SKELID- tiludronate disodium tablet
sanofi-aventis U.S. LLC
----------
SKELID®
(tiludronate disodium)
DESCRIPTION
SKELID is a bisphosphonate characterized by a (4-chlorophenylthio) group on the carbon atom of the basic P-C-P structure common to all bisphosphonates. Its generic name is tiludronate disodium. Tiludronate disodium is the hydrated hemihydrate form of the disodium salt of tiludronic acid. Its chemical name is [[(4-Chlorophenyl) thio]methylene]bis[phosphonic acid], disodium salt, and its structural formula is as follows:

tiludronate disodium
(molecular weight 380.6)
SKELID tablets for oral administration contain 240 mg tiludronate disodium, which is the molar equivalent of 200 mg tiludronic acid. SKELID tablets also contain sodium lauryl sulfate, hydroxypropyl methylcellulose 2910, crospovidone, magnesium stearate, and lactose monohydrate.
CLINICAL PHARMACOLOGY
Mechanism of Action
In vitro studies indicate that tiludronate disodium acts primarily on bone through a mechanism that involves inhibition of osteoclastic activity with a probable reduction in the enzymatic and transport processes that lead to resorption of the mineralized matrix.
Bone resorption occurs following recruitment, activation, and polarization of osteoclasts. Tiludronate disodium appears to inhibit osteoclasts through at least two mechanisms: disruption of the cytoskeletal ring structure, possibly by inhibition of protein-tyrosine-phosphatase, thus leading to detachment of osteoclasts from the bone surface and the inhibition of the osteoclastic proton pump.
Pharmacokinetics
Absorption
Relative to an intravenous (IV) reference dose, the mean oral bioavailability of tiludronate disodium in healthy male subjects was 6% after an oral dose equivalent to 400 mg tiludronic acid administered after an overnight fast and 4 hours before a standard breakfast. In single-dose studies, bioavailability was reduced by 90% when an oral dose equivalent to 400 mg tiludronic acid was administered with, or 2 hours after, a standard breakfast compared to the same dose administered after an overnight fast and 4 hours before a standard breakfast. However, in clinical studies, efficacy was seen when SKELID was dosed at least 2 hours before or after meals.
After administration of a single dose equivalent to 400 mg tiludronic acid to healthy male subjects, tiludronic acid was rapidly absorbed with peak plasma concentrations of approximately 3 mg/L occurring within 2 hours. In pagetic patients, after repeated administration of doses equivalent to 400 mg/day tiludronic acid (2 hours before or 2 hours after a meal) for durations of 12 days to 12 weeks, average plasma concentrations of tiludronic acid occurring between 1 and 2 hours after dosing ranged between 1 and 4.6 mg/L.
Distribution
Animal pharmacology studies in rats demonstrate that tiludronic acid is widely distributed to bone and soft tissues. Over a period of days, loss of drug occurs from most tissues with the exception of bone and cartilage. Tiludronate is then slowly released from bone with a half-life in rats of 30 days or longer depending on the status of bone turnover.
After oral administration of doses equivalent to 400 mg/day tiludronic acid to nonpagetic patients with osteoarthrosis, the steady state in bone was not reached after 30 days of dosing. At plasma concentrations between 1 and 10 mg/L, tiludronic acid was approximately 90% bound to human serum protein (mainly albumin).
Metabolism
In laboratory animals, tiludronic acid undergoes little if any metabolism. In vitro, tiludronic acid is not metabolized in human liver microsomes and hepatocytes.
Elimination
The principal route of elimination of tiludronic acid is in the urine. After IV administration to healthy volunteers, approximately 60% of the dose was excreted in the urine as tiludronic acid within 13 days. Renal clearance is dose independent and is approximately 10 mL/min in healthy subjects. In pagetic patients treated with doses equivalent to 400 mg/day tiludronic acid for 12 days, the mean apparent plasma elimination half-life was approximately 150 hours. The elimination rate from human bone is unknown.
Special Populations
Geriatric
No dosage adjustment in elderly patients is necessary. Plasma concentrations of tiludronic acid were higher in elderly pagetic patients (≥65 years of age); however, this difference was not clinically significant.
Gender
There were no clinically significant differences in plasma concentrations after repeated administration of tiludronate disodium to male and female pagetic patients.
Renal Insufficiency
SKELID is not recommended for patients with severe renal failure (creatinine clearance <30 mL/min) due to lack of clinical experience. After a single oral dose equivalent to 400 mg tiludronic acid, subjects with creatinine clearance between 11 and 18 mL/min had Cmax values (approximately 3 mg/L) in the range of healthy volunteers. However, the plasma elimination half-life was approximately 205 hours, which is longer than that observed in pagetic patients after repeated doses (150 hours) and healthy subjects after single doses (50 hours). These values were obtained in a cross-study comparison between healthy volunteers and pagetic patients.
Hepatic Insufficiency
No dosage adjustment is needed. Since tiludronate undergoes little or no metabolism, no studies were conducted in subjects with hepatic insufficiency.
Drug-Drug Interactions
(See also PRECAUTIONS, Drug Interactions.) The bioavailability of SKELID is decreased 80% by calcium, when calcium and SKELID are administered at the same time, and 60% by some aluminum- or magnesium-containing antacids, when administered 1 hour before SKELID. Aspirin may decrease bioavailability of SKELID by up to 50% when taken 2 hours after SKELID. The bioavailability of SKELID is increased 2–4 fold by indomethacin and is not significantly altered by coadministration of diclofenac. The pharmacokinetic parameters of digoxin are not significantly modified by SKELID coadministration. In vitro studies show that tiludronate disodium does not displace warfarin from its binding site on protein.
| Summary of Pharmacokinetic Parameters in the Normal Population |
|
|---|---|
| Parameter | Mean (SD) |
|
|
| Absolute bioavailability of two 200-mg tablets taken 4 hrs before standard breakfast | 6% (2%)* |
| Time to peak plasma concentration (taken 4 hrs before first meal of day, n=151) | 1.5 (0.9) hr |
| Maximum plasma concentration after a single 400-mg dose (taken 4 hrs before first meal of day, n=151) | 2.66 (1.22) mg/L |
| Renal clearance after IV administration of 20-mg dose | 0.54 (0.14) L/hr |
Pharmacodynamics
Paget's disease of bone is a chronic, focal skeletal disorder characterized by greatly increased and disorderly bone remodeling. Excessive osteoclastic bone resorption is followed by osteoblastic new bone formation, leading to the replacement of the normal bone architecture by disorganized, enlarged, and weakened bone structure.
Clinical manifestations of Paget's disease range from no symptoms to severe bone pain, bone deformity, pathological fractures, and neurological and other complications. Serum alkaline phosphatase, the most frequently used biochemical index of disease activity, provides an objective measure of disease severity and response to therapy.
In pagetic patients treated with SKELID 400 mg/day for 3 months, changes in urinary hydroxyproline, a biochemical marker of bone resorption, and in serum alkaline phosphatase, a marker of bone formation, indicate a reduction toward normal in the rate of bone turnover. In addition, reduced numbers of osteoclasts by histomorphometric analysis and radiological improvement of lytic lesions indicate that SKELID can suppress the pagetic disease process.
Clinical Studies
The efficacy of SKELID 400 mg/day treatment was demonstrated in two randomized, double-blind, placebo-controlled multicenter studies and one positive-controlled study. All three studies included male and female patients with Paget's disease of the bone (radiograph examination and level of serum alkaline phosphatase [SAP] at least twice the upper normal limit). In one placebo-controlled study, conducted in North America, patients were randomly assigned to receive a daily dose of placebo or 200 or 400 mg/day SKELID for 3 months followed by an additional 12 weeks without treatment. A second placebo-controlled study of similar design was conducted in the UK.
A positive-controlled study was conducted in Europe with treatment groups of 400 mg/day SKELID for 3 months with a 3-month treatment-free follow-up, 400 mg/day SKELID for 6 months, and 400 mg/day etidronate for 6 months. In all of these studies, the efficacy of SKELID was primarily assessed by SAP activity after 3 and 6 months.
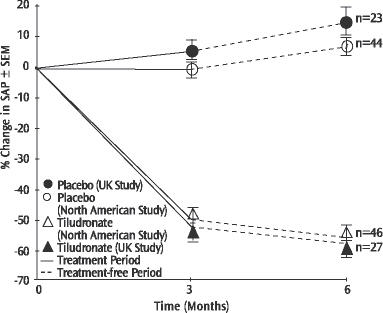
Figure 1
In the placebo-controlled trials, suppression of SAP levels was statistically significantly greater with 400 mg/day SKELID both at the end of treatment (3 months) and on follow-up (6 months) than with placebo (See Figure 1). The proportion of patients demonstrating at least a 50% reduction in SAP at 3 months with 400 mg/day SKELID was 61% in the North American study and 52% in the UK study.
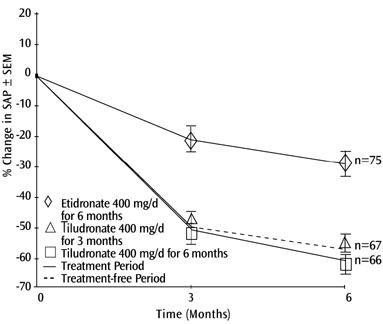
Figure 2
In the positive-controlled trial, six months after the start of dosing, the decrease in SAP levels in patients who ceased dosing after a 3-month course of SKELID was significantly greater than with 6 months of etidronate 400 mg/day, and was equivalent to levels in patients who completed a 6-month course of SKELID (See Figure 2).
Treatment effects of SKELID were similar, regardless of pagetic patients' baseline SAP level, gender or age in the population studied.
Histomorphometry of the bone was studied in 19 pagetic and 29 nonpagetic patients. Bone biopsy results in nonpagetic bone confirmed that SKELID did not impair bone remodeling or induce a significant decline in bone turnover. Results obtained in pagetic and nonpagetic bone indicated no evidence of osteomalacia or accumulation of unmineralized osteoid, and there was no reduction in the mineralization rate.
INDICATIONS AND USAGE
SKELID is indicated for treatment of Paget's disease of bone (osteitis deformans).
Treatment is indicated in patients with Paget's disease of bone (1) who have a level of serum alkaline phosphatase (SAP) at least twice the upper limit of normal, or (2) who are symptomatic, or (3) who are at risk for future complications of their disease.
CONTRAINDICATIONS
SKELID is contraindicated in individuals with known hypersensitivity to any component of this product.
Inability to stand or sit upright for at least 30 minutes.
WARNINGS
SKELID, like other bisphosphonates administered orally, may cause local irritation of the upper gastrointestinal mucosa. Because of these possible irritant effects and a potential for worsening of the underlying disease, caution should be used when SKELID is given to patients with active upper gastrointestinal problems (such as known Barrett's esophagus, dysphagia, other esophageal diseases, gastritis, duodenitis, or ulcers).
Esophageal adverse experiences, such as esophagitis, esophageal ulcers and esophageal erosions, occasionally with bleeding and rarely followed by esophageal stricture or perforation, have been reported in patients receiving treatment with oral bisphosphonates. In some cases, these have been severe and required hospitalization. Physicians should therefore be alert to any signs or symptoms signaling a possible esophageal reaction and patients should be instructed to discontinue SKELID and seek medical attention if they develop dysphagia, odynophagia, retrosternal pain or new or worsening heartburn.
The risk of severe esophageal adverse experiences appears to be greater in patients who lie down after taking oral bisphosphonates and/or who fail to swallow it with the recommended full glass (6–8oz) of water, and/or who continue to take oral bisphosphonates after developing symptoms suggestive of esophageal irritation. Therefore, it is very important that the full dosing instructions are provided to, and understood by, the patient (see DOSAGE AND ADMINISTRATION). In patients who cannot comply with dosing instructions due to mental or physical disability, therapy with SKELID should be used under appropriate supervision.
There have been post-marketing reports of gastric and duodenal ulcers with oral bisphosphonate use, some severe and with complications, although no increased risk was observed in controlled clinical trials.
PRECAUTIONS
General
SKELID is not recommended for patients with severe renal failure, for example, those with creatinine clearance <30 mL/min (see CLINICAL PHARMACOLOGY, Renal Insufficiency).
Osteonecrosis, primarily in the jaw, has been reported in patients treated with bisphosphonates. Most cases have been in cancer patients undergoing dental procedures, but some have occurred in patients with postmenopausal osteoporosis or other diagnoses. Known risk factors for osteonecrosis include a diagnosis of cancer, concomitant therapies (e.g., chemotherapy, radiotherapy, corticosteroids), and co-morbid disorders (e.g., anemia, coagulopathy, infection, pre-existing dental disease). Most reported cases have been in patients treated with bisphosphonates intravenously but some have been in patients treated orally.
For patients who develop osteonecrosis of the jaw (ONJ) while on bisphosphonate therapy, dental surgery may exacerbate the condition. For patients requiring dental procedures, there are no data available to suggest whether discontinuation of bisphosphonate treatment reduces the risk of ONJ. Clinical judgement of the treating physician should guide the management plan of each patient based on individual benefit/risk assessment.
Musculoskeletal Pain
In post marketing experience, severe and occasionally incapaciting bone, joint, and/or muscle pain has been reported in patients taking bisphosphonates. However, such reports have been infrequent. This category of drugs includes SKELID. The time to onset of symptoms varied from one day to several months after starting the drug. Most patients had relief of symptoms after stopping. A subset had recurrence of symptoms when rechallenged with the same drug or another bisphosphonate.
Information for Patients
Patients receiving SKELID should be instructed to:
- Take SKELID with 6 to 8 ounces of plain water.
- Not lie down for at least 30 minutes after taking this medication.
- SKELID should not be taken within 2 hours of food.
- Maintain adequate vitamin D and calcium intake.
- Calcium supplements, aspirin, and indomethacin should not be taken within 2 hours before or 2 hours after SKELID.
- Aluminum- or magnesium-containing antacids, if needed, should be taken at least 2 hours after taking SKELID.
Drug Interactions
The bioavailability of SKELID is decreased 80% by calcium, when calcium and SKELID are administered at the same time, and 60% by some aluminum- or magnesium-containing antacids, when administered 1 hour before SKELID. Aspirin may decrease bioavailability of SKELID by up to 50% when taken 2 hours after SKELID. The bioavailability of SKELID is increased 2–4 fold by indomethacin but is not significantly altered by coadministration of diclofenac. The pharmacokinetic parameters of digoxin are not significantly modified by SKELID coadministration. In vitro studies show that tiludronate does not displace warfarin from its binding site on protein.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not yet been completed.
Tiludronate was not genotoxic in the following assays: an in vitro microbial mutagenesis assay with and without metabolic activation, a human lymphocyte assay, a yeast cell assay for forward mutation and mitotic crossing over, or the in vivo mouse micronucleus test.
Tiludronate had no effect on rat fertility (male or female) at exposures up to two times the 400 mg/day human dose, based on surface area, mg/m2 (75 mg/kg/day tiludronic acid dose).
Pregnancy
Pregnancy Category C
In a teratology study in rabbits dosed during days 6–18 of gestation at 42 mg/kg/day and 130 mg/kg/day (2 and 5 times the 400 mg/day human dose based on body surface area), there was dose-related scoliosis likely attributable to the pharmacologic properties of the drug.
Mice receiving 375 mg/kg/day tiludronic acid (7 times the 400 mg/day human dose based on body surface area, mg/m2) for days 6–15 of gestation showed slight maternal toxicity (decreased body weight gain), increased post-implantation loss, decreased number of fetuses/dam, and decreased fetus body weight. Uncommon malformations of the paw (shortened or missing digits, blood blisters between or in place of digits) were present in six fetuses at 375 mg/kg/day, all from the same litter.
Maternal toxicity (decreased body weight) was also observed in a teratology study in rats dosed during days 6–18 of gestation at 375 mg/kg/day tiludronic acid (10 times the 400 mg/day human dose based on body surface area, mg/m2). There were reduced percent implantations, increased postimplantation loss, and increased intra-uterine deaths in the rats. There were no teratogenic effects on fetuses.
Protracted parturition and maternal death, presumably due to hypocalcemia, occurred at 75 mg/kg/day tiludronic acid (two times the 400 mg/day human dose based on body surface area, mg/m2) when rats were treated from day 15 of gestation to day 25 postpartum.
There are no adequate and well-controlled studies in pregnant women. SKELID should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Bisphosphonates are incorporated into the bone matrix, from where they are gradually released over periods of weeks to years. The extent of bisphosphonate incorporation into adult bone, and hence, the amount available for release back into the systemic circulation, is directly related to the total dose and duration of bisphosphonate use. Although there are no data on fetal risk in humans, bisphosphonates do cause fetal harm in animals, and animal data suggest that uptake of bisphosphonates into fetal bone is greater than into maternal bone. Therefore, there is a theoretical risk of fetal harm (e.g., skeletal and other abnormalities) if a woman becomes pregnant after completing a course of bisphosphonate therapy. The impact of variables such as time between cessation of bisphosphonate therapy to conception, the particular bisphosphonate used, and the route of administration (intravenous versus oral) on this risk has not been established.
ADVERSE REACTIONS
The safety of SKELID has been studied in more than 1100 patients, and the adverse experience profile is similar between controlled and uncontrolled clinical trials. Adverse events occurring in placebo-controlled trials of pagetic patients treated with SKELID 400 mg/day are presented in the table below.
The most frequently occurring adverse events in patients who received SKELID 400 mg/day were in the gastrointestinal body system: nausea (9.3%), diarrhea (9.3%), and dyspepsia (5.3%).
Adverse events associated with SKELID usually have been mild, and generally have not required discontinuation of therapy. In two placebo-controlled trials, 1.3% of patients receiving 400 mg SKELID and 5.4% of patients receiving placebo discontinued therapy due to any clinical adverse event.
| SKELID 400 mg/day (n=75) | Placebo (n=74) |
|
|---|---|---|
| BODY AS A WHOLE | ||
| Pain | 21.3 | 23.0 |
| Back Pain | 8.0 | 8.1 |
| Accidental Injury | 4.0 | 2.7 |
| Influenza-like Symptoms | 4.0 | 5.4 |
| Chest Pain | 2.7 | 0 |
| Peripheral Edema | 2.7 | 1.4 |
| CARDIOVASCULAR, GENERAL | ||
| Dependent Edema | 2.7 | 0 |
| Central and Peripheral Nervous Systems | ||
| Headache | 6.7 | 12.2 |
| Dizziness | 4.0 | 6.8 |
| Paresthesia | 4.0 | 0 |
| ENDOCRINE | ||
| Hyperparathyroidism | 2.7 | 0 |
| GASTROINTESTINAL | ||
| Diarrhea | 9.3 | 4.1 |
| Nausea | 9.3 | 5.4 |
| Dyspepsia | 5.3 | 8.1 |
| Vomiting | 4.0 | 0 |
| Flatulence | 2.7 | 0 |
| Tooth Disorder | 2.7 | 1.4 |
| Metabolic and Nutritional | ||
| Vitamin D Deficiency | 2.7 | 2.7 |
| Musculoskeletal System | ||
| Arthralgia | 2.7 | 5.4 |
| Arthrosis | 2.7 | 0 |
| Resistance Mechanism | ||
| Infection | 2.7 | 0 |
| Respiratory System | ||
| Rhinitis | 5.3 | 0 |
| Sinusitis | 5.3 | 1.4 |
| Upper Respiratory Tract Infection | 5.3 | 14.9 |
| Coughing | 2.7 | 2.7 |
| Pharyngitis | 2.7 | 1.4 |
| Skin and Appendage | ||
| Rash | 2.7 | 1.4 |
| Skin Disorder | 2.7 | 1.4 |
| Vision | ||
| Cataract | 2.7 | 0 |
| Conjunctivitis | 2.7 | 0 |
| Glaucoma | 2.7 | 0 |
Other adverse events not listed in the table above but reported in ≥1% of pagetic patients treated with SKELID in all clinical trials of at least one month duration, regardless of dose and causality assessment, are listed below. The adverse event terms within each body system are listed in the order of decreasing frequency occurring in the population.
Body as a Whole: Asthenia, syncope, fatigue
Cardiovascular: Hypertension
Central and Peripheral Nervous Systems: Vertigo, involuntary muscle contractions
Gastrointestinal: Abdominal pain, constipation, dry mouth, gastritis
Musculoskeletal: Fracture pathological
Psychiatric: Anorexia, somnolence, anxiety, nervousness, insomnia
Respiratory System: Bronchitis
Skin and Appendages: Pruritus, increased sweating
Urinary System: Urinary tract infection
Vascular (extracardiac): Flushing
Stevens-Johnson type syndrome has been observed rarely; the causality relationship of this to SKELID has not been established.
OVERDOSAGE
Based on the known action of tiludronate, hypocalcemia is a potential consequence of SKELID overdose. In one patient with hypercalcemia of malignancy, intravenous administration of high doses of SKELID (800 mg/day total dose, 6 mg/kg/day for 2 days) was associated with acute renal failure and death.
No specific information is available on the treatment of overdose with SKELID. Dialysis would not be beneficial. Standard medical practices may be used to manage renal insufficiency or hypocalcemia, if signs of these develop.
DOSAGE AND ADMINISTRATION
A single 400-mg daily oral dose of SKELID, taken with 6 to 8 ounces of plain water only, should be administered for a period of 3 months. Beverages other than plain water (including mineral water), food (see below), and some medications (see PRECAUTIONS, Drug Interactions) are likely to reduce the absorption of SKELID (see CLINICAL PHARMACOLOGY, Pharmacokinetics).
Patients should not lie down for at least 30 minutes after taking this medication. In patients who cannot comply with dosing instructions due to mental or physical disability, therapy with SKELID should be used under appropriate supervision (See WARNINGS).
SKELID should not be taken within 2 hours of food.
Calcium or mineral supplements should be taken at least 2 hours before or two hours after SKELID. Aluminum- or magnesium-containing antacids, if needed, should be taken at least two hours after taking SKELID.
SKELID should not be taken within 2 hours of indomethacin.
Following therapy, allow an interval of 3 months to assess response. Specific data regarding retreatment are limited, although results from uncontrolled studies indicate favorable biochemical improvement similar to initial SKELID treatment.
HOW SUPPLIED
SKELID is supplied as white to practically white, biconvex round tablets containing 240 mg tiludronate disodium, which is the molar equivalent of 200 mg tiludronic acid. SKELID tablets are engraved with "S.W" on one side and "200" on the other side and packaged in foil strips in cartons of 56 tablets per carton (0024-1800-16).
| SKELID
tiludronate disodium tablet |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Labeler - sanofi-aventis U.S. LLC (824676584) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Sanofi Winthrop Industrie | 763683216 | MANUFACTURE(0024-1800) | |