Label: CLONIDINE HYDROCHLORIDE tablet






-
NDC Code(s):
68001-237-00,
68001-237-03,
68001-238-00,
68001-238-03, view more68001-239-00
- Packager: BluePoint Laboratories
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated November 21, 2023
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
- SPL UNCLASSIFIED SECTION
-
DESCRIPTION
Clonidine hydrochloride, USP is a centrally acting alpha-agonist hypotensive agent available as tablets for oral administration in three dosage strengths: 0.1 mg, 0.2 mg, and 0.3 mg. The 0.1 mg tablet is equivalent to 0.087 mg of the free base.
The following inactive ingredients are contained in these products: corn starch, D&C Yellow #10 Aluminum Lake, FD&C Yellow #6 Aluminum Lake (Sunset Yellow Lake), lactose monohydrate, magnesium stearate, and sodium starch glycolate.
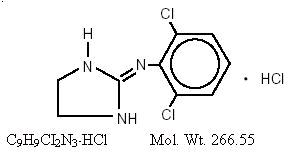
Clonidine hydrochloride, USP is an imidazoline derivative and exists as a mesomeric compound. The chemical name is 2-(2,6-dichlorophenylamino)-2-imidazoline hydrochloride. The following is the structural formula:
Clonidine hydrochloride, USP is an odorless, bitter, white, crystalline substance soluble in water and alcohol.
-
CLINICAL PHARMACOLOGY
Clonidine stimulates alpha-adrenoreceptors in the brain stem. This action results in reduced sympathetic outflow from the central nervous system and in decreases in peripheral resistance, renal vascular resistance, heart rate, and blood pressure. Clonidine hydrochloride tablets act relatively rapidly. The patient’s blood pressure declines within 30 to 60 minutes after an oral dose, the maximum decrease occurring within 2 to 4 hours. Renal blood flow and glomerular filtration rate remain essentially unchanged. Normal postural reflexes are intact; therefore, orthostatic symptoms are mild and infrequent.
Acute studies with clonidine hydrochloride in humans have demonstrated a moderate reduction (15% to 20%) of cardiac output in the supine position with no change in the peripheral resistance: at a 45 degree tilt there is a smaller reduction in cardiac output and a decrease of peripheral resistance. During long term therapy, cardiac output tends to return to control values, while peripheral resistance remains decreased. Slowing of the pulse rate has been observed in most patients given clonidine, but the drug does not alter normal hemodynamic response to exercise.
Tolerance to the antihypertensive effect may develop in some patients, necessitating a reevaluation of therapy.
Other studies in patients have provided evidence of a reduction in plasma renin activity and in the excretion of aldosterone and catecholamines. The exact relationship of these pharmacologic actions to the antihypertensive effect of clonidine has not been fully elucidated.
Clonidine acutely stimulates growth hormone release in both children and adults, but does not produce a chronic elevation of growth hormone with long-term use.
Pharmacokinetics
The pharmacokinetics of clonidine is dose-proportional in the range of 100 mcg to 600 mcg. The absolute bioavailability of clonidine on oral administration is 70% to 80%. Peak plasma clonidine levels are attained in approximately 1 to 3 hours.
Following intravenous administration, clonidine displays biphasic disposition with a distribution half-life of about 20 minutes and an elimination half-life ranging from 12 to 16 hours. The half-life increases up to 41 hours in patients with severe impairment of renal function. Clonidine crosses the placental barrier. It has been shown to cross the blood-brain barrier in rats.
Following oral administration about 40% to 60% of the absorbed dose is recovered in the urine as unchanged drug in 24 hours. About 50% of the absorbed dose is metabolized in the liver. Neither food nor the race of the patient influences the pharmacokinetics of clonidine.
The antihypertensive effect is reached at plasma concentrations between about 0.2 and 2.0 ng/mL in patients with normal excretory function. A further rise in the plasma levels will not enhance the antihypertensive effect.
- INDICATIONS AND USAGE
-
CONTRAINDICATIONS
Clonidine hydrochloride tablets should not be used in patients with known hypersensitivity to clonidine (see PRECAUTIONS).
-
WARNINGS
Withdrawal
Patients should be instructed not to discontinue therapy without consulting their physician. Sudden cessation of clonidine treatment has, in some cases, resulted in symptoms such as nervousness, agitation, headache, and tremor accompanied or followed by a rapid rise in blood pressure and elevated catecholamine concentrations in the plasma. The likelihood of such reactions to discontinuation of clonidine therapy appears to be greater after administration of higher doses or continuation of concomitant beta-blocker treatment and special caution is therefore advised in these situations. Rare instances of hypertensive encephalopathy, cerebrovascular accidents and death have been reported after clonidine withdrawal. When discontinuing therapy with clonidine hydrochloride tablets, the physician should reduce the dose gradually over 2 to 4 days to avoid withdrawal symptomatology.
An excessive rise in blood pressure following discontinuation of clonidine hydrochloride tablets therapy can be reversed by administration of oral clonidine hydrochloride or by intravenous phentolamine. If therapy is to be discontinued in patients receiving a beta-blocker and clonidine concurrently, the beta-blocker should be withdrawn several days before the gradual discontinuation of clonidine hydrochloride tablets.
Because children commonly have gastrointestinal illnesses that lead to vomiting, they may be particularly susceptible to hypertensive episodes resulting from abrupt inability to take medication.
-
PRECAUTIONS
General
In patients who have developed localized contact sensitization to transdermal clonidine, continuation of transdermal clonidine or substitution of oral clonidine hydrochloride therapy may be associated with the development of a generalized skin rash.
In patients who develop an allergic reaction to transdermal clonidine, substitution of oral clonidine hydrochloride may also elicit an allergic reaction (including generalized rash, urticaria, or angioedema).
The sympatholytic action of clonidine may worsen sinus node dysfunction and atrioventricular (AV) block, especially in patients taking other sympatholytic drugs. There are postmarketing reports of patients with conduction abnormalities and/or taking other sympatholytic drugs who developed severe bradycardia requiring IV atropine, IV isoproterenol and temporary cardiac pacing while taking clonidine.
In hypertension caused by pheochromocytoma, no therapeutic effect of clonidine hydrochloride tablets can be expected.
Perioperative Use
Administration of clonidine hydrochloride tablets should be continued to within 4 hours of surgery and resumed as soon as possible thereafter. Blood pressure should be carefully monitored during surgery and additional measures to control blood pressure should be available if required.
Information for Patients
Patients should be cautioned against interruption of clonidine hydrochloride tablets therapy without their physician’s advice.
Since patients may experience a possible sedative effect, dizziness, or accommodation disorder with use of clonidine, caution patients about engaging in activities such as driving a vehicle or operating appliances or machinery. Also, inform patients that this sedative effect may be increased by concomitant use of alcohol, barbiturates, or other sedating drugs.
Patients who wear contact lenses should be cautioned that treatment with clonidine hydrochloride tablets may cause dryness of eyes.
Drug Interactions
Clonidine may potentiate the CNS-depressive effects of alcohol, barbiturates or other sedating drugs. If a patient receiving clonidine hydrochloride is also taking tricyclic antidepressants, the hypotensive effect of clonidine may be reduced, necessitating an increase in the clonidine dose.
If a patient receiving clonidine is also taking neuroleptics, orthostatic regulation disturbances (e.g., orthostatic hypotension, dizziness, fatigue) may be induced or exacerbated.
Monitor heart rate in patients receiving clonidine concomitantly with agents known to affect sinus node function or AV nodal conduction, e.g., digitalis, calcium channel blockers, and beta-blockers. Sinus bradycardia resulting in hospitalization and pacemaker insertion has been reported in association with the use of clonidine concomitantly with diltiazem or verapamil.
Amitriptyline in combination with clonidine enhances the manifestation of corneal lesions in rats (see Toxicology).
Based on observations in patients in a state of alcoholic delirium it has been suggested that high intravenous doses of clonidine may increase the arrhythmogenic potential (QT-prolongation, ventricular fibrillation) of high intravenous doses of haloperidol. Causal relationship and relevance for clonidine oral tablets have not been established.
Toxicology
In several studies with oral clonidine hydrochloride, a dose-dependent increase in the incidence and severity of spontaneous retinal degeneration was seen in albino rats treated for six months or longer. Tissue distribution studies in dogs and monkeys showed a concentration of clonidine in the choroid.
In view of the retinal degeneration seen in rats, eye examinations were performed during clinical trials in 908 patients before, and periodically after, the start of clonidine therapy. In 353 of these 908 patients, the eye examinations were carried out over periods of 24 months or longer. Except for some dryness of the eyes, no drug-related abnormal ophthalmological findings were recorded and, according to specialized tests such as electroretinography and macular dazzle, retinal function was unchanged.
In combination with amitriptyline, clonidine hydrochloride administration led to the development of corneal lesions in rats within 5 days.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Chronic dietary administration of clonidine was not carcinogenic to rats (132 weeks) or mice (78 weeks) dosed, respectively, at up to 46 or 70 times the maximum recommended daily human dose as mg/kg (9 or 6 times the MRDHD on a mg/m 2basis). There was no evidence of genotoxicity in the Ames test for mutagenicity or mouse micronucleus test for clastogenicity.
Fertility of male or female rats was unaffected by clonidine doses as high as 150 mcg/kg (approximately 3 times MRDHD). In a separate experiment, fertility of female rats appeared to be affected at dose levels of 500 mcg/kg to 2000 mcg/kg (10 to 40 times the oral MRDHD on a mg/kg basis; 2 to 8 times the MRDHD on a mg/m 2 basis).
Pregnancy
Teratogenic Effects
Reproduction studies performed in rabbits at doses up to approximately 3 times the oral maximum recommended daily human dose (MRDHD) of clonidine hydrochloride tablets produced no evidence of a teratogenic or embryotoxic potential in rabbits. In rats, however, doses as low as 1/3 the oral MRDHD (1/15 the MRDHD on a mg/m 2 basis) of clonidine were associated with increased resorptions in a study in which dams were treated continuously from 2 months prior to mating. Increased resorptions were not associated with treatment at the same time or at higher dose levels (up to 3 times the oral MRDHD) when the dams were treated on gestation days 6 to 15. Increases in resorption were observed at much higher dose levels (40 times the oral MRDHD on a mg/kg basis; 4 to 8 times the MRDHD on a mg/m 2 basis) in mice and rats treated on gestation days 1 to 14 (lowest dose employed in the study was 500 mcg/kg).
No adequate, well-controlled studies have been conducted in pregnant women. Clonidine crosses the placental barrier (see CLINICAL PHARMACOLOGY, Pharmacokinetics). Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed.
Nursing Mothers
As clonidine hydrochloride is excreted in human milk, caution should be exercised when clonidine hydrochloride tablets are administered to a nursing woman.
Pediatric Use
Safety and effectiveness in pediatric patients have not been established in adequate and well-controlled trials (see WARNINGS, Withdrawal).
-
ADVERSE REACTIONS
Most adverse effects are mild and tend to diminish with continued therapy. The most frequent (which appear to be dose-related) are dry mouth, occurring in about 40 of 100 patients; drowsiness, about 33 in 100; dizziness, about 16 in 100; constipation and sedation, each about 10 in 100.
The following less frequent adverse experiences have also been reported in patients receiving clonidine hydrochloride tablets, but in many cases patients were receiving concomitant medication and a causal relationship has not been established.
Body as a Whole: Fatigue, fever, headache, pallor, weakness, and withdrawal syndrome. Also reported were a weakly positive Coombs’ test and increased sensitivity to alcohol.
Cardiovascular: Bradycardia, congestive heart failure, electrocardiographic abnormalities (i.e., sinus node arrest, junctional bradycardia, high degree AV block and arrhythmias), orthostatic symptoms, palpitations, Raynaud’s phenomenon, syncope, and tachycardia. Cases of sinus bradycardia and atrioventricular block have been reported, both with and without the use of concomitant digitalis.
Central Nervous System: Agitation, anxiety, delirium, delusional perception, hallucinations (including visual and auditory), insomnia, mental depression, nervousness, other behavioral changes, paresthesia, restlessness, sleep disorder, and vivid dreams or nightmares.
Dermatological: Alopecia, angioneurotic edema, hives, pruritus, rash, and urticaria.
Gastrointestinal: Abdominal pain, anorexia, constipation, hepatitis, malaise, mild transient abnormalities in liver function tests, nausea, parotitis, pseudo-obstruction (including colonic pseudo-obstruction), salivary gland pain, and vomiting.
Genitourinary: Decreased sexual activity, difficulty in micturition, erectile dysfunction, loss of libido, nocturia, and urinary retention.
Hematologic: Thrombocytopenia.
Metabolic: Gynecomastia, transient elevation of blood glucose or serum creatine phosphokinase, and weight gain.
Musculoskeletal: Leg cramps and muscle or joint pain.
Oro-otolaryngeal: Dryness of the nasal mucosa.
Ophthalmological: Accommodation disorder, blurred vision, burning of the eyes, decreased lacrimation, and dryness of eyes.
To report SUSPECTED ADVERSE EVENTS, contact Teva at 1-888-838-2872 or FDA at 1-800-FDA-1088 or http://www.fda.gov/medwatch for voluntary reporting of adverse reactions.
-
OVERDOSAGE
Hypertension may develop early and may be followed by hypotension, bradycardia, respiratory depression, hypothermia, drowsiness, decreased or absent reflexes, weakness, irritability and miosis. The frequency of CNS depression may be higher in children than adults. Large overdoses may result in reversible cardiac conduction defects or dysrhythmias, apnea, coma and seizures. Signs and symptoms of overdose generally occur within 30 minutes to two hours after exposure. As little as 0.1 mg of clonidine has produced signs of toxicity in children.
There is no specific antidote for clonidine overdosage. Clonidine overdosage may result in the rapid development of CNS depression; therefore, induction of vomiting with ipecac syrup is not recommended. Gastric lavage may be indicated following recent and/or large ingestions. Administration of activated charcoal and/or a cathartic may be beneficial. Supportive care may include atropine sulfate for bradycardia, intravenous fluids and/or vasopressor agents for hypotension and vasodilators for hypertension. Naloxone may be a useful adjunct for the management of clonidine-induced respiratory depression, hypotension and/or coma; blood pressure should be monitored since the administration of naloxone has occasionally resulted in paradoxical hypertension. Tolazoline administration has yielded inconsistent results and is not recommended as first-line therapy. Dialysis is not likely to significantly enhance the elimination of clonidine.
The largest overdose reported to date involved a 28-year old male who ingested 100 mg of clonidine hydrochloride powder. This patient developed hypertension followed by hypotension, bradycardia, apnea, hallucinations, semicoma, and premature ventricular contractions. The patient fully recovered after intensive treatment. Plasma clonidine levels were 60 ng/mL after 1 hour, 190 ng/mL after 1.5 hours, 370 ng/mL after 2 hours, and 120 ng/mL after 5.5 and 6.5 hours. In mice and rats, the oral LD 50 of clonidine is 206 and 465 mg/kg, respectively.
-
DOSAGE AND ADMINISTRATION
Adults: The dose of clonidine hydrochloride tablets must be adjusted according to the patient’s individual blood pressure response. The following is a general guide to its administration.
Initial Dose: 0.1 mg tablet twice daily (morning and bedtime). Elderly patients may benefit from a lower initial dose.
Maintenance Dose: Further increments of 0.1 mg per day may be made at weekly intervals if necessary until the desired response is achieved. Taking the larger portion of the oral daily dose at bedtime may minimize transient adjustment effects of dry mouth and drowsiness. The therapeutic doses most commonly employed have ranged from 0.2 mg to 0.6 mg per day given in divided doses.
Studies have indicated that 2.4 mg is the maximum effective daily dose, but doses as high as this have rarely been employed.
Renal Impairment: Patients with renal impairment may benefit from a lower initial dose. Patients should be carefully monitored. Since only a minimal amount of clonidine is removed during routine hemodialysis, there is no need to give supplemental clonidine following dialysis.
For questions regarding this product, call Teva at 1-888-838-2872.
-
HOW SUPPLIED
Clonidine hydrochloride tablets, USP are supplied as follows:
0.1 mg — Each orange, round tablet imprinted with
 and 127 on one side and bisect on the other side contains 0.1 mg of clonidine hydrochloride, USP and is supplied in bottles of 100 (NDC 68001-237-00) and 500 (NDC 68001-237-03).
and 127 on one side and bisect on the other side contains 0.1 mg of clonidine hydrochloride, USP and is supplied in bottles of 100 (NDC 68001-237-00) and 500 (NDC 68001-237-03).
0.2 mg — Each orange, round tablet imprinted with
 on one side and 128 and bisect on the other side contains 0.2 mg of clonidine hydrochloride, USP and is supplied in bottles of 100 (NDC 68001-238-00) and 500 (NDC 68001-238-03).
on one side and 128 and bisect on the other side contains 0.2 mg of clonidine hydrochloride, USP and is supplied in bottles of 100 (NDC 68001-238-00) and 500 (NDC 68001-238-03).
0.3 mg — Each orange, round tablet imprinted with
 on one side and 129 and bisect on the other side contains 0.3 mg of clonidine hydrochloride, USP and is supplied in bottles of 100 (NDC 68001-239-00).
on one side and 129 and bisect on the other side contains 0.3 mg of clonidine hydrochloride, USP and is supplied in bottles of 100 (NDC 68001-239-00).
Dispense in a tight, light-resistant container as defined in the USP.
Store at 25°C (77ºF); excursions permitted to 15º to 30ºC (59º to 86ºF) [see USP Controlled Room Temperature].
Manufactured In Croatia By:
Pliva Hrvatska d.o.o.
Zagreb, CroatiaFor BluePoint Laboratories
Rev. A 9/2022
- PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
- PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
- PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
CLONIDINE HYDROCHLORIDE
clonidine hydrochloride tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:68001-237 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength CLONIDINE HYDROCHLORIDE (UNII: W76I6XXF06) (CLONIDINE - UNII:MN3L5RMN02) CLONIDINE HYDROCHLORIDE 0.1 mg Inactive Ingredients Ingredient Name Strength STARCH, CORN (UNII: O8232NY3SJ) FD&C YELLOW NO. 6 (UNII: H77VEI93A8) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) D&C YELLOW NO. 10 ALUMINUM LAKE (UNII: CQ3XH3DET6) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) Product Characteristics Color orange Score 2 pieces Shape ROUND Size 6mm Flavor Imprint Code R127 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:68001-237-00 100 in 1 BOTTLE; Type 0: Not a Combination Product 01/03/1995 2 NDC:68001-237-03 500 in 1 BOTTLE; Type 0: Not a Combination Product 01/03/1995 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA070974 01/03/1995 CLONIDINE HYDROCHLORIDE
clonidine hydrochloride tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:68001-238 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength CLONIDINE HYDROCHLORIDE (UNII: W76I6XXF06) (CLONIDINE - UNII:MN3L5RMN02) CLONIDINE HYDROCHLORIDE 0.2 mg Inactive Ingredients Ingredient Name Strength STARCH, CORN (UNII: O8232NY3SJ) D&C YELLOW NO. 10 ALUMINUM LAKE (UNII: CQ3XH3DET6) FD&C YELLOW NO. 6 (UNII: H77VEI93A8) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) Product Characteristics Color orange Score 2 pieces Shape ROUND Size 7mm Flavor Imprint Code R128 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:68001-238-00 100 in 1 BOTTLE; Type 0: Not a Combination Product 01/03/1995 2 NDC:68001-238-03 500 in 1 BOTTLE; Type 0: Not a Combination Product 01/03/1995 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA070975 01/03/1995 CLONIDINE HYDROCHLORIDE
clonidine hydrochloride tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:68001-239 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength CLONIDINE HYDROCHLORIDE (UNII: W76I6XXF06) (CLONIDINE - UNII:MN3L5RMN02) CLONIDINE HYDROCHLORIDE 0.3 mg Inactive Ingredients Ingredient Name Strength STARCH, CORN (UNII: O8232NY3SJ) D&C YELLOW NO. 10 ALUMINUM LAKE (UNII: CQ3XH3DET6) FD&C YELLOW NO. 6 (UNII: H77VEI93A8) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) Product Characteristics Color orange Score 2 pieces Shape ROUND Size 9mm Flavor Imprint Code R129 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:68001-239-00 100 in 1 BOTTLE; Type 0: Not a Combination Product 01/03/1995 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA070976 01/03/1995 Labeler - BluePoint Laboratories (985523874)