Label: TOREMIFENE CITRATE tablet

- NDC Code(s): 69539-152-30
- Packager: MSN LABORATORIES PRIVATE LIMITED
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated August 20, 2020
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use TOREMIFENE CITRATE TABLETS safely and effectively. See full prescribing information for TOREMIFENE CITRATE TABLETS.
TOREMIFENE CITRATE tablets, for oral administration
Initial U.S. Approval: [1997]
WARNING: QT PROLONGATION
Toremifene citrate has been shown to prolong the QTc interval in a dose- and concentration-related manner [see Clinical Pharmacology (12.2)].Prolongation of the QT interval can result in a type of ventricular tachycardia called Torsade de pointes, which may result in syncope, seizure, and/or death. Toremifene should not be prescribed to patients with congenital/acquired QT prolongation, uncorrected hypokalemia or uncorrected hypomagnesemia. Drugs known to prolong the QT interval and strong CYP3A4 inhibitors should be avoided [see Warnings and Precautions (5.1)].INDICATIONS AND USAGE
Toremifene citrate tablets are estrogen agonist/antagonist indicated for the treatment of metastatic breast cancer in postmenopausal women with estrogen-receptor positive or unknown tumors. (1)
DOSAGE AND ADMINISTRATION
- 60 mg once daily, orally (2)
DOSAGE FORMS AND STRENGTHS
- 60 mg tablet is white to off white colored, round shaped, uncoated tablets, debossed with “MT” on one side and plain on other side. (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Prolongation of the QT Interval(5.1)
- Heptatotoxicty (5.2)
- Hypercalcemia and Tumor Flare (5.3)
- Risk of Uterine Malignancy (5.4)
- General (5.5)
- Laboratory Tests (5.6)
- Pregnancy: Fetal harm may occur when administered to a pregnant woman. Women should be advised not to become pregnant when taking toremifene citrate. (5.7, 8.1)
- Women of Childbearing Potential: Use effective nonhormonal contraception during toremifene citrate therapy. (5.8)
ADVERSE REACTIONS
Most common adverse reactions are hot flashes, sweating, nausea and vaginal discharge. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact MSN Pharmaceuticals Inc. at 1-855-668-2369 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.DRUG INTERACTIONS
- Drugs that decrease renal calcium excretion, e.g., thiazide diuretics, may increase the risk of hypercalcemia in patients receiving toremifene citrate. (7.1)
- Agents that prolong QT should be avoided. (7.2)
- Coadministration with a strong CYP3A4 inducer may result in a relevant decrease in toremifene citrate exposure and should be avoided. (7.3)
- Coadministration with a strong CYP3A4 inhibitor can result in a relevant increase in toremifene citrate exposure and should be avoided. (7.4)
- CYP2C9 substrates with a narrow therapeutic index such as warfarin or phenytoin with toremifene citrate should be used with caution and require careful monitoring. (7.6)
USE IN SPECIFIC POPULATIONS
- Nursing Mothers: Discontinue drug or nursing taking into account the importance of the drug to the mother. (8.2)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 7/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: QT PROLONGATION
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
4.1 Hypersensitivity to the Drug
4.2 QT Prolongation, Hypokalemia, Hypomagnesemia
5 WARNINGS AND PRECAUTIONS
5.1 Prolongation of the QT Interval
5.2 Hepatotoxicity
5.3 Hypercalcemia and Tumor Flare
5.4 Risk of Uterine Malignancy
5.5 General
5.6 Laboratory Tests
5.7 Use in Pregnancy
5.8 Women of Childbearing Potential
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Post-marketing Experience
7 DRUG INTERACTIONS
7.1 Drugs that Decrease Renal Calcium Excretion
7.2 Agents that Prolong QT
7.3 Effect of Strong CYP3A4 Inducers on Toremifene
7.4 Effect of Strong CYP3A4 Inhibitors on Toremifene
7.5 Effect of Toremifene on CYP3A4 Substrates
7.6 Effect of Toremifene on CYP2C9 Substrates
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Nursing Mothers
8.3 Pediatric Use
8.4 Geriatric Use
8.5 Renal Impairment
8.6 Hepatic Impairment
8.7 Race
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, and Impairment of Fertility
14 CLINICAL STUDIES
16 SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: QT PROLONGATION
WARNING: QT PROLONGATION
Toremifene citrate has been shown to prolong the QTc interval in a dose- and concentration-related manner [see Clinical Pharmacology (12.2)].Prolongation of the QT interval can result in a type of ventricular tachycardia called Torsade de pointes, which may result in syncope, seizure, and/or death. Toremifene should not be prescribed to patients with congenital/acquired QT prolongation, uncorrected hypokalemia or uncorrected hypomagnesemia. Drugs known to prolong the QT interval and strong CYP3A4 inhibitors should be avoided[see Warnings and Precautions (5.1)]. - 1 INDICATIONS AND USAGE
- 2 DOSAGE AND ADMINISTRATION
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Prolongation of the QT Interval
Toremifene has been shown to prolong the QTc interval in a dose- and concentration-related manner[see Clinical Pharmacology (12.2)] .Prolongation of the QT interval can result in a type of ventricular tachycardia called Torsade de pointes, which may result in syncope, seizure, and/or death.
Toremifene should be avoided in patients with long QT syndrome. Caution should be exercised in patients with congestive heart failure, hepatic impairment and electrolyte abnormalities. Hypokalemia or hypomagnesemia must be corrected prior to initiating toremifene and these electrolytes should be monitored periodically during therapy. Drugs that prolong the QT interval should be avoided. In patients at increased risk, electrocardiograms (ECGs) should be obtained at baseline and as clinically indicated [see Drug Interactions (7.2) and Clinical Pharmacology (12.2)].5.2 Hepatotoxicity
Hepatotoxicity, both increases in the serum concentration for grade 3 and 4 transaminitis and hyperbilirubinemia, including jaundice, hepatitis, and non-alcoholic fatty liver disease, have also been reported in clinical trials and postmarketing with toremifene citrate. Liver function tests should be performed periodically. [see Adverse Reactions (6.1), Post-marketing Experience (6.2)]
5.3 Hypercalcemia and Tumor Flare
As with other antiestrogens, hypercalcemia and tumor flare have been reported in some breast cancer patients with bone metastases during the first weeks of treatment with toremifene citrate. Tumor flare is a syndrome of diffuse musculoskeletal pain and erythema with increased size of tumor lesions that later regress. It is often accompanied by hypercalcemia. Tumor flare does not imply failure of treatment or represent tumor progression. If hypercalcemia occurs, appropriate measures should be instituted and, if hypercalcemia is severe, toremifene citrate treatment should be discontinued.
5.4 Risk of Uterine Malignancy
Endometrial cancer, endometrial hypertrophy, hyperplasia, and uterine polyps have been reported in some patients treated with toremifene citrate. Endometrial hyperplasia of the uterus was observed in animals treated with toremifene [see Nonclinical Toxicology (13.1)]. Long-term use of toremifene citrate has not been established in patients with pre-existing endometrial hyperplasia. All patients should have baseline and annual gynecological examinations. In particular, patients at high risk of endometrial cancer should be closely monitored.
5.5 General
Patients with a history of thromboembolic diseases should generally not be treated with toremifene citrate. Patients with bone metastases should be monitored closely for hypercalcemia during the first weeks of treatment [see Warnings and Precautions (5.2)].
Leukopenia and thrombocytopenia have been reported rarely; leukocyte and platelet counts should be monitored when using toremifene citrate in patients with leukopenia and thrombocytopenia.5.6 Laboratory Tests
Periodic complete blood counts, calcium levels, and liver function tests should be obtained.
5.7 Use in Pregnancy
Based on its mechanism of action in humans and findings of increased pregnancy loss and fetal malformation in animal studies, toremifene citrate can cause fetal harm when administered to a pregnant woman. Toremifene caused embryo-fetal toxicities at maternal doses that were lower than the 60 mg daily recommended human dose on a mg/m2 basis. There are no adequate and well-controlled studies in pregnant women using toremifene citrate. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the fetus [see Use in Specific Populations (8.1)].
-
6 ADVERSE REACTIONS
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
6.1 Clinical Trials Experience
Adverse drug reactions are principally due to the antiestrogenic actions of toremifene citrate and typically occur at the beginning of treatment.
The incidences of the following eight clinical toxicities were prospectively assessed in the North American Study. The incidence reflects the toxicities that were considered by the investigator to be drug related or possibly drug related.
North American Study
FAR60
TAM20
n = 221
n = 215
Hot Flashes
35%
30%
Sweating
20%
17%
Nausea
14%
15%
Vaginal Discharge
13%
16%
Dizziness
9%
7%
Edema
5%
5%
Vomiting
4%
2%
Vaginal Bleeding
2%
4%
Approximately 1% of patients receiving toremifene citrate (n = 592) in the three controlled studies discontinued treatment as a result of adverse reactions (nausea and vomiting, fatigue, thrombophlebitis, depression, lethargy, anorexia, ischemic attack, arthritis, pulmonary embolism, and myocardial infarction).
Serious adverse reactions occurring in at least 1% of patients receiving toremifene citrate in the three major trials are listed in the table below.
Three prospective, randomized, controlled clinical studies (North American, Eastern European, and Nordic) were conducted. The patients were randomized to parallel groups receiving toremifene citrate 60 mg (FAR60) or tamoxifen 20 mg (TAM20) in the North American Study or tamoxifen 40 mg (TAM40) in the Eastern European and Nordic studies. The North American and Eastern European studies also included high-dose toremifene arms of 200 and 240 mg daily, respectively [see Clinical Studies (14)].
Adverse Reactions
North American
Eastern European
Nordic
FAR60
TAM20
FAR60
TAM40
FAR60
TAM40
n=221(%)
n=215(%)
n=157(%)
n=149(%)
n=214(%)
n=201(%)
Cardiac
Cardiac Failure
2
(1)
1
(<1)
-
1
(<1)
2
(1)
3
(1.5)
Myocardial Infarction
2
(1)
3
(1.5)
1
(<1)
2
(1)
-
1
(<1)
Arrhythmia
-
-
-
-
3
(1.5)
1
(<1)
Angina Pectoris
-
-
1
(<1)
-
1
(<1)
2
(1)
Ocular*
Cataracts
22
(10)
16
(7.5)
-
-
-
5
(3)
Dry Eyes
20
(9)
16
(7.5)
-
-
-
-
Abnormal Visual Fields
8
(4)
10
(5)
-
-
-
1
(<1)
Corneal Keratopathy
4
(2)
2
(1)
-
-
-
-
Glaucoma
3
(1.5)
2
(1)
1
(<1)
-
-
1
(<1)
Abnormal
-
-
-
-
3
(1.5)
-
Vision/Diplopia
Thromboembolic
Pulmonary Embolism
4
(2)
2
(1)
1
(<1)
-
-
1
(<1)
Thrombophlebitis
-
2
(1)
1
(<1)
1
(<1)
4
(2)
3
(1.5)
Thrombosis
-
1
(<1)
1
(<1)
-
3
(1.5)
4
(2)
CVA/TIA
1
(<1)
-
-
1
(<1)
4
(2)
4
(2)
Elevated Liver Tests**
AST
11
(5)
4
(2)
30
(19)
22
(15)
32
(15)
35
(17)
Alkaline Phosphatase
41
(19)
24
(11)
16
(10)
13
(9)
18
(8)
31
(15)
Bilirubin
3
(1.5)
4
(2)
2
(1)
1
(<1)
2
(1)
3
(1.5)
Hypercalcemia
6
(3)
6
(3)
1
(<1)
-
-
-
* Most of the ocular abnormalities were observed in the North American Study in which on-study and biannual ophthalmic examinations were performed. No cases of retinopathy were observed in any arm.
** Elevated defined as follows: North American Study: AST >100 IU/L; alkaline phosphatase >200 IU/L; bilirubin > 2 mg/dL. Eastern European and Nordic studies: AST, alkaline phosphatase, and bilirubin – WHO Grade 1 (1.25 times the upper limit of normal).
Other adverse reactions included leukopenia and thrombocytopenia, skin discoloration or dermatitis, constipation, dyspnea, paresis, tremor, vertigo, pruritus, anorexia, reversible corneal opacity (corneal verticulata), asthenia, alopecia, depression, jaundice, and rigors.
The incidence of AST elevations was greater in the 200 and 240 mg toremifene citrate dose arms than in the tamoxifen arms. Higher doses of toremifene citrate were also associated with an increase in nausea.
Approximately 4% of patients were withdrawn for toxicity from the high-dose toremifene citrate treatment arms. Reasons for withdrawal included hypercalcemia, abnormal liver function tests, and one case each of toxic hepatitis, depression, dizziness, incoordination, ataxia, blurry vision, diffuse dermatitis, and a constellation of symptoms consisting of nausea, sweating, and tremor.6.2 Post-marketing Experience
The following adverse reactions were identified during post approval use of toremifene citrate. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Adverse reactions reported during post approval use of toremifene citrate have been consistent with clinical trial experience. The most frequently reported adverse reactions related to toremifene citrate use since market introduction include hot flash, sweating, nausea, and vaginal discharge.
Hepatototoxicity [see Warnings and Precautions (5.2)]
Risk of Uterine Malignancy[see Warnings and Precautions (5.4)] -
7 DRUG INTERACTIONS
7.1 Drugs that Decrease Renal Calcium Excretion
Drugs that decrease renal calcium excretion, e.g., thiazide diuretics, may increase the risk of hypercalcemia in patients receiving toremifene citrate.
7.2 Agents that Prolong QT
The administration of toremifene citrate with agents that have demonstrated QT prolongation as one of their pharmacodynamic effects should be avoided. Should treatment with any of these agents be required, it is recommended that therapy with toremifene citrate be interrupted. If interruption of treatment with toremifene citrate is not possible, patients who require treatment with a drug that prolongs QT should be closely monitored for prolongation of the QT interval. Agents generally accepted to prolong QT interval include Class 1A (e.g., quinidine, procainamide, disopyramide) and Class III (e.g., amiodarone, sotalol, ibutilide, dofetilide) antiarrhythmics; certain antipsychotics (e.g., thioridazine, haloperidol); certain antidepressants (e.g., venlafaxine, amitriptyline); certain antibiotics (e.g., erythromycin, clarithromycin, levofloxacin, ofloxacin); and certain anti-emetics (e.g., ondansetron, granisetron). In patients at increased risk, electrocardiograms (ECGs) should be obtained and patients monitored as clinically indicated [see Boxed Warningand Warnings and Precautions (5.1)].
7.3 Effect of Strong CYP3A4 Inducers on Toremifene
Strong CYP3A4 enzyme inducers, such as dexamethasone, phenytoin, carbamazepine, rifampin, rifabutin, phenobarbital, St. John’s Wort, lower the steady-state concentration of toremifene in serum.
7.4 Effect of Strong CYP3A4 Inhibitors on Toremifene
In a study of 18 healthy subjects, 80 mg toremifene once daily coadministered with 200 mg of ketoconazole twice daily increased the toremifene Cmax and AUC by 1.4- and 2.9-fold, respectively. N-demethyltoremifene Cmax and AUC were reduced by 56% and 20%, respectively.
The administration of toremifene citrate with agents that are strong CYP3A4 inhibitors (e.g., ketoconazole, itraconazole, clarithromycin, atazanavir, indinavir, nefazodone, nelfinavir, ritonavir, saquinavir, telithromycin, and voriconazole) increase the steady-state concentration in serum and should be avoided. Grapefruit juice may also increase plasma concentrations of toremifene and should be avoided. Should treatment with any of these agents be required, it is recommended that therapy with toremifene citrate be interrupted. If interruption of treatment with toremifene citrate is not possible, patients who require treatment with a drug that strongly inhibits CYP3A4 should be closely monitored for prolongation of the QT interval [see Boxed Warningand Warnings and Precautions (5.1)].7.5 Effect of Toremifene on CYP3A4 Substrates
In a study of 20 healthy subjects, 2 mg midazolam once daily (days 6 and 18) coadministered with toremifene as a 480 mg loading dose followed by 80 mg once daily for 16 days. Following coadministration on days 6 and 18 relevant increases in midazolam and α-hydroxymidazolam Cmax and AUC were not observed. Following coadministration on day 18 midazolam and α-hydroxymidazolam Cmax and AUC were reduced by less than 20%.
Clinically relevant exposure changes in sensitive substrates due to inhibition or induction of CYP3A4 by toremifene appear unlikely.7.6 Effect of Toremifene on CYP2C9 Substrates
In a study of 20 healthy subjects, 500 mg tolbutamide once daily (days 7 and 19) coadministered with toremifene as a 480 mg loading dose followed by 80 mg once daily for 16 days. Following coadministration on days 7 and 19 plasma tolbutamide Cmax and AUC were increased by less than 30%. A reduction of similar magnitude was observed for hydroxytolbutamide and carboxytolbutamide Cmax and AUC.
Toremifene is a weak inhibitor of CYP2C9. Concomitant use of CYP2C9 substrates with a narrow therapeutic index such as warfarin or phenytoin with toremifene citrate should be done with caution and requires careful monitoring (e.g., substrate concentrations (if possible), appropriate laboratory markers, and signs and symptoms of increased exposure). -
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category D [see Warnings and Precautions (5.6).]
Based on its mechanism of action in humans and findings of increased pregnancy loss and fetal malformation in animal studies, toremifene citrate can cause fetal harm when administered to a pregnant woman. Toremifene caused embryo-fetal toxicities at maternal doses that were lower than the 60 mg daily recommended human dose on a mg/m2 basis. There are no adequate and well-controlled studies in pregnant women using toremifene citrate. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the fetus.
In animal studies, toremifene crossed the placenta and accumulated in the rodent fetus. Administration of toremifene to pregnant rats during organogenesis at doses of approximately 6% the daily maximum recommended human dose of 60 mg (on a mg/m2 basis) resulted in signs of maternal toxicity and increased preimplantation loss, increased resorptions, reduced fetal weight, and fetal anomalies. Fetal anomalies include malformation of limbs, incomplete ossification, misshapen bones, ribs/spine anomalies, hydroureter, hydronephrosis, testicular displacement, and subcutaneous edema. Maternal toxicity may have contributed to these adverse embryo-fetal effects. Similar embryo-fetal toxicities occurred in rabbits that received toremifene at doses approximately 40% the daily recommended human dose of 60 mg (on a mg/m2 basis). Findings in rabbits included increased preimplantation loss, increased resorptions, and fetal anomalies, including incomplete ossification and anencephaly.
Animal doses resulting in embryo-fetal toxicities were ≥1.0 mg/kg/day in rats and ≥1.25 mg/kg/day in rabbits.
In rodent models of fetal reproductive tract development, toremifene produced inhibition of uterine development in female pups similar to effects seen with diethylstilbestrol (DES) and tamoxifen. The clinical relevance of these changes is not known. Neonatal rodent studies have not been conducted to assess the potential for toremifene to cause other DES-like effects in offspring (i.e., vaginal adenosis). Vaginal adenosis in animals occurred following treatment with other drugs of this class and has been observed in women exposed to diethylstilbestrol in utero.8.2 Nursing Mothers
It is not known if toremifene is excreted in human milk. Toremifene is excreted in the milk of lactating rats. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from toremifene citrate, a decision should be made to either discontinue nursing or discontinue the drug, taking into account the importance of the drug to the mother.
8.4 Geriatric Use
The pharmacokinetics of toremifene were studied in 10 healthy young males and 10 elderly females following a single 120 mg dose under fasting conditions. Increases in the elimination half-life (4.2 versus 7.2 days) and the volume of distribution (457 versus 627 L) of toremifene were seen in the elderly females without any change in clearance or AUC.
The median ages in the three controlled studies ranged from 60 to 66 years. No significant age-related differences in toremifene citrate effectiveness or safety were noted.8.5 Renal Impairment
The pharmacokinetics of toremifene and N-demethyltoremifene were similar in normals and in patients with impaired kidney function.
8.6 Hepatic Impairment
The mean elimination half-life of toremifene was increased by less than twofold in 10 patients with hepatic impairment (cirrhosis or fibrosis) compared to subjects with normal hepatic function. The pharmacokinetics of N-demethyltoremifene were unchanged in these patients. Ten patients on anticonvulsants (phenobarbital, clonazepam, phenytoin, and carbamazepine) showed a twofold increase in clearance and a decrease in the elimination half-life of toremifene.
-
10 OVERDOSAGE
Lethality was observed in rats following single oral doses that were ≥1000 mg/kg (about 150 times the recommended human dose on a mg/m2 basis) and was associated with gastric atony/dilatation leading to interference with digestion and adrenal enlargement.
Vertigo, headache, and dizziness were observed in healthy volunteer studies at a daily dose of 680 mg for 5 days. The symptoms occurred in two of the five subjects during the third day of the treatment and disappeared within 2 days of discontinuation of the drug. No immediate concomitant changes in any measured clinical chemistry parameters were found. In a study in postmenopausal breast cancer patients, toremifene 400 mg/m2/day caused dose-limiting nausea, vomiting, and dizziness, as well as reversible hallucinations and ataxia in one patient.
Theoretically, overdose may be manifested as an increase of antiestrogenic effects, such as hot flashes; estrogenic effects, such as vaginal bleeding; or nervous system disorders, such as vertigo, dizziness, ataxia, and nausea. There is no specific antidote and the treatment is symptomatic. -
11 DESCRIPTION
Toremifene citrate tablets for oral administration each contain 88.5 mg of toremifene citrate, which is equivalent to 60 mg toremifene.
Toremifene citrate tablets are estrogen agonist/antagonist. The chemical name of toremifene is: 2-{p-[(Z)-4-chloro-1,2-diphenyl-1-butenyl]phenoxy}-N,N-dimethylethylamine citrate(1:1).The structural formula is:
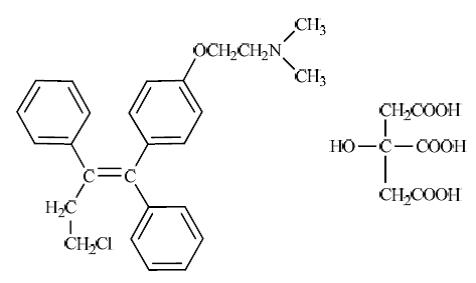
and the molecular formula is C26H28ClNO . C6H8O7. The molecular weight of toremifene citrate is 598.10. The pKa is 7.76. Freely soluble in dimethyl sulphoxide, sparingly soluble in methanol and insoluble in water.
Toremifene citrate tablets are available only as tablets for oral administration. Inactive ingredients: colloidal silicon dioxide, corn starch, lactose monohydrate, magnesium stearate, microcrystalline cellulose, povidone K 30, and sodium starch glycolate. -
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Toremifene is a nonsteroidal triphenylethylene derivative. Toremifene binds to estrogen receptors and may exert estrogenic, antiestrogenic, or both activities, depending upon the duration of treatment, animal species, gender, target organ, or endpoint selected. In general, however, nonsteroidal triphenylethylene derivatives are predominantly antiestrogenic in rats and humans and estrogenic in mice. In rats, toremifene causes regression of established dimethylbenzanthracene (DMBA)-induced mammary tumors. The antitumor effect of toremifene in breast cancer is believed to be mainly due to its antiestrogenic effects, i.e., its ability to compete with estrogen for binding sites in the cancer, blocking the growth-stimulating effects of estrogen in the tumor.
12.2 Pharmacodynamics
Toremifene causes a decrease in the estradiol-induced vaginal cornification index in some postmenopausal women, indicative of its antiestrogenic activity. Toremifene also has estrogenic activity as shown by decreases in serum gonadotropin concentrations (FSH and LH).
Effects on Cardiac Electrophysiology
The effect of 20 mg, 80 mg, and 300 mg of toremifene on QT interval was evaluated in a double-blind, randomized study in healthy male subjects aged 18 to 45 years. The QT interval was measured at steady state of toremifene (Day 5 of dosing), including the time of peak plasma concentration (Tmax), at 13 time points (4 ECGs/time point) over 24 hours post dose in a time matched analysis. The 300 mg dose of toremifene (approximately five times the highest recommended dose 60 mg) was chosen because this dose produces exposure to toremifene that will cover the expected exposures that may result from potential drug interactions and hepatic impairment [see Drug Interactions (7.2)].
Dose and concentration-related increases in the QTc interval and T wave changes were observed (see Table 1). These effects are believed to be caused by toremifene and N-demethyltoremifene. Toremifene had no effects on heart rate, PR and QRS interval duration [see Boxed Warningand Warnings and Precautions (5.1)].
Table 1: QTc Prolongation in Healthy Male Volunteers
Treatment Arm
Mean (90% CI)
ΔΔQTc, ms
ΔQTc > 60 ms (n, %)
QTc > 500 ms (n, %)
Toremifene 20 mg (N = 47)
7 (0.9, 13.6)
0
0
Toremifene 80 mg (N = 47)
26 (21.1, 31.2)
2 (4.3%)
0
Toremifene 300 mg (N = 48)
65 (60.1, 69.2)
43 (89.6%)
5 (10.4%)
12.3 Pharmacokinetics
Absorption – Toremifene is well absorbed after oral administration and absorption is not influenced by food. Peak plasma concentrations are obtained within 3 hours. Toremifene displays linear pharmacokinetics after single oral doses of 10 to 680 mg. After multiple dosing, dose proportionality was observed for doses of 10 to 400 mg. Steady state concentrations were reached in about 4-6 weeks.
Distribution – Toremifene has an apparent volume of distribution of 580 L and binds extensively (>99.5%) to serum proteins, mainly albumin.
Metabolism – Toremifene is extensively metabolized, principally by CYP3A4 to N-demethyltoremifene which is also antiestrogenic but with weak in vivo antitumor potency. Serum concentrations of N-demethyltoremifene are 2 to 4 times higher than toremifene at steady state.
Following multiple dosing with toremifene in 20 healthy volunteers, plasma toremifene exposure was lower on Day 17 compared to Day 5 by approximately 14%. N-demethyltoremifene exposure was higher on Day 17 compared to Day 5 by approximately 80%. Based on these data and an in vitro induction study in human hepatocytes, auto- induction of CYP3A4 by toremifene is likely. The effect of auto-induction on efficacy was likely captured following prolonged dosing in the clinical studies.
Elimination – The plasma concentration time profile of toremifene declines biexponentially after absorption with a mean distribution half-life of about 4 hours and an elimination half-life of about 5 days. Elimination half-lives of major metabolites, N-demethyltoremifene and (Deaminohydroxy) toremifene, were 6 and 4 days, respectively. Mean total clearance of toremifene was approximately 5 L/h. Toremifene is eliminated as metabolites primarily in the feces, with about 10% excreted in the urine during a 1-week period. Elimination of toremifene is slow, in part because of enterohepatic circulation.
Renal insufficiency – The pharmacokinetics of toremifene and N-demethyltoremifene were similar in normals and patients with impaired kidney function.
Hepatic insufficiency – The mean elimination half-life of toremifene was increased by less than twofold in 10 patients with hepatic impairment (cirrhosis or fibrosis) compared to subjects with normal hepatic function. The pharmacokinetics of N-demethyltoremifene were unchanged in these patients. Ten patients on anticonvulsants (phenobarbital, clonazepam, phenytoin, and carbamazepine) showed a twofold increase in clearance and a decrease in the elimination half-life of toremifene.
Geriatric patients – The pharmacokinetics of toremifene were studied in 10 healthy young males and 10 elderly females following a single 120 mg dose under fasting conditions. Increases in the elimination half-life (4.2 versus 7.2 days) and the volume of distribution (457 versus 627 L) of toremifene were seen in the elderly females without any change in clearance or AUC. The median ages in the three controlled studies ranged from 60 to 66 years. No significant age-related differences in toremifene citrate effectiveness or safety were noted.
Food – The rate and extent of absorption of toremifene citrate are not influenced by food; thus toremifene citrate may be taken with or without food.
Race – The pharmacokinetics of toremifene in patients of different races has not been studied. Fourteen percent of patients in the North American Study were non-Caucasian. No significant race-related differences in toremifene citrate effectiveness or safety were noted. -
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, and Impairment of Fertility
Conventional carcinogenesis studies in rats at doses of 0.12 to 12 mg/kg/day (approximately 1/50 to 2 times the daily maximum recommended human dose of 60 mg, on a mg/m2 basis) for up to 2 years did not show evidence of carcinogenicity. Studies in mice at doses of 1.0 to 30.0 mg/kg/day (approximately 1/15 to 2 times the daily maximum recommended human dose of 60 mg, on a mg/m2 basis) for up to 2 years revealed increased incidence of ovarian and testicular tumors and increased incidence of osteoma and osteosarcoma. The significance of the mouse findings is uncertain because of the different role of estrogens in mice and the estrogenic effect of toremifene in mice. An increased incidence of ovarian and testicular tumors in mice has also been observed with other human estrogen agonists/antagonists that have primarily estrogenic activity in mice. Endometrial hyperplasia of the uterus was observed in monkeys following 52 weeks of treatment at ≥1 mg/kg and in dogs following 16 weeks of treatment at ≥3 mg/kg with toremifene (approximately 1/3 and 1.4 times, respectively, the daily maximum recommended human dose of 60 mg, on a mg/m2 basis).
Toremifene has not been shown to be mutagenic in in vitro tests (Ames and E. coli bacterial tests). Toremifene is clastogenic in vitro (chromosomal aberrations and micronuclei formation in human lymphoblastoid MCL-5 cells) and in vivo (chromosomal aberrations in rat hepatocytes).
Toremifene produced impairment of fertility and conception in male and female rats at doses ≥25.0 and 0.14 mg/kg/day, respectively (approximately 4 times and 1/50 the daily maximum recommended human dose of 60 mg, on a mg/m2 basis). At these doses, sperm counts, fertility index, and conception rate were reduced in males with atrophy of seminal vesicles and prostate. In females, fertility and reproductive indices were markedly reduced with increased pre- and post-implantation loss. In addition, offspring of treated rats exhibited depressed reproductive indices. Toremifene produced ovarian atrophy in dogs administered doses ≥3 mg/kg/day (approximately 1.5 times the daily maximum recommended human dose of 60 mg, on a mg/m2 basis) for 16 weeks. Cystic ovaries and reduction in endometrial stromal cellularity were observed in monkeys at doses ≥1 mg/kg/day (about 1/3 the daily maximum recommended human dose of 60 mg, on a mg/m2 basis) for 52 weeks. -
14 CLINICAL STUDIES
Three prospective, randomized, controlled clinical studies (North American, Eastern European, and Nordic) were conducted to evaluate the efficacy of toremifene citrate for the treatment of breast cancer in postmenopausal women. The patients were randomized to parallel groups receiving toremifene citrate 60 mg (FAR60) or tamoxifen 20 mg (TAM20) in the North American Study or tamoxifen 40 mg (TAM40) in the Eastern European and Nordic studies. The North American and Eastern European studies also included high-dose toremifene arms of 200 and 240 mg daily, respectively. The studies included postmenopausal patients with estrogen-receptor (ER) positive or estrogen-receptor (ER) unknown metastatic breast cancer. The patients had at least one measurable or evaluable lesion. The primary efficacy variables were response rate (RR) and time to progression (TTP). Survival (S) was also determined. Ninety-five percent confidence intervals (95% CI) were calculated for the difference in RR between FAR60 and TAM groups and the hazard ratio (relative risk for an unfavorable event, such as disease progression or death) between TAM and FAR60 for TTP and S.
Two of the 3 studies showed similar results for all effectiveness endpoints. However, the Nordic Study showed a longer time to progression for tamoxifen (see table).
Clinical Studies
Study
North American
Eastern European
Nordic
Treatment Group
FAR60
TAM20
FAR60
TAM40
FAR60
TAM40
No. Patients
221
215
157
149
214
201
Responses
CR1+ PR2
14 + 33
11 + 30
7 + 25
3 + 28
19 + 48
19 + 56
RR3(CR + PR)%
21.3
19.1
20.4
20.8
31.3
37.3
Difference in RR
2.2
-0.4
-6.095% CI4for Difference in RR
-5.8 to 10.2
-9.5 to 8.6
-15.1 to 3.1
Time to Progression (TTP)
Median TTP (mo.)
5.6
5.8
4.9
5.0
7.3
10.2
Hazard Ratio (TAM/FAR)
1.01
1.02
0.8095% CI4 for Hazard Ratio (%)
0.81 to 1.26
0.79 to 1.31
0.64 to 1.00
Survival (S)
Median S (mo.)
33.6
34.0
25.4
23.4
33.0
38.7
Hazard Ratio (TAM/FAR)
0.94
0.96
0.9495% CI4 for Hazard Ratio(%)
0.74 to 1.24
0.72 to 1.28
0.73 to 1.22
1CR = complete response; 2PR = partial response; 3RR = response rate; 4CI = confidence interval
The high-dose groups, toremifene 200 mg daily in the North American Study and 240 mg daily in the Eastern European Study, were not superior to the lower toremifene dose groups, with response rates of 22.6% and 28.7%, median times to progression of 5.6 and 6.1 months, and median survivals of 30.1 and 23.8 months, respectively. The median treatment duration in the three pivotal studies was 5 months (range 4.2-6.3 months). -
16 SUPPLIED/STORAGE AND HANDLING
Toremifene citrate tablets, containing toremifene citrate in an amount equivalent to 60 mg of toremifene, are white to off white colored, round shaped, uncoated tablets, debossed with “MT” on one side and plain on other side and free from physical defects.
Toremifene citrate tablets are available as:
NDC 69539-152-30 bottles of 30
Store at 25°C (77°F).
Excursions permitted to 15-30°C (59-86°F)
[See USP Controlled Room Temperature.]
Protect from heat and light. -
17 PATIENT COUNSELING INFORMATION
Vaginal bleeding has been reported in patients using toremifene citrate. Patients should be informed about this and instructed to contact their physician if such bleeding or other gynecological symptoms (changes in vaginal discharge, pelvic pain or pressure) occur. Patients should have a gynecological examination prior to initiation of therapy and at regular intervals while on therapy.
Liver disorders including transaminits grade 3 and 4, hyperbilirubinemia with jaundice have been reported in patients using toremifene citrate. Patients should have liver function tests performed periodically while on therapy.
Toremifene citrate may harm the fetus and increase the risk for pregnancy loss [see Warnings and Precautions (5.6) and Use in Specific Populations (8.1)].
Premenopausal women using toremifene citrate should use nonhormonal contraception during treatment and should be apprised of the potential hazard to the fetus should pregnancy occur [see Warnings and Precautions (5.7)].
Patients with bone metastases should be informed about the typical signs and symptoms of hypercalcemia and instructed to contact their physician for further assessment if such signs or symptoms occur.
Patients who must take medications known to prolong the QT interval, or potent CYP3A4 inhibitors, should be informed of the effect of toremifene on QT interval. Toremifene has been shown to prolong the QTc interval in a dose-related manner [see Boxed Warning, Warnings and Precautions (5.1),and Clinical Pharmacology (12.2)].
Specific interactions with foods that inhibit CYP3A4, including grapefruit juice, have not been studied but may increase toremifene concentrations. Patients should avoid grapefruit products and other foods that are known to inhibit CYP3A4 during toremifene citrate treatment.
Certain other medicines, including over-the-counter medications or herbal supplements (such as St. John’s Wort) and toremifene, can reduce concentrations of co-administered drugs [see Drug Interactions (7.3)].
Manufactured by:
MSN Laboratories Private Limited
Telangana – 509 228,
INDIA
Distributed by:
MSN Pharmaceuticals Inc.
Piscataway, NJ 08854 -3714
Issued on: 07/2019 - PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
TOREMIFENE CITRATE
toremifene citrate tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:69539-152 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TOREMIFENE CITRATE (UNII: 2498Y783QT) (TOREMIFENE - UNII:7NFE54O27T) TOREMIFENE 60 mg Inactive Ingredients Ingredient Name Strength CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) STARCH, CORN (UNII: O8232NY3SJ) POVIDONE K30 (UNII: U725QWY32X) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) MAGNESIUM STEARATE (UNII: 70097M6I30) Product Characteristics Color WHITE (white to off-white) Score no score Shape ROUND Size 9mm Flavor Imprint Code MT Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:69539-152-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 08/20/2020 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA212818 08/20/2020 Labeler - MSN LABORATORIES PRIVATE LIMITED (650786952) Establishment Name Address ID/FEI Business Operations MSN LABORATORIES PRIVATE LIMITED 650786952 ANALYSIS(69539-152) , MANUFACTURE(69539-152)
