RIMANTADINE HYDROCHLORIDE- rimantadine hydrochloride tablet, film coated
Bryant Ranch Prepack
----------
Rimantadine Hydrochloride Tablets
(100 mg)
Rx only
DESCRIPTION
Rimantadine hydrochloride is a synthetic antiviral drug available as a 100 mg film-coated tablet. Each film-coated tablet contains 100 mg of rimantadine hydrochloride. In addition, each tablet contains the following inactive ingredients: hydroxypropyl methylcellulose, magnesium stearate, microcrystalline cellulose, purified water, sodium starch glycolate and FD&C Yellow No. 6 Lake. Film coating material, Opadry (YS-1-19025-A), contains hypromellose and polyethylene glycol.
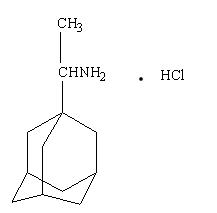
Rimantadine hydrochloride is a white to off-white crystalline powder which is freely soluble in water (50 mg/mL at 20°C). Chemically, rimantadine hydrochloride is alpha-methyltricyclo-[3.3.1.1/3.7]decane-1-methanamine hydrochloride, with a molecular formula of C12H21N•HCl, a molecular weight of 215.77 and the following structural formula:
CLINICAL PHARMACOLOGY
Mechanism of Action
The mechanism of action of rimantadine is not fully understood. Rimantadine appears to exert its inhibitory effect early in the viral replicative cycle, possibly inhibiting the uncoating of the virus. Genetic studies suggest that a virus protein specified by the virion M2 gene plays an important role in the susceptibility of influenza A virus to inhibition by rimantadine.
Microbiology
Rimantadine inhibits the replication in cell culture of influenza A virus isolates from each of the three antigenic subtypes, i.e., H1N1, H2N2 and H3N2, that have been isolated from man. Rimantadine has little or no activity against influenza B virus (Ref. 1, 2). Rimantadine does not appear to interfere with the immunogenicity of inactivated influenza A vaccine.
A quantitative relationship between the susceptibility in cell culture of influenza A virus to rimantadine and clinical response to therapy has not been established.
Susceptibility test results, expressed as the concentration of the drug required to inhibit virus replication by 50% or more in a cell culture system, vary greatly (from 19 nM to 93 µM) depending upon the assay protocol used, size of the virus inoculum, isolates of the influenza A virus strains tested, and the cell types used (Ref. 2).
Resistance
Influenza A virus isolates resistant to rimantadine have been selected in cell culture and in vivo as a result of treatment. Rimantadine-resistant strains of influenza A virus have emerged among freshly isolated strains in closed settings where rimantadine has been used. Resistant viruses have been shown to be transmissible and to cause typical influenza illness (Ref. 3, 9). Substitutions at any one of five amino acid positions in the transmembrane domain of M2 confer resistance to rimantadine. The most common substitution causing resistance among influenza A (H1N1) and A (H3N2) is S31N. Other less common substitutions that cause resistance include substitutions A30F, V27A, V30A, and L26F.
Rimantadine resistance has been observed in circulating seasonal influenza and pandemic isolates from individuals who have not received rimantadine. Swine-origin influenza A (H1N1) (S-OIV) viruses that were resistant to rimantadine have been shown to contain the S31N substitution. Existing primers used for detection of adamantine resistance in seasonal viruses do not work with all tested S-OIVs (Ref. 11). The CDC should be consulted for questions regarding resistance to rimantadine in circulating influenza strains.
Cross-Resistance
Cross-resistance among the adamantanes, rimantadine and amantadine, has been observed. Resistance to rimantadine confers cross-resistance to amantadine and vice-versa. Substitutions that confer resistance to rimantadine include (most frequently) M2 S31N, as well as the less common changes V27A, V30A, L26F and A30T (Ref. 10).
Pharmacokinetics
Although the pharmacokinetic profile of rimantadine hydrochloride has been described, no pharmacodynamic data establishing a correlation between plasma concentration and its antiviral effect are available.
Rimantadine hydrochloride is absorbed after oral administration. The mean ± SD peak plasma concentration after a single 100 mg dose of rimantadine hydrochloride was 74 ± 22 ng/mL (range: 45 to 138 ng/mL). The time to peak concentration was 6 ± 1 hours in healthy adults (age 20 to 44 years). The single dose elimination half-life in this population was 25.4 ± 6.3 hours (range: 13 to 65 hours). The single dose elimination half-life in a group of healthy 71 to 79 year-old subjects was 32 ± 16 hours (range: 20 to 65 hours).
After the administration of rimantadine 100 mg twice daily to healthy volunteers (age 18 to 70 years) for 10 days, area under the curve (AUC) values were approximately 30% greater than predicted from a single dose. Plasma trough levels at steady state ranged between 118 and 468 ng/mL. In these patients no age-related differences in pharmacokinetics were detected. However, in a comparison of three groups of healthy older subjects (age 50 to 60, 61 to 70 and 71 to 79 years), the 71 to 79 year-old group had average AUC values, peak concentrations and elimination half-life values at steady state that were 20 to 30% higher than the other two groups. Steady-state concentrations in elderly nursing home patients (age 68 to 102 years) were 2- to 4-fold higher than those seen in healthy young and elderly adults.
The pharmacokinetic profile of rimantadine in children has not been established.
Following oral administration, rimantadine is extensively metabolized in the liver with less than 25% of the dose excreted in the urine as unchanged drug. Three hydroxylated metabolites have been found in plasma. These metabolites, an additional conjugated metabolite and parent drug account for 74 ± 10% (n=4) of a single 200 mg dose of rimantadine excreted in urine over 72 hours.
In a group (n=14) of patients with chronic liver disease, the majority of whom were stabilized cirrhotics, the pharmacokinetics of rimantadine were not appreciably altered following a single 200 mg oral dose compared to six healthy subjects who were sex, age and weight matched to six of the patients with liver disease. After administration of a single 200 mg dose to patients (n=10) with severe hepatic dysfunction, AUC was approximately 3-fold larger, elimination half-life was approximately 2-fold longer and apparent clearance was about 50% lower when compared to historic data from healthy subjects.
Rimantadine pharmacokinetics were evaluated following administration of 100 mg rimantadine hydrochloride twice daily for 14 days to subjects with mild (creatinine clearance [CrCl] 50 mL/min to 80 mL/min), moderate ([CrCl] 30 mL/min to 49 mL/min), and severe ([CrCl] 5 mL/min to 29 mL/min) renal impairment and to healthy subjects (CrCl > 80 mL/min). There were no clinically relevant differences in rimantadine Cmax, Cmin, and AUC0-τ between subjects with mild or moderate renal impairment compared to healthy subjects. In subjects with severe renal impairment, rimantadine Cmax, Cmin, and AUC0-τ on Day 14 increased by 75%, 82%, and 81%, respectively, compared to healthy subjects. The rimantadine elimination half-life was slightly prolonged (increase of 18% or less) in subjects with mild and moderate renal impairment but increased by 49% in subjects with severe renal impairment compared to healthy subjects.
After a single 200 mg oral dose of rimantadine was given to eight hemodialysis patients (CrCl 0 mL/min to 10 mL/min), there was a 1.6-fold increase in the elimination half-life and a 40% decrease in apparent clearance compared to age-matched healthy subjects. Hemodialysis did not contribute to the clearance of rimantadine.
The in vitro human plasma protein binding of rimantadine is about 40% over typical plasma concentrations. Albumin is the major binding protein.
INDICATIONS AND USAGE
Rimantadine hydrochloride tablet is indicated for the prophylaxis and treatment of illness caused by various strains of influenza A virus in adults (17 years and older).
Rimantadine hydrochloride tablet is indicated for prophylaxis against influenza A virus in children (1 year to 16 years of age).
Prophylaxis
In controlled studies of children (1 year to 16 years of age), healthy adults (17 years and older), and elderly patients (65 years of age and older), rimantadine hydrochloride has been shown to be safe and effective in preventing signs and symptoms of infection caused by various strains of influenza A virus. Since rimantadine hydrochloride does not completely prevent the host immune response to influenza A infection, individuals who take this drug may still develop immune responses to natural disease or vaccination and may be protected when later exposed to antigenically-related viruses. Following vaccination during an influenza outbreak, rimantadine hydrochloride prophylaxis should be considered for the 2 to 4 week time period required to develop an antibody response. However, the safety and effectiveness of rimantadine hydrochloride prophylaxis have not been demonstrated for longer than 6 weeks.
Treatment
Rimantadine hydrochloride therapy should be considered for adults (17 years and older) who develop an influenza-like illness during known or suspected influenza A infection in the community. When administered within 48 hours after onset of signs and symptoms of infection caused by influenza A virus strains, rimantadine hydrochloride has been shown to reduce the duration of fever and systemic symptoms.
The following points should be considered before initiating treatment or prophylaxis with rimantadine hydrochloride:
- Rimantadine hydrochloride is not a substitute for early vaccination on an annual basis as recommended by the Centers for Disease Control and Prevention Advisory Committee on Immunization Practices.
- Influenza viruses change over time. Emergence of resistance mutations could decrease drug effectiveness. Other factors (for example, changes in viral virulence) might also diminish clinical benefit of antiviral drugs. Prescribers should consider available information on influenza drug susceptibility patterns and treatment effects when deciding whether to use rimantadine hydrochloride.
CONTRAINDICATIONS
Rimantadine hydrochloride is contraindicated in patients with known hypersensitivity to drugs of the adamantane class, including rimantadine and amantadine.
PRECAUTIONS
General
An increased incidence of seizures has been reported in patients with a history of epilepsy who received the related drug amantadine. In clinical trials of rimantadine hydrochloride, the occurrence of seizure-like activity was observed in a small number of patients with a history of seizures who were not receiving anticonvulsant medication while taking rimantadine hydrochloride. If seizures develop, rimantadine hydrochloride should be discontinued.
The safety and pharmacokinetics of rimantadine in hepatic insufficiency have only been evaluated after single dose administration. In a study of 14 subjects with chronic liver disease (mostly stabilized cirrhotics), no alterations in the pharmacokinetics were observed after the administration of a single dose of rimantadine. However, the apparent clearance of rimantadine following a single dose to 10 patients with severe liver dysfunction was 50% lower than reported for healthy subjects. Because of the potential for accumulation of rimantadine and its metabolites in plasma, caution should be exercised when patients with hepatic insufficiency are treated with rimantadine.
Following multiple-dose administration of rimantadine, there were no clinically relevant differences in rimantadine systemic exposure between subjects with mild or moderate renal impairment compared to healthy subjects. In subjects with severe renal impairment, rimantadine systemic exposure increased by 81%, compared with healthy subjects. Because of the potential for increased accumulation of rimantadine metabolites in renally impaired subjects, caution should be exercised when these patients are treated with rimantadine.
Transmission of rimantadine resistant virus should be considered when treating patients whose contacts are at high risk for influenza A illness. Influenza A virus strains resistant to rimantadine can emerge during treatment and such resistant strains have been shown to be transmissible and to cause typical influenza illness (Ref. 3). Although the frequency, rapidity, and clinical significance of the emergence of drug-resistant virus are not yet established, several small studies have demonstrated that 10% to 30% of patients with initially sensitive virus, upon treatment with rimantadine, shed rimantadine resistant virus. (Ref. 3, 4, 5, 6)
Clinical response to rimantadine, although slower in those patients who subsequently shed resistant virus, was not significantly different from those who did not shed resistant virus. (Ref. 3) No data are available in humans that address the activity or effectiveness of rimantadine therapy in subjects infected with resistant virus.
Serious bacterial infections may begin with influenza-like symptoms or may coexist with or occur as complications during the course of influenza. Rimantadine hydrochloride has not been shown to prevent such complications.
Drug Interactions
Acetaminophen: Rimantadine hydrochloride, 100 mg, was given twice daily for 13 days to 12 healthy volunteers. On day 11, acetaminophen (650 mg four times daily) was started and continued for 8 days. The pharmacokinetics of rimantadine were assessed on days 11 and 13. Co-administration with acetaminophen reduced the peak concentration and AUC values for rimantadine by approximately 11%.
Aspirin: Rimantadine hydrochloride, 100 mg, was given twice daily for 13 days to 12 healthy volunteers. On day 11, aspirin (650 mg, four times daily) was started and continued for 8 days. The pharmacokinetics of rimantadine were assessed on days 11 and 13. Peak plasma concentrations and AUC of rimantadine were reduced approximately 10% in the presence of aspirin.
Cimetidine: When a single 100 mg dose of rimantadine hydrochloride was administered with steady-state cimetidine (300 mg four times a day), there were no statistically significant differences in rimantadine Cmax or AUC between rimantadine hydrochloride alone and rimantadine hydrochloride in the presence of cimetdine.
Live Attenuated Influenza Vaccine (LAIV): The concurrent use of rimantadine hydrochloride with live attenuated intranasal influenza vaccine has not been evaluated. However, because of potential interference between these products, the live attenuated intranasal influenza vaccine should not be administered until 48 hours after cessation of rimantadine hydrochloride and rimantadine hydrochloride should not be administered until two weeks after the administration of live attenuated intranasal influenza vaccine unless medically indicated. The concern about potential interference arises principally from the potential for antiviral drugs to inhibit replication of live vaccine virus.
Carcinogenesis, Mutagenesis, and Impairment of Fertility
Carcinogenesis: Oral administration of rimantadine to rats for 2 years at doses up to 100 mg/kg/d [approximately 11 to 14 times the maximum recommended human dose (MRHD) based on AUC] showed no evidence of increased tumor incidence.
Mutagenesis: No mutagenic effects were seen when rimantadine was evaluated in several standard assays for mutagenicity.
Impairment of Fertility: A reproduction study in male and female rats did not show detectable impairment of fertility at dosages up to 60 mg/kg/day (3 times the MRHD based on mg/m2).
Pregnancy
Teratogenic Effects: Pregnancy Category C. There are no adequate and well-controlled studies in pregnant women. Rimantadine is reported to cross the placenta in mice. Rimantadine has been shown to be embryotoxic in rats when given at a dose of 200 mg/kg/d (11 times the MRHD based on mg/m2). At this dose the embryotoxic effect consisted of increased fetal resorption in rats; this dose also produced a variety of maternal effects including ataxia, tremors, convulsions and significantly reduced weight gain. No embryotoxicity was observed when rabbits were given doses up to 50 mg/kg/d (approximately 0.1 times the MRHD based on AUC), but evidence of a developmental abnormality in the form of a change in the ratio of fetuses with 12 or 13 ribs were noted. This ratio is normally about 50:50 in a litter but was 80:20 after rimantadine treatment. However, in a repeat embryofetal toxicity study in rabbits at doses up to 50 mg/kg/d (approximately 0.1 times the MRHD based on AUC), this abnormality was not observed.
Nonteratogenic Effects: Rimantadine was administered to pregnant rats in a peri- and postnatal reproduction toxicity study at doses of 30, 60 and 120 mg/kg/d (1.7, 3.4 and 6.8 times the MRHD based on mg/m2). Maternal toxicity during gestation was noted at the two higher doses of rimantadine, and at the highest dose, 120 mg/kg/day, there was an increase in pup mortality during the first 2 to 4 days postpartum. Decreased fertility of the F1 generation was also noted for the two higher doses.
For these reasons, rimantadine hydrochloride should be used during pregnancy only if the potential benefit justifies the risk to the fetus.
Nursing Mothers
Rimantadine hydrochloride should not be administered to nursing mothers because of the adverse effects noted in offspring of rats treated with rimantadine during the nursing period. Rimantadine is concentrated in rat milk in a dose-related manner: 2 to 3 hours following administration of rimantadine, rat breast milk levels were approximately twice those observed in the serum.
Pediatric Use
In children (1 year to 16 years of age), rimantadine hydrochloride is recommended for the prophylaxis of influenza A. The safety and effectiveness of rimantadine hydrochloride in the treatment of symptomatic influenza infection in children (1 year to 16 years of age) have not been established. Prophylaxis studies with rimantadine hydrochloride have not been performed in children below the age of 1 year.
ADVERSE REACTIONS
In 1,027 patients treated with rimantadine hydrochloride in controlled clinical trials at the recommended dose of 200 mg daily, the most frequently reported adverse events involved the gastrointestinal and nervous systems.
Incidence >1%: Adverse events reported most frequently (1 to 3%) at the recommended dose in controlled clinical trials are shown in the table below.
| Rimantadine (n=1027) | Control (n=986) |
|
|---|---|---|
| Nervous System | ||
| Insomnia | 2.1% | 0.9% |
| Dizziness | 1.9% | 1.1% |
| Headache | 1.4% | 1.3% |
| Nervousness | 1.3% | 0.6% |
| Fatigue | 1.0% | 0.9% |
| Gastrointestinal System | ||
| Nausea | 2.8% | 1.6% |
| Vomiting | 1.7% | 0.6% |
| Anorexia | 1.6% | 0.8% |
| Dry mouth | 1.5% | 0.6% |
| Abdominal Pain | 1.4% | 0.8% |
| Body as a Whole | ||
| Asthenia | 1.4% | 0.5% |
Less frequent adverse events (0.3 to 1%) at the recommended dose in controlled clinical trials were: Gastrointestinal System: diarrhea, dyspepsia; Nervous System: impairment of concentration, ataxia, somnolence, agitation, depression; Skin and Appendages: rash; Hearing and Vestibular: tinnitus; Respiratory: dyspnea.
Additional adverse events (less than 0.3%) reported at recommended doses in controlled clinical trials were: Nervous System: gait abnormality, euphoria, hyperkinesia, tremor, hallucination, confusion, convulsions; Respiratory: bronchospasm, cough; Cardiovascular: pallor, palpitation, hypertension, cerebrovascular disorder, cardiac failure, pedal edema, heart block, tachycardia, syncope; Reproduction: non-puerperal lactation; Special Senses: taste loss/change, parosmia.
Rates of adverse events, particularly those involving the gastrointestinal and nervous systems, increased significantly in controlled studies using higher than recommended doses of rimantadine hydrochloride. In most cases, symptoms resolved rapidly with discontinuation of treatment. In addition to the adverse events reported above, the following were also reported at higher than recommended doses: increased lacrimation, increased micturition frequency, fever, rigors, agitation, constipation, diaphoresis, dysphagia, stomatitis, hypesthesia and eye pain.
Adverse Reactions in Trials of Rimantadine and Amantadine: In a six-week prophylaxis study of 436 healthy adults comparing rimantadine with amantadine and placebo, the following adverse reactions were reported with an incidence >1%.
| Rimantadine 200 mg/day (n=145) | Placebo (n=143) | Amantadine 200 mg/day (n=148) |
|
|---|---|---|---|
| Nervous System | |||
| Insomia | 3.4% | 0.7% | 7.0% |
| Nervousness | 2.1% | 0.7% | 2.8% |
| Impaired Concentration |
2.1% |
1.4% |
2.1% |
| Dizziness | 0.7% | 0.0% | 2.1% |
| Depression | 0.7% | 0.7% | 3.5% |
| Total % of subjects with adverse reactions |
6.9% |
4.1% |
14.7% |
| Total % of subjects withdrawn due to adverse reactions |
6.9% |
3.4% |
14.0% |
Geriatric Use
Approximately 200 patients over the age of 65 were evaluated for safety in controlled clinical trials with rimantadine hydrochloride. Geriatric subjects who received either 200 mg or 400 mg of rimantadine daily for 1 to 50 days experienced considerably more central nervous system and gastrointestinal adverse events than comparable geriatric subjects receiving placebo. Central nervous system events including dizziness, headache, anxiety, asthenia, and fatigue, occurred up to two times more often in subjects treated with rimantadine than in those treated with placebo. Gastrointestinal symptoms, particularly nausea, vomiting, and abdominal pain occurred at least twice as frequently in subjects receiving rimantadine than in those receiving placebo. The gastrointestinal symptoms appeared to be dose related. In patients over 65, the recommended dose is 100 mg, daily (see CLINCAL PHARMACOLOGY and DOSAGE AND ADMINISTRATION).
To report SUSPECTED ADVERSE REACTIONS, contact Amneal Pharmaceuticals at 1-877-835-5472 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
OVERDOSAGE
As with any overdose, supportive therapy should be administered as indicated. Overdoses of a related drug, amantadine, have been reported with adverse reactions consisting of agitation, hallucinations, cardiac arrhythmia and death. The administration of intravenous physostigmine (a cholinergic agent) at doses of 1 to 2 mg in adults (Ref. 7) and 0.5 mg in children (Ref. 8) repeated as needed as long as the dose did not exceed 2 mg/hour has been reported anecdotally to be beneficial in patients with central nervous system effects from overdoses of amantadine.
DOSAGE AND ADMINISTRATION
For Prophylaxis in Adults and Children
Adults (17 years and older)
The recommended adult dose of rimantadine hydrochloride is 100 mg twice a day. Study durations ranged from 11 days to 6 weeks in adult and elderly patients. In patients with severe hepatic dysfunction, severe renal impairment (CrCl 5 to 29 mL/min) or renal failure (CrCl ≤ 10 mL/min) and in elderly nursing home patients, a dose reduction to 100 mg daily is recommended. Because of the potential for accumulation of rimantadine metabolites during multiple dosing, patients with hepatic or renal impairment should be monitored for adverse effects.
Children (1 year to 16 years of age)
- Study duration ranged from 5 weeks to 6 weeks in pediatric subjects.
- In children 1 year to 9 years of age, rimantadine hydrochloride should be administered once a day, at a dose of 5 mg/kg but not exceeding 150 mg.
- For children 10 years of age or older, use the adult dose.
(see Directions for Compounding of an Oral Suspension from Rimantadine Hydrochloride Tablets to prepare an oral suspension for administration to children and patients with difficulty swallowing tablets).
For Treatment in Adults
Adults (17 years and older)
The recommended adult dose of rimantadine hydrochloride is 100 mg twice a day for 7 days. In patients with severe hepatic dysfunction, severe renal impairment (CrCl 5 to 29 mL/min) or renal failure (CrCl ≤ 10 mL/min) and elderly nursing home patients, a dose reduction to 100 mg daily is recommended. Because of the potential for accumulation of rimantadine metabolites during multiple dosing, patients with hepatic or renal impairment should be monitored for adverse effects. Rimantadine hydrochloride therapy should be initiated as soon as possible, preferably within 48 hours after onset of signs and symptoms of influenza A infection. Therapy should be continued for approximately seven days from the initial onset of symptoms.
Directions for the Compounding of an Oral Suspension from Rimantadine Hydrochloride Tablets (Final Concentration = 10 mg/mL)1
These directions are provided for use only during emergency situations, for patients who have difficulty swallowing tablets or where lower doses are needed. The pharmacist may compound a suspension (10 mg/mL) from rimantadine hydrochloride tablets, 100 mg using Ora-Sweet®†. Other vehicles have not been studied.
To make an oral suspension (10 mg/mL) from 100 mg rimantadine hydrochloride tablets, you will need the following:
- 100 mg tablets of rimantadine hydrochloride
- Ora-Sweet® (a vehicle manufactured by Paddock Laboratories)
- a graduated cylinder
- a mortar and pestle
- an Amber Glass or Polyethylene terephthalate plastic (PET) bottle
- a funnel (optional)
Compounding Procedures
A 100 mg tablet of rimantadine hydrochloride is required for each 10 mL of compounded oral suspension to make a concentration of 10 mg/mL.
A compounded oral suspension is stable for 14 days. Therefore, the maximum amount of oral suspension that can be dispensed to a patient should not exceed a 14 day supply.
Step A: Guidance for how to determine the Number of Tablets and Total Volume needed to compound a 10 mg/mL oral suspension for each patient.
1. Verify the prescribed dose is correct.
2. Calculate the mg amount of rimantadine hydrochloride needed for the duration of therapy.
(Daily Dose) × (Number of days) = (mg of rimantadine hydrochloride)
For example, 75 mg/day × 10 days = 750 mg
3. Round up the mg of rimantadine hydrochloride amount to the next 100 mg designation.
For example, Round up 750 mg to 800 mg
4. Calculate the Number of 100 mg tablets that are required for the compounded oral suspension.
(Rounded mg of rimantadine hydrochloride) ÷ (100 mg/tablet) = (Number of tablets)
For example, 800 mg ÷ 100 mg/tablet = 8 tablets
5. Calculate the Total Volume of compounded oral suspension (10 mg/mL) (Rounded mg of rimantadine hydrochloride) ÷ (10 mg/mL) = (Total Volume)
For example, 800 mg ÷ 10 mg/mL = 80 mL
Step B: Once the total Number of Tablets and Volume are determined then follow the procedures below for compounding the oral suspension (10 mg/mL) from rimantadine hydrochloride tablets 100 mg
Verify your calculations before you begin to compound an oral suspension.
A 100 mg tablet of rimantadine hydrochloride is required for each 10 mL of compounded oral suspension to make a concentration of 10 mg/mL.
1. Place the required number of rimantadine hydrochloride 100 mg tablets into a clean mortar of sufficient size to contain the tablets and volume of vehicle,
Ora-Sweet® used in Step 3.
2. Grind the tablets and triturate to a fine powder using a pestle. Powder on the sides of the mortar or pestle should be removed using a spatula and
incorporated into the trituration throughout the process.
3. Slowly add approximately one-third (1/3) of the total volume of vehicle to the mortar while triturating until a uniform suspension is achieved.
4. Transfer the suspension to an amber glass or a PET plastic bottle. Other types of bottles, such as non-PET plastic or uncolored bottles, have not been
evaluated and should not be used. A funnel may be used to eliminate any spillage.
5. Slowly add the second one-third (1/3) of the total volume of vehicle to the mortar, rinse the pestle and mortar by a triturating motion and transfer the
contents into the bottle.
6. Repeat the rinsing (Step 5) with the remaining one-third (1/3) of the vehicle, transferring the remaining contents to the fullest extent possible.
Verify that the suspension is at the desired total volume or add additional vehicle if needed.
7. Close the bottle using a child-resistant cap.
8. Shake well to ensure homogeneous suspension. (Note: The active drug, rimantadine hydrochloride readily dissolves in the specified vehicle. The
suspension is caused by some of the inert ingredients of rimantadine hydrochloride tablets 100 mg which are insoluble in this vehicle.)
Labeling and Dispensing Information for the Compounded Oral Suspension
1. Include an ancillary label on the bottle indicating "Shake Gently Before Use". This compounded suspension should be gently shaken prior to administration to
minimize the tendency for air entrapment with the Ora-Sweet® preparation. The need to shake the compounded oral suspension gently prior to administration
should be reviewed with the parent or guardian when the suspension is dispensed.
2. Provide an oral dosing device (a graduated oral syringe or spoon) that will measure the prescribed dose (in mL). If possible, mark or highlight the graduation
corresponding to the appropriate dose on the oral syringe or spoon for each patient.
3. Include an Expiration Date label according to storage condition (see below) and a "Discard any Unused Portion" label to the bottle. Instruct the parent or
guardian that any remaining material following completion of therapy or after the expiration date on the label must be discarded.
STORAGE OF THE PHARMACY-COMPOUNDED SUSPENSION
Room Temperature: Stable for 14 days when stored in ambient room temperature conditions. Other storage conditions have not been studied.
Note: The storage conditions are based on stability studies of compounded oral suspensions, using the above mentioned vehicle, which was placed in amber glass and PET plastic bottles at 25°C (77°F). Stability studies have not been conducted with other vehicles or bottle types.
HOW SUPPLIED
Product: 63629-3552
NDC: 63629-3552-1 30 TABLET, FILM COATED in a BOTTLE
NDC: 63629-3552-2 10 TABLET, FILM COATED in a BOTTLE
REFERENCES
- Belshe RB, Burk B, Newman F, et al. J Infect Dis. 1989; 159(3):430-435.
- Sim IS, Cerruti RL, Connell EV. J Respir Dis. 1989 (Suppl): S46-S51.
- Hayden FG, Belshe RB, Clover RD, et al. N Engl J Med. 1989; 321(25), 1696-1702.
- Hall CB, Dolin R, Gala CL, et al. Pediatrics. 1987; 80(2): 275-282.
- Thompson J, Fleet W, Lawrence E, et al. J Med Virol. 1987; 21(3): 249-255.
- Belshe RB, Smith MH, Hall CB, et al. J Virol. 1988; 62(5): 1508-1512.
- Casey DE. N Engl J Med. 1978; 298(9):516.
- Berkowitz CD. J Pediatr. 1979; 95(1): 144-145.
- Hayden FG, Sperber SJ, Belshe RB, et al. Antimicrob Agents Chemother. 1991; 35(9): 1741-1747.
- Deyde VM, Xu X, Bright RA, et al. J Infect Dis. 2007; 196(2): 249-257.
- CDC. MMWR Morb Mortal Wkly Rep. 2009; 58(16): 433-435.
† Ora-Sweet® is a registered trademark of Paddock Laboratories.

| RIMANTADINE HYDROCHLORIDE
rimantadine hydrochloride tablet, film coated |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Bryant Ranch Prepack (171714327) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Bryant Ranch Prepack | 171714327 | REPACK(63629-3552) , RELABEL(63629-3552) | |