REBETOL- ribavirin capsule
REBETOL- ribavirin liquid
Merck Sharp & Dohme LLC
----------
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use REBETOL safely and effectively. See full prescribing information for REBETOL.
REBETOL® (ribavirin USP) capsules, for oral use REBETOL® (ribavirin USP) oral solution Initial U.S. Approval: 1998 WARNING: EMBRYO-FETAL TOXICITY, HEMOLYTIC ANEMIA, and MONOTHERAPY NOT RECOMMENDEDSee full prescribing information for complete boxed warning.
INDICATIONS AND USAGEREBETOL is a nucleoside analogue indicated in combination with interferon alfa-2b (pegylated and nonpegylated) for the treatment of Chronic Hepatitis C (CHC) in patients 3 years of age or older with compensated liver disease. (1.1)
DOSAGE AND ADMINISTRATIONREBETOL is administered according to body weight. (2.1, 2.2, 2.3) Dose reduction or discontinuation is recommended in patients experiencing certain adverse reactions or renal dysfunction. (2.5, 2.6, 12.3) CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
Patients exhibiting the following conditions should be closely monitored and may require dose reduction or discontinuation of therapy:
ADVERSE REACTIONSHemolytic anemia occurred in more than 10% of adult patients receiving REBETOL/PegIntron or INTRON A combination therapy. (6.1) Most common adverse reactions (40% or greater) in adult patients receiving REBETOL/PegIntron or INTRON A combination therapy are injection site reaction, fatigue/asthenia, headache, rigors, fevers, nausea, myalgia and anxiety/emotional lability/irritability. (6.1) Most common adverse reactions (greater than 25%) in pediatric patients receiving REBETOL/PegIntron therapy are: pyrexia, headache, neutropenia, fatigue, anorexia, injection site erythema, and vomiting. (6.1) To report SUSPECTED ADVERSE REACTIONS, contact Merck Sharp & Dohme Corp., a subsidiary of Merck & Co. Inc., at 1-877-888-4231 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch . DRUG INTERACTIONSNucleoside analogues: Closely monitor for toxicities. Discontinue nucleoside reverse transcriptase inhibitors or reduce dose or discontinue interferon, ribavirin or both with worsening toxicities. (7.2) USE IN SPECIFIC POPULATIONSSee 17 for PATIENT COUNSELING INFORMATION and Medication Guide. Revised: 3/2022 |
FULL PRESCRIBING INFORMATION
WARNING: EMBRYO-FETAL TOXICITY, HEMOLYTIC ANEMIA, and MONOTHERAPY NOT RECOMMENDED
- Significant teratogenic and embryocidal effects have been demonstrated in all animal species exposed to ribavirin. In addition, ribavirin has a multiple-dose half-life of 12 days and may persist in non-plasma compartments for as long as 6 months. Therefore, REBETOL therapy is contraindicated in women who are pregnant and in the male partners of women who are pregnant. Avoid pregnancy and use effective contraception during therapy and for 9 months after completion of treatment in female patients and for 6 months in female partners of male patients who are taking REBETOL therapy. [see Contraindications (4), Warnings and Precautions (5.1), Use in Specific Populations (8.1, 8.3), and Nonclinical Toxicology (13.1)].
- Hemolytic anemia has been reported with ribavirin therapy. The anemia associated with REBETOL therapy may result in worsening of cardiac disease that has led to fatal and nonfatal myocardial infarctions. Patients with a history of significant or unstable cardiac disease should not be treated with REBETOL [see Dosage and Administration (2.5), Warnings and Precautions (5.2), and Adverse Reactions (6.1)].
- REBETOL monotherapy is not effective for the treatment of chronic hepatitis C virus infection and should not be used alone for this indication [see Warnings and Precautions (5.10)].
1 INDICATIONS AND USAGE
1.1 Chronic Hepatitis C (CHC)
REBETOL® (ribavirin) in combination with interferon alfa-2b (pegylated and nonpegylated) is indicated for the treatment of Chronic Hepatitis C (CHC) in patients 3 years of age and older with compensated liver disease [see Warnings and Precautions (5.9, 5.10), and Use in Specific Populations (8.4)].
The following points should be considered when initiating REBETOL combination therapy with PegIntron® or INTRON A®:
- Combination therapy with REBETOL/PegIntron is preferred over REBETOL/INTRON A as this combination provides substantially better response rates [see Clinical Studies (14)].
- Patients with the following characteristics are less likely to benefit from re-treatment after failing a course of therapy: previous nonresponse, previous pegylated interferon treatment, significant bridging fibrosis or cirrhosis, and genotype 1 infection [see Clinical Studies (14)].
- No safety and efficacy data are available for treatment duration lasting longer than one year.
2 DOSAGE AND ADMINISTRATION
2.1 General Dosing Information
Do not open, crush or break REBETOL capsules. REBETOL should be taken with food [see Clinical Pharmacology (12.3)].
2.2 REBETOL/PegIntron Combination Therapy
Adult Patients
The recommended dose of REBETOL when used in combination with PegIntron is 800 mg to 1,400 mg based on patient body weight in two divided doses (see Table 1). Refer to PegIntron labeling for PegIntron dosing information.
Duration of Treatment – Interferon Alpha-naïve Patients
The treatment duration for patients with genotype 1 is 48 weeks. Discontinuation of therapy should be considered in patients who do not achieve at least a 2 log10 drop or loss of hepatitis C virus (HCV)-RNA at 12 weeks, or if HCV-RNA remains detectable after 24 weeks of therapy. Patients with genotype 2 and 3 should be treated for 24 weeks.
Duration of Treatment – Re-treatment with PegIntron/REBETOL of Prior Treatment Failures
The treatment duration for patients who previously failed therapy is 48 weeks, regardless of HCV genotype. Re-treated patients who fail to achieve undetectable HCV-RNA at Week 12 of therapy, or whose HCV-RNA remains detectable after 24 weeks of therapy, are highly unlikely to achieve SVR and discontinuation of therapy should be considered [see Clinical Studies (14.1)].
| Body Weight (kg) | REBETOL Daily Dose | REBETOL Number of Capsules |
|---|---|---|
| Less than 66 | 800 mg/day | 2 × 200-mg capsules AM 2 × 200-mg capsules PM |
| 66-80 | 1,000 mg/day | 2 × 200-mg capsules AM 3 × 200-mg capsules PM |
| 81-105 | 1,200 mg/day | 3 × 200-mg capsules AM 3 × 200-mg capsules PM |
| Greater than 105 | 1,400 mg/day | 3 × 200-mg capsules AM 4 × 200-mg capsules PM |
Pediatric Patients
Dosing of REBETOL in pediatric patients is determined by body weight. The recommended dose of REBETOL when used in combination with PegIntron in pediatric patients ages 3-17 years is 15 mg/kg/day in two divided doses (see Table 2). Refer to PegIntron labeling for PegIntron dosing information. The treatment duration for patients with genotype 1 is 48 weeks. Patients with genotype 2 and 3 should be treated for 24 weeks.
| Body Weight (kg) | REBETOL Daily Dose | REBETOL Number of Capsules |
|---|---|---|
|
||
| Less than 47 | 15 mg/kg/day | Use REBETOL Oral Solution* |
| 47-59 | 800 mg/day | 2 × 200-mg capsules AM 2 × 200-mg capsules PM |
| 60-73 | 1,000 mg/day | 2 × 200-mg capsules AM 3 × 200-mg capsules PM |
| Greater than 73 | 1,200 mg/day | 3 × 200-mg capsules AM 3 × 200-mg capsules PM |
2.3 REBETOL/INTRON A Combination Therapy
Adults
Duration of Treatment – Interferon Alpha-naïve Patients
The recommended dose of REBETOL when used in combination with INTRON A depends on the patient's body weight (see Table 3). Refer to Intron A labeling for interferon dosing information. The recommended duration of treatment for patients previously untreated with interferon is 24 to 48 weeks. The duration of treatment should be individualized to the patient depending on baseline disease characteristics, response to therapy, and tolerability of the regimen [see Indications and Usage (1.1), Adverse Reactions (6.1), and Clinical Studies (14)]. After 24 weeks of treatment, virologic response should be assessed. Treatment discontinuation should be considered in any patient who has not achieved an HCV-RNA below the limit of detection of the assay by 24 weeks. There are no safety and efficacy data for treatment duration lasting longer than 48 weeks in the previously untreated patient population.
Duration of Treatment – Re-treatment with INTRON A/REBETOL in Relapse Patients
In patients who relapse following nonpegylated interferon monotherapy, the recommended duration of treatment is 24 weeks.
| Body Weight | REBETOL Capsules |
|---|---|
| At least 75 kg | 2 × 200-mg capsules AM 3 × 200-mg capsules PM daily orally |
| Greater than 75 kg | 3 × 200-mg capsules AM 3 × 200-mg capsules PM daily orally |
Pediatrics The recommended dose of REBETOL when used in combination with INTRON A is 15 mg/kg per day orally in two divided doses (see Table 2). Refer to Intron A labeling for interferon dosing information.
The recommended duration of treatment is 48 weeks for pediatric patients with genotype 1. After 24 weeks of treatment, virologic response should be assessed. Treatment discontinuation should be considered in any patient who has not achieved an HCV-RNA below the limit of detection of the assay by this time. The recommended duration of treatment for pediatric patients with genotype 2 and 3 is 24 weeks.
2.4 Testing Prior to Initiation of REBETOL
The following laboratory tests are recommended in all patients treated with REBETOL prior to initiation of treatment and periodically thereafter.
- Standard hematologic tests - including hemoglobin (pretreatment, Week 2 and Week 4 of therapy, and as clinically appropriate [see Warnings and Precautions (5.2, 5.6)], complete and differential white blood cell counts, and platelet count.
- Blood chemistries - liver function tests and TSH.
- Pregnancy - in women of childbearing potential.
- ECG [see Warnings and Precautions (5.2)].
2.5 Dose Modifications
If severe adverse reactions or laboratory abnormalities develop during REBETOL combination therapy, modify or discontinue the dose until the adverse reaction abates or decreases in severity (see Table 4) [see Warnings and Precautions (5)]. If intolerance persists after dose adjustment, combination therapy should be discontinued. Refer to PegIntron labeling for additional information regarding dose reduction of PegIntron.
Dose reduction in pediatric patients is accomplished by modifying the recommended REBETOL dose from the original starting dose of 15 mg/kg daily in a two-step process to 12 mg/kg/day, then to 8 mg/kg/day, if needed (see Table 4).
REBETOL is contraindicated in patients with creatinine clearance less than 50 mL/min [see Contraindications (4)]. Patients with impaired renal function and those over the age of 50 should be carefully monitored with respect to development of anemia [see Warnings and Precautions (5.2), Use in Specific Populations (8.5), and Clinical Pharmacology (12.3)].
REBETOL should be administered with caution to patients with pre-existing cardiac disease. Assess cardiovascular status before initiation of treatment and during therapy. If there is any deterioration of cardiovascular status, discontinue combination therapy [see Warnings and Precautions (5.2)].
In patients with a history of stable cardiovascular disease, a permanent dose reduction is required if the hemoglobin decreases by 2 g/dL or more during any 4-week period. If the hemoglobin level remains below 12 g/dL after 4 weeks on a reduced dose, discontinue combination therapy.
Modify or discontinue REBETOL dosing in any patient whose hemoglobin level falls below 10 g/dL (see Table 4) [see Warnings and Precautions (5.2)].
| Laboratory Parameters | Reduce REBETOL Daily Dose (see note 1) if: | Reduce PegIntron or INTRON A Dose (see note 2) if: | Discontinue Therapy if: |
|---|---|---|---|
|
Note 1: Adult patients: 1st dose reduction of ribavirin is by 200 mg/day (except in patients receiving the 1,400 mg, dose reduction should be by 400 mg/day). If needed, 2nd dose reduction of ribavirin is by an additional 200 mg/day. Patients whose dose of ribavirin is reduced to 600 mg daily receive one 200 mg capsule in the morning and two 200 mg capsules in the evening. Pediatric patients: 1st dose reduction of ribavirin is to 12 mg/kg/day, 2nd dose reduction of ribavirin is to 8 mg/kg/day. |
|||
|
Note 2: Adult patients treated with REBETOL and PegIntron: 1st dose reduction of PegIntron is to 1 mcg/kg/week. If needed, 2nd dose reduction of PegIntron is to 0.5 mcg/kg/week. Pediatric patients treated with REBETOL and PegIntron: 1st dose reduction of PegIntron is to 40 mcg/m2/week, 2nd dose reduction of PegIntron is to 20 mcg/m2/week. For patients on REBETOL/INTRON A combination therapy: reduce INTRON A dose by 50%. |
|||
|
|||
| WBC | N/A | 1.0 to <1.5 × 109/L | <1.0 × 109/L |
| Neutrophils | N/A | 0.5 to <0.75 × 109/L | <0.5 × 109/L |
| Platelets | N/A | 25 to < 50 × 109/L (adults) | <25 × 109/L (adults) |
| N/A | 50 to <70 × 109/L (pediatrics) | <50 × 109/L (pediatrics) | |
| Creatinine | N/A | N/A | >2 mg/dL (pediatrics) |
| Hemoglobin in patients without history of cardiac disease | 8.5 to <10 g/dL | N/A | <8.5 g/dL |
| Reduce REBETOL Dose by 200 mg/day and PegIntron or INTRON A Dose by Half if: | |||
| Hemoglobin in patients with history of stable cardiac disease*† | ≥2 g/dL decrease in hemoglobin during any four-week period during treatment | <8.5 g/dL or <12 g/dL after four weeks of dose reduction | |
Refer to labeling for INTRON A or PegIntron for additional information about how to reduce an INTRON A or PegIntron dose.
2.6 Discontinuation of Dosing
Adults In HCV genotype 1, interferon-alfa-naïve patients receiving PegIntron in combination with ribavirin, discontinue therapy if there is not at least a 2 log10 drop or loss of HCV-RNA at 12 weeks of therapy, or if HCV-RNA levels remain detectable after 24 weeks of therapy. Regardless of genotype, previously treated patients who have detectable HCV-RNA at Week 12 or 24 are highly unlikely to achieve SVR and discontinuation of therapy should be considered.
4 CONTRAINDICATIONS
REBETOL combination therapy is contraindicated in:
- pregnancy. REBETOL may cause fetal harm when administered to a pregnant woman. REBETOL is contraindicated in women who are pregnant or planning to become pregnant. If a patient becomes pregnant while taking REBETOL, the patient should be apprised of the potential hazard to the fetus [see Warnings and Precautions (5.1), and Use in Specific Populations (8.1, 8.3)].
- men whose female partners are pregnant [see Use in Specific Populations (8.3)]
- patients with known hypersensitivity reactions such as Stevens-Johnson syndrome, toxic, epidermal necrolysis, and erythema multiforme to ribavirin or any component of the product
- patients with autoimmune hepatitis
- patients with hemoglobinopathies (e.g., thalassemia major, sickle-cell anemia)
- patients with creatinine clearance less than 50 mL/min [see Clinical Pharmacology (12.3)]
- when coadministered with didanosine because exposure to the active metabolite of didanosine (dideoxyadenosine 5'-triphosphate) is increased. Fatal hepatic failure, as well as peripheral neuropathy, pancreatitis, and symptomatic hyperlactatemia/lactic acidosis, has been reported in patients receiving didanosine in combination with ribavirin [see Drug Interactions (7.1)].
5 WARNINGS AND PRECAUTIONS
5.1 Embryo-Fetal Toxicity
REBETOL capsules and oral solution may cause birth defects, miscarriage or stillbirth. REBETOL therapy should not be started until a report of a negative pregnancy test has been obtained immediately prior to planned initiation of therapy. Female patients should use effective contraception and have periodic monitoring with pregnancy tests during treatment and during the 9-month period after treatment has been stopped. Male patients and their female partners should use effective contraception during treatment and during the 6-month period after treatment has been stopped. Extreme care must be taken to avoid pregnancy in female patients and in female partners of male patients. REBETOL has demonstrated significant teratogenic and embryocidal effects in all animal species tested. These effects occurred at doses as low as one twentieth of the recommended human dose of ribavirin. [see Boxed Warning, Contraindications (4), and Use in Specific Populations (8.1, 8.3)].
5.2 Anemia
Hemolytic anemia was observed in approximately 10% of REBETOL/INTRON A-treated subjects in clinical trials. The anemia associated with REBETOL occurs within 1 to 2 weeks of initiation of therapy. Because the initial drop in hemoglobin may be significant, obtain hemoglobin or hematocrit levels before the start of treatment and at Week 2 and Week 4 of therapy, or more frequently if clinically indicated. Patients should then be followed as clinically appropriate [see Dosage and Administration (2.5, 2.6)].
Fatal and nonfatal myocardial infarctions have been reported in patients with anemia caused by REBETOL. Patients should be assessed for underlying cardiac disease before initiation of ribavirin therapy. Patients with pre-existing cardiac disease should have electrocardiograms administered before treatment and should be appropriately monitored during therapy. If there is any deterioration of cardiovascular status, therapy should be suspended or discontinued [see Dosage and Administration (2.5, 2.6)]. Because cardiac disease may be worsened by drug-induced anemia, patients with a history of significant or unstable cardiac disease should not use REBETOL.
5.3 Pancreatitis
Suspend REBETOL and INTRON A or PegIntron combination therapy in patients with signs and symptoms of pancreatitis and discontinue in patients with confirmed pancreatitis.
5.4 Pulmonary Disorders
Pulmonary symptoms, including dyspnea, pulmonary infiltrates, pneumonitis, pulmonary hypertension, and pneumonia, have been reported during REBETOL with alpha interferon combination therapy; occasional cases of fatal pneumonia have occurred. In addition, sarcoidosis or the exacerbation of sarcoidosis has been reported. If there is evidence of pulmonary infiltrates or pulmonary function impairment, closely monitor the patient, and if appropriate, discontinue combination therapy.
5.5 Ophthalmologic Disorders
Ribavirin is used in combination therapy with INTRON A or PegIntron. Refer to labeling for PegIntron for additional information.
5.6 Laboratory Tests
PegIntron in combination with ribavirin may cause severe decreases in neutrophil and platelet counts, and hematologic, endocrine (e.g., TSH), and hepatic abnormalities.
Obtain hematology and blood chemistry testing in patients on PegIntron/REBETOL combination therapy before the start of treatment and then periodically thereafter. In the adult clinical trial, complete blood counts (including hemoglobin, neutrophil, and platelet counts) and chemistries (including AST, ALT, bilirubin, and uric acid) were measured during the treatment period at Weeks 2, 4, 8, 12, and then at 6-week intervals, or more frequently if abnormalities developed. In pediatric subjects, the same laboratory parameters were evaluated with additional assessment of hemoglobin at treatment Week 6. TSH levels were measured every 12 weeks during the treatment period. HCV-RNA should be measured periodically during treatment [see Dosage and Administration (2)].
5.7 Dental and Periodontal Disorders
Dental and periodontal disorders have been reported in patients receiving ribavirin and interferon or peginterferon combination therapy. In addition, dry mouth could have a damaging effect on teeth and mucous membranes of the mouth during long-term treatment with the combination of REBETOL and pegylated or nonpegylated interferon alfa-2b. Advise patients to brush their teeth thoroughly twice daily and have regular dental examinations. If vomiting occurs, advise patients to rinse out their mouth thoroughly afterwards.
5.8 Concomitant Administration of Azathioprine
Pancytopenia (marked decreases in red blood cells, neutrophils, and platelets) and bone marrow suppression have been reported in the literature to occur within 3 to 7 weeks after the concomitant administration of pegylated interferon/ribavirin and azathioprine. In this limited number of patients (n=8), myelotoxicity was reversible within 4 to 6 weeks upon withdrawal of both HCV antiviral therapy and concomitant azathioprine and did not recur upon reintroduction of either treatment alone. Discontinue PegIntron, REBETOL, and azathioprine for pancytopenia, and do not reintroduce pegylated interferon/ribavirin with concomitant azathioprine [see Drug Interactions (7.4)].
5.9 Impact on Growth in Pediatric Patients
Data on the effects of PegIntron and REBETOL on growth come from an open-label study in subjects 3 through 17 years of age, in which weight and height changes were compared to US normative population data. In general, the weight and height gain of pediatric subjects treated with PegIntron and REBETOL lagged behind that predicted by normative population data for the entire length of treatment. Severely inhibited growth velocity (less than 3rd percentile) was observed in 70% of the subjects while on treatment. Following treatment, rebound growth and weight gain occurred in most subjects. Long-term follow-up data in pediatric subjects, however, indicates that PegIntron in combination therapy with REBETOL may induce a growth inhibition that results in reduced adult height in some patients [see Adverse Reactions (6.1)].
Similarly, an impact on growth was seen in subjects after treatment with REBETOL and INTRON A combination therapy for one year. In a long-term follow-up trial of a limited number of these subjects, combination therapy resulted in reduced final adult height in some subjects [see Adverse Reactions (6.1)].
5.10 Not Recommended for Monotherapy and Risks Associated with Combination Therapy
Based on results of clinical trials, ribavirin monotherapy is not effective for the treatment of chronic hepatitis C virus infection; therefore, REBETOL capsules or oral solution must not be used alone. The safety and efficacy of REBETOL capsules and oral solution have only been established when used together with INTRON A or PegIntron (not other interferons) as combination therapy.
The safety and efficacy of REBETOL with INTRON A or PegIntron combination therapy for the treatment of HIV infection, adenovirus, RSV, parainfluenza, or influenza infections have not been established. REBETOL capsules should not be used for these indications.
There are significant adverse reactions caused by REBETOL/INTRON A or PegIntron combination therapy, including severe depression and suicidal or homicidal ideation, hemolytic anemia, suppression of bone marrow function, autoimmune and infectious disorders, pulmonary dysfunction, pancreatitis, and diabetes. Suicidal ideation or attempts occurred more frequently among pediatric patients, primarily adolescents, compared to adult patients (2.4% versus 1%) during treatment and off-therapy follow-up. Labeling for INTRON A and PegIntron should be reviewed in their entirety for additional safety information prior to initiation of combination treatment.
6 ADVERSE REACTIONS
The following serious adverse drug reactions are discussed in other sections of the labeling:
- Embryo-Fetal Toxicity [see Warnings and Precautions (5.1)]
- Anemia [see Warnings and Precautions (5.2)]
- Pancreatitis [see Warnings and Precautions (5.3)]
- Pulmonary Disorders [see Warnings and Precautions (5.4)]
- Ophthalmic Disorders [see Warnings and Precautions (5.5)]
- Dental and Periodontal Disorders [see Warnings and Precautions (5.7)]
- Impact on Growth in Pediatric Patients [see Warnings and Precautions (5.9)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Clinical trials with REBETOL in combination with PegIntron or INTRON A have been conducted in over 7,800 subjects from 3 to 76 years of age.
The primary toxicity of ribavirin is hemolytic anemia. Reductions in hemoglobin levels occurred within the first 1 to 2 weeks of oral therapy. Cardiac and pulmonary reactions associated with anemia occurred in approximately 10% of patients [see Warnings and Precautions (5.2)].
Greater than 96% of all subjects in clinical trials experienced one or more adverse reactions. The most commonly reported adverse reactions in adult subjects receiving PegIntron or INTRON A in combination with REBETOL were injection site inflammation/reaction, fatigue/asthenia, headache, rigors, fevers, nausea, myalgia and anxiety/emotional lability/irritability. The most common adverse reactions in pediatric subjects, ages 3 and older, receiving REBETOL in combination with PegIntron or INTRON A were pyrexia, headache, neutropenia, fatigue, anorexia, injection site erythema, and vomiting.
The Adverse Reactions section references the following clinical trials:
- REBETOL/PegIntron Combination therapy trials:
- Clinical Study 1 – evaluated PegIntron monotherapy (not further described in this label; see labeling for PegIntron for information about this trial).
- Study 2 – evaluated REBETOL 800 mg/day flat dose in combination with 1.5 mcg/kg/week PegIntron or with INTRON A.
- Study 3 – evaluated PegIntron/weight-based REBETOL in combination with PegIntron/flat dose REBETOL regimen.
- Study 4 – compared two PegIntron (1.5 mcg/kg/week and 1 mcg/kg/week) doses in combination with REBETOL and a third treatment group receiving Pegasys® (180 mcg/week)/Copegus® (1000-1200 mg/day).
- Study 5 – evaluated PegIntron (1.5 mcg/kg/week) in combination with weight-based REBETOL in prior treatment failure subjects.
- PegIntron/REBETOL Combination Therapy in Pediatric Patients
- REBETOL/INTRON A Combination Therapy trials for adults and pediatrics
Serious adverse reactions have occurred in approximately 12% of subjects in clinical trials with PegIntron with or without REBETOL [see Boxed Warning, Warnings and Precautions (5)]. The most common serious events occurring in subjects treated with PegIntron and REBETOL were depression and suicidal ideation [see Warnings and Precautions (5.10)], each occurring at a frequency of less than 1%. Suicidal ideation or attempts occurred more frequently among pediatric patients, primarily adolescents, compared to adult patients (2.4% versus 1%) during treatment and off-therapy follow-up [see Warnings and Precautions (5.10)]. The most common fatal reaction occurring in subjects treated with PegIntron and REBETOL was cardiac arrest, suicidal ideation, and suicide attempt [see Warnings and Precautions (5.10)], all occurring in less than 1% of subjects.
Adverse Reaction - REBETOL/PegIntron Combination Therapy
Adult Subjects
Adverse reactions that occurred in the clinical trial at greater than 5% incidence are provided by treatment group from the REBETOL/PegIntron Combination Therapy (Study 2) in Table 5.
| Adverse Reactions | Percentage of Subjects Reporting Adverse Reactions* | Adverse Reactions | Percentage of Subjects Reporting Adverse Reactions* | ||
|---|---|---|---|---|---|
| PegIntron 1.5 mcg/kg/ REBETOL | INTRON A/REBETOL | PegIntron 1.5 mcg/kg/ REBETOL | INTRON A/REBETOL | ||
| (N=511) | (N=505) | (N=511) | (N=505) | ||
|
|||||
| Application Site | Musculoskeletal | ||||
| Injection Site Inflammation | 25 | 18 | Myalgia | 56 | 50 |
| Injection Site Reaction | 58 | 36 | Arthralgia | 34 | 28 |
| Autonomic Nervous System | Musculoskeletal Pain | 21 | 19 | ||
| Dry Mouth | 12 | 8 | Psychiatric | ||
| Increased Sweating | 11 | 7 | Insomnia | 40 | 41 |
| Flushing | 4 | 3 | Depression | 31 | 34 |
| Body as a Whole | Anxiety/Emotional Lability/Irritability | 47 | 47 | ||
| Fatigue/Asthenia | 66 | 63 | Concentration Impaired | 17 | 21 |
| Headache | 62 | 58 | Agitation | 8 | 5 |
| Rigors | 48 | 41 | Nervousness | 6 | 6 |
| Fever | 46 | 33 | Reproductive, Female | ||
| Weight Loss | 29 | 20 | Menstrual Disorder | 7 | 6 |
| Right Upper Quadrant Pain | 12 | 6 | Resistance Mechanism | ||
| Chest Pain | 8 | 7 | Viral Infection | 12 | 12 |
| Malaise | 4 | 6 | Fungal Infection | 6 | 1 |
| Central/Peripheral Nervous System | Respiratory System | ||||
| Dizziness | 21 | 17 | Dyspnea | 26 | 24 |
| Endocrine | Coughing | 23 | 16 | ||
| Hypothyroidism | 5 | 4 | Pharyngitis | 12 | 13 |
| Gastrointestinal | Rhinitis | 8 | 6 | ||
| Nausea | 43 | 33 | Sinusitis | 6 | 5 |
| Anorexia | 32 | 27 | Skin and Appendages | ||
| Diarrhea | 22 | 17 | Alopecia | 36 | 32 |
| Vomiting | 14 | 12 | Pruritus | 29 | 28 |
| Abdominal Pain | 13 | 13 | Rash | 24 | 23 |
| Dyspepsia | 9 | 8 | Skin Dry | 24 | 23 |
| Constipation | 5 | 5 | Special Senses, Other | ||
| Hematologic Disorders | Taste Perversion | 9 | 4 | ||
| Neutropenia | 26 | 14 | Vision Disorders | ||
| Anemia | 12 | 17 | Vision Blurred | 5 | 6 |
| Leukopenia | 6 | 5 | Conjunctivitis | 4 | 5 |
| Thrombocytopenia | 5 | 2 | |||
| Liver and Biliary System | |||||
| Hepatomegaly | 4 | 4 | |||
Table 6 summarizes the treatment-related adverse reactions in Study 4 that occurred at a greater than or equal to 10% incidence.
| Study 4 Percentage of Subjects Reporting Treatment-Related Adverse Reactions |
|||
|---|---|---|---|
| Adverse Reactions | PegIntron 1.5 mcg/kg with REBETOL | PegIntron 1 mcg/kg with REBETOL | Pegasys 180 mcg with Copegus |
| (N=1019) | (N=1016) | (N=1035) | |
| Fatigue | 67 | 68 | 64 |
| Headache | 50 | 47 | 41 |
| Nausea | 40 | 35 | 34 |
| Chills | 39 | 36 | 23 |
| Insomnia | 38 | 37 | 41 |
| Anemia | 35 | 30 | 34 |
| Pyrexia | 35 | 32 | 21 |
| Injection Site Reactions | 34 | 35 | 23 |
| Anorexia | 29 | 25 | 21 |
| Rash | 29 | 25 | 34 |
| Myalgia | 27 | 26 | 22 |
| Neutropenia | 26 | 19 | 31 |
| Irritability | 25 | 25 | 25 |
| Depression | 25 | 19 | 20 |
| Alopecia | 23 | 20 | 17 |
| Dyspnea | 21 | 20 | 22 |
| Arthralgia | 21 | 22 | 22 |
| Pruritus | 18 | 15 | 19 |
| Influenza-like Illness | 16 | 15 | 15 |
| Dizziness | 16 | 14 | 13 |
| Diarrhea | 15 | 16 | 14 |
| Cough | 15 | 16 | 17 |
| Weight Decreased | 13 | 10 | 10 |
| Vomiting | 12 | 10 | 9 |
| Unspecified Pain | 12 | 13 | 9 |
| Dry Skin | 11 | 11 | 12 |
| Anxiety | 11 | 11 | 10 |
| Abdominal Pain | 10 | 10 | 10 |
| Leukopenia | 9 | 7 | 10 |
The incidence of serious adverse reactions was comparable in all trials. In Study 2, the incidence of serious adverse reactions was 17% in the PegIntron/REBETOL groups compared to 14% in the INTRON A/REBETOL group. In Study 3, there was a similar incidence of serious adverse reactions reported for the weight-based REBETOL group (12%) and for the flat-dose REBETOL regimen.
In many but not all cases, adverse reactions resolved after dose reduction or discontinuation of therapy. Some subjects experienced ongoing or new serious adverse reactions during the 6-month follow-up period. In Study 2, many subjects continued to experience adverse reactions several months after discontinuation of therapy. By the end of the 6-month follow-up period, the incidence of ongoing adverse reactions by body class in the PegIntron 1.5/REBETOL group was 33% (psychiatric), 20% (musculoskeletal), and 10% (for endocrine and for GI). In approximately 10 to 15% of subjects, weight loss, fatigue, and headache had not resolved.
There have been 28 subject deaths that occurred during treatment or follow-up in Studies 2, 3, and 4. In Study 2, there was 1 suicide in a subject receiving PegIntron/REBETOL combination therapy; and 1 subject death in the INTRON A/REBETOL group (motor vehicle accident). In Study 3, there were 14 deaths, 2 of which were probable suicides and 1 was an unexplained death in a person with a relevant medical history of depression. In Study 4, there were 12 deaths, 6 of which occurred in subjects who received PegIntron/REBETOL combination therapy, 5 in the PegIntron 1.5 mcg/REBETOL arm (N=1019) and 1 in the PegIntron 1 mcg/REBETOL arm (N=1016), and 6 of which occurred in subjects receiving Pegasys/Copegus (N=1035); there were 3 suicides that occurred during the off treatment follow-up period in subjects who received PegIntron (1.5 mcg/kg)/REBETOL combination therapy.
In Studies 1 and 2, 10 to 14% of subjects receiving PegIntron, alone or in combination with REBETOL, discontinued therapy compared with 6% treated with INTRON A alone and 13% treated with INTRON A in combination with REBETOL. In Study 3, 15% of subjects receiving PegIntron in combination with weight-based REBETOL and 14% of subjects receiving PegIntron with flat-dose REBETOL discontinued therapy due to an adverse reaction. The most common reasons for discontinuation were related to known interferon effects of psychiatric, systemic (e.g., fatigue, headache), or gastrointestinal adverse reactions. In Study 4, 13% of subjects in the PegIntron 1.5 mcg/REBETOL arm, 10% in the PegIntron 1 mcg/REBETOL arm, and 13% in the Pegasys 180 mcg/Copegus arm discontinued due to adverse events.
In Study 2, dose reductions for REBETOL were similar across all three groups [see Clinical Studies (14.1)], 33 to 35%. The most common reasons for dose modifications were neutropenia (18%), or anemia (9%) (see Laboratory Values). Other common reasons included depression, fatigue, nausea, and thrombocytopenia. In Study 3, dose modifications due to adverse reactions occurred more frequently with weight-based REBETOL dosing compared to flat dosing (29% and 23%, respectively). In Study 4, 16% of subjects had a dose reduction of PegIntron to 1 mcg/kg in combination with REBETOL, with an additional 4% requiring the second dose reduction of PegIntron to 0.5 mcg/kg due to adverse events compared to 15% of subjects in the Pegasys/Copegus arm, who required a dose reduction to 135 mcg/week with Pegasys, with an additional 7% in the Pegasys/Copegus arm requiring a second dose reduction to 90 mcg/week with Pegasys.
In the PegIntron/REBETOL combination trials the most common adverse reactions were psychiatric, which occurred among 77% of subjects in Study 2 and 68% to 69% of subjects in Study 3. These psychiatric adverse reactions included most commonly depression, irritability, and insomnia, each reported by approximately 30% to 40% of subjects in all treatment groups. Suicidal behavior (ideation, attempts, and suicides) occurred in 2% of all subjects during treatment or during follow-up after treatment cessation [see Warnings and Precautions (5)]. In Study 4, psychiatric adverse reactions occurred in 58% of subjects in the PegIntron 1.5 mcg/REBETOL arm, 55% of subjects in the PegIntron 1 mcg/REBETOL arm, and 57% of subjects in the Pegasys 180 mcg/Copegus arm.
In Study 2, PegIntron/REBETOL combination therapy induced fatigue or headache in approximately two-thirds of subjects, with fever or rigors in approximately half of the subjects. The severity of some of these systemic symptoms (e.g., fever and headache) tended to decrease as treatment continued.
Subjects receiving REBETOL/PegIntron as re-treatment after failing a previous interferon combination regimen reported adverse reactions similar to those previously associated with this regimen during clinical trials of treatment-naïve subjects.
Pediatric Subjects
In general, the adverse reaction profile in the pediatric population was similar to that observed in adults. In the pediatric trial, the most prevalent adverse reactions were pyrexia (80%), headache (62%), neutropenia (33%), fatigue (30%), anorexia (29%), injection-site erythema (29%) and vomiting (27%). The majority of adverse reactions were mild or moderate in severity. Severe adverse reactions were reported in 7% (8/107) of all subjects and included injection site pain (1%), pain in extremity (1%), headache (1%), neutropenia (1%), and pyrexia (4%). Important adverse reactions that occurred in this subject population were nervousness (7%; 7/107), aggression (3%; 3/107), anger (2%; 2/107), and depression (1%; 1/107). Five subjects received levothyroxine treatment, three with clinical hypothyroidism and two with asymptomatic TSH elevations. Weight and height gain of pediatric subjects treated with PegIntron plus REBETOL lagged behind that predicted by normative population data for the entire length of treatment. Severely inhibited growth velocity (less than 3rd percentile) was observed in 70% of the subjects while on treatment.
Dose modifications of PegIntron and/or ribavirin were required in 25% of subjects due to treatment-related adverse reactions, most commonly for anemia, neutropenia and weight loss. Two subjects (2%; 2/107) discontinued therapy as the result of an adverse reaction.
Adverse reactions that occurred with a greater than or equal to 10% incidence in the pediatric trial subjects are provided in Table 7.
| System Organ Class Preferred Term | All Subjects (N=107) |
|---|---|
| Blood and Lymphatic System Disorders | |
| Neutropenia | 33% |
| Anemia | 11% |
| Leukopenia | 10% |
| Gastrointestinal Disorders | |
| Abdominal Pain | 21% |
| Abdominal Pain Upper | 12% |
| Vomiting | 27% |
| Nausea | 18% |
| General Disorders and Administration Site Conditions | |
| Pyrexia | 80% |
| Fatigue | 30% |
| Injection-site Erythema | 29% |
| Chills | 21% |
| Asthenia | 15% |
| Irritability | 14% |
| Investigations | |
| Weight Loss | 19% |
| Metabolism and Nutrition Disorders | |
| Anorexia | 29% |
| Decreased Appetite | 22% |
| Musculoskeletal and Connective Tissue Disorders | |
| Arthralgia | 17% |
| Myalgia | 17% |
| Nervous System Disorders | |
| Headache | 62% |
| Dizziness | 14% |
| Skin and Subcutaneous Tissue Disorders | |
| Alopecia | 17% |
Ninety-four of 107 subjects enrolled in a 5-year follow-up trial. The long-term effects on growth were less in subjects treated for 24 weeks than in those treated for 48 weeks. Twenty-four percent of subjects (11/46) treated for 24 weeks and 40% of subjects (19/48) treated for 48 weeks had a >15 percentile height-for-age decrease from pre-treatment baseline to the end of 5-year follow-up. Eleven percent of subjects (5/46) treated for 24 weeks and 13% of subjects (6/48) treated for 48 weeks had a >30 percentile height-for-age decrease from pre-treatment baseline to the end of the 5-year follow-up. While observed across all age groups, the highest risk for reduced height at the end of long-term follow-up appeared to be initiation of combination therapy during the years of expected peak growth velocity [see Warnings and Precautions (5.9)].
Laboratory Values
Adult and Pediatric Subjects
The adverse reaction profile in Study 3, which compared PegIntron/weight-based REBETOL combination to a PegIntron/flat dose REBETOL regimen, revealed an increased rate of anemia with weight-based dosing (29% vs. 19% for weight-based vs. flat dose regimens, respectively). However, the majority of cases of anemia were mild and responded to dose reductions.
Changes in selected laboratory values during treatment in combination with REBETOL treatment are described below. Decreases in hemoglobin, leukocytes, neutrophils, and platelets may require dose reduction or permanent discontinuation from therapy [see Dosage and Administration (2.5)]. Changes in selected laboratory values during therapy are described in Table 8. Most of the changes in laboratory values in the PegIntron/REBETOL trial with pediatrics were mild or moderate.
| Laboratory Parameters* | Percentage of Subjects | ||
|---|---|---|---|
| Adults (Study 2) | Pediatrics | ||
| PegIntron/ REBETOL (N=511) | INTRON A/ REBETOL (N=505) | PegIntron/REBETOL (N=107)* |
|
| Hemoglobin (g/dL) | |||
| 9.5 to <11.0 | 26 | 27 | 30 |
| 8.0 to <9.5 | 3 | 3 | 2 |
| 6.5-7.9 | 0.2 | 0.2 | - |
| Leukocytes (× 109/L) | |||
| 2.0-2.9 | 46 | 41 | 39 |
| 1.5 to <2.0 | 24 | 8 | 3 |
| 1.0-1.4 | 5 | 1 | - |
| Neutrophils (× 109/L) | |||
| 1.0-1.5 | 33 | 37 | 35 |
| 0.75 to <1.0 | 25 | 13 | 26 |
| 0.5 to <0.75 | 18 | 7 | 13 |
| <0.5 | 4 | 2 | 3 |
| Platelets (× 109/L) | |||
| 70-100 | 15 | 5 | 1 |
| 50 to <70 | 3 | 0.8 | - |
| 30-49 | 0.2 | 0.2 | - |
| 25 to <50 | - | - | 1 |
| Total Bilirubin | (mg/dL) | (µmole/L) | |
| 1.5-3.0 | 10 | 13 | - |
| 1.26-2.59 × ULN† | - | - | 7 |
| 3.1-6.0 | 0.6 | 0.2 | - |
| 2.6-5 × ULN† | - | - | - |
| 6.1-12.0 | 0 | 0.2 | - |
| ALT (U/L) | |||
| 2 × Baseline | 0.6 | 0.2 | 1 |
| 2.1-5 × Baseline | 3 | 1 | 5 |
| 5.1-10 × Baseline | 0 | 0 | 3 |
Hemoglobin. In Study 2, hemoglobin levels decreased to less than 11 g/dL in about 30% of subjects. In Study 3, 47% of subjects receiving weight-based dosing of REBETOL and 33% on flat-dose REBETOL had decreases in hemoglobin levels to less than 11 g/dL. Reductions in hemoglobin to less than 9 g/dL occurred more frequently in subjects receiving weight-based dosing compared to flat dosing (4% and 2%, respectively). In Study 2, dose modification was required in 9% and 13% of subjects in the PegIntron/REBETOL and INTRON A/REBETOL groups. In Study 4, subjects receiving PegIntron (1.5 mcg/kg)/REBETOL had decreases in hemoglobin levels to between 8.5 to less than 10 g/dL (28%) and to less than 8.5 g/dL (3%), whereas in patients receiving Pegasys 180 mcg/Copegus these decreases occurred in 26% and 4% of subjects, respectively. On average, hemoglobin levels became stable by treatment Weeks 4-6. The typical pattern observed was a decrease in hemoglobin levels by treatment Week 4 followed by stabilization and a plateau, which was maintained to the end of treatment [see Dosage and Administration (2.5)].
Neutrophils. In Study 2, decreases in neutrophil counts were observed in a majority of adult subjects treated with PegIntron/REBETOL (85%) and INTRON A/REBETOL (60%). Severe, potentially life-threatening neutropenia (less than 0.5 × 109/L) occurred in approximately 4% of subjects treated with PegIntron/REBETOL and 2% of subjects treated with INTRON A/REBETOL. Eighteen percent of subjects receiving PegIntron/REBETOL required modification of interferon dosage. Few subjects (less than 1%) required permanent discontinuation of treatment. Neutrophil counts generally returned to pre-treatment levels 4 weeks after cessation of therapy [see Dosage and Administration (2.5)].
Platelets. In Study 2, platelet counts decreased to less than 100,000/mm3 in approximately 20% of subjects treated with PegIntron alone or with REBETOL and in 6% of adult subjects treated with INTRON A/REBETOL. Severe decreases in platelet counts (less than 50,000/mm3) occur in less than 4% of adult subjects. In Study 2, 1% or 3% of subjects required dose modification of INTRON A or PegIntron, respectively. Platelet counts generally returned to pretreatment levels 4 weeks after the cessation of therapy [see Dosage and Administration (2.5)].
Thyroid Function. In Study 2, clinically apparent thyroid disorders occurred among subjects treated with either INTRON A or PegIntron (with or without REBETOL) at a similar incidence (5% for hypothyroidism and 3% for hyperthyroidism). Subjects developed new onset TSH abnormalities while on treatment and during the follow-up period. At the end of the follow-up period, 7% of subjects still had abnormal TSH values.
Adverse Reactions with REBETOL/INTRON A Combination Therapy
Adult Subjects
In clinical trials, 19% and 6% of previously untreated and relapse subjects, respectively, discontinued therapy due to adverse reactions in the combination arms compared to 13% and 3% in the interferon-only arms. Selected treatment-related adverse reactions that occurred in the US trials with incidence 5% or greater are provided by treatment group (see Table 9). In general, the selected treatment-related adverse reactions were reported with lower incidence in the international trials as compared to the US trials, except for asthenia, influenza-like symptoms, nervousness, and pruritus.
Pediatric Subjects
In clinical trials of 118 pediatric subjects 3 to 16 years of age, 6% discontinued therapy due to adverse reactions. Dose modifications were required in 30% of subjects, most commonly for anemia and neutropenia. In general, the adverse-reaction profile in the pediatric population was similar to that observed in adults. Injection site disorders, fever, anorexia, vomiting, and emotional lability occurred more frequently in pediatric subjects compared to adult subjects. Conversely, pediatric subjects experienced less fatigue, dyspepsia, arthralgia, insomnia, irritability, impaired concentration, dyspnea, and pruritus compared to adult subjects. Selected treatment-related adverse reactions that occurred with incidence 5% or greater among all pediatric subjects who received the recommended dose of REBETOL/INTRON A combination therapy are provided in Table 9.
| Subjects Reporting Adverse Reactions* | Percentage of Subjects | ||||||
|---|---|---|---|---|---|---|---|
| US Previously Untreated Study | US Relapse Study | Pediatric Subjects | |||||
| 24 weeks of treatment | 48 weeks of treatment | 24 weeks of treatment | 48 weeks of treatment | ||||
| INTRON A/ REBETOL (N=228) | INTRON A/ Placebo (N=231) | INTRON A/ REBETOL (N=228) | INTRON A/ Placebo (N=225) | INTRON A/ REBETOL (N=77) | INTRON A/ Placebo (N=76) | INTRON A/ REBETOL (N=118) |
|
|
|||||||
| Application Site Disorders | |||||||
| Injection Site Inflammation | 13 | 10 | 12 | 14 | 6 | 8 | 14 |
| Injection Site Reaction | 7 | 9 | 8 | 9 | 5 | 3 | 19 |
| Body as a Whole - General Disorders | |||||||
| Headache | 63 | 63 | 66 | 67 | 66 | 68 | 69 |
| Fatigue | 68 | 62 | 70 | 72 | 60 | 53 | 58 |
| Rigors | 40 | 32 | 42 | 39 | 43 | 37 | 25 |
| Fever | 37 | 35 | 41 | 40 | 32 | 36 | 61 |
| Influenza-like Symptoms | 14 | 18 | 18 | 20 | 13 | 13 | 31 |
| Asthenia | 9 | 4 | 9 | 9 | 10 | 4 | 5 |
| Chest Pain | 5 | 4 | 9 | 8 | 6 | 7 | 5 |
| Central & Peripheral Nervous System Disorders | |||||||
| Dizziness | 17 | 15 | 23 | 19 | 26 | 21 | 20 |
| Gastrointestinal System Disorders | |||||||
| Nausea | 38 | 35 | 46 | 33 | 47 | 33 | 33 |
| Anorexia | 27 | 16 | 25 | 19 | 21 | 14 | 51 |
| Dyspepsia | 14 | 6 | 16 | 9 | 16 | 9 | <1 |
| Vomiting | 11 | 10 | 9 | 13 | 12 | 8 | 42 |
| Musculoskeletal System Disorders | |||||||
| Myalgia | 61 | 57 | 64 | 63 | 61 | 58 | 32 |
| Arthralgia | 30 | 27 | 33 | 36 | 29 | 29 | 15 |
| Musculoskeletal Pain | 20 | 26 | 28 | 32 | 22 | 28 | 21 |
| Psychiatric Disorders | |||||||
| Insomnia | 39 | 27 | 39 | 30 | 26 | 25 | 14 |
| Irritability | 23 | 19 | 32 | 27 | 25 | 20 | 10 |
| Depression | 32 | 25 | 36 | 37 | 23 | 14 | 13 |
| Emotional Lability | 7 | 6 | 11 | 8 | 12 | 8 | 16 |
| Concentration Impaired | 11 | 14 | 14 | 14 | 10 | 12 | 5 |
| Nervousness | 4 | 2 | 4 | 4 | 5 | 4 | 3 |
| Respiratory System Disorders | |||||||
| Dyspnea | 19 | 9 | 18 | 10 | 17 | 12 | 5 |
| Sinusitis | 9 | 7 | 10 | 14 | 12 | 7 | <1 |
| Skin and Appendages Disorders | |||||||
| Alopecia | 28 | 27 | 32 | 28 | 27 | 26 | 23 |
| Rash | 20 | 9 | 28 | 8 | 21 | 5 | 17 |
| Pruritus | 21 | 9 | 19 | 8 | 13 | 4 | 12 |
| Special Senses, Other Disorders | |||||||
| Taste Perversion | 7 | 4 | 8 | 4 | 6 | 5 | <1 |
During a 48-week course of therapy there was a decrease in the rate of linear growth (mean percentile assignment decrease of 7%) and a decrease in the rate of weight gain (mean percentile assignment decrease of 9%). A general reversal of these trends was noted during the 24-week post-treatment period. Long-term data in a limited number of patients, however, suggests that combination therapy may induce a growth inhibition that results in reduced final adult height in some patients [see Warnings and Precautions (5.9)].
Laboratory Values
Changes in selected hematologic values (hemoglobin, white blood cells, neutrophils, and platelets) during therapy are described below (see Table 10).
Hemoglobin. Hemoglobin decreases among subjects receiving REBETOL therapy began at Week 1, with stabilization by Week 4. In previously untreated subjects treated for 48 weeks, the mean maximum decrease from baseline was 3.1 g/dL in the US trial and 2.9 g/dL in the international trial. In relapse subjects, the mean maximum decrease from baseline was 2.8 g/dL in the US trial and 2.6 g/dL in the international trial. Hemoglobin values returned to pretreatment levels within 4 to 8 weeks of cessation of therapy in most subjects.
Bilirubin and Uric Acid. Increases in both bilirubin and uric acid, associated with hemolysis, were noted in clinical trials. Most changes were moderate and reversed within 4 weeks after treatment discontinuation. This observation occurred most frequently in subjects with a previous diagnosis of Gilbert's syndrome. This has not been associated with hepatic dysfunction or clinical morbidity.
| Percentage of Subjects | |||||||
|---|---|---|---|---|---|---|---|
| US Previously Untreated Study | US Relapse Study | Pediatric Subjects | |||||
| 24 weeks of treatment | 48 weeks of treatment | 24 weeks of treatment | 48 weeks of treatment | ||||
| INTRON A/ REBETOL (N=228) | INTRON A/ Placebo (N=231) | INTRON A/REBETOL (N=228) | INTRON A/ Placebo (N=225) | INTRON A/ REBETOL (N=77) | INTRON A/ Placebo (N=76) | INTRON A/ REBETOL (N=118) |
|
| Hemoglobin (g/dL) | |||||||
| 9.5 to 10.9 | 24 | 1 | 32 | 1 | 21 | 3 | 24 |
| 8.0 to 9.4 | 5 | 0 | 4 | 0 | 4 | 0 | 3 |
| 6.5 to 7.9 | 0 | 0 | 0 | 0.4 | 0 | 0 | 0 |
| <6.5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Leukocytes (× 109/L) | |||||||
| 2.0 to 2.9 | 40 | 20 | 38 | 23 | 45 | 26 | 35 |
| 1.5 to 1.9 | 4 | 1 | 9 | 2 | 5 | 3 | 8 |
| 1.0 to 1.4 | 0.9 | 0 | 2 | 0 | 0 | 0 | 0 |
| <1.0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Neutrophils (× 109/L) | |||||||
| 1.0 to 1.49 | 30 | 32 | 31 | 44 | 42 | 34 | 37 |
| 0.75 to 0.99 | 14 | 15 | 14 | 11 | 16 | 18 | 15 |
| 0.5 to 0.74 | 9 | 9 | 14 | 7 | 8 | 4 | 16 |
| <0.5 | 11 | 8 | 11 | 5 | 5 | 8 | 3 |
| Platelets (× 109/L) | |||||||
| 70 to 99 | 9 | 11 | 11 | 14 | 6 | 12 | 0.8 |
| 50 to 69 | 2 | 3 | 2 | 3 | 0 | 5 | 2 |
| 30 to 49 | 0 | 0.4 | 0 | 0.4 | 0 | 0 | 0 |
| <30 | 0.9 | 0 | 1 | 0.9 | 0 | 0 | 0 |
| Total Bilirubin (mg/dL) | |||||||
| 1.5 to 3.0 | 27 | 13 | 32 | 13 | 21 | 7 | 2 |
| 3.1 to 6.0 | 0.9 | 0.4 | 2 | 0 | 3 | 0 | 0 |
| 6.1 to 12.0 | 0 | 0 | 0.4 | 0 | 0 | 0 | 0 |
| >12.0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
6.2 Postmarketing Experiences
The following adverse reactions have been identified and reported during post approval use of REBETOL in combination with INTRON A or PegIntron. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood and Lymphatic System disorders
Pure red cell aplasia, aplastic anemia
Ear and Labyrinth disorders
Hearing disorder, vertigo
Respiratory, Thoracic and Mediastinal disorders
Pulmonary hypertension
Eye disorders
Serous retinal detachment
Endocrine disorders
Diabetes
7 DRUG INTERACTIONS
7.1 Didanosine
Exposure to didanosine or its active metabolite (dideoxyadenosine 5'-triphosphate) is increased when didanosine is coadministered with ribavirin, which could cause or worsen clinical toxicities; therefore, coadministration of REBETOL capsules or oral solution and didanosine is contraindicated [see Contraindications (4)]. Reports of fatal hepatic failure, as well as peripheral neuropathy, pancreatitis, and symptomatic hyperlactatemia/lactic acidosis have been reported in clinical trials.
7.2 Nucleoside Analogues
Hepatic decompensation (some fatal) has occurred in cirrhotic HIV/HCV co-infected patients receiving combination antiretroviral therapy for HIV and interferon alpha and ribavirin. Patients receiving interferon with ribavirin and nucleoside reverse transcriptase inhibitors (NRTIs) should be closely monitored for treatment-associated toxicities, especially hepatic decompensation and anemia. Discontinuation of NRTIs should be considered as medically appropriate (see labeling for individual NRTI product). Dose reduction or discontinuation of interferon, ribavirin, or both should also be considered if worsening clinical toxicities are observed, including hepatic decompensation (e.g., Child-Pugh greater than 6).
Ribavirin may antagonize the cell culture antiviral activity of stavudine and zidovudine against HIV. Ribavirin has been shown in cell culture to inhibit phosphorylation of lamivudine, stavudine, and zidovudine, which could lead to decreased antiretroviral activity. However, in a study with another pegylated interferon in combination with ribavirin, no pharmacokinetic (e.g., plasma concentrations or intracellular triphosphorylated active metabolite concentrations) or pharmacodynamic (e.g., loss of HIV/HCV virologic suppress) interaction was observed when ribavirin and lamivudine (n=18), stavudine (n=10), or zidovudine (n=6) were coadministered as part of a multidrug regimen in HIV/HCV co-infected subjects. Concomitant use of ribavirin with any of these drugs should be done with caution.
7.3 Drugs Metabolized by Cytochrome P-450
Results of in vitro studies using both human and rat liver microsome preparations indicated little or no cytochrome P-450 enzyme-mediated metabolism of ribavirin, with minimal potential for P-450 enzyme-based drug interactions.
No pharmacokinetic interactions were noted between INTRON A and REBETOL capsules in a multiple-dose pharmacokinetic study.
7.4 Azathioprine
The use of ribavirin for the treatment of chronic hepatitis C in patients receiving azathioprine has been reported to induce severe pancytopenia and may increase the risk of azathioprine-related myelotoxicity. Inosine monophosphate dehydrogenase (IMDH) is required for one of the metabolic pathways of azathioprine. Ribavirin is known to inhibit IMDH, thereby leading to accumulation of an azathioprine metabolite, 6-methylthioinosine monophosphate (6-MTITP), which is associated with myelotoxicity (neutropenia, thrombocytopenia, and anemia). Patients receiving azathioprine with ribavirin should have complete blood counts, including platelet counts, monitored weekly for the first month, twice monthly for the second and third months of treatment, then monthly or more frequently if dosage or other therapy changes are necessary [see Warnings and Precautions (5.8)].
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
REBETOL is contraindicated for use in pregnant women and in men whose female partners are pregnant [see Contraindications (4)]. Based on animal data, ribavirin use in pregnancy may be associated with birth defects. Data from the Ribavirin Pregnancy Registry are insufficient to identify a drug-associated risk of birth defects, miscarriage, or adverse maternal or fetal outcomes (see Data). Ribavirin is known to accumulate in intracellular components from where it is cleared very slowly. In animal studies, ribavirin exposure was shown to have teratogenic and/or embryocidal effects (see Data).
All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage is 2-4% and 15-20%, respectively.
Data
Human Data
Available data from the Ribavirin Pregnancy Registry on 88 live births from pregnancies in women directly exposed and 98 live births from pregnancies in women indirectly exposed (by a male partner) to ribavirin during pregnancy or during the 6 months prior to pregnancy show a higher rate of birth defects (9.09% and 6.12%, respectively) compared to a background birth defect rate of 2.72% in the Metropolitan Atlanta Congenital Defects Program (MACDP) birth defects surveillance system. No pattern of birth defects can be identified from these reports. The miscarriage rate was approximately 21%. The current sample size is insufficient for reaching definitive conclusions based on statistical analysis. Trends suggesting a common etiology or relationship with ribavirin exposure were not observed. Methodologic limitations of the Ribavirin Pregnancy Registry include the use of MACDP as the external comparator group. Limitations of using an external comparator include differences in methodology and populations, as well as confounding due to the underlying disease and comorbidities.
Animal Data
Embryotoxicity/teratogenicity studies with ribavirin were conducted in rats (oral doses of 0.3, 1.0 and 10 mg/kg on Gestation Days 6-15) and rabbits (oral dose of 0.1, 0.3 and 1.0 mg/kg on Gestation Days 6-18). Ribavirin demonstrated significant embryocidal and teratogenic effects at doses well below the recommended human dose in all animal species in which adequate studies have been conducted. Malformations of the skull, palate, eye, jaw, limbs, skeleton, and gastrointestinal tract were noted. The incidence and severity of teratogenic effects increased with escalation of the drug dose. Survival of fetuses and offspring was reduced [see Contraindications (4) and Warnings and Precautions (5.1)].
8.2 Lactation
Risk Summary
There are no data on the presence of ribavirin in human milk or the effects on the breastfed infant or milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for REBETOL and any potential adverse effects on the breastfed infant from REBETOL or from the underlying maternal condition.
8.3 Females and Males of Reproductive Potential
REBETOL may cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Pregnancy Testing
REBETOL therapy should not be started until a report of a negative pregnancy test has been obtained immediately prior to planned initiation of treatment. Patients should have periodic pregnancy tests during treatment and during the 9-month period after treatment has been stopped [see Warnings and Precautions (5.1)].
Contraception
Female patients of reproductive potential should use effective contraception during treatment and for 9 months post-therapy.
Male patients and their female partners should use effective contraception during treatment with REBETOL and for the 6-month post-therapy period [see Warnings and Precautions (5.1)].
Infertility
Based on animal data, REBETOL may impair male fertility. In animal studies, these effects were mostly reversible within a few months after drug cessation [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
Safety and effectiveness of REBETOL in combination with PegIntron has not been established in pediatric patients below the age of 3 years. For treatment with REBETOL/INTRON A, evidence of disease progression, such as hepatic inflammation and fibrosis, as well as prognostic factors for response, HCV genotype and viral load should be considered when deciding to treat a pediatric patient. The benefits of treatment should be weighed against the observed safety findings.
Long-term follow-up data in pediatric subjects indicates that REBETOL in combination with PegIntron or with INTRON A may induce a growth inhibition that results in reduced height in some patients [see Warnings and Precautions (5.9) and Adverse Reactions (6.1)].
Suicidal ideation or attempts occurred more frequently among pediatric patients, primarily adolescents, compared to adult patients (2.4% vs. 1%) during treatment and off-therapy follow-up [see Warnings and Precautions (5.10)]. As in adult patients, pediatric patients experienced other psychiatric adverse reactions (e.g., depression, emotional lability, somnolence), anemia, and neutropenia [see Warnings and Precautions (5.2)].
Juvenile Animal Toxicity Data
In a study in which rat pups were dosed postnatally with ribavirin at doses of 10, 25, and 50 mg/kg/day, drug-related deaths occurred at 50 mg/kg (at rat pup plasma concentrations below human plasma concentrations at the human therapeutic dose) between study Days 13 and 48. Rat pups dosed from postnatal Days 7 through 63 demonstrated a minor, dose-related decrease in overall growth at all doses, which was subsequently manifested as slight decreases in body weight, crown-rump length, and bone length. These effects showed evidence of reversibility, and no histopathological effects on bone were observed. No ribavirin effects were observed regarding neurobehavioral or reproductive development.
8.5 Geriatric Use
Clinical trials of REBETOL combination therapy did not include sufficient numbers of subjects aged 65 and over to determine if they respond differently from younger subjects.
REBETOL is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients often have decreased renal function, care should be taken in dose selection. Renal function should be monitored and dosage adjustments made accordingly. REBETOL should not be used in patients with creatinine clearance less than 50 mL/min [see Contraindications (4)].
In general, REBETOL capsules should be administered to elderly patients cautiously, starting at the lower end of the dosing range, reflecting the greater frequency of decreased hepatic and cardiac function, and of concomitant disease or other drug therapy. In clinical trials, elderly subjects had a higher frequency of anemia (67%) than younger patients (28%) [see Warnings and Precautions (5.2)].
8.6 Organ Transplant Recipients
The safety and efficacy of INTRON A and PegIntron alone or in combination with REBETOL for the treatment of hepatitis C in liver or other organ transplant recipients have not been established. In a small (n=16) single-center, uncontrolled case experience, renal failure in renal allograft recipients receiving interferon alpha and ribavirin combination therapy was more frequent than expected from the center's previous experience with renal allograft recipients not receiving combination therapy. The relationship of the renal failure to renal allograft rejection is not clear.
10 OVERDOSAGE
There is limited experience with overdosage. Acute ingestion of up to 20 g of REBETOL capsules, INTRON A ingestion of up to 120 million units, and subcutaneous doses of INTRON A up to 10 times the recommended doses have been reported. Primary effects that have been observed are increased incidence and severity of the adverse reactions related to the therapeutic use of INTRON A and REBETOL. However, hepatic enzyme abnormalities, renal failure, hemorrhage, and myocardial infarction have been reported with administration of single subcutaneous doses of INTRON A that exceed dosing recommendations.
There is no specific antidote for INTRON A or REBETOL overdose, and hemodialysis and peritoneal dialysis are not effective for treatment of overdose of these agents.
11 DESCRIPTION
REBETOL (ribavirin), is a synthetic nucleoside analogue (purine analogue). The chemical name of ribavirin is 1-β-D-ribofuranosyl-1H-1,2,4-triazole-3-carboxamide and has the following structural formula (see Figure 1).
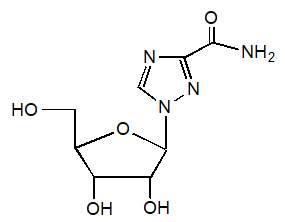
Ribavirin is a white, crystalline powder. It is freely soluble in water and slightly soluble in anhydrous alcohol. The empirical formula is C8H12N4O5 and the molecular weight is 244.21.
REBETOL capsules consist of a white powder in a white, opaque, gelatin capsule. Each capsule contains 200 mg ribavirin and the inactive ingredients microcrystalline cellulose, lactose monohydrate, croscarmellose sodium, and magnesium stearate. The capsule shell consists of gelatin, sodium lauryl sulfate, silicon dioxide, and titanium dioxide. The capsule is printed with edible blue pharmaceutical ink which is made of shellac, anhydrous ethyl alcohol, isopropyl alcohol, n-butyl alcohol, propylene glycol, ammonium hydroxide, and FD&C Blue #2 aluminum lake.
REBETOL oral solution is a clear, colorless to pale or light-yellow bubble gum-flavored liquid. Each milliliter of the solution contains 40 mg of ribavirin and the inactive ingredients sucrose, glycerin, sorbitol, propylene glycol, sodium citrate, citric acid, sodium benzoate, natural and artificial flavor for bubble gum #15864, and water.
12 CLINICAL PHARMACOLOGY
12.3 Pharmacokinetics
Single- and multiple-dose pharmacokinetic properties in adults are summarized in Table 11. Ribavirin was rapidly and extensively absorbed following oral administration. However, due to first-pass metabolism, the absolute bioavailability averaged 64% (44%). There was a linear relationship between dose and AUCtf (AUC from time zero to last measurable concentration) following single doses of 200 to 1200 mg ribavirin. The relationship between dose and Cmax was curvilinear, tending to asymptote above single doses of 400 to 600 mg.
Upon multiple oral dosing, based on AUC12hr, a 6-fold accumulation of ribavirin was observed in plasma. Following oral dosing with 600 mg twice daily, steady-state was reached by approximately 4 weeks, with mean steady-state plasma concentrations of 2200 ng/mL (37%). Upon discontinuation of dosing, the mean half-life was 298 (30%) hours, which probably reflects slow elimination from nonplasma compartments.
Effect of Antacid on Absorption of Ribavirin: Coadministration of REBETOL capsules with an antacid containing magnesium, aluminum, and simethicone resulted in a 14% decrease in mean ribavirin AUCtf. The clinical relevance of results from this single-dose study is unknown.
| Parameter | REBETOL | ||
|---|---|---|---|
| Single-Dose 600 mg Oral Solution (N=14) | Single-Dose 600 mg Capsules (N=12) | Multiple-Dose 600 mg Capsules twice daily (N=12) |
|
| Tmax (hr) | 1.00 (34) | 1.7 (46)* | 3 (60) |
| Cmax (ng/mL) | 872 (42) | 782 (37) | 3680 (85) |
| AUCtf (ng∙hr/mL) | 14,098 (38) | 13,400 (48) | 228,000 (25) |
| T1/2 (hr) | 43.6 (47) | 298 (30) | |
| Apparent Volume of Distribution (L) | 2825 (9)† | ||
| Apparent Clearance (L/hr) | 38.2 (40) | ||
| Absolute Bioavailability | 64% (44)‡ | ||
Tissue Distribution: Ribavirin transport into nonplasma compartments has been most extensively studied in red blood cells and has been identified to be primarily via an es-type equilibrative nucleoside transporter. This type of transporter is present on virtually all cell types and may account for the extensive volume of distribution. Ribavirin does not bind to plasma proteins.
Metabolism and Excretion: Ribavirin has two pathways of metabolism: (i) a reversible phosphorylation pathway in nucleated cells; and (ii) a degradative pathway involving deribosylation and amide hydrolysis to yield a triazole carboxylic acid metabolite. Ribavirin and its triazole carboxamide and triazole carboxylic acid metabolites are excreted renally. After oral administration of 600 mg of 14C-ribavirin, approximately 61% and 12% of the radioactivity was eliminated in the urine and feces, respectively, in 336 hours. Unchanged ribavirin accounted for 17% of the administered dose.
Special Populations:
Renal Dysfunction
The pharmacokinetics of ribavirin were assessed after administration of a single oral dose (400 mg) of ribavirin to non-HCV-infected subjects with varying degrees of renal dysfunction. The mean AUCtf value was threefold greater in subjects with creatinine clearance values between 10 to 30 mL/min when compared to control subjects (creatinine clearance greater than 90 mL/min). In subjects with creatinine clearance values between 30 to 60 mL/min, AUCtf was twofold greater when compared to control subjects. The increased AUCtf appears to be due to reduction of renal and nonrenal clearance in these subjects. Phase 3 efficacy trials included subjects with creatinine clearance values greater than 50 mL/min. The multiple-dose pharmacokinetics of ribavirin cannot be accurately predicted in patients with renal dysfunction. Ribavirin is not effectively removed by hemodialysis. Patients with creatinine clearance less than 50 mL/min should not be treated with REBETOL [see Contraindications (4)].
Hepatic Dysfunction
The effect of hepatic dysfunction was assessed after a single oral dose of ribavirin (600 mg). The mean AUCtf values were not significantly different in subjects with mild, moderate, or severe hepatic dysfunction (Child-Pugh Classification A, B, or C) when compared to control subjects. However, the mean Cmax values increased with severity of hepatic dysfunction and was twofold greater in subjects with severe hepatic dysfunction when compared to control subjects.
Gender
There were no clinically significant pharmacokinetic differences noted in a single-dose trial of 18 male and 18 female subjects.
Pediatric Patients
Multiple-dose pharmacokinetic properties for REBETOL capsules and INTRON A in pediatric subjects with chronic hepatitis C between 5 and 16 years of age are summarized in Table 12. The pharmacokinetics of REBETOL and INTRON A (dose-normalized) are similar in adults and pediatric subjects.
Complete pharmacokinetic characteristics of REBETOL oral solution have not been determined in pediatric subjects. Ribavirin Cmin values were similar following administration of REBETOL oral solution or REBETOL capsules during 48 weeks of therapy in pediatric subjects (3 to 16 years of age).
| Parameter | REBETOL 15 mg/kg/day as 2 divided doses (N=17) | INTRON A 3 MIU/m2 three times weekly (N=54) |
|---|---|---|
| Note: numbers in parenthesis indicate % coefficient of variation. | ||
| Tmax (hr) | 1.9 (83) | 5.9 (36) |
| Cmax (ng/mL) | 3275 (25) | 51 (48) |
| AUC* | 29,774 (26) | 622 (48) |
| Apparent Clearance L/hr/kg | 0.27 (27) | ND† |
A clinical trial in pediatric subjects with chronic hepatitis C between 3 and 17 years of age was conducted in which pharmacokinetics for PegIntron and REBETOL (capsules and oral solution) were evaluated. In pediatric subjects receiving body surface area-adjusted dosing of PegIntron at 60 mcg/m2/week, the log transformed ratio estimate of exposure during the dosing interval was predicted to be 58% [90% CI: 141%, 177%] higher than observed in adults receiving 1.5 mcg/kg/week. The pharmacokinetics of REBETOL (dose-normalized) in this trial were similar to those reported in a prior study of REBETOL in combination with INTRON A in pediatric subjects and in adults.
Effect of Food on Absorption of Ribavirin
Both AUCtf and Cmax increased by 70% when REBETOL capsules were administered with a high-fat meal (841 kcal, 53.8 g fat, 31.6 g protein, and 57.4 g carbohydrate) in a single-dose pharmacokinetic study [see Dosage and Administration (2)].
12.4 Microbiology
Mechanism of Action
The mechanism by which ribavirin contributes to its antiviral efficacy in the clinic is not fully understood. Ribavirin has direct antiviral activity in cell culture against many RNA viruses. Ribavirin increases the mutation frequency in the genomes of several RNA viruses and ribavirin triphosphate inhibits HCV polymerase in a biochemical reaction.
Antiviral Activity in Cell Culture
The antiviral activity of ribavirin in the HCV replicon is not well understood and has not been defined because of the cellular toxicity of ribavirin. Direct antiviral activity has been observed in cell culture of other RNA viruses. The anti-HCV activity of interferon was demonstrated in cell culture using self-replicating HCV RNA (HCV replicon cells) or HCV infection.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Ribavirin did not cause an increase in any tumor type when administered for 6 months in the transgenic p53 deficient mouse model at doses up to 300 mg/kg (estimated human equivalent of 25 mg/kg based on body surface area adjustment for a 60 kg adult; approximately 1.9 times the maximum recommended human daily dose). Ribavirin was noncarcinogenic when administered for 2 years to rats at doses up to 40 mg/kg (estimated human equivalent of 5.71 mg/kg based on body surface area adjustment for a 60 kg adult).
Mutagenesis
Ribavirin demonstrated increased incidences of mutation and cell transformation in multiple genotoxicity assays. Ribavirin was active in the Balb/3T3 In Vitro Cell Transformation Assay. Mutagenic activity was observed in the mouse lymphoma assay, and at doses of 20 to 200 mg/kg (estimated human equivalent of 1.67 to 16.7 mg/kg, based on body surface area adjustment for a 60 kg adult; 0.1 to 1 times the maximum recommended human 24-hour dose of ribavirin) in a mouse micronucleus assay. A dominant lethal assay in rats was negative, indicating that if mutations occurred in rats they were not transmitted through male gametes.
Impairment of Fertility
In studies in mice to evaluate the time course and reversibility of ribavirin-induced testicular degeneration at doses of 15 to 150 mg/kg/day (estimated human equivalent of 1.25 to 12.5 mg/kg/day, based on body surface area adjustment for a 60-kg adult; 0.1-0.8 times the maximum human 24-hour dose of ribavirin) administered for 3 or 6 months, abnormalities in sperm occurred. Upon cessation of treatment, recovery from ribavirin-induced testicular toxicity was mostly apparent within 1 or 2 spermatogenesis cycles.
13.2 Animal Toxicology and Pharmacology
Long-term studies in the mouse and rat [18 to 24 months; doses of 20 to 75 and 10 to 40 mg/kg/day, respectively (estimated human equivalent doses of 1.67 to 6.25 and 1.43 to 5.71 mg/kg/day, respectively, based on body surface area adjustment for a 60 kg adult; approximately 0.1 to 0.4 times the maximum human 24-hour dose of ribavirin)] have demonstrated a relationship between chronic ribavirin exposure and increased incidences of vascular lesions (microscopic hemorrhages) in mice. In rats, retinal degeneration occurred in controls, but the incidence was increased in ribavirin-treated rats.
14 CLINICAL STUDIES
Clinical Study 1 evaluated PegIntron monotherapy. See PegIntron labeling for information about this trial.
14.1 REBETOL/PegIntron Combination Therapy
Adult Subjects
Study 2
A randomized trial compared treatment with two PegIntron/REBETOL regimens [PegIntron 1.5 mcg/kg subcutaneously once weekly/REBETOL 800 mg orally daily (in divided doses); PegIntron 1.5 mcg/kg subcutaneously once weekly for 4 weeks then 0.5 mcg/kg subcutaneously once weekly for 44 weeks/REBETOL 1000 or 1200 mg orally daily (in divided doses)] with INTRON A [3 MIU subcutaneously three times weekly/REBETOL 1000 or 1200 mg orally daily (in divided doses)] in 1,530 adults with chronic hepatitis C. Interferon-naïve subjects were treated for 48 weeks and followed for 24 weeks post-treatment. Eligible subjects had compensated liver disease, detectable HCV-RNA, elevated ALT, and liver histopathology consistent with chronic hepatitis.
Response to treatment was defined as undetectable HCV-RNA at 24 weeks post-treatment (see Table 13). The response rate to the PegIntron 1.5 mcg/kg and ribavirin 800 mg dose was higher than the response rate to INTRON A/REBETOL (see Table 13). The response rate to PegIntron 1.5→0.5 mcg/kg/REBETOL was essentially the same as the response to INTRON A/REBETOL (data not shown).
| PegIntron 1.5 mcg/kg once weekly REBETOL 800 mg once daily | INTRON A 3 MIU three times weekly REBETOL 1000/1200 mg once daily |
|
|---|---|---|
|
||
| Overall response*,† | 52% (264/511) | 46% (231/505) |
| Genotype 1 | 41% (141/348) | 33% (112/343) |
| Genotype 2-6 | 75% (123/163) | 73% (119/162) |
Subjects with viral genotype 1, regardless of viral load, had a lower response rate to PegIntron (1.5 mcg/kg)/REBETOL (800 mg) compared to subjects with other viral genotypes. Subjects with both poor prognostic factors (genotype 1 and high viral load) had a response rate of 30% (78/256) compared to a response rate of 29% (71/247) with INTRON A/REBETOL combination therapy.
Subjects with lower body weight tended to have higher adverse-reaction rates [see Adverse Reactions (6.1)] and higher response rates than subjects with higher body weights. Differences in response rates between treatment arms did not substantially vary with body weight.
Treatment response rates with PegIntron/REBETOL combination therapy were 49% in men and 56% in women. Response rates were lower in African American and Hispanic subjects and higher in Asians compared to Caucasians. Although African Americans had a higher proportion of poor prognostic factors compared to Caucasians, the number of non-Caucasians studied (11% of the total) was insufficient to allow meaningful conclusions about differences in response rates after adjusting for prognostic factors in this trial.
Liver biopsies were obtained before and after treatment in 68% of subjects. Compared to baseline, approximately two-thirds of subjects in all treatment groups were observed to have a modest reduction in inflammation.
Study 3
In a large United States community-based trial, 4,913 subjects with chronic hepatitis C were randomized to receive PegIntron 1.5 mcg/kg subcutaneously once weekly in combination with a REBETOL dose of 800 to 1400 mg (weight-based dosing [WBD]) or 800 mg (flat) orally daily (in divided doses) for 24 or 48 weeks based on genotype. Response to treatment was defined as undetectable HCV-RNA (based on an assay with a lower limit of detection of 125 IU/mL) at 24 weeks post-treatment.
Treatment with PegIntron 1.5 mcg/kg and REBETOL 800 to 1400 mg resulted in a higher sustained virologic response compared to PegIntron in combination with a flat 800 mg daily dose of REBETOL. Subjects weighing greater than 105 kg obtained the greatest benefit with WBD, although a modest benefit was also observed in subjects weighing greater than 85 to 105 kg (see Table 14). The benefit of WBD in subjects weighing greater than 85 kg was observed with HCV genotypes 1-3. Insufficient data were available to reach conclusions regarding other genotypes. Use of WBD resulted in an increased incidence of anemia [see Adverse Reactions (6.1)].
| Treatment Group | Subject Baseline Weight | |||
|---|---|---|---|---|
| <65 kg (<143 lb) | 65-85 kg (143-188 lb) | >85-105 kg (>188-231 lb) | >105 kg (>231 lb) |
|
|
||||
| WBD* | 50% (173/348) | 45% (449/994) | 42% (351/835) | 47% (138/292) |
| Flat | 51% (173/342) | 44% (443/1011) | 39% (318/819) | 33% (91/272) |
A total of 1,552 subjects weighing greater than 65 kg in Study 3 had genotype 2 or 3 and were randomized to 24 or 48 weeks of therapy. No additional benefit was observed with the longer treatment duration.
Study 4
A large randomized trial compared the safety and efficacy of treatment for 48 weeks with two PegIntron/REBETOL regimens [PegIntron 1.5 mcg/kg and 1 mcg/kg subcutaneously once weekly both in combination with REBETOL 800 to 1400 mg PO daily (in two divided doses)] and Pegasys 180 mcg subcutaneously once weekly in combination with Copegus 1000 to 1200 mg PO daily (in two divided doses) in 3,070 treatment-naïve adults with chronic hepatitis C genotype 1. In this trial, lack of early virologic response (undetectable HCV-RNA or greater than or equal to 2 log10 reduction from baseline) by treatment Week 12 was the criterion for discontinuation of treatment. SVR was defined as undetectable HCV-RNA (Roche COBAS TaqMan assay, a lower limit of quantitation of 27 IU/mL) at 24 weeks post-treatment (see Table 15).
| % (number) of Subjects | ||
|---|---|---|
| PegIntron 1.5 mcg/kg/REBETOL | PegIntron 1 mcg/kg/REBETOL | Pegasys 180 mcg/Copegus |
| 40 (406/1019) | 38 (386/1016) | 41 (423/1035) |
Overall SVR rates were similar among the three treatment groups. Regardless of treatment group, SVR rates were lower in subjects with poor prognostic factors. Subjects with poor prognostic factors randomized to PegIntron (1.5 mcg/kg)/REBETOL or Pegasys/Copegus, however, achieved higher SVR rates compared to similar subjects randomized to PegIntron 1 mcg/kg/REBETOL. For the PegIntron 1.5 mcg/kg and REBETOL dose, SVR rates for subjects with and without the following prognostic factors were as follows: cirrhosis (10% vs. 42%), normal ALT levels (32% vs. 42%), baseline viral load greater than 600,000 IU/mL (35% vs. 61%), 40 years of age and older (38% vs. 50%), and African American race (23% vs. 44%). In subjects with undetectable HCV-RNA at treatment Week 12 who received PegIntron (1.5 mcg/kg)/REBETOL, the SVR rate was 81% (328/407).
Study 5 - REBETOL/PegIntron Combination Therapy in Prior Treatment Failures
In a noncomparative trial, 2,293 subjects with moderate to severe fibrosis who failed previous treatment with combination alpha interferon/ribavirin were re-treated with PegIntron, 1.5 mcg/kg subcutaneously, once weekly, in combination with weight adjusted ribavirin. Eligible subjects included prior nonresponders (subjects who were HCV-RNA positive at the end of a minimum 12 weeks of treatment) and prior relapsers (subjects who were HCV-RNA negative at the end of a minimum 12 weeks of treatment and subsequently relapsed after post-treatment follow-up). Subjects who were negative at Week 12 were treated for 48 weeks and followed for 24 weeks post-treatment. Response to treatment was defined as undetectable HCV-RNA at 24 weeks post-treatment (measured using a research-based test, limit of detection 125 IU/mL). The overall response rate was 22% (497/2293) (99% CI: 19.5, 23.9). Subjects with the following characteristics were less likely to benefit from re-treatment: previous nonresponse, previous pegylated interferon treatment, significant bridging fibrosis or cirrhosis, and genotype 1 infection.
The re-treatment sustained virologic response rates by baseline characteristics are summarized in Table 16.
| HCV Genotype/ Metavir Fibrosis Score | Overall SVR by Previous Response and Treatment | |||
|---|---|---|---|---|
| Nonresponder | Relapser | |||
| interferon alfa/ribavirin % (number of subjects) | peginterferon (2a and 2b combined)/ribavirin % (number of subjects) | interferon alfa/ribavirin % (number of subjects) | peginterferon (2a and 2b combined)/ribavirin % (number of subjects) |
|
| Overall | 18 (158/903) | 6 (30/476) | 43 (130/300) | 35 (113/344) |
| HCV 1 | 13 (98/761) | 4 (19/431) | 32 (67/208) | 23 (56/243) |
| F2 | 18 (36/202) | 6 (7/117) | 42 (33/79) | 32 (23/72) |
| F3 | 16 (38/233) | 4 (4/112) | 28 (16/58) | 21 (14/67) |
| F4 | 7 (24/325) | 4 (8/202) | 26 (18/70) | 18 (19/104) |
| HCV 2/3 | 49 (53/109) | 36 (10/28) | 67 (54/81) | 57 (52/92) |
| F2 | 68 (23/34) | 56 (5/9) | 76 (19/25) | 61 (11/18) |
| F3 | 39 (11/28) | 38 (3/8) | 67 (18/27) | 62 (18/29) |
| F4 | 40 (19/47) | 18 (2/11) | 59 (17/29) | 51 (23/45) |
| HCV 4 | 17 (5/29) | 7 (1/15) | 88 (7/8) | 50 (4/8) |
Achievement of an undetectable HCV-RNA at treatment Week 12 was a strong predictor of SVR. In this trial, 1,470 (64%) subjects did not achieve an undetectable HCV-RNA at treatment Week 12, and were offered enrollment into long-term treatment trials, due to an inadequate treatment response. Of the 823 (36%) subjects who were HCV-RNA undetectable at treatment Week 12, those infected with genotype 1 had an SVR of 48% (245/507), with a range of responses by fibrosis scores (F4-F2) of 39-55%. Subjects infected with genotype 2/3 who were HCV-RNA undetectable at treatment Week 12 had an overall SVR of 70% (196/281), with a range of responses by fibrosis scores (F4-F2) of 60-83%. For all genotypes, higher fibrosis scores were associated with a decreased likelihood of achieving SVR.
Pediatric Subjects
Previously untreated pediatric subjects 3 to 17 years of age with compensated chronic hepatitis C and detectable HCV-RNA were treated with REBETOL 15 mg/kg per day and PegIntron 60 mcg/m2 once weekly for 24 or 48 weeks based on HCV genotype and baseline viral load. All subjects were to be followed for 24 weeks post-treatment. A total of 107 subjects received treatment, of which 52% were female, 89% were Caucasian, and 67% were infected with HCV Genotype 1. Subjects infected with Genotypes 1, 4 or Genotype 3 with HCV-RNA greater than or equal to 600,000 IU/mL received 48 weeks of therapy while those infected with Genotype 2 or Genotype 3 with HCV-RNA less than 600,000 IU/mL received 24 weeks of therapy. The trial results are summarized in Table 17.
| Genotype | All Subjects N=107 |
|
|---|---|---|
| 24 Weeks | 48 Weeks | |
| Virologic Response N*,† (%) | Virologic Response N*,† (%) |
|
|
||
| All | 26/27 (96.3) | 44/80 (55.0) |
| 1 | - | 38/72 (52.8) |
| 2 | 14/15 (93.3) | - |
| 3‡ | 12/12 (100) | 2/3 (66.7) |
| 4 | - | 4/5 (80.0) |
14.2 REBETOL/INTRON A Combination Therapy
Adult Subjects
Previously Untreated Subjects
Adults with compensated chronic hepatitis C and detectable HCV-RNA (assessed by a central laboratory using a research-based RT-PCR assay) who were previously untreated with alpha interferon therapy were enrolled into two multicenter, double-blind trials (US and international) and randomized to receive REBETOL capsules 1200 mg/day (1000 mg/day for subjects weighing less than or equal to 75 kg) and INTRON A 3 MIU three times weekly or INTRON A and placebo for 24 or 48 weeks followed by 24 weeks of off-therapy follow-up. The international trial did not contain a 24-week INTRON A and placebo treatment arm. The US trial enrolled 912 subjects who, at baseline, were 67% male, 89% Caucasian with a mean Knodell HAI score (I+II+III) of 7.5, and 72% genotype 1. The international trial, conducted in Europe, Israel, Canada, and Australia, enrolled 799 subjects (65% male, 95% Caucasian, mean Knodell score 6.8, and 58% genotype 1).
Trial results are summarized in Table 18.
| US Trial | International Trial | ||||||
|---|---|---|---|---|---|---|---|
| 24 weeks of treatment | 48 weeks of treatment | 24 weeks of treatment | 48 weeks of treatment | ||||
| INTRON A/ REBETOL (N=228) | INTRON A/ Placebo (N=231) | INTRON A/ REBETOL (N=228) | INTRON A/ Placebo (N=225) | INTRON A/ REBETOL (N=265) | INTRON A/ REBETOL (N=268) | INTRON A/ Placebo (N=266) |
|
|
|||||||
| Virologic Response | |||||||
| Responder† | 65 (29) | 13 (6) | 85 (37) | 27 (12) | 86 (32) | 113 (42) | 46 (17) |
| Nonresponder | 147 (64) | 194 (84) | 110 (48) | 168 (75) | 158 (60) | 120 (45) | 196 (74) |
| Missing Data | 16 (7) | 24 (10) | 33 (14) | 30 (13) | 21 (8) | 35 (13) | 24 (9) |
| Histologic Response | |||||||
| Improvement‡ | 102 (45) | 77 (33) | 96 (42) | 65 (29) | 103 (39) | 102 (38) | 69 (26) |
| No improvement | 77 (34) | 99 (43) | 61 (27) | 93 (41) | 85 (32) | 58 (22) | 111 (41) |
| Missing Data | 49 (21) | 55 (24) | 71 (31) | 67 (30) | 77 (29) | 108 (40) | 86 (32) |
Of subjects who had not achieved HCV-RNA below the limit of detection of the research-based assay by Week 24 of REBETOL/INTRON A treatment, less than 5% responded to an additional 24 weeks of combination treatment.
Among subjects with HCV Genotype 1 treated with REBETOL/INTRON A therapy who achieved HCV-RNA below the detection limit of the research-based assay by 24 weeks, those randomized to 48 weeks of treatment had higher virologic responses compared to those in the 24-week treatment group. There was no observed increase in response rates for subjects with HCV non-genotype 1 randomized to REBETOL/INTRON A therapy for 48 weeks compared to 24 weeks.
Relapse Subjects
Subjects with compensated chronic hepatitis C and detectable HCV-RNA (assessed by a central laboratory using a research-based RT-PCR assay) who had relapsed following one or two courses of interferon therapy (defined as abnormal serum ALT levels) were enrolled into two multicenter, double-blind trials (US and international) and randomized to receive REBETOL 1200 mg/day (1000 mg/day for subjects weighing ≤75 kg) and INTRON A 3 MIU three times weekly or INTRON A and placebo for 24 weeks followed by 24 weeks of off-therapy follow-up. The US trial enrolled 153 subjects who, at baseline, were 67% male, 92% Caucasian with a mean Knodell HAI score (I+II+III) of 6.8, and 58% genotype 1. The international trial, conducted in Europe, Israel, Canada, and Australia, enrolled 192 subjects (64% male, 95% Caucasian, mean Knodell score 6.6, and 56% genotype 1). Trial results are summarized in Table 19.
| US Trial | International Trial | |||
|---|---|---|---|---|
| INTRON A/ REBETOL (N=77) | INTRON A/ Placebo (N=76) | INTRON A/ REBETOL (N=96) | INTRON A/ Placebo (N=96) |
|
|
||||
| Virologic Response | ||||
| Responder† | 33 (43) | 3 (4) | 46 (48) | 5 (5) |
| Nonresponder | 36 (47) | 66 (87) | 45 (47) | 91 (95) |
| Missing Data | 8 (10) | 7 (9) | 5 (5) | 0 (0) |
| Histologic Response | ||||
| Improvement‡ | 38 (49) | 27 (36) | 49 (51) | 30 (31) |
| No improvement | 23 (30) | 37 (49) | 29 (30) | 44 (46) |
| Missing Data | 16 (21) | 12 (16) | 18 (19) | 22 (23) |
Virologic and histologic responses were similar among male and female subjects in both the previously untreated and relapse trials.
Pediatric Subjects
Pediatric subjects 3 to 16 years of age with compensated chronic hepatitis C and detectable HCV-RNA (assessed by a central laboratory using a research-based RT-PCR assay) were treated with REBETOL 15 mg/kg per day and INTRON A 3 MIU/m2 three times weekly for 48 weeks followed by 24 weeks of off-therapy follow-up. A total of 118 subjects received treatment, of which 57% were male, 80% Caucasian, and 78% genotype 1. Subjects less than 5 years of age received REBETOL oral solution and those 5 years of age or older received either REBETOL oral solution or capsules.
Trial results are summarized in Table 20.
| INTRON A 3 MIU/m2 three times weekly/ REBETOL 15 mg/kg/day | |
|---|---|
| Overall Response† (N=118) | 54 (46) |
| Genotype 1 (N=92) | 33 (36) |
| Genotype non-1 (N=26) | 21 (81) |
Subjects with viral genotype 1, regardless of viral load, had a lower response rate to INTRON A/REBETOL combination therapy compared to subjects with genotype non-1, 36% vs. 81%. Subjects with both poor prognostic factors (genotype 1 and high viral load) had a response rate of 26% (13/50).
16 HOW SUPPLIED/STORAGE AND HANDLING
REBETOL 200 mg Capsules are white, opaque capsules with REBETOL, 200 mg, and the Schering Corporation logo imprinted on the capsule shell; the capsules are packaged in a bottle containing 56 capsules (NDC 0085-1351-05), 70 capsules (NDC 0085-1385-07), and 84 capsules (NDC 0085-1194-03). The bottle of REBETOL Capsules should be stored at 25°C (77°F); excursions permitted to 15-30°C (59-86°F) [see USP Controlled Room Temperature].
REBETOL Oral Solution 40 mg per mL is a clear, colorless to pale or light-yellow bubble gum-flavored liquid and it is packaged in 4-oz amber glass bottles (100 mL/bottle) with child-resistant closures (NDC 0085-1318-01). REBETOL Oral Solution should be stored between 2-8°C (36-46°F) or at 25°C (77°F); excursions permitted to 15-30°C (59-86°F) [see USP Controlled Room Temperature].
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Anemia
The most common adverse experience occurring with REBETOL capsules is anemia, which may be severe [see Warnings and Precautions (5.2) and Adverse Reactions (6)]. Advise patients that laboratory evaluations are required prior to starting therapy and periodically thereafter [see Dosage and Administration (2.4)]. Advise patients to be well hydrated, especially during the initial stages of treatment.
Embryo-Fetal Toxicity
Inform females of reproductive potential and pregnant women that REBETOL capsules and oral solution may cause birth defects, miscarriage, and stillbirth. Advise females of reproductive potential that they must have a pregnancy test prior to initiating treatment and periodically during therapy. Advise female patients of reproductive potential to use effective contraception during treatment with REBETOL and for 9 months post therapy and male patients with female partners of reproductive potential to use effective contraception during treatment with REBETOL and for 6 months post therapy. Advise patients to notify the physician immediately in the event of a pregnancy [see Contraindications (4), Warnings and Precautions (5.1), and Use in Specific Populations (8.1, 8.3)].
Missed Dose
Inform patients that in the event a dose is missed, the missed dose should be taken as soon as possible during the same day. Patients should not double the next dose. Advise patients to contact their healthcare provider if they have questions.
Dental and Periodontal Disorders
Advise patients to brush their teeth thoroughly twice daily and have regular dental examinations. If vomiting occurs, advise patients to rinse out their mouth thoroughly afterwards [see Warnings and Precautions (5.7)].
REBETOL Oral Solution manufactured for:
Merck Sharp & Dohme Corp., a subsidiary of
MERCK & CO., INC.
Whitehouse Station, NJ 08889, USA
Manufactured by:
Famar Montréal Inc.
Pointe-Claire, Quebec H9R 1B4, Canada
REBETOL Capsules manufactured by:
Merck Sharp & Dohme Corp., a subsidiary of
MERCK & CO., INC.
Whitehouse Station, NJ 08889, USA
For patent information: www.merck.com/product/patent/home.html
Copyright © 2003-2022 Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc.
All rights reserved.
Trademarks depicted herein are the property of their respective owners.
uspi-mk8908-mtl-2203r035
| This Medication Guide has been approved by the U.S. Food and Drug Administration. | Revised: 03/2022 |
| MEDICATION GUIDE
REBETOL® (REB-eh-tol) (ribavirin) capsules and oral solution |
|
|
What is the most important information I should know about REBETOL? |
|
|
|
| What is REBETOL?
REBETOL is a medicine used with either interferon alfa-2b or peginterferon alfa-2b to treat chronic (lasting a long time) hepatitis C infection in people 3 years and older with liver disease. It is not known if REBETOL use for longer than 1 year is safe and will work. It is not known if REBETOL use in children younger than 3 years old is safe and will work. |
|
| Who should not take REBETOL? See "What is the most important information I should know about REBETOL?" Do not take REBETOL if you have:
|
|
| What should I tell my healthcare provider before taking REBETOL? Before you take REBETOL, tell your healthcare provider if you have or ever had:
Especially tell your healthcare provider if you take didanosine or a medicine that contains azathioprine. Know the medicines you take. Keep a list of them to show your healthcare provider or pharmacist when you get a new medicine. |
|
How should I take REBETOL?
|
|
| What are the possible side effects of REBETOL? REBETOL may cause serious side effects, including: See "What is the most important information I should know about REBETOL?"
|
|
The most common side effects of REBETOL in adults include:
|
|
The most common side effects of REBETOL in children include:
|
|
| These are not all the possible side effects of REBETOL. For more information ask your healthcare provider or pharmacist. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|
How should I store REBETOL?
|
|
| General information about the safe and effective use of REBETOL.
It is not known if treatment with REBETOL will cure hepatitis C virus infections or prevent cirrhosis, liver failure, or liver cancer that can be caused by hepatitis C virus infections. It is not known if taking REBETOL will prevent you from infecting another person with the hepatitis C virus. Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use REBETOL for a condition for which it was not prescribed. Do not give REBETOL to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about REBETOL that is written for health professionals. |
|
| What are the ingredients in REBETOL? Active ingredients: ribavirin Inactive ingredients: REBETOL Capsules: microcrystalline cellulose, lactose monohydrate, croscarmellose sodium, and magnesium stearate. The capsule shell consists of gelatin, sodium lauryl sulfate, silicon dioxide, and titanium dioxide. The capsule is printed with edible blue pharmaceutical ink which is made of shellac, anhydrous ethyl alcohol, isopropyl alcohol, n-butyl alcohol, propylene glycol, ammonium hydroxide, and FD&C Blue #2 aluminum lake. REBETOL Oral Solution: sucrose, glycerin, sorbitol, propylene glycol, sodium citrate, citric acid, sodium benzoate, natural and artificial flavor for bubble gum # 15864, and water. REBETOL Oral Solution manufactured for: Merck Sharp & Dohme Corp., a subsidiary of MERCK & CO., INC. Whitehouse Station, NJ 08889, USA Manufactured by: Famar Montréal Inc., Pointe-Claire, Quebec H9R 1B4, Canada REBETOL Capsules manufactured by: Merck Sharp & Dohme Corp., a subsidiary of MERCK & CO., INC. Whitehouse Station, NJ 08889, USA For patent information: www.merck.com/product/patent/home.html Copyright © 2003-2022 Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc. All rights reserved. usmg-mk8908-mtl-2203r023 |
|
PRINCIPAL DISPLAY PANEL - 56 Capsule Label
NDC 0085-1351-05
REBETOL®
(Ribavirin USP)
200 mg Capsules
Rx only
Each capsule contains:
Ribavirin USP 200 mg
56 capsules
ATTENTION PHARMACIST:
Each patient is required to
receive a Medication Guide.

PRINCIPAL DISPLAY PANEL - 70 Capsule Label
NDC 0085-1385-07
REBETOL®
(Ribavirin USP)
200 mg Capsules
Rx only
Each capsule contains:
Ribavirin USP 200 mg
70 capsules
ATTENTION PHARMACIST:
Each patient is required to
receive a Medication Guide.

| REBETOL
ribavirin capsule |
||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||
| REBETOL
ribavirin capsule |
||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||
| REBETOL
ribavirin capsule |
||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||
| REBETOL
ribavirin liquid |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Merck Sharp & Dohme LLC (118446553) |