LAMIVUDINE- lamivudine tablet, film coated
AvPAK
----------
HIGHLIGHTS OF PRESCRIBING INFORMATIONLamivudine Tablets
These highlights do not include all the information needed to use lamivudine safely and effectively. See full prescribing information for lamivudine. Initial U.S. Approval: 1995
WARNING: LACTIC ACIDOSIS, POSTTREATMENT EXACERBATIONS OF HEPATITS B IN CO-INFECTED PATIENTS, DIFFERENT FORMULATIONS OF LAMIVUDINE
|
FULL PRESCRIBING INFORMATION
WARNING: RISK OF LACTIC ACIDOSIS, EXACERBATIONS OF HEPATITIS B IN CO- INFECTED PATIENTS UPON DISCONTINUATION OF LAMIVUDINE, DIFFERENT FORMULATIONS OF LAMIVUDINE.
Lactic Acidosis and Severe Hepatomegaly: Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogues alone or in combination, including lamivudine and other antiretrovirals. Suspend treatment if clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity occur [see Warnings and Precautions (5.1)].
Exacerbations of Hepatitis B: Severe acute exacerbations of hepatitis B have been reported in patients who are co-infected with hepatitis B virus (HBV) and human immunodeficiency virus (HIV-l) and have discontinued lamivudine. Hepatic function should be monitored closely with both clinical and laboratory follow-up for at least several months in patients who discontinue lamivudine and are co-infected with HIV-l and HBV. If appropriate, initiation of anti-hepatitis B therapy may be warranted [see Warnings and Precautions (5.2)] .
Important Differences Among Lamivudine-Containing Products: Lamivudine Tablets (used to treat HIV-l infection) contain a higher dose of the active ingredient (lamivudine) than EPIVIR-HBV® Tablets (used to treat chronic HBV infection). Patients with HIV-l infection should receive only dosage forms appropriate for treatment of HIV-1 [see Warnings and Precautions (5.2)] .
1 INDICATIONS & USAGE
Lamivudine Tablet is a nucleoside analogue indicated in combination with other antiretroviral agents for the treatment of human immunodeficiency virus (HIV-l) infection. Limitation of use: The dosage of this product is for HIV-1and not for HBV.
2 DOSAGE & ADMINISTRATION
2.1 Adults and Adolescents >16 years of age
The recommended oral dose of lamivudine in HIV-1-infected adults and adolescents >16 years of age is 300 mg daily, administered as either 150 mg twice daily or 300 mg once daily, in combination with other antiretroviral agents. If lamivudine is administered to a patient infected with HIV-1 and HBV, the dosage indicated for HIV-l therapy should be used as part of an appropriate combination regimen [see Warnings and Precautions (5.2)] .
2.2 Pediatric Patients
Lamivudine is also available as a scored tablet for HIV-l-infected pediatric patients who weigh ≥14 kg for whom a solid dosage form is appropriate. Before prescribing lamivudine tablets, children should be assessed for the ability to swallow tablets. If a child is unable to reliably swallow lamivudine tablets, the oral solution formulation should be prescribed. The recommended oral dosage of lamivudine tablets for HIV-1-infected pediatric patients is presented in Table 1.
Table 1. Dosing Recommendations for Lamivudine Tablets in Pediatric Patients
|
Weight (kg) | Dosage Regimen Using Scored 150 mg Tablet
| Total
Daily Dose |
|
| AM Dose
|
|||
|
14 to 21 | ½ tablet (75 mg)
| ½ tablet (75 mg)
| 150 mg
|
|
>21 to <30 | ½ tablet (75 mg)
| 1 tablet (150 mg)
| 225 mg
|
|
≥30 | 1 tablet (150 mg)
| 1 tablet (150 mg)
| 300 mg
|
2.3 Patients With Renal Impairment
Dosing of lamivudine is adjusted in accordance with renal function. Dosage adjustments are listed in Table 2
[see
Clinical Pharmacology (12.3)].
Table 2. Adjustment of Dosage of Lamivudine in Adults and Adolescents (≥30 kg) in Accordance With Creatinine Clearance
| Creatinine Clearance (mL/min)
| Recommended Dosage of Lamivudine
|
| ≥50
| 150 mg twice daily or 300 mg once daily
|
| 30-49
| 150 mg once daily
|
| 15-29
| 150 mg first dose, then 100 mg once daily
|
| 5-14
| 150 mg first dose, then 50 mg once daily
|
| <5
| 50 mg first dose, then 25 mg once daily
|
No additional dosing of lamivudine is required after routine (4-hour) hemodialysis or peritoneal dialysis.
Although there are insufficient data to recommend a specific dose adjustment of lamivudine in pediatric patients with renal impairment, a reduction in the dose and/or an increase in the dosing interval should be considered.
3 DOSAGE FORMS & STRENGTHS
•
Lamivudine Scored Tablets
150 mg, are white capsule shaped, biconvex, scored film coated tablets debossed with ‘J’ on one side and ‘16’ on the other side, 1 and 6 seperated by a score line.
•
Lamivudine Tablets
300 mg, are white capsule shaped, biconvex, film coated tablets debossed with ‘17’ on one side and ‘J’ on the other side.
4 CONTRAINDICATIONS
Lamivudine tablets are contraindicated in patients with previously demonstrated clinically significant hypersensitivity (e.g., anaphylaxis) to any of the components of the products.
5 WARNINGS AND PRECAUTIONS
5.1 Lactic Acidosis/Severe Hepatomegaly With Steatosis
Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogues alone or in combination, including lamivudine and other antiretrovirals. A majority of these cases have been in women. Obesity and prolonged nucleoside exposure may be risk factors. Particular caution should be exercised when administering lamivudine to any patient with known risk factors for liver disease; however, cases also have been reported in patients with no known risk factors. Treatment with lamivudine should be suspended in any patient who develops clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity (which may include hepatomegaly and steatosis even in the absence of marked transaminase elevations).
5.2 Patients With HIV-1 and Hepatitis B Virus Co-infection
Posttreatment Exacerbations of Hepatitis:
In clinical trials in non-HIV-1- infected patients treated with lamivudine for chronic hepatitis B, clinical and laboratory evidence of exacerbations of hepatitis have occurred after discontinuation of lamivudine. These exacerbations have been detected primarily by serum ALT elevations in addition to re-emergence of HBV DNA. Although most events appear to have been self-limited, fatalities have been reported in some cases. Similar events have been reported from postmarketing experience after changes from lamivudine-containing HIV-1 treatment regimens to non-lamivudine-containing regimens in patients infected with both HIV-1 and HBV. The causal relationship to discontinuation of lamivudine treatment is unknown. Patients should be closely monitored with both clinical and laboratory follow-up for at least several months after stopping treatment. There is insufficient evidence to determine whether re-initiation of lamivudine alters the course of posttreatment exacerbations of hepatitis.
Important Differences Among Lamivudine-Containing Products:
Lamivudine Tablets contain a higher dose of the same active ingredient (lamivudine) than EPIVIR- HBV Tablets. EPIVIR-HBV was developed for patients with chronic hepatitis B. The formulation and dosage of lamivudine in EPIVIR-HBV are not appropriate for patients co-infected with HIV-1 and HBV. Safety and efficacy of lamivudine have not been established for treatment of chronic hepatitis B in patients co-infected with HIV-1 and HBV. If treatment with EPIVIR-HBV is prescribed for chronic hepatitis B for a patient with unrecognized or untreated HIV-1 infection, rapid emergence of HIV-1 resistance is likely to result because of the subtherapeutic dose and the inappropriateness of monotherapy HIV-1 treatment. If a decision is made to administer lamivudine to patients co-infected with HIV-1 and HBV, lamivudine tablets, COMBIVIR
® (lamivudine/zidovudine) Tablets, EPZICOM
®(abacavir sulfate and lamivudine) Tablets, or TRIZIVIR
®(abacavir sulfate, lamivudine, and zidovudine) Tablets should be used as part of an appropriate combination regimen.
Emergence of Lamivudine-Resistant HBV:
In non-HIV-l-infected patients treated with lamivudine for chronic hepatitis B, emergence of lamivudine-resistant HBV has been detected and has been associated with diminished treatment response (see full prescribing information for EPIVIR-HBV for additional information). Emergence of hepatitis B virus variants associated with resistance to lamivudine has also been reported in HIV-1-infected patients who have received lamivudine-containing antiretroviral regimens in the presence of concurrent infection with hepatitis B virus.
5.3 Use With Other Lamivudine- and Emtricitabine-Containing Products
Lamivudine should not be administered concomitantly with other lamivudine- containing products including EPIVIR-HBV Tablets,COMBIVIR (lamivudine/zidovudine) Tablets, EPZICOM (abacavir sulfate and lamivudine) Tablets, or TRIZIVIR (abacavir sulfate, lamivudine, and zidovudine) or emtricitabine-containing products, including ATRIPLA ® (efavirenz, emtricitabine, and tenofovir), EMTRIVA ®(emtricitabine), or TRUVADA ® (emtricitabine and tenofovir), or COMPLERA TM (rilpivirine/emtricitabine/tenofovir).
5.4 Use With Interferon- and Ribavirin-Based Regimens
In vitro studies have shown ribavirin can reduce the phosphorylation of pyrimidine nucleoside analogues such as lamivudine. Although no evidence of a pharmacokinetic or pharmacodynamic interaction (e.g., loss of HIV-l/HCV virologic suppression) was seen when ribavirin was coadministered with lamivudine in HIV-l/HCV co-infected patients [see Clinical Pharmacology (12.3)], hepatic decompensation (some fatal) has occurred in HIV-l/HCV co-infected patients receiving combination antiretroviral therapy for HIV -1 and interferon alfa with or without ribavirin. Patients receiving interferon alfa with or without ribavirin and lamivudine should be closely monitored for treatment-associated toxicities, especially hepatic decompensation. Discontinuation of lamivudine should be considered as medically appropriate. Dose reduction or discontinuation of interferon alfa, ribavirin, or both should also be considered if worsening clinical toxicities are observed, including hepatic decompensation (e.g., Child-Pugh >6). See the complete prescribing information for interferon and ribavirin.
5.5 Pancreatitis
In pediatric patients with a history of prior antiretroviral nucleoside exposure, a history of pancreatitis, or other significant risk factors for the development of pancreatitis, lamivudine should be used with caution. Treatment with lamivudine should be stopped immediately if clinical signs, symptoms, or laboratory abnormalities suggestive of pancreatitis occur [see Adverse Reactions (6.1)] .
5.6 Immune Reconstitution Syndrome
Immune reconstitution syndrome has been reported in patients treated with combination antiretrovira1 therapy, including lamivudine. During the initial phase of combination antiretroviral treatment, patients whose immune system responds may develop an inflammatory response to indolent or residual opportunistic infections (such as
Mycobacterium avium infection, cytomegalovirus,
Pneumocystis jirovecii pneumonia [PCP], or tuberculosis), which may necessitate further evaluation and treatment.
Autoimmune disorders (such as Graves’ disease, polymyositis, and Guillain-Barré syndrome) have also been reported to occur in the setting of immune reconstitution, however, the time to onset is more variable, and can occur many months after initiation of treatment.
5.7 Fat Redistribution
Redistribution/accumulation of body fat including central obesity, dorsocervical fat enlargement (buffalo hump), peripheral wasting, facial wasting, breast enlargement, and "cushingoid appearance" have been observed in patients receiving antiretroviral therapy. The mechanism and long-term consequences of these events are currently unknown. A causal relationship has not been established.
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the labeling:
• Lactic acidosis and severe hepatomegaly with steatosis
[see
Boxed Warning,
Warnings and Precautions (5.1)].
• Severe acute exacerbations of hepatitis B
[see
Boxed Warning,
Warnings and Precautions (5.2)].
• Hepatic decompensation in patients co-infected with HIV-l and Hepatitis C
[see
Warnings and Precautions (5.4)].
• Pancreatitis
[see
Warnings and Precautions (5.5)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adults - Clinical Trials in HIV-1: The safety profile of lamivudine in adults is primarily based on 3,568 HIV-l-infected patients in 7 clinical trials.
The most common adverse reactions are headache, nausea, malaise, fatigue, nasal signs and symptoms, diarrhea and cough.
Selected clinical adverse reactions of in ≥5% of patients during therapy with lamivudine 150 mg twice daily plus zidovudine 200 mg 3 times daily for up to 24 weeks are listed in Table 3.
Table 3. Selected Clinical Adverse Reactions (≥5% Frequency) in Four Controlled Clinical Trials (NUCA3001, NUCA3002, NUCB3001, NUCB3002)
| Adverse Reaction |
Lamivudine 150 mg Twice Daily plus Zidovudine (n = 251) |
Zidovudine a (n = 230) |
| Body as a Whole | ||
| Headache | 35% | 27% |
| Malaise & fatigue | 27% | 23% |
| Fever or chills | 10% | 12% |
| Digestive | ||
| Nausea | 33% | 29% |
| Diarrhea | 18% | 22% |
| Nausea & vomiting | 13% | 12% |
| Anorexia and/or decreased appetite | 10% | 7% |
| Abdominal pain | 9% | 11% |
| Abdominal cramps | 6% | 3% |
| Dyspepsia | 5% | 5% |
| Nervous System | ||
| Neuropathy | 12% | 10% |
| Insomnia & other sleep disorders | 11% | 7% |
| Dizziness | 10% | 4% |
| Depressive Disorders | 9% | 4% |
| Respiratory | ||
| Nasal signs & symptoms | 20% | 11% |
| Cough | 18% | 13% |
| Skin | ||
| Skin rashes | 9% | 6% |
| Musculoskeletal | ||
| Musculoskeletal pain | 12% | 10% |
| Myalgia | 8% | 6% |
| Arthralgia | 5% | 5% |
aEither zidovudine monotherapy or zidovudine in combination with zalcitabine.
Pancreatitis: Pancreatitis was observed in 9 out of 2,613 adult patients (0.3%) who received lamivudine in the controlled clinical trials EPV20001, NUCA3001, NUCB3001, NUCA3002, NUCB3002, and NUCB3007
[see Warnings and Precautions (5.5)].
Lamivudine 300 mg Once Daily
: The types and frequencies of clinical adverse reactions reported in patients receiving lamivudine 300 mg once daily or lamivudine
150 mg twice daily (in 3-drug combination regimens in EPV20001 and EPV40001) for48 weeks were similar.
Selected laboratory abnormalities observed during therapy are summarized in Table 4.
Table 4.Frequencies of Selected Grade 3 to 4 Laboratory Abnormalities in Adults in Four 24-Week Surrogate Endpoint Studies (NUCA3001, NUCA3002, NUCB3001, NUCB3002) and a Clinical End point Study (NUCB3007)
| Test
(Threshold Level) | 24-Week Surogate Endpoint Studies a | Clinical Endpoint Study a | ||
| Lamivudine plus Zidovudine | Zidovudine b | Lamivudine plus Current Therapy | Placebo plus Current Therapy c | |
| Absolute neutrophil count
(<750/mm 3) | 7.2% | 5.4% | 15% | 13% |
| Hemoglobin (<8.0 g/dL) | 2.9% | 1.8% | 2.2% | 3.4% |
| Platelets (<50,000/mm 3) | 0.4% | 1.3% | 2.8% | 3.8% |
| ALT (>5.0 x ULN) | 3.7% | 3.6% | 3.8% | 1.9% |
| AST (>5.0 x ULN) | 1.7% | 1.8% | 4.0% | 2.1% |
| Bilirubin (>2.5 x ULN) | 0.8% | 0.4% | ND | ND |
| Amylase (>2.0 x ULN) | 4.2% | 1.5% | 2.2% | 1.1% |
a The median duration on study was 12 months.
b Either zidovudine monotherapy or zidovudine in combination with zalcitabine.
c Current therapy was either zidovudine, zidovudine plus didonasine, or zidovudine plus zalcitabine
ULN = Upper limit of normal. ND = Not done.
The frequencies of selected laboratory abnormalities reported in patients receiving lamivudine 300 mg once daily or lamivudine 150 mg twice daily (in 3-drug combination regimens in EPV20001 and EPV40001) were similar.
Pediatric Patients - Clinical Trials in HIV-1:
Selected clinical adverse reactions and physical findings with a ≥5% frequency during therapy with lamivudine 4 mg/kg twice daily plus zidovudine 160 mg/m
2 3 times daily in therapy-naive (≤56 days of antiretroviral therapy) pediatric patients are listed in Table 5.
Table 5. Seleted Clinical Adverse Reactions and Physical Findings (≥5% frequency) in Pediatric Patients in Study ACTG300
| Adverse Reaction
| Lamivudine plus Zidovudine (n=236)
| Didanosine (n=235)
|
| Body as a whole
| ||
| Fever
| 25%
| 32%
|
| Digestive
| ||
| Hepatomegaly
| 11%
| 11%
|
| Nausea & vomiting
| 8%
| 7%
|
| Diarrhea
| 8%
| 6%
|
| Stomatitis
| 6%
| 12%
|
| Splenomegaly
| 5%
| 8%
|
| Respiratory
| ||
| Cough
| 15%
| 18%
|
| Abnormal breath sounds/wheezling
| 7%
| 9%
|
| Ear, Nose, and Throat
| ||
| Signs or symptoms of ears
a
| 7%
| 6%
|
| Nasal discharge or congestion
| 8%
| 11%
|
| Other
| ||
| Skin rashes
| 12%
| 14%
|
| Lymphadenopathy
| 9%
| 11%
|
aIncludes pain, discharge, erythema, or swelling of an ear.
Pancreatitis: Pancreatitis, which has been fatal in some cases, has been observed in antiretroviral nucleoside-experienced pediatric patients receiving lamivudine alone or in combination with other antiretroviral agents. In an open-label dose-escalation study (NUCA2002), 14 patients (14%) developed pancreatitis while receiving monotherapy with lamivudine. Three of these patients died of complications of pancreatitis. In a second open-label study (NUCA2005), 12 patients (18%) developed pancreatitis. In Study ACTG300, pancreatitis was not observed in 236 patients randomized to lamivudine plus zidovudine. Pancreatitis was observed in 1 patient in this study who received open- label lamivudine in combination with zidovudine and ritonavir following discontinuation of didanosine monotherapy
[see
Warnings and Precautions (5.5)].
Paresthesias and Peripheral Neuropathies: Paresthesias and peripheral neuropathies were reported in 15 patients (15%) in Study NUCA2002, 6 patients (9%) in Study NUCA2005, and 2 patients (<1%) in Study ACTG300.
Selected laboratory abnormalities experienced by therapy-naive (≤56 days of antiretroviral therapy) pediatric patients are listed in Table 6.
Table 6. Frequencies of Selected Grade 3 to 4 Laboratory Abnormalities in Pediatric Patients in Study ACTG300
| Test (Threshold Level) | Lamivudine plus Zidovudine | Didanosine |
| Absolute neutrophil count (<400/mm 3) | 8% | 3% |
| Hemoglobin (<7.0 g/dL) | 4% | 2% |
| Platelets (<50,000/mm 3) | 1% | 3% |
| ALT (>10 x ULN) | 1% | 3% |
| AST (>10 x ULN) | 2% | 4% |
| Lipase (>2.5 x ULN) | 3% | 3% |
| Total Amylase (>2.5 x ULN) | 3% | 3% |
ULN = Upper limit of normal.
Neonates - Clinical Trials in HIV-1: Limited short-term safety information is available from 2 small, uncontrolled studies in South Africa in neonates receiving lamivudine with or without zidovudine for the first week of life following maternal treatment starting at Week 38 or 36 of gestation
[see
Clinical Pharmacology (12.3)].
Selected adverse reactions reported in these neonates included increased liver function tests,anemia, diarrhea, electrolyte disturbances, hypoglycemia, jaundice and hepatomegaly,rash,respiratory infections, and sepsis; 3 neonates died (1from gastroenteritis with acidosis and convulsions, 1 from traumatic injury,and 1 from unknown causes). Two other nonfatal gastroenteritis or diarrhea cases were reported, including 1 with convulsions; 1 infant had transient renal insufficiency associated with dehydration. The absence of control groups limits assessments of causality, but it should be assumed that perinatally exposed infants may be at risk for adverse reactions comparable to those reported in pediatric and adult HIV-1-infected patients treated with lamivudine-containing combination regimens. Long-term effects of in utero and infant lamivudine exposure are not known.
6.2 Postmarketing Experience
In addition to adverse reactions reported from clinical trials, the following adverse reactions have been reported during postmarketing use of lamivudine. Because these reactions are reported voluntarily from a population of unknown size, estimates of frequency cannot be made. These reactions have been chosen for inclusion due to a combination of their seriousness, frequency of reporting, or potential causal connection to lamivudine.
Body as a Whole: Redistribution/accumulation of body fat
[see
Warnings and Precautions (5.7)]
.
Endocrine and Metabolic: Hyperglycemia.
General: Weakness.
Hemic and Lymphatic: Anemia (including pure red cell aplasia and severe anemias progressing on therapy).
Hepatic and Pancreatic: Lactic acidosis and hepatic steatosis, posttreatment exacerbation of hepatitis B
[see
Boxed Warning,
Warnings and Precautions (5.1,
5.2)]
.
Hypersensitivity: Anaphylaxis, urticaria.
Musculoskeletal: Muscle weakness, CPK elevation, rhabdomyolysis.
Skin: Alopecia, pruritus
7 DRUG INTERACTIONS
Lamivudine is predominantly eliminated in the urine by active organic cationic secretion. The possibility of interactions with other drugs administered concurrently should be considered, particularly when their main route of elimination is active renal secretion via the organic cationic transport system (e.g., trimethoprim). No data are available regarding interactions with other drugs that have renal clearance mechanisms similar to that of lamivudine.
7.1 Interferon- and Ribavirin-Based Regimens
Although no evidence of a pharmacokinetic or pharmacodynamic interaction (e.g., loss of HIV-1/HCV virologic suppression) was seen when ribavirin was coadministered with lamivudine in HIV-l/HCV co-infected patients, hepatic decompensation (some fatal) has occurred in HIV–l/HCV co-infected patients receiving combination antiretroviral therapy for HIV-1 and interferon alfa with or without ribavirin [see Warnings and Precautions (5.4), Clinical Pharmacology (12.3)] .
7.2 Zalcitabine
Lamivudine and zalcitabine may inhibit the intracellular phosphorylation of one another. Therefore, use of lamivudine in combination with zalcitabine is not recommended.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Teratogenic Effects: Pregnancy Category C. There are no adequate and well- controlled studies of lamivudine in pregnant women. Animal reproduction studies in rats and rabbits revealed no evidence of teratogenicity. Increased early embryolethality occurred in rabbits at exposure levels similar to those in humans. Lamivudine should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Lamivudine pharmacokinetics were studied in pregnant women during 2 clinical studies conducted in South Africa. The study assessed pharmacokinetics in: 16 women at 36 weeks gestation using 150 mg lamivudine twice daily with zidovudine, 10 women at 38 weeks gestation using 150 mg lamivudine twice daily with zidovudine, and 10 women at 38 weeks gestation using lamivudine 300 mg twice daily without other antiretrovirals. These studies were not designed or powered to provide efficacy information. Lamivudine pharmacokinetics in the pregnant women were similar to those seen in non-pregnant adults and in postpartum women. Lamivudine concentrations were generally similar in maternal, neonatal, and umbilical cord serum samples. In a subset of subjects, lamivudine amniotic fluid specimens were collected following natural rupture of membranes. Amniotic fluid concentrations of lamivudine were typically 2 times greater than maternal serum levels and ranged from 1.2 to 2.5 mcg/mL (150 mg twice daily) and 2.1 to 5.2 mcg/mL (300 mg twice daily). It is not known whether risks of adverse events associated with lamivudine are altered in pregnant women compared with other HIV-1- infected patients.
Animal reproduction studies performed at oral doses up to 130 and 60 times the adult dose in rats and rabbits, respectively, revealed no evidence of teratogenicity due to lamivudine. Increased early embryolethality occurred in rabbits at exposure levels similar to those in humans. However, there was no indication of this effect in rats at exposure levels up to 35 times those in humans. Based on animal studies, lamivudine crosses the placenta and is transferred to the fetus.
[see
Non clinical Toxicology (13.2)].
Antiretroviral Pregnancy Registry: To monitor maternal-fetal outcomes of pregnant women exposed to lamivudine, a Pregnancy Registry has been established. Physicians are encouraged to register patients by calling 1-800-258-4263.
8.3 Nursing Mothers
The Centers for Disease Control and Prevention recommend that HIV-1-infected mothers in the United States not breastfeed their infants to avoid risking postnatal transmission of HIV-l infection. Because of the potential for serious adverse reactions in nursing infants and HIV-l transmission, mothers should be instructed not to breastfeed if they are receiving lamivudine.
Lamivudine is excreted into human milk. Samples of breast milk obtained from 20 mothers receiving lamivudine monotherapy (300 mg twice daily) or combination therapy (150 mg lamivudine twice daily and 300 mg zidovudine twice daily) had measurable concentrations of lamivudine.
8.4 Pediatric Use
The safety and effectiveness of twice-daily lamivudine in combination with other antiretroviral agents have been established in pediatric patients 3 months and older [see Adverse Reactions (6.1), Clinical Pharmacology (12.3), Clinical Studies (14.2)].
8.5 Geriatric Use
Clinical studies of lamivudine did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. In general, dose selection for an elderly patient should be cautious, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy. In particular, because lamivudine is substantially excreted by the kidney and elderly patients are more likely to have decreased renal function, renal function should be monitored and dosage adjustments should be made accordingly [see Dosage and Administration (2.3), Clinical Pharmacology (12.3)] .
8.6 Patients With Impaired Renal Function
Reduction of the dosage of lamivudine is recommended for patients with impaired renal function [see Dosage and Administration (2.3), Clinical Pharmacology (12.3)]
10 OVERDOSAGE
There is no known antidote for lamivudine. One case of an adult ingesting 6 g of lamivudine was reported; there were no clinical signs or symptoms noted and hematologic tests remained normal. Two cases of pediatric overdose were reported in Study ACTG300. One case involved a single dose of 7 mg/kg of lamivudine; the second case involved use of 5 mg/kg of lamivudine twice daily for 30 days. There were no clinical signs or symptoms noted in either case. Because a negligible amount of lamivudine was removed via (4-hour) hemodialysis, continuous ambulatory-peritoneal dialysis, and automated peritoneal dialysis, it is not known if continuous hemodialysis would provide clinical benefit in a lamivudine overdose event. If overdose occurs, the patient should be monitored, and standard supportive treatment applied as required.
11 DESCRIPTION
Lamivudine USP (also known as 3TC), a synthetic nucleoside analogue with activity against HIV-1 and HBV. The chemical name of lamivudine USP is 2(1H) - Pyrimidinone, 4-amino-1- [2- (hydroxymethyl)-1,3-oxathio-lan-5-yl], (2R-cis)-. It has a molecular formula of C 8H 11N 3O 3S and a molecular weight of 229.26. It has the following structural formula:
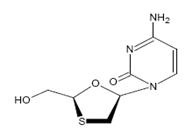
Lamivudine USP is a white to an off white solid and soluble in water.
Lamivudine tablets are for oral administration. Each film-coated tablet contains 150 mg or 300 mg of lamivudine USP and the following inactive ingredients: crospovidone, isomalt, isopropyl alcohol, magnesium stearate and methylene chloride. The tablets are coated with opadry white containing hypromellose, polyethylene glycol, polysorbate 80 and titanium dioxide.
12 CLINICAL PHARMACOLOGY
12.3 Pharmacokinetics
Pharmacokinetics in Adults: The pharmacokinetic properties of lamivudine have been studied in asymptomatic, HIV-l-infected adult patients after administration of single intravenous (IV) doses ranging from 0.25 to 8 mg/kg, as well as single and multiple (twice-daily regimen) oral doses ranging from 0.25 to 10 mg/kg.
The pharmacokinetic properties of lamivudine have also been studied as single and multiple oral doses ranging from 5 mg to 600 mg/day administered to HBV-infected patients.
The steady-state pharmacokinetics properties of the lamivudine 300-mg tablet once daily for 7 days compared with the lamivudine 150-mg tablet twice daily for 7 days were assessed in a crossover study in 60 healthy volunteers. Lamivudine 300 mg once daily resulted in lamivudine exposures that were similar to lamivudine 150 mg twice daily with respect to plasma AUC
24,.ss; however, C
max,ss was 66% higher and the trough value was 53% lower compared with the 150-mg twice-daily regimen. Intracellular lamivudine triphosphate exposures in peripheral blood mononuclear cells were also similar with respect to AUC
24,ss and C
max24,ss; however, trough values were lower compared with the 150-mg twice-daily regimen. Inter-subject variability was greater for intracellular lamivudine triphosphate concentrations versus lamivudine plasma trough concentrations. The clinical significance of observed differences for both plasma lamivudine concentrations and intracellular lamivudine triphosphate concentrations is not known.
Absorption and Bioavailability: Lamivudine was rapidly absorbed after oral administration in HIV-1-infected patients. Absolute bioavailability in 12 adult patients was 86%±16% (mean ± SD) for the 150-mg tablet and 87%±13% for the oral solution. After oral administration of 2 mg/kg twice a day to 9 adults with HIV-1, the peak serum lamivudine concentration (C
max) was 1.5 ±0.5 mcg/mL (mean ± SD). The area under the plasma concentration versus time curve (AUC) and C
max increased in proportion to oral dose over the range from 0.25 to 10 mg/kg.
The accumulation ratio of lamivudine in HIV-1-positive asymptomatic adults with normal renal function was 1.50 following 15 days of oral administration of 2 mg/kg twice daily.
Effects of Food on Oral Absorption: An investigational 25-mg dosage form of lamivudine was administered orally to 12 asymptomatic, HIV-1-infected patients on 2 occasions, once in the fasted state and once with food (1,099 kcal; 75 grams fat, 34 grams protein, 72 grams carbohydrate). Absorption of lamivudine was slower in the fed state (Tmax: 3.2 ±1.3 hours) compared with the fasted state (Tmax: 0.9 ± 0.3 hours); C
max in the fed state was 40%±23% (mean ± SD) lower than in the fasted state. There was no significant difference in systemic exposure (AUC¥) in the fed and fasted states;therefore, lamivudine tablets may be administered with or without food.
Distribution: The apparent volume of distribution after IV administration of lamivudine to 20 patients was 1.3± 0.4 L/kg, suggesting that lamivudine distributes into extravascular spaces. Volume of distribution was independent of dose and did not correlate with body weight.
Binding of lamivudine to human plasma proteins is low (<36%). In vitro studies showed that over the concentration range of 0.1 to 100 mcg/mL, the amount of lamivudine associated with erythrocytes ranged from 53% to 57% and was independent of concentration.
Metabolism: Metabolism of lamivudine is a minor route of elimination. In man, the only known metabolite of lamivudine is the trans-sulfoxide metabolite. Within
12 hours after a single oral dose of lamivudine in 6 HIV-l-infected adults, 5.2% ± 1.4% (mean ± SD) of the dose was excreted as the trans-sulfoxide metabolite in the urine. Serum concentrations of this metabolite have not been determined.
Elimination: The majority of lamivudine is eliminated unchanged in urine by active organic cationic secretion. In 9 healthy subjects given a single 300-mg oral dose of lamivudine, renal clearance was 199.7 ± 56.9 mL/min (mean ± SD). In 20 HIV-l-infected patients given a single IV dose, renal clearance was 280.4 ± 75.2 mL/min (mean ± SD), representing 71% ± 16% (mean ±SD) of total clearance of lamivudine.
In most single-dose studies in HIV-1-infected patients, HBV-infected patients, or healthy subjects with serum sampling for 24 hours after dosing, the observed mean elimination half-life (t1/2) ranged from 5 to 7 hours. In HIV-1-infected patients, total clearance was 398.5 ± 69.1 mL/min (mean ± SD). Oral clearance and elimination half- life were independent of dose and body weight over an oral dosing range of 0.25 to10 mg/kg.
Special Populations: Renal Impairment: The pharmacokinetic properties of lamivudine have been determined in a small group of HIV-1-infected adults with impaired renal function (Table 7).
Table 7. Pharmacokinetic Parameters (Mean ± SD) After a Single 300-mg Oral Dose of Lamivudine in 3 Groups of Adults With Varying Degrees of Renal Function
| Parameter | Creatitine Clearance Criterion (Number of Subjects) | ||
| >60 mL/min | 10-30 mL/min | <10 mL/min | |
| Creatitine clearance | 111± 14 | 28 ± 8 | 6 ± 2 |
| C max (mcg/mL) | 2.6 ± 0.5 | 3.6 ± 0.8 | 5.8 ± 1.2 |
| AUC∞ (mcg.hr/mL) | 11.0 ± 1.7 | 48.0 ± 19 | 157± 74 |
| Cl/F (mL/min) | 464 ± 76 | 114 ± 34 | 36 ± 11 |
Exposure (AUC∞), C
max, and half-life increased with diminishing renal function (as expressed by creatinine clearance). Apparent total oral clearance (Cl/F) of lamivudine decreased as creatinine clearance decreased. Tmax was not significantly affected by renal function. Based on these observations, it is recommended that the dosage of lamivudine be modified in patients with renal impairment
[see
Dosage and Administration (2.3)].
Based on a study in otherwise healthy subjects with impaired renal function, hemodialysis increased lamivudine clearance from a mean of 64 to 88 mL/min; however, the length of time of hemodialysis (4 hours) was insufficient to significantly alter mean lamivudine exposure after a single dose administration. Continuous ambulatory peritoneal dialysis and automated peritoneal dialysis have negligible effects on lamivudine clearance. Therefore, it is recommended, following correction of dose for creatinine clearance, that no additional dose modification be made after routine hemodialysis or peritoneal dialysis.
It is not known whether lamivudine can be removed by continuous (24-hour) hemodialysis.
The effects of renal impairment on lamivudine pharmacokinetics in pediatric patients are not known.
Hepatic Impairment: The pharmacokinetic properties of lamivudine have been determined in adults with impaired hepatic function. Pharmacokinetic parameters were not altered by diminishing hepatic function; therefore, no dose adjustment for lamivudine is required for patients with impaired hepatic function. Safety and efficacy of lamivudine have not been established in the presence of decompensate liver disease.
Pediatric Patients: In Study NUCA2002, pharmacokinetic properties of lamivudine were assessed in a subset of 57 HIV-1-infected pediatric patients (age range:
4.8 months to 16 years, weight range: 5 to 66 kg) after oral and IV administration of 1, 2,4, 8, 12, and 20 mg/kg/day. The mechanism for the diminished absolute bioavailabilty of lamivudine in infants and children is unknown.
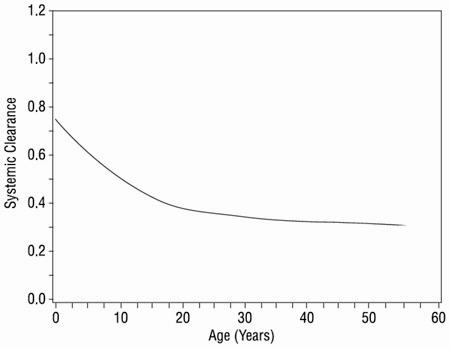
Systemic clearance decreased with increasing age in pediatric patients, as shown in Figure 1.
Figure 1. Systemic Clearance (L/hr•kg) of Lamivudine in Relation to Age
After oral administration of lamivudine 4 mg/kg twice daily to 11 pediatric patients ranging from 4 months to 14 years of age, C
max was 1.1 ± 0.6 mcg/mL and half- life was 2.0 ± 0.6 hours. (In adults with similar blood sampling, the half-life was 3.7 ± 1 hours.) Total exposure to lamivudine, as reflected by mean AUC values, was comparable between pediatric patients receiving an 8-mg/kg/day dose and adults receiving a 4-mg/kg/day dose.
Distribution of lamivudine into cerebrospinal fluid (CSF) was assessed in 38 pediatric patients after multiple oral dosing with lamivudine. CSF samples were collected between 2 and 4 hours postdose. At the dose of 8 mg/kg/day, CSF lamivudine concentrations in 8 patients ranged from 5.6% to 30.9% (mean ± SD of 14.2% ± 7.9%) of the concentration in a simultaneous serum sample, with CSF lamivudine concentrations ranging from 0.04 to 0.3 mcg/mL.
Limited, uncontrolled pharmacokinetic and safety data are available from administration of lamivudine (and zidovudine) to 36 infants up to 1 week of age in 2 studies in South Africa. In these studies, lamivudine clearance was substantially reduced in 1-week-old neonates relative to pediatric patients (>3 months of age) studied previously. There is insufficient information to establish the time course of changes in clearance between the immediate neonatal period and the age-ranges >3 months old
[see Adverse Reactions (6.1)].
Geriatric Patients: The pharmacokinetics of lamivudine after administration of lamivudine to patients over 65 years of age have not been studied
[see Use in Specific Populations (8.5)].
Gender:
There are no significant gender differences in lamivudine pharmacokinetics.
Race: There are no significant racial differences in lamivudine pharmacokinetics.
Drug Interactions: Interferon Alfa
: There was no significant pharmacokinetic interaction between lamivudine and interferon alfa in a study of 19 healthy male subjects
[see Warnings and Precautions (5.4)].
Ribavirin: In vitro
data indicate ribavirin reduces phosphorylation of lamivudine, stavudine, and zidovudine. However, no pharmacokinetic (e.g., plasma concentrations or intracellular triphosphorylated active metabolite concentrations) or pharmacodynamic (e.g., loss of HIV-l/HCV virologic suppression) interaction was observed when ribavirin and lamivudine (n = 18), stavudine (n = 10), or zidovudine (n = 6) were coadministered as part of a multi-drug regimen to HIV-l/HCV co-infected patients
[see
Warnings and Precautions (5.4)].
Trimethoprim/Sulfamethoxazole: Lamivudine and TMP/SMX were co administered to 14 HIV-1-positive patients in a single-center, open-label, randomized, crossover study. Each patient received treatment with a single 300-mg dose of lamivudine and TMP160 mg/SMX 800 mg once a day for 5 days with concomitant administration of lamivudine 300 mg with the fifth dose in a crossover design. Coadministration of TMP/SMX with lamivudine resulted in an increase of 43% ± 23% (mean ± SD) in lamivudine AUC¥, a decrease of 29% ± 13% in lamivudine oral clearance, and a decrease of 30% ± 36% in lamivudine renal clearance. The pharmacokinetic properties of TMP and SMX were not altered by coadministration with lamivudine
[see
Drug Interactions (7.3)].
Zidovudine: No clinically significant alterations in lamivudine or zidovudine pharmacokinetics were observed in 12 asymptomatic HIV-l-infected adult patients given a single dose of zidovudine (200 mg) in combination with multiple doses of lamivudine (300 mg q 12 hr)
[see
Drug Interactions (7.4)].
12.4 Microbiology
Mechanism of Action: Intracellularly, lamivudine is phosphorylated to its active 5’-triphosphate metabolite, lamivudine triphosphate (3TC-TP). The principal mode of action of 3TC-TP is the inhibition of HIV-1 reverse transcriptase (RT) via DNA chain termination after incorporation of the nucleotide analogue into viral DNA. 3TC-TP is a weak inhibitor of mammalian DNA polymerases α, β, and γ.
Antiviral Activity: The antiviral activity of lamivudine against HIV-l was assessed in a number of cell lines (including monocytes and fresh human peripheral blood lymphocytes) using standard susceptibility assays. EC50 values (50% effective concentrations) were in the range of 0.003 to 15µM (l µM = 0.23 mcg/mL). HIV-l from therapy-naive subjects with no amino acid substitutions associated with resistance gavemedian EC
50 values of 0.429 µM (range: 0.200 to 2.007 µM) from Virco (n = 92 baseline samples from COLA40263) and 2.35 µM (1.37 to 3.68 µM) from Monogram Biosciences (n = 135 baseline samples from ESS30009). The EC
50 values of lamivudine against different HIV-l clades (A-G) ranged from 0.001 to 0.120 µM, and against HIV-2 isolates from 0.003 to 0.120 µM in peripheral blood mononuclear cells. Ribavirin (50 µM) decreased the anti-HIV-1 activity of lamivudine by 3.5 fold in MT-4 cells. In HIV-1- infected MT-4 cells, lamivudine in combination with zidovudine at various ratios exhibited synergistic antiretroviral activity. Please see the full prescribing information for EPIVIR-HBV for information regarding the inhibitory activity of lamivudine against HBV.
Resistance: Lamivudine-resistant variants of HIV-1 have been selected in cell culture. Genotypic analysis showed that the resistance was due to a specific amino acid substitution in the HIV-1 reverse transcriptase at codon 184 changing the methionine to either isoleucine or valine (M184V/I).
HIV-1 strains resistant to both lamivudine and zidovudine have been isolated from patients. Susceptibility of clinical isolates to lamivudine and zidovudine was monitored in controlled clinical trials. In patients receiving lamivudine monotherapy or combination therapy with lamivudine plus zidovudine, HIV-1 isolates from most patients became phenotypically and genotypically resistant to lamivudine within 12 weeks. In some patients harboring zidovudine-resistant virus at baseline, phenotypic sensitivity to zidovudine was restored by 12 weeks of treatment with lamivudine and zidovudine. Combination therapy with lamivudine plus zidovudine delayed the emergence of mutations conferring resistance to zidovudine.
Lamivudine-resistant HBV isolates develop substitutions (rtM204V/I) in the YMDD motif of the catalytic domain of the viral reverse transcriptase. rtM204V/I substitutions are frequently accompanied by other substitutions (rtV173L, rtL180M) which enhance the level of lamivudine resistance or act as compensatory mutations improving replication efficiency. Other substitutions detected in lamivudine-resistant HBV isolates include: rtL80I and rtA181T. Similar HBV mutants have been reported in HIV-1-infected patients who received lamivudine-containing antiretroviral regimens in the presence of concurrent infection with hepatitis B virus
[see
Warnings and Precautions (5.2)].
Cross-Resistance: Lamivudine-resistant HIV-1 mutants were cross-resistant to didanosine (ddI) and zalcitabine (ddC). In some patients treated with zidovudine plus didanosine or zalcitabine, isolates resistant to multiple reverse transcriptase inhibitors, including lamivudine, have emerged.
Genotypic and Phenotypic Analysis of On-Therapy HIV-1 Isolates From Patients With Virologic Failure: Study EPV20001: Fifty-three of 554 (10%) patients enrolled in EPV20001 were identified as virological failures (plasma HIV-l RNA level ≥400 copies/mL) by Week 48. Twenty-eight patients were randomized to the lamivudine once-daily treatment group and 25 to the lamivudine twice-daily treatment group. The median baseline plasma HIV-l RNA levels of patients in the lamivudine once-daily group and lamivudine twice-daily group were 4.9 log10 copies/mL and 4.6 log10 copies/mL, respectively.
Genotypic analysis of on-therapy isolates from 22 patients identified as virologic failures in the lamivudine once-daily group showed that isolates from 0/22 patients contained treatment-emergent amino acid substitutions associated with zidovudine resistance (M41L, D67N, K70R, L210W, T215Y/F, or K219Q/E), isolates from 10/22 patients contained treatment-emergent amino acid substitutions associated with efavirenz resistance (L100I, K101E, K103N, V108I, or Y181C), and isolates from 8/22 patients contained a treatment-emergent lamivudine resistance-associated substitution (M184I or M184V).
Genotypic analysis of on-therapy isolates from patients (n = 22) in the lamivudine twice-daily treatment group showed that isolates from 1/22 patients contained treatment-emergent zidovudine resistance substitutions, isolates from 7/22 contained treatment-emergent efavirenz resistance substitutions, and isolates from 5/22 contained treatment-emergent lamivudine resistance substitutions.
Phenotypic analysis of baseline-matched on-therapy HIV-l isolates from patients (n =13) receiving lamivudine once daily showed that isolates from 12/13 patients were susceptible to zidovudine; isolates from 8/13 patients exhibited a 25- to 295-fold decrease in susceptibility to efavirenz, and isolates from 7/13 patients showed an 85- to 299-fold decrease in susceptibility to lamivudine.
Phenotypic analysis of baseline-matched on-therapy HIV-1 isolates from patients (n=13) receiving lamivudine twice daily showed that isolates from all 13 patients were susceptible to zidovudine; isolates from 3/13 patients exhibited a 21- to 342-fold decrease in susceptibility to efavirenz, and isolates from 4/13 patients exhibited a 29- to 159-fold decrease in susceptibility to lamivudine.
Study EPV40001: Fifty patients received zidovudine 300 mg twice daily plus abacavir 300 mg twice daily plus lamivudine 300 mg once daily and 50 patients received zidovudine 300 mg plus abacavir 300 mg plus lamivudine 150 mg all twice daily. The median baseline plasma HIV-1 RNA levels for patients in the 2 groups were 4.79 log10 copies/mL and 4.83 log10 copies/mL, respectively. Fourteen of 50 patients in the lamivudine once-daily treatment group and 9 of 50 patients in the lamivudine twice-daily group were identified as virologic failures.
Genotypic analysis of on-therapy HIV-1 isolates from patients (n = 9) in the lamivudine once-daily treatment group showed that isolates from 6 patients had an abacavir and/or lamivudine resistance-associated substitution M184V alone. On-therapy isolates from patients (n = 6) receiving lamivudine twice daily showed that isolates from 2 patients had M184 and isolates from 2 patients harbored the M184Vsubstitution in combination with zidovudine resistance-associated amino acid substitutions.
Phenotypic analysis of on-therapy isolates from patients (n = 6) receiving lamivudine once daily showed that HIV-l isolates from 4 patients exhibited a 32- to
53-fold decrease in susceptibility to lamivudine. HIV-1 isolates from these 6 patients were susceptible to zidovudine.
Phenotypic analysis of on-therapy isolates from patients (n = 4) receiving lamivudine twice daily showed that HIV-l isolates from 1 patient exhibited a 45-fold decrease in susceptibility to lamivudine and a 4.5-fold decrease in susceptibility to zidovudine.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis & Mutagenesis & Impairment Of Fertility
Long-term carcinogenicity studies with lamivudine in mice and rats showed no evidence of carcinogenic potential at exposures up to 10 times (mice) and 58 times (rats) those observed in humans at the recommended therapeutic dose for HIV-l infection. Lamivudine was not active in a microbial mutagenicity screen or an in vitro cell transformation assay, but showed weak in vitro mutagenic activity in a cytogenetic assay using cultured human lymphocytes and in the mouse lymphoma assay. However, lamivudine showed no evidence of in vivo genotoxic activity in the rat at oral doses of up to 2,000 mg/kg, producing plasma levels of 35 to 45 times those in humans at the recommended dose for HIV-l infection. In a study of reproductive performance, lamivudine administered to rats at doses up to 4,000 mg/kg/day, producing plasma levels 47 to 70 times those in humans, revealed no evidence of impaired fertility and no effect on the survival, growth, and development to weaning of the offspring.
13.2 Reproductive Toxicology Studies
Reproduction studies have been performed in rats and rabbits at orally administered doses up to 4,000 mg/kg/day and 1,000 mg/kg/day, respectively, producing plasma levels up to approximately 35 times that for the adult HIV dose. No evidence of teratogenicity due to lamivudine was observed. Evidence of early embryolethality was seen in the rabbit at exposure levels similar to those observed in humans, but there was no indication of this effect in the rat at exposure levels up to 35 times those in humans. Studies in pregnant rats and rabbits showed that lamivudine is transferred to the fetus through the placenta.
14 CLINICAL STUDIES
The use of lamivudine is based on the results of clinical studies in HIV-1-infected patients in combination regimens with other antiretroviral agents. Information from trials with clinical endpoints or a combination of CD4+ cell counts and HIV-l RNA measurements is included below as documentation of the contribution of lamivudine to a combination regimen in controlled trials.
14.1 Adults
Clinical Endpoint Study: NUCB3007 (CAESAR) was a multi-center, double-blind, placebo-controlled study comparing continued current therapy (zidovudine alone [62% of patients] or zidovudine with didanosine or zalcitabine [38% of patients]) to the addition of lamivudine or lamivudine plus an investigational non-nucleoside reverse transcriptase inhibitor (NNRTI), randomized 1:2:1. A total of 1,816 HIV-l-infected adults with 25 to 250 CD4+ cells/mm 3 (median = 122 cells/mm 3) at baseline were enrolled: median age was 36 years, 87% were male, 84% were nucleoside-experienced, and 16% were therapy-naive. The median duration on study was 12 months. Results are summarized in Table 8.
Table 8. Number of Patients (%) With at Least One HIV-1 Disease Progression Event or Death
|
Endpoint | Current Therapy
(n= 460) | Lamivudine plus Current Therapy
(n=896) | Lamivudine plus an NNRTI
a plus Current Therapy (n=460)
|
| HIV-1 progression or death
| 90 (19.6%)
| 86 (9.6%)
| 41 (8.9%)
|
| Death
| 27 (5.9%)
| 23 (2.6%)
|
14 (3.0%) |
a An investigational non-nucleoside reverse transcriptase inhibitor not approved in the United States
Surrogate Endpoint Studies:
Dual Nucleoside Analogue Studies: Principal clinical trials in the initial development of lamivudine compared lamivudine/zidovudine combinations with zidovudine monotherapy or with zidovudine plus zalcitabine. These studies demonstrated the antiviral effect of lamivudine in a 2-drug combination. More recent uses of lamivudine in treatment of HIV-1 infection incorporate it into multiple- drug regimens containing at least 3 antiretroviral drugs for enhanced viral suppression.
Dose Regimen Comparison Surrogate Endpoint Studies in Therapy-Naive Adults: EPV20001 was a multi-center, double-blind, controlled study in which patients were randomized 1:1 to receive lamivudine 300 mg once daily or lamivudine 150 mg twice daily, in combination with zidovudine 300 mg twice daily and efavirenz 600 mg once daily. A total of 554 antiretroviral treatment-naive HIV-1-infected adults enrolled: male (79%), Caucasian (50%), median age of 35 years, baseline CD4+ cell counts of 69 to 1,089 cells/mm
3 (median = 362 cells/mm
3), and median baseline plasma HIV-1 RNA of 4.66 log
10 copies/mL. Outcomes of treatment through 48 weeks are summarized in Figure 2 and Table 9.
Figure 2. Virologic Response Through Week 48, EPV20001
ab (Intent-to-Treat)
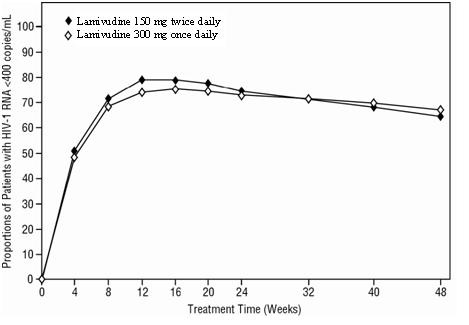
a Roche AMPLICOR HIV-1 MONITOR.
b Responders at each visit are patients who had achieved and maintained
HIV-1 RNA<400 copies/mL without discontinuation by that visit.
Table 9. Outcomes of Randomized Treatment Through 48 Weeks (Intent-to- Treat)
| Outcome
| Lamivudine300mg
Once Daily plus Zidovudine plus Efavirenz (n = 278) | Lamivudine150mg
Twice Daily plus Zidovudine plus Efavirenz (n = 276) |
| Responder
a
| 67%
| 65%
|
| Virologic failure
b
| 8%
| 8%
|
| Discontinued due to clinical progression
| <1%
| 0%
|
| Discontinued due to adverse events
| 6%
| 12%
|
| Discontinued due to other reasons
c
| 18%
| 14%
|
a Achieved confirmed plasma HIV-1 RNA <400 copies/mL and maintained through 48 weeks.
b Achieved suppression but rebounded by Week 48, discontinued due to virologic failure, insufficient viral response according to the investigator, or never suppressed through
Week 48.
c Includes consent withdrawn, lost to followup, protocol violation, data outside the study-defined schedule, and randomized but never initiated treatment.
The proportions of patients with HIV-l RNA <50 copies/mL (via Roche Ultrasensitive assay) through Week 48 were 61% for patients receiving lamivudine 300 mg once daily and 63% for patients receiving lamivudine 150 mg twice daily. Median increases in CD4+ cell counts were 144 cells/mm
3 at Week 48 in patients receiving lamivudine 300 mg once daily and 146 cells/mm
3 for patients receiving lamivudine 150 mg twice daily.
A small, randomized, open-label pilot study, EPV40001, was conducted in Thailand. A total of 159 treatment-naive adult patients (male 32%, Asian 100%, median age 30 years, baseline median CD4+ cell count 380 cells/mm
3, median plasma HIV-1 RNA 4.8 log
10 copies/mL) were enrolled. Two of the treatment arms in this study provided a comparison between lamivudine 300 mg once daily (n = 54) and lamivudine 150 mg twice daily (n = 52), each in combination with zidovudine 300 mg twice daily and abacavir 300 mg twice daily. In intent-to-treat analyses of 48-week data, the proportions of patients with HIV-1 RNA below 400 copies/mL were 61% (33/54) in the group randomized to once-daily lamivudine and 75% (39/52) in the group randomized to receive all 3 drugs twice daily; the proportions with HIV-l RNA below 50 copies/mL were 54% (29/54) in the once-daily lamivudine group and 67% (35/52) in the all-twice- daily group; and the median increases in CD4+ cell counts were 166 cells/mm
3 in the once-daily lamivudine group and 216 cells/mm
3 in the all-twice-daily group.
14.2 Pediatric Patients
Clinical Endpoint Study: ACTG300 was a multi-center, randomized, double-blind study that provided for comparison of lamivudine plus RETROVIR (zidovudine) with didanosine monotherapy. A total of 471 symptomatic, HIV-1-infected therapy-naive (≤56 days of antiretroviral therapy) pediatric patients were enrolled in these 2 treatment arms. The median age was 2.7 years (range: 6 weeks to 14 years), 58% were female, and 86% were non-Caucasian. The mean baseline CD4+ cell count was 868 cells/mm 3 (mean: 1,060 cells/mm3 and range: 0 to 4,650 cells/mm3 for patients ≤5 years of age; mean: 419 cells/mm 3 and range: 0 to 1,555 cells/mm 3 for patients >5 years of age) and the mean baseline plasma HIV-1 RNA was 5.0 log 10 copies/mL. The median duration on study was 10.1 months for the patients receiving lamivudine plus zidovudine and 9.2 months for patients receiving didanosine monotherapy. Results are summarized in Table 10.
Table 10. Number of Patients (%) Reaching a Primary Clinical Endpoint (Disease Progression or-Death)
| Endpoint
| Lamivudine plus
Zidovudine (n = 236) | Didanosine
(n = 235) |
| HIV-1 disease progression or death (total)
| 15 (6.4%)
| 37 (15.7%)
|
| Physical growth failure
| 7 (3.0%)
| 6 (2.6%)
|
| Central nervous system deterioration
| 4 (1.7%)
| 12 (5.1%)
|
| CDC Clinical Category C
| 2 (0.8%)
| 8 (3.4%)
|
| Death
| 2 (0.8%)
| 11 (4.7%)
|
16 HOW SUPPLIED/STORAGE AND HANDLING
Lamivudine Scored Tablets, 150 mg
White capsule shaped, biconvex, scored film coated tablets debossed with ‘J’ on one side and ‘16’ on the other side, 1 and 6 seperated by a score line.
NDC 50268-459-15 10 tablets per card, 5 cards per carton
Lamivudine Tablets, 300 mg
White capsule shaped, biconvex, film coated tablets debossed with ‘17’ on one side and ‘J’ on the other side.
NDC 50268-460-13 10 tablets per card, 3 cards per carton
Dispensed in blister punch material. For Institutional Use Only.
Store at 20° to 25°C (68° to 77°F) [see USP Controlled Room Temperature]. Preserve in well-closed, light-resistant containers.
17 PATIENT COUNSELING INFORMATION
17.1 Advice for the Patient
Lactic Acidosis/Hepatomegaly: Patients should be informed that some HIV medicines, including lamivudine, can cause a rare, but serious condition called lactic acidosis with liver enlargement (hepatomegaly) [see
Warnings and Precautions (5.1)].
HIV-1/HBV Co-infection:
Patients co-infected with HIV-1 and HBV should be informed that deterioration of liver disease has occurred in some cases when treatment with lamivudine was discontinued. Patients should be advised to discuss any changes in regimen with their physician [see
Warnings and Precautions (5.2)].
Differences in Formulations of Lamivudine:
Patients should be advised that lamivudine tablets contain a higher dose of the same active ingredient (lamivudine) as EPIVIR-HBV Tablets. If a decision is made to include lamivudine in the HIV-1 treatment regimen of a patient co-infected with HIV-1 and HBV, the formulation and dosage of lamivudine in EPIVIR (not EPIVIR-HBV) should be used [see
Warnings and Precautions (5.2)].
Use With Other Lamivudine- and Emtricitabine-Containing Products:
Lamivudine should not be coadministered with drugs containing lamivudine or emtricitabine, including COMBIVIR (lamivudine/zidovudine) Tablets, EPZICOM (abacavir sulfate and lamivudine) Tablets, TRIZIVIR (abacavir sulfate, lamivudine, and zidovudine), ATRIPLA (efavirenz, emtricitabine, and tenofovir), EMTRIVA (emtricitabine), TRUVADA (emtricitabine and tenofovir), or COMPLERA (rilpivirine/emtricitabine/tenofovir) [see
Warnings and Precautions (5.3)].
HIV-1/HCV Co-Infection:
Patients with HIV-1/HCV co-infection should be informed that hepatic decompensation (some fatal) has occurred in HIV-1/HCV co-infected patients receiving combination antiretroviral therapy for HIV-1 and interferon alfa with or without ribavirin [see
Warnings and Precautions (5.4)].
Risk of Pancreatitis:
Parents or guardians should be advised to monitor pediatric patients for signs and symptoms of pancreatitis [see
Warnings and Precautions (5.5)].
Redistribution/Accumulation of Body Fat: Patients should be informed that redistribution or accumulation of body fat may occur in patients receiving antiretroviral therapy, including lamivudine, and that the cause and long-term health effects of these conditions are not known at this time [see
Warnings and Precautions (5.7)].
Information About HIV-1 Infection: Lamivudine is not a cure for HIV-1 infection and patients may continue to experience illnesses associated with HIV-1 infection, including opportunistic infections. Patients should remain under the care of a physician when using lamivudine.
Patients should be advised to avoid doing things that can spread HIV-1 infection to others.
• Do not share needles or other injection equipment.
• Do not share personal items that can have blood or body fluids on them, like toothbrushes and razor blades.
•
Do not have any kind of sex without protection. Always practice safe sex by using a latex or polyurethane condom or other barrier method to lower the chance of sexual contact
with semen, vaginal secretions, or blood.
• Do not breastfeed. Lamivudine is excreted in human breast milk. Mothers with HIV-1 should not breastfeed because HIV-1 can be passed to the baby in the breast milk.
Patients should be informed to take all HIV medications exactly as prescribed.
COMBIVIR, EPZICOM and TRIZIVIR are registered trademarks of ViiV Healthcare.
The other brands listed are trademarks of their respective owners and are not trademarks of Hetero Labs Limited. The makers of these brands are not affiliated with and do not endorse Hetero Labs Limited or its products.
Manufactured for:
AvKARE, Inc.
Pulaski, TN 38478
Mfg. Rev. 12/11
AV 07/15 (P)
AvPAK
PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
NDC 50268-459-15
Lamivudine Tablets150mg
Rx Only
50 Tablets (5 X 10) Unit Dose
5026845915
NDC 50268-459-15
Lamivudine Tablets150mg
Rx Only
50 Tablets (5 X 10) Unit Dose
5026845915
Each film coated tablet contains 150 mg of lamivudine USP.
USUAL DOSAGE: See prescribing information for Dosage and Administration.
Store at 20° to 25°C (68° to 77°F) [see USP Controlled Room Temperature].
Contact Number: 1-855-361-3993
KEEP OUT OF THE REACH OF CHILDREN.
Manufactured for:
AvKARE, Inc.
Pulaski, TN 38478
AvPAK
A PRODUCT OF AvKARE
Mfg. Lic. No. 50/MN/AP/2009/F/G AV 07/15 (P)

NDC 50268-460-13
Lamivudine Tablets300mg
Rx Only
30 Tablets (3 X 10) Unit Dose
5026846013
NDC 50268-460-13
Lamivudine Tablets300mg
Rx Only
30 Tablets (3 X 10) Unit Dose
5026846013
Each film coated tablet contains 300 mg of lamivudine USP.
USUAL DOSAGE: See prescribing information for Dosage and Administration.
Store at 20° to 25°C (68° to 77°F) [see USP Controlled Room Temperature].
Contact Number: 1-855-361-3993
KEEP OUT OF THE REACH OF CHILDREN.
Manufactured for:
AvKARE, Inc.
Pulaski, TN 38478
AvPAK
A PRODUCT OF AvKARE
Mfg. Lic. No. 50/MN/AP/2009/F/G AV 07/15 (P)

| LAMIVUDINE
lamivudine tablet, film coated |
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
| LAMIVUDINE
lamivudine tablet, film coated |
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
| Labeler - AvPAK (832926666) |