | ELOXATIN + FU/LV N=150 | ELOXATIN N=153 | FU/LV N=142 |
|---|---|---|---|
| All Grades (%) | All Grades (%) | All Grades (%) |
|
| Gastrointestinal | |||
| Constipation | 32 | 31 | 23 |
| Dyspepsia | 14 | 7 | 10 |
| Taste Perversion | 13 | 5 | 1 |
| Mucositis | 7 | 2 | 10 |
| Flatulence | 5 | 3 | 6 |
| Constitutional Symptoms/Pain/Ocular/Visual | |||
| Headache | 17 | 13 | 8 |
| Arthralgia | 10 | 7 | 10 |
| Epistaxis | 9 | 2 | 1 |
| Abnormal Lacrimation | 7 | 1 | 6 |
| Rigors | 7 | 9 | 6 |
| Allergy/Immunology | |||
| Rhinitis | 15 | 6 | 4 |
| Allergic Reaction | 10 | 3 | 1 |
| Rash | 9 | 5 | 5 |
| Neurology | |||
| Dizziness | 13 | 7 | 8 |
| Insomnia | 9 | 11 | 4 |
| Dermatology/Skin | |||
| Hand-Foot Syndrome | 11 | 1 | 13 |
| Flushing | 10 | 3 | 2 |
| Alopecia† | 7 | 3 | 3 |
| Pulmonary | |||
| Upper Respiratory Tract Infection | 10 | 7 | 4 |
| Pharyngitis | 9 | 2 | 10 |
| Cardiovascular | |||
| Peripheral Edema | 10 | 5 | 11 |
| Hepatic/Metabolic/Laboratory/Renal | |||
| Hematuria | 6 | 0 | 4 |
| Dysuria | 6 | 1 | 1 |
Clinically relevant adverse reactions in greater than or equal to 2% and less than 5% of the patients in the ELOXATIN and fluorouracil/leucovorin combination arm (listed in decreasing order of frequency) were: anxiety, myalgia, erythematous rash, increased sweating, conjunctivitis, weight decrease, dry mouth, rectal hemorrhage, depression, ataxia, ascites, hemorrhoids, muscle weakness, nervousness, tachycardia, abnormal micturition frequency, dry skin, pruritus, hemoptysis, purpura, vaginal hemorrhage, melena, somnolence, pneumonia, proctitis, involuntary muscle contractions, intestinal obstruction, gingivitis, tenesmus, hot flashes, enlarged abdomen, and urinary incontinence.
| Laboratory-Related Adverse Reaction | ELOXATIN and FU/LV N=150 | ELOXATIN N=153 | FU/LV N=142 |
|||
|---|---|---|---|---|---|---|
| All Grades (%) | Grades 3–4 (%) | All Grades (%) | Grades 3–4 (%) | All Grades (%) | Grades 3–4 (%) |
|
| Hematology | ||||||
| Anemia | 81 | 2 | 64 | 1 | 68 | 2 |
| Leukopenia | 76 | 19 | 13 | 0 | 34 | 1 |
| Neutropenia | 73 | 44 | 7 | 0 | 25 | 5 |
| Thrombocytopenia | 64 | 4 | 30 | 3 | 20 | 0 |
| Hepatic | ||||||
| Increased ALT* | 31 | 0 | 36 | 1 | 28 | 3 |
| Increased AST† | 47 | 0 | 54 | 4 | 39 | 2 |
| Increased Bilirubin | 13 | 1 | 13 | 5 | 22 | 6 |
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of ELOXATIN. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- General: angioedema, anaphylactic shock
- Cardiovascular: QT prolongation leading to ventricular arrhythmias, including fatal torsade de pointes; bradyarrhythmia
- Neurological: loss of deep tendon reflexes, dysarthria, Lhermitte's sign, cranial nerve palsies, fasciculations, convulsion
- Hearing and vestibular system: deafness
- Infections: septic shock, including fatal outcomes
- Infusion-related reactions and hypersensitivity reactions: laryngospasm
- Hepatic and gastrointestinal: severe diarrhea/vomiting resulting in hypokalemia, colitis (including Clostridium difficile diarrhea), metabolic acidosis, ileus, intestinal obstruction, pancreatitis, sinusoidal obstruction syndrome, perisinusoidal fibrosis which rarely may progress, focal nodular hyperplasia, esophagitis
- Musculoskeletal and connective tissue: rhabdomyolysis, including fatal outcomes
- Platelet, bleeding, and clotting disorders: immuno-allergic thrombocytopenia, prolonged prothrombin time and INR in patients receiving anticoagulants
- Blood disorders: secondary leukemia
- Red blood cell: hemolytic uremic syndrome, immuno-allergic hemolytic anemia
- Renal: acute tubular necrosis, acute interstitial nephritis, acute renal failure
- Respiratory: interstitial lung diseases (sometimes fatal) and pneumonia (including fatal outcomes)
- Vision: decrease of visual acuity, visual field disturbance, optic neuritis and transient vision loss (reversible following treatment discontinuation)
- Injury, poisoning, and procedural complications: fall-related injuries
7 DRUG INTERACTIONS
7.1 Drugs that Prolong the QT Interval
QT interval prolongation and ventricular arrhythmias can occur with ELOXATIN [see Warnings and Precautions (5.7)]. Avoid coadministration of ELOXATIN with medicinal products with a known potential to prolong the QT interval.
7.2 Use with Nephrotoxic Products
Because platinum-containing species are eliminated primarily through the kidney, clearance of these products may be decreased by coadministration of potentially nephrotoxic compounds [see Clinical Pharmacology (12.3)]. Avoid coadministration of ELOXATIN with medicinal products known to cause nephrotoxicity.
7.3 Use with Anticoagulants
Prolonged prothrombin time and INR occasionally associated with hemorrhage have been reported in patients who received ELOXATIN with fluorouracil/leucovorin while on anticoagulants [see Warnings and Precautions (5.9), Adverse Reactions (6.2)]. Increase frequency of monitoring in patients who are receiving ELOXATIN with fluorouracil/leucovorin and oral anticoagulants.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on its direct interaction with DNA, ELOXATIN can cause fetal harm when administered to a pregnant woman. The available human data do not establish the presence or absence of major birth defects or miscarriage related to the use of ELOXATIN. Reproductive toxicity studies demonstrated adverse effects on embryo-fetal development in rats at maternal doses that were below the recommended human dose based on body surface area (see Data). Advise a pregnant woman of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal data
Pregnant rats were administered oxaliplatin at less than one-tenth the recommended human dose based on body surface area during gestation days (GD)1–5 (preimplantation), GD 6–10, or GD 11–16 (during organogenesis). Oxaliplatin caused developmental mortality (increased early resorptions) when administered on days GD 6–10 and GD 11–16 and adversely affected fetal growth (decreased fetal weight, delayed ossification) when administered on days GD 6–10.
8.2 Lactation
Risk Summary
There are no data on the presence of oxaliplatin or its metabolites in human or animal milk or its effects on the breastfed infant or on milk production. Because of the potential for serious adverse reactions in breastfed infants, advise women not to breastfeed during treatment with ELOXATIN and for 3 months after the final dose.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Verify pregnancy status in females of reproductive potential prior to initiating ELOXATIN [see Use in Specific Populations (8.1)].
Contraception
ELOXATIN can cause embryo-fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Females
Advise female patients of reproductive potential to use effective contraception while receiving ELOXATIN and for 9 months after the final dose.
Males
Based on its mechanism action as a genotoxic drug, advise males with female partners of reproductive potential to use effective contraception while receiving ELOXATIN and for 6 months after the final dose [see Nonclinical Toxicology (13.1)].
Infertility
Based on animal studies, ELOXATIN may impair fertility in males and females [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of ELOXATIN in pediatrics have not been established. Safety and effectiveness were assessed across 4 open-label studies in 235 patients aged 7 months to 22 years with solid tumors.
In a multicenter, open-label, non-comparative, non-randomized study (ARD5531), oxaliplatin was administered to 43 patients with refractory or relapsed malignant solid tumors, mainly neuroblastoma and osteosarcoma. The dose limiting toxicity (DLT) was sensory neuropathy at a dose of 110 mg/m2. The main adverse reactions were: paresthesia (60%, grade 3–4: 7%), fever (40%, grade 3–4: 7%), and thrombocytopenia (40%, grade 3–4: 27%). No responses were observed.
In an open-label non-randomized study (DFI7434), oxaliplatin was administered to 26 pediatric patients with metastatic or unresectable solid tumors, mainly neuroblastoma and ganglioneuroblastoma. The DLT was sensory neuropathy at a dose of 160 mg/m2. No responses were observed.
In an open-label, single-agent study (ARD5021), oxaliplatin was administered to 43 pediatric patients with recurrent or refractory embryonal CNS tumors. The most common adverse reactions reported were: leukopenia (67%, grade 3–4: 12%), anemia (65%, grade 3–4: 5%), thrombocytopenia (65%, grade 3–4: 26%), vomiting (65%, grade 3–4: 7%), neutropenia (58%, grade 3–4: 16%), and sensory neuropathy (40%, grade 3–4: 5%).
In an open-label single-agent study (ARD5530), oxaliplatin was administered to 123 pediatric patients with recurrent solid tumors, including neuroblastoma, osteosarcoma, Ewing sarcoma or peripheral PNET, ependymoma, rhabdomyosarcoma, hepatoblastoma, high grade astrocytoma, brain stem glioma, low grade astrocytoma, malignant germ cell tumor and other tumors. The most common adverse reactions reported were: sensory neuropathy (52%, grade 3–4: 12%), thrombocytopenia (37%, grade 3–4: 17%), anemia (37%, grade 3–4: 9%), vomiting (26%, grade 3–4: 4%), increased ALT (24%, grade 3–4: 6%), increased AST (24%, grade 3–4: 2%), and nausea (23%, grade 3–4: 3%).
The pharmacokinetic parameters of ultrafiltrable platinum were evaluated in 105 pediatric patients during the first cycle. The mean clearance in pediatric patients estimated by the population pharmacokinetic analysis was 4.7 L/h (%CV, 41%). Mean platinum pharmacokinetic parameters in ultrafiltrate were Cmax of 0.75 ± 0.24 mcg/mL, AUC0–48h of 7.52 ± 5.07 mcg∙h/mL and AUCinf of 8.83 ± 1.57 mcg∙h/mL at 85 mg/m2 of oxaliplatin and Cmax of 1.10 ± 0.43 mcg/mL, AUC0–48h of 9.74 ± 2.52 mcg∙h/mL and AUCinf of 17.3 ± 5.34 mcg∙h/mL at 130 mg/m2 of oxaliplatin.
8.5 Geriatric Use
In the adjuvant treatment trial [see Clinical Studies (14.1)], 400 patients who received ELOXATIN with fluorouracil/leucovorin were greater than or equal to 65 years. The effect of ELOXATIN in patients greater than or equal to 65 years was not conclusive. Patients greater than or equal to 65 years receiving ELOXATIN experienced more diarrhea and grade 3–4 neutropenia (45% vs 39%) compared to patients less than 65 years.
In the previously untreated advanced colorectal cancer trial [see Clinical Studies (14.2)], 99 patients who received ELOXATIN with fluorouracil and leucovorin were greater than or equal to 65 years. The same efficacy improvements in response rate, time to tumor progression, and overall survival were observed in the greater than or equal to 65 years patients as in the overall study population. Adverse reactions were similar in patients less than 65 and greater than or equal to 65 years, but older patients may have been more susceptible to diarrhea, dehydration, hypokalemia, leukopenia, fatigue, and syncope.
In the previously treated advanced colorectal cancer trial [see Clinical Studies (14.3)], 55 patients who received ELOXATIN with fluorouracil and leucovorin were greater than or equal to 65 years. No overall differences in effectiveness were observed between these patients and younger adults. Adverse reactions were similar in patients less than 65 and greater than or equal to 65 years, but older patients may have been more susceptible to diarrhea, dehydration, hypokalemia, and fatigue.
No significant effect of age on the clearance of ultrafiltrable platinum has been observed [see Clinical Pharmacology (12.3)].
8.6 Patients with Renal Impairment
The AUC of unbound platinum in plasma ultrafiltrate was increased in patients with renal impairment [see Clinical Pharmacology (12.3)]. No dose reduction is recommended for patients with mild (creatinine clearance 50 to 79 mL/min) or moderate (creatinine clearance 30 to 49 mL/min) renal impairment, calculated by Cockcroft-Gault equation. Reduce the dose of ELOXATIN in patients with severe renal impairment (creatinine clearance less than 30 mL/min) [see Dosage and Administration (2.3)].
10 OVERDOSAGE
The maximum dose of oxaliplatin that has been administered in a single infusion is 825 mg. Several cases of overdoses have been reported with ELOXATIN. Adverse reactions observed following an overdosage were grade 4 thrombocytopenia (less than 25,000/mm3) without bleeding, anemia, sensory neuropathy (including paresthesia, dysesthesia, laryngospasm and facial muscle spasms), gastrointestinal disorders (including nausea, vomiting, stomatitis, flatulence, abdomen enlarged and grade 4 intestinal obstruction), grade 4 dehydration, dyspnea, wheezing, chest pain, respiratory failure, severe bradycardia, and death.
Closely monitor patients suspected of receiving an overdose, including for the adverse reactions described above and administer appropriate supportive treatment.
11 DESCRIPTION
Oxaliplatin is a platinum-based drug with the molecular formula C8H14N2O4Pt and the chemical name of cis-[(1 R,2 R)-1,2-cyclohexanediamine-N,N′] [oxalato(2-)-O,O′] platinum. Oxaliplatin is an organoplatinum complex in which the platinum atom is complexed with 1,2-diaminocyclohexane (DACH) and with an oxalate ligand as a leaving group.
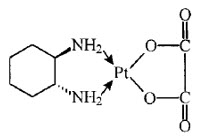
The molecular weight is 397.3. Oxaliplatin is slightly soluble in water at 6 mg/mL, very slightly soluble in methanol, and practically insoluble in ethanol and acetone.
ELOXATIN (oxaliplatin) injection, for intravenous use is supplied in vials containing 50 mg or 100 mg of oxaliplatin as a sterile, preservative-free, aqueous solution at a concentration of 5 mg/mL. Water for Injection, USP is present as an inactive ingredient.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Oxaliplatin undergoes nonenzymatic conversion in physiologic solutions to active derivatives via displacement of the labile oxalate ligand. Several transient reactive species are formed, including monoaquo and diaquo DACH platinum, which covalently bind with macromolecules. Both inter- and intrastrand Pt-DNA crosslinks are formed. Crosslinks are formed between the N7 positions of two adjacent guanines (GG), adjacent adenine-guanines (AG), and guanines separated by an intervening nucleotide (GNG). These crosslinks inhibit DNA replication and transcription. Cytotoxicity is cell-cycle nonspecific.
In vivo studies have shown antitumor activity of oxaliplatin against colon carcinoma. In combination with fluorouracil, oxaliplatin exhibits in vitro and in vivo antiproliferative activity greater than either compound alone in several tumor models (HT29 [colon], GR [mammary], and L1210 [leukemia]).
12.2 Pharmacodynamics
A pharmacodynamic relationship between platinum ultrafiltrate levels and clinical safety and effectiveness has not been established.
12.3 Pharmacokinetics
The reactive oxaliplatin derivatives are present as a fraction of the unbound platinum in plasma ultrafiltrate. After a single 2-hour intravenous infusion of ELOXATIN at a dose of 85 mg/m2, pharmacokinetic parameters expressed as ultrafiltrable platinum were Cmax of 0.814 mcg/mL and volume of distribution of 440 L.
Interpatient and intrapatient variability in ultrafiltrable platinum exposure (AUC0–48hr) assessed over 3 cycles was 23% and 6%, respectively.
Distribution
At the end of a 2-hour infusion of ELOXATIN, approximately 15% of the administered platinum is present in the systemic circulation. The remaining 85% is rapidly distributed into tissues or eliminated in the urine. The decline of ultrafiltrable platinum levels following ELOXATIN administration is triphasic, including two distribution phases (t1/2α; 0.43 hours and t1/2β; 16.8 hours).
In patients, plasma protein binding of platinum is irreversible and is greater than 90%. The main binding proteins are albumin and gamma-globulins.
Platinum also binds irreversibly and accumulates (approximately 2-fold) in erythrocytes, where it appears to have no relevant activity. No platinum accumulation was observed in plasma ultrafiltrate following 85 mg/m2 every two weeks.
Elimination
The decline of ultrafiltrable platinum concentrations from plasma is characterized by a long terminal elimination phase (t1/2γ; 391 hours).
Metabolism
Oxaliplatin undergoes rapid and extensive nonenzymatic biotransformation. There is no evidence of cytochrome P450-mediated metabolism in vitro.
Up to 17 platinum-containing derivatives have been observed in plasma ultrafiltrate samples from patients, including several cytotoxic species (monochloro DACH platinum, dichloro DACH platinum, and monoaquo and diaquo DACH platinum) and a number of noncytotoxic, conjugated species.
Excretion
The major route of platinum elimination is renal excretion. At five days after a single 2-hour infusion of ELOXATIN, urinary elimination accounted for about 54% of the platinum eliminated, with fecal excretion accounting for only about 2%. Platinum was cleared from plasma at a rate (10–17 L/h) that was similar to or exceeded the average human glomerular filtration rate (GFR; 7.5 L/h). The renal clearance of ultrafiltrable platinum is significantly correlated with GFR.
Special Populations
Patients with renal impairment
Patients with normal function (CLcr greater than 80 mL/min) and patients with mild (CLcr=50–80 mL/min) and moderate (CLcr equal to 30–49 mL/min) renal impairment received ELOXATIN 85 mg/m2 and those with severe (CLcr less than 30 mL/min) renal impairment received ELOXATIN 65 mg/m2. Mean dose adjusted AUC of unbound platinum was 40%, 95%, and 342% higher for patients with mild, moderate, and severe renal impairment, respectively, compared to patients with normal renal function. Mean dose adjusted Cmax of unbound platinum appeared to be similar among the normal, mild and moderate renal function groups, but was 38% higher in the severe group than in the normal group [see Dosage and Administration (2.3)].
Drug Interaction Studies
No pharmacokinetic interaction between ELOXATIN 85 mg/m2 and infusional fluorouracil has been observed in patients treated every 2 weeks, but increases of fluorouracil plasma concentrations by approximately 20% have been observed with doses of 130 mg/m2 of ELOXATIN administered every 3 weeks.
In vitro platinum was not displaced from plasma proteins by the following medications: erythromycin, salicylate, sodium valproate, granisetron, and paclitaxel.
In vitro oxaliplatin does not inhibit human cytochrome P450 isoenzymes.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term animal studies have not been performed to evaluate the carcinogenic potential of oxaliplatin. Oxaliplatin was not mutagenic to bacteria (Ames test) but was mutagenic to mammalian cells in vitro (L5178Y mouse lymphoma assay). Oxaliplatin was clastogenic both in vitro (chromosome aberration in human lymphocytes) and in vivo (mouse bone marrow micronucleus assay).
In a fertility study, male rats were given oxaliplatin at 0, 0.5, 1, or 2 mg/kg/day for five days every 21 days for a total of three cycles prior to mating with females that received two cycles of oxaliplatin on the same schedule. A dose of 2 mg/kg/day (less than one-seventh the recommended human dose on a body surface area basis) did not affect pregnancy rate, but resulted in 97% postimplantation loss (increased early resorptions, decreased live fetuses, decreased live births), and delayed growth (decreased fetal weight).
Testicular damage, characterized by degeneration, hypoplasia, and atrophy, was observed in dogs administered oxaliplatin at 0.75 mg/kg/day (approximately one-sixth of the recommended human dose on a body surface area basis) × 5 days every 28 days for three cycles. A no effect level was not identified.
14 CLINICAL STUDIES
14.1 Adjuvant Treatment with ELOXATIN in Combination with Fluorouracil and Leucovorin
The efficacy of ELOXATIN in combination with fluorouracil (FU)/leucovorin (LV) was evaluated in an international, multicenter, randomized (1:1) trial (The Multicenter International Study of Oxaliplatin/5-Fluorouracil/Leucovorin in the Adjuvant Treatment of Colon Cancer [MOSAIC], NCT00275210) in patients with stage II (Dukes' B2) or III (Dukes' C) colon cancer who had undergone complete resection of the primary tumor. Patients were randomized to receive ELOXATIN with fluorouracil/leucovorin or fluorouracil/leucovorin alone for a total of 6 months (i.e., 12 cycles). Table 14 shows the dosing regimens for the two arms.
Eligible patients were between 18 and 75 years of age, had histologically proven stage II (T3–T4 N0 M0; Dukes' B2) or III (any T N1–2 M0; Dukes' C) colon carcinoma (with the inferior pole of the tumor above the peritoneal reflection, i.e., greater than or equal to 15 cm from the anal margin) and had undergone (within 7 weeks prior to randomization) complete resection of the primary tumor without gross or microscopic evidence of residual disease and carcino-embyrogenic antigen (CEA) less than 10 ng/mL. Additional eligibility criteria were no prior chemotherapy, immunotherapy or radiotherapy; Eastern Cooperative Oncology Group performance status of 0, 1, or 2 (Karnofsky Performance Status greater than or equal to 60%); no pre-existing neuropathy; and absolute neutrophil count (ANC) greater than or equal to 1.5 × 109/L, platelets greater than or equal to 100 × 109/L, serum creatinine less than or equal to 1.25 × upper limit normal (ULN), total bilirubin less than 2 × ULN, and aspartate transaminase (AST)/alanine transaminase (ALT) less than 2 × ULN. The major efficacy outcome was 3-year disease-free survival (DFS).
| Treatment Arm | Dose | Regimen |
|---|---|---|
| ELOXATIN + FU/LV (FOLFOX4) (N=1123) | Day 1: ELOXATIN: 85 mg/m2 (2-hour infusion) + LV: 200 mg/m2 (2-hour infusion), followed by FU: 400 mg/m2 (bolus), 600 mg/m2 (22-hour infusion) Day 2: LV: 200 mg/m2 (2-hour infusion), followed by FU: 400 mg/m2 (bolus), 600 mg/m2 (22-hour infusion) | every 2 weeks 12 cycles |
| FU/LV (N=1123) | Day 1: LV: 200 mg/m2 (2-hour infusion), followed by FU: 400 mg/m2 (bolus), 600 mg/m2 (22-hour infusion) Day 2: LV: 200 mg/m2 (2-hour infusion), followed by FU: 400 mg/m2 (bolus), 600 mg/m2 (22-hour infusion) | every 2 weeks 12 cycles |
There were 2246 patients enrolled, of whom 1347 (60%) had Stage III disease. Tables 15 and 16 show the baseline characteristics and exposure to ELOXATIN.
| ELOXATIN + Infusional FU/LV N=1123 | Infusional FU/LV N=1123 |
|
|---|---|---|
| Sex: Male (%) | 56.1 | 52.4 |
| Female (%) | 43.9 | 47.6 |
| Median age (years) | 61.0 | 60.0 |
| <65 years of age (%) | 64.4 | 66.2 |
| ≥65 years of age (%) | 35.6 | 33.8 |
| KPS (%) | ||
| 100 | 29.7 | 30.5 |
| 90 | 52.2 | 53.9 |
| 80 | 4.4 | 3.3 |
| 70 | 13.2 | 11.9 |
| ≤60 | 0.6 | 0.4 |
| Primary site (%) | ||
| Colon including cecum | 54.6 | 54.4 |
| Sigmoid | 31.9 | 33.8 |
| Recto sigmoid | 12.9 | 10.9 |
| Other including rectum | 0.6 | 0.9 |
| Bowel obstruction (%) | ||
| Yes | 17.9 | 19.3 |
| Perforation (%) | ||
| Yes | 6.9 | 6.9 |
| Stage at Randomization (%) | ||
| II (T=3,4 N=0, M=0) | 40.1 | 39.9 |
| III (T=any, N=1,2, M=0) | 59.6 | 59.3 |
| IV (T=any, N=any, M=1) | 0.4 | 0.8 |
| Staging – T (%) | ||
| T1 | 0.5 | 0.7 |
| T2 | 4.5 | 4.8 |
| T3 | 76.0 | 75.9 |
| T4 | 19.0 | 18.5 |
| Staging – N (%) | ||
| N0 | 40.2 | 39.9 |
| N1 | 39.4 | 39.4 |
| N2 | 20.4 | 20.7 |
| Staging – M (%) | ||
| M1 | 0.4 | 0.8 |
| ELOXATIN + Infusional FU/LV N=1108 | Infusional FU/LV N=1111 |
|
|---|---|---|
| Median Relative Dose Intensity (%) | ||
| FU | 84.4 | 97.7 |
| ELOXATIN | 80.5 | N/A |
| Median Number of Cycles | 12 | 12 |
| Median Number of Cycles with ELOXATIN | 11 | N/A |
The median duration of follow-up was approximately 77 months. In the overall and the stage III colon cancer populations, DFS was statistically significantly improved in the ELOXATIN-containing arm compared to fluorouracil/leucovorin alone; however, a statistically significant improvement in DFS was not observed in Stage II patients. No significant differences in overall survival (OS) were detected in the overall population or those with Stage III disease. Table 17 and Figures 1 and 2 summarize the 5-year DFS rates in the overall randomized population and in patients with stage II and III disease based on an intention-to-treat (ITT) analysis.
| Parameter | ELOXATIN + Infusional FU/LV | Infusional FU/LV |
|---|---|---|
| A hazard ratio of less than 1 favors ELOXATIN + Infusional FU/LV Data cut off for disease-free survival June 1, 2006 |
||
| Overall | ||
| Number of patients | 1123 | 1123 |
| Number of events – relapse or death (%) | 304 (27.1) | 360 (32.1) |
| 5-yr Disease-free survival % (95% CI) | 73.3 (70.7, 76.0) | 67.4 (64.6, 70.2) |
| Hazard ratio (95% CI) | 0.80 (0.68, 0.93) | |
| Stratified Log rank test | p=0.003 | |
| Stage III (Dukes' C) | ||
| Number of patients | 672 | 675 |
| Number of events – relapse or death (%) | 226 (33.6) | 271 (40.1) |
| 5-yr Disease-free survival % (95% CI) | 66.4 (62.7, 70.0) | 58.9 (55.2, 62.7) |
| Hazard ratio (95% CI) | 0.78 (0.65, 0.93) | |
| Log rank test | p=0.005 | |
| Stage II (Dukes' B2) | ||
| Number of patients | 451 | 448 |
| Number of events – relapse or death (%) | 78 (17.3) | 89 (19.9) |
| 5-yr Disease-free survival % (95% CI) | 83.7 (80.2, 87.1) | 79.9 (76.2, 83.7) |
| Hazard ratio (95% CI) | 0.84 (0.62, 1.14) | |
| Log rank test | p=0.258 | |
Figure 1: Kaplan-Meier Curves of Disease-Free Survival (cutoff: 1 June 2006) in Adjuvant Treatment Trial – ITT Population
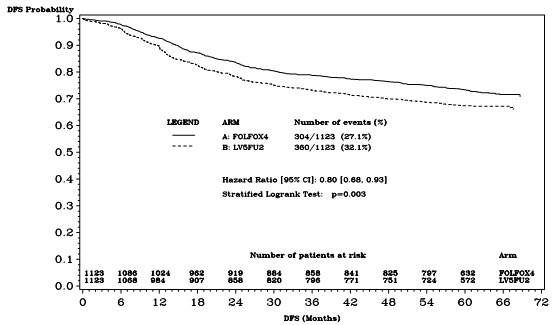
Figure 2: Kaplan-Meier Curves of Disease-Free Survival in Stage III Patients (cutoff: 1 June 2006) in Adjuvant Treatment Trial – ITT Population
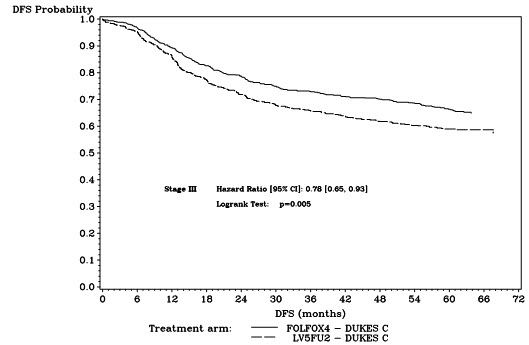
Table 18 summarizes the OS results in the overall randomized population and in patients with stage II and III disease, based on the ITT analysis.
| Parameter | ELOXATIN + Infusional FU/LV | Infusional FU/LV |
|---|---|---|
| A hazard ratio of less than 1 favors ELOXATIN + Infusional FU/LV Data cut off for overall survival January 16, 2007 |
||
| Overall | ||
| Number of patients | 1123 | 1123 |
| Number of death events (%) | 245 (21.8) | 283 (25.2) |
| Hazard ratio (95% CI) | 0.84 (0.71 , 1.00) | |
| Stage III (Dukes' C) | ||
| Number of patients | 672 | 675 |
| Number of death events (%) | 182 (27.1) | 220 (32.6) |
| Hazard ratio (95% CI) | 0.80 (0.65 , 0.97) | |
| Stage II (Dukes' B2) | ||
| Number of patients | 451 | 448 |
| Number of death events (%) | 63 (14.0) | 63 (14.1) |
| Hazard ratio (95% CI) | 1.00 (0.70, 1.41) | |
14.2 Previously Untreated Advanced Colorectal Cancer
The efficacy of ELOXATIN in combination with fluorouracil (FU)/leucovorin (LV) was evaluated in a North American, multicenter, open-label, randomized, active-controlled trial (A Randomized Phase III Trial of Three Different Regimens of CPT-11 Plus 5-Fluorouracil and Leucovorin Compared to 5-Fluorouracil and Leucovorin in Patients with Advanced Adenocarcinoma of the Colon and Rectum; NCT00003594). The trial included 7 arms at different times during its conduct, four of which were closed due to either changes in the standard of care, toxicity, or simplification. During the trial, the control arm was changed to irinotecan with fluorouracil/leucovorin.
The results reported below compared the efficacy of ELOXATIN with fluorouracil/leucovorin and ELOXATIN with irinotecan to an approved control regimen of irinotecan with fluorouracil/leucovorin in 795 concurrently randomized patients previously untreated for locally advanced or metastatic colorectal cancer. Table 19 presents the dosing regimens for the three arms. After completion of enrollment, the dose of irinotecan with fluorouracil/leucovorin was decreased due to toxicity.
Eligible patients were at least 18 years of age; had known locally advanced, locally recurrent, or metastatic colorectal adenocarcinoma not curable by surgery or amenable to radiation therapy; with an Eastern Cooperative Oncology Group (ECOG) performance status ≤0, 1, or 2. Patients had to have absolute neutrophil count (ANC) greater than or equal to 1.5 × 109/L, platelets greater than or equal to 100 × 109/L, hemoglobin greater than or equal to 9.0 g/dL, creatinine less than or equal to 1.5 × upper limit of normal (ULN), total bilirubin less than or equal to 1.5 mg/dL, aspartate transaminase (AST) less than or equal to 5 × ULN, and alkaline phosphatase less than or equal to 5 × ULN. Patients may have received adjuvant treatment for resected Stage II or III disease without recurrence within 12 months. Randomization was stratified by ECOG performance status (0, 1 vs 2), prior adjuvant chemotherapy (yes vs no), prior immunotherapy (yes vs no), and age (less than 65 vs greater than or equal to 65 years). Although no post study treatment was specified in the protocol, 65% to 72% of patients received additional post study chemotherapy after study treatment discontinuation on all arms. Fifty-eight percent of patients on the ELOXATIN with fluorouracil/leucovorin arm received an irinotecan-containing regimen and 23% of patients on the irinotecan with fluorouracil/leucovorin arm received an oxaliplatin-containing regimen. The main efficacy outcome measure was 3-year disease-free survival (DFS) and additional efficacy outcome measures were overall survival (OS).
| Treatment Arm | Dose | Regimen |
|---|---|---|
| ELOXATIN + FU/LV (FOLFOX4) (N=267) | Day 1: ELOXATIN: 85 mg/m2 (2-hour infusion) + LV 200 mg/m2 (2-hour infusion), followed by FU: 400 mg/m2 (bolus), 600 mg/m2 (22-hour infusion) Day 2: LV 200 mg/m2 (2-hour infusion), followed by FU: 400 mg/m2 (bolus), 600 mg/m2 (22-hour infusion) | every 2 weeks |
| Irinotecan + FU/LV (IFL) (N=264) | Day 1: irinotecan 125 mg/m2 as a 90-min infusion + LV 20 mg/m2 as a 15-min infusion or intravenous push, followed by FU 500 mg/m2 intravenous bolus weekly × 4 | every 6 weeks |
| ELOXATIN + Irinotecan (IROX) (N=264) | Day 1: ELOXATIN: 85 mg/m2 intravenous (2-hour infusion) + irinotecan 200 mg/m2 intravenous over 30 minutes | every 3 weeks |
Table 20 presents the baseline characteristics.
| ELOXATIN + FU/LV N=267 | Irinotecan + FU/LV N=264 | ELOXATIN + Irinotecan N=264 |
|
|---|---|---|---|
| Sex: Male (%) | 58.8 | 65.2 | 61.0 |
| Female (%) | 41.2 | 34.8 | 39.0 |
| Median age (years) | 61.0 | 61.0 | 61.0 |
| <65 years of age (%) | 61 | 62 | 63 |
| ≥65 years of age (%) | 39 | 38 | 37 |
| ECOG (%) | |||
| 0–1 | 94.4 | 95.5 | 94.7 |
| 2 | 5.6 | 4.5 | 5.3 |
| Involved organs (%) | |||
| Colon only | 0.7 | 0.8 | 0.4 |
| Liver only | 39.3 | 44.3 | 39.0 |
| Liver + other | 41.2 | 38.6 | 40.9 |
| Lung only | 6.4 | 3.8 | 5.3 |
| Other (including lymph nodes) | 11.6 | 11.0 | 12.9 |
| Not reported | 0.7 | 1.5 | 1.5 |
| Prior radiation (%) | 3.0 | 1.5 | 3.0 |
| Prior surgery (%) | 74.5 | 79.2 | 81.8 |
| Prior adjuvant (%) | 15.7 | 14.8 | 15.2 |
The median number of cycles administered per patient was 10 (23.9 weeks) for the ELOXATIN plus fluorouracil/leucovorin regimen, 4 (23.6 weeks) for the irinotecan plus fluorouracil/leucovorin regimen, and 7 (21.0 weeks) for the ELOXATIN plus irinotecan regimen.
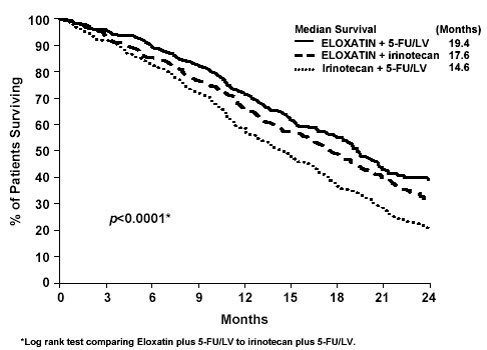
Patients who received ELOXATIN with fluorouracil/leucovorin had a significantly longer time to tumor progression based on investigator assessment, longer OS, and a significantly higher confirmed response rate based on investigator assessment compared to patients who received irinotecan with fluorouracil/leucovorin. Efficacy results are summarized in Table 21 and Figure 3.
| ELOXATIN + FU/LV N=267 | Irinotecan + FU/LV N=264 | ELOXATIN + Irinotecan N=264 |
|
|---|---|---|---|
| The numbers in the response rate and TTP analysis are based on unblinded investigator assessment. | |||
| Survival (ITT) | |||
| Number of deaths (%) | 155 (58.1) | 192 (72.7) | 175 (66.3) |
| Median survival (months) | 19.4 | 14.6 | 17.6 |
| Hazard ratio (95% CI)* | 0.65 (0.53, 0.80)† | - | |
| P-value | <0.0001† | - | |
| TTP (ITT, investigator assessment) | |||
| Percentage of progressors | 82.8 | 81.8 | 89.4 |
| Median TTP (months) | 8.7 | 6.9 | 6.5 |
| Hazard ratio (95% CI)* | 0.74 (0.61, 0.89)† | - | |
| P-value | 0.0014† | - | |
| Response Rate (investigator assessment)‡ | |||
| Patients with measurable disease | 210 | 212 | 215 |
| Complete response, N (%) | 13 (6.2) | 5 (2.4) | 7 (3.3) |
| Partial response, N (%) | 82 (39.0) | 64 (30.2) | 67 (31.2) |
| Complete and partial response, N (%) | 95 (45.2) | 69 (32.5) | 74 (34.4) |
| 95% CI | (38.5, 52.0) | (26.2, 38.9) | (28.1, 40.8) |
| P-value | 0.0080† | - | |
Figure 3: Kaplan-Meier Curves for Overall Survival in Previously Untreated Advanced Colorectal Cancer Trial
In descriptive subgroup analyses, the improvement in overall survival (OS) for ELOXATIN with fluorouracil/leucovorin compared to irinotecan with fluorouracil/leucovorin appeared to be maintained across age groups, prior adjuvant treatment, number of organs involved and both sexes; however, the effect appeared larger among women than men.
14.3 Previously Treated Advanced Colorectal Cancer
The efficacy of ELOXATIN in combination with fluorouracil (FU)/leucovorin (LV) was evaluated in a multicenter, open-label, randomized, three-arm controlled trial was conducted in the US and Canada in patients with advanced colorectal cancer who had relapsed/progressed during or within 6 months of first-line treatment with bolus fluorouracil/leucovorin and irinotecan (A multicenter, open-label, randomized, three-arm study of 5-fluorouracil (5-FU) + leucovorin (LV) or oxaliplatin or a combination of 5-FU/LV + oxaliplatin as second-line treatment of metastatic colorectal carcinoma: NCT00008281). Patients were randomized to one of three regimens; the dosing regimens are presented in Table 22. Eligible patients were at least 18 years of age, had unresectable, measurable, histologically proven colorectal adenocarcinoma, with a Karnofsky performance status (KPS) greater than 50%. Patients had to have aspartate transaminase (AST), alanine transaminase (ALT) and alkaline phosphatase less than or equal to 2× upper limit of normal (ULN), unless liver metastases were present and documented at baseline by CT or MRI scan, in which case less than or equal to 5× ULN was permitted. Prior radiotherapy was permitted if it had been completed at least 3 weeks before randomization. The main efficacy outcome measure was 3-year disease-free survival (DFS) and an additional outcome measure was overall survival (OS).
| Treatment Arm | Dose | Regimen |
|---|---|---|
| ELOXATIN + FU/LV (N=152) | Day 1: ELOXATIN: 85 mg/m2 (2-hour infusion) + LV 200 mg/m2 (2-hour infusion), followed by FU: 400 mg/m2 (bolus), 600 mg/m2 (22-hour infusion) Day 2: LV 200 mg/m2 (2-hour infusion), followed by FU: 400 mg/m2 (bolus), 600 mg/m2 (22-hour infusion) | every 2 weeks |
| FU/LV (N=151) | Day 1: LV 200 mg/m2 (2-hour infusion), followed by FU: 400 mg/m2 (bolus), 600 mg/m2 (22-hour infusion) Day 2: LV 200 mg/m2 (2-hour infusion), followed by FU: 400 mg/m2 (bolus), 600 mg/m2 (22-hour infusion) | every 2 weeks |
| ELOXATIN (N=156) | Day 1: ELOXATIN 85 mg/m2 (2-hour infusion) | every 2 weeks |
Patients must have had at least one unidimensional lesion measuring greater than or equal to 20 mm using conventional CT or MRI scans or greater than or equal to 10 mm using a spiral CT scan. Tumor response and progression were assessed every 3 cycles (6 weeks) using the Response Evaluation Criteria in Solid Tumors (RECIST) until radiological documentation of progression or for 13 months following the first dose of study drug(s), whichever came first. Confirmed responses were based on two tumor assessments separated by at least 4 weeks. Baseline characteristics are shown in Table 23.
| ELOXATIN + FU/LV N=152 | ELOXATIN N=156 | FU/LV N=151 |
|
|---|---|---|---|
| Sex: Male (%) | 57.2 | 60.9 | 54.3 |
| Female (%) | 42.8 | 39.1 | 45.7 |
| Median age (years) | 59.0 | 61.0 | 60.0 |
| Range | 22–88 | 27–79 | 21–80 |
| Race (%) | |||
| Caucasian | 88.8 | 84.6 | 87.4 |
| Black | 5.9 | 7.1 | 7.9 |
| Asian | 2.6 | 2.6 | 1.3 |
| Other | 2.6 | 5.8 | 3.3 |
| KPS (%) | |||
| 70–100 | 95.4 | 92.3 | 94.7 |
| 50–60 | 2.0 | 4.5 | 2.6 |
| Not reported | 2.6 | 3.2 | 2.6 |
| Prior radiotherapy (%) | 25.0 | 19.2 | 25.2 |
| Prior pelvic radiation (%) | 21.1 | 13.5 | 18.5 |
| Number of metastatic sites (%) | |||
| 1 | 25.7 | 31.4 | 27.2 |
| ≥2 | 74.3 | 67.9 | 72.2 |
| Liver involvement (%) | |||
| Liver only | 18.4 | 25.6 | 22.5 |
| Liver + other | 53.3 | 59.0 | 60.3 |
The median number of cycles administered per patient was 6 for the ELOXATIN and fluorouracil/leucovorin combination and 3 each for fluorouracil/leucovorin alone and ELOXATIN alone. Patients treated with the combination of ELOXATIN and fluorouracil/leucovorin had an increased response rate compared to patients given fluorouracil/leucovorin or oxaliplatin alone. Efficacy results are summarized in Tables 24 and 25.
| Best Response | ELOXATIN + FU/LV N=152 | ELOXATIN N=156 | FU/LV N=151 |
|---|---|---|---|
| Complete Response | 0 | 0 | 0 |
| Partial Response | 13 (9%) | 2 (1%) | 0 |
| P-value | 0.0002 FU/LV vs ELOXATIN + FU/LV | ||
| 95% CI | 4.6%, 14.2% | 0.2%, 4.6% | 0, 2.4% |
| Arm | ELOXATIN + FU/LV N=152 | ELOXATIN N=156 | FU/LV N=151 |
|---|---|---|---|
|
|||
| Number of progressors | 50 | 101 | 74 |
| Number of patients with no radiological evaluation beyond baseline | 17 (11%) | 16 (10%) | 22 (15%) |
| Median TTP (months) | 4.6 | 1.6 | 2.7 |
| 95% CI | 4.2, 6.1 | 1.4, 2.7 | 1.8, 3.0 |
At the time of the interim analysis 49% of the radiographic progression events had occurred. In this interim analysis an estimated 2-month increase in median time to radiographic progression was observed compared to fluorouracil/leucovorin alone.
16 HOW SUPPLIED/STORAGE AND HANDLING
ELOXATIN (oxaliplatin) injection is supplied in clear, glass, single-dose vials with gray elastomeric stoppers and aluminum flip-off seals containing 50 mg or 100 mg of oxaliplatin as a clear, colorless, sterile, preservative-free, aqueous solution at a concentration of 5 mg/mL.
NDC 0024-0590-10: 50 mg single-dose vial with green flip-off seal individually packaged in a carton.
NDC 0024-0591-20: 100 mg single-dose vial with dark blue flip-off seal individually packaged in a carton.
Store at 25°C (77°F); excursions permitted to 15°C to 30°C (59°F to 86°F). Do not freeze and protect from light (keep in original outer carton). Discard unused portion.
ELOXATIN is a cytotoxic drug. Follow applicable special handling and disposal procedures.1 The use of gloves is recommended. If a solution of ELOXATIN contacts the skin, wash the skin immediately and thoroughly with soap and water. If ELOXATIN contacts the mucous membranes, flush thoroughly with water.
17 PATIENT COUNSELING INFORMATION
Hypersensitivity Reactions
Advise patients of the potential risk of hypersensitivity and that ELOXATIN is contraindicated in patients with a history of hypersensitivity reactions to oxaliplatin and other platinum-based drugs. Instruct patients to seek immediate medical attention for signs of severe hypersensitivity reaction such as chest tightness; shortness of breath; wheezing; dizziness or faintness; or swelling of the face, eyelids, or lips [see Warnings and Precautions (5.1)].
Peripheral Sensory Neuropathy
Advise patients of the risk of acute reversible or persistent-type neurosensory toxicity. Advise patients to avoid cold drinks, use of ice, and exposure of skin to cold temperature or cold objects [see Warnings and Precautions (5.2)].
Myelosuppression
Inform patients that ELOXATIN can cause low blood cell counts and the need for frequent monitoring of blood cell counts. Advise patients to contact their healthcare provider immediately for bleeding, fever, particularly if associated with persistent diarrhea, or symptoms of infection develop [see Warnings and Precautions (5.3)].
Posterior Reversible Encephalopathy Syndrome
Advise patients of the potential effects of vision abnormalities, in particular transient vision loss (reversible following therapy discontinuation), which may affect the patients' ability to drive and use machines [see Warnings and Precautions (5.4)].
Pulmonary Toxicity
Advise patients to report immediately to their healthcare provider any persistent or recurrent respiratory symptoms, such as non-productive cough and dyspnea [see Warnings and Precautions (5.5)].
Hepatotoxicity
Advise patients to report signs or symptoms of hepatic toxicity to their healthcare provider [see Warnings and Precautions (5.6)].
QT Interval Prolongation
Advise patients that ELOXATIN can cause QTc interval prolongation and to inform their physician if they have any symptoms, such as syncope [see Warnings and Precautions (5.7)].
Rhabdomyolysis
Advise patients to contact their healthcare provider immediately for new or worsening signs or symptoms of muscle toxicity, dark urine, decreased urine output, or the inability to urinate [see Warnings and Precautions (5.8)].
Hemorrhage
Advise patients that ELOXATIN may increase the risk of bleeding and to promptly inform their healthcare provider of any bleeding episodes [see Warnings and Precautions (5.9)].
Embryo-Fetal Toxicity
Advise females of reproductive potential of the potential risk to a fetus. Advise females to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.10), Use in Specific Populations (8.1)].
Advise females of reproductive potential to use effective contraception during treatment with ELOXATIN and for 9 months after the final dose [see Use in Specific Populations (8.3)].
Advise male patients with female partners of reproductive potential to use effective contraception during treatment with ELOXATIN and for 6 months after the final dose [see Use in Specific Populations (8.3), Nonclinical Toxicology (13.1)].
Lactation
Advise women not to breastfeed during treatment with ELOXATIN and for 3 months after the final dose [see Use in Specific Populations (8.2)].
Infertility
Advise females and males of reproductive potential that ELOXATIN may impair fertility [see Use in Specific Populations (8.3), Nonclinical Toxicology (13.1)].
Gastrointestinal
Advise patients to contact their healthcare provider for persistent vomiting, diarrhea, or signs of dehydration [see Adverse Reactions (6.1)].
Drug Interactions
Inform patients about the risk of drug interactions and the importance of providing a list of prescription and nonprescription drugs to their healthcare provider [see Drug Interactions (7)].
| This Patient Information has been approved by the U.S. Food and Drug Administration. | Revised: June 2023 | ||
| Patient Information ELOXATIN® (eh-LOX-ah-tin) (oxaliplatin) injection, for intravenous use |
|||
| What is the most important information I should know about ELOXATIN? ELOXATIN can cause serious allergic reactions, including allergic reactions that can lead to death. ELOXATIN is a platinum-based medicine. Serious allergic reactions including death can happen in people who take ELOXATIN and who have had previous allergic reactions to platinum-based medicines. Serious allergic reactions can happen within a few minutes of your ELOXATIN infusion or any time during your treatment with ELOXATIN. Get emergency help right away if you:
|
|||
|
|
||
| See "What are the possible side effects of ELOXATIN?" for information about other serious side effects. | |||
| What is ELOXATIN?
ELOXATIN is an anti-cancer (chemotherapy) medicine that is used with other anti-cancer medicines called fluorouracil and leucovorin to treat people with:
|
|||
| Do not receive ELOXATIN if you are allergic to oxaliplatin or any of the ingredients in ELOXATIN or if you are allergic to other platinum-based medicines. See the end of this leaflet for a complete list of the ingredients in ELOXATIN. Ask your doctor if you are not sure if you have taken a platinum-based medicine. |
|||
Before receiving ELOXATIN, tell your doctor about all of your medical conditions, including if you:
Know the medicines you take. Keep a list of them and show it to your doctor and pharmacist when you get a new medicine. |
|||
How will I receive ELOXATIN?
You will not get ELOXATIN on Day 2. Leucovorin and fluorouracil will be given the same way as on Day 1. The fluorouracil will be given through your IV with a pump. If you have any problems with the pump or the tube, call your doctor, your nurse, or the person who is responsible for your pump. Do not let anyone other than a healthcare provider touch your infusion pump or tubing. |
|||
What should I avoid while receiving ELOXATIN?
Talk with your doctor and nurse about your level of activity during treatment with ELOXATIN. Follow their instructions. |
|||
| What are the possible side effects of ELOXATIN? ELOXATIN can cause serious side effects, including:
|
|||
|
|
||
|
|||
|
|
||
|
|||
|
|
||
| The most common side effects of ELOXATIN include: | |||
|
|
||
| ELOXATIN may cause fertility problems in males and females. Talk to your doctor if this is a concern for you. Tell your doctor if you have any side effect that bothers you or that does not go away. These are not all the possible side effects of ELOXATIN. For more information, ask your doctor or pharmacist. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|||
How can I reduce the side effects caused by cold temperatures?
|
|||
| General information about the safe and effective use of ELOXATIN.
Medicines are sometimes prescribed for purposes other than those listed in the Patient Information leaflet. You can ask your doctor or pharmacist for information about ELOXATIN that is written for health professionals. |
|||
| What are the ingredients in ELOXATIN?
Active ingredient: oxaliplatin Inactive ingredient: water for injection Manufactured by: sanofi-aventis U.S. LLC, Bridgewater, NJ 08807, A SANOFI COMPANY ©2023 sanofi-aventis U.S. LLC |
|||
PRINCIPAL DISPLAY PANEL - 50 mg Carton
NDC 0024-0590-10
50 mg
Eloxatin®
(OXALIplatin injection) 5 mg/mL
INJECTION
50 mg
FOR INTRAVENOUS USE ONLY
SINGLE USE VIAL ONLY
Sterile Aqueous Solution - Preservative Free
See package insert for further
required dilution.
DO NOT MIX OR ADD TO
SODIUM CHLORIDE/
CHLORIDE-CONTAINING
SOLUTIONS
Rx only
SANOFI
PRINCIPAL DISPLAY PANEL - 100 mg Carton
NDC 0024-0591-20
100 mg
Eloxatin®
(OXALIplatin injection) 5 mg/mL
INJECTION
100 mg
FOR INTRAVENOUS USE ONLY
SINGLE USE VIAL ONLY
Sterile Aqueous Solution - Preservative Free
See package insert for further
required dilution.
DO NOT MIX OR ADD TO
SODIUM CHLORIDE/
CHLORIDE-CONTAINING
SOLUTIONS
Rx only
SANOFI
| ELOXATIN
oxaliplatin injection, solution, concentrate |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| ELOXATIN
oxaliplatin injection, solution, concentrate |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - sanofi-aventis U.S. LLC (824676584) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| sanofi-aventis Deutschland GmbH | 313218430 | MANUFACTURE(0024-0590, 0024-0591) , ANALYSIS(0024-0590, 0024-0591) , PACK(0024-0590, 0024-0591) , LABEL(0024-0590, 0024-0591) | |