FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
ZYKADIA® is indicated for the treatment of adult patients with metastatic non-small cell lung cancer (NSCLC) whose tumors are anaplastic lymphoma kinase (ALK)-positive as detected by an FDA-approved test [see Dosage and Administration (2.1)].
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
Select patients for treatment of metastatic NSCLC with ZYKADIA based on the presence of ALK positivity in tumor specimens [see Clinical Studies (14.1)].
Information on FDA-approved tests for the detection of ALK rearrangements in NSCLC is available at: http://www.fda.gov/CompanionDiagnostics.
2.2 Recommended Dosage
The recommended dosage of ZYKADIA is 450 mg orally once daily with food until disease progression or unacceptable toxicity [see Clinical Pharmacology (12.3)].
If a dose of ZYKADIA is missed, make up that dose unless the next dose is due within 12 hours.
If vomiting occurs during the course of treatment, do not administer an additional dose and continue with the next scheduled dose of ZYKADIA.
2.3 Dosage Modifications for Adverse Reactions
| Dose Reduction | Recommended Dosage |
| First-dose reduction | 300 mg taken orally once daily with food |
| Second-dose reduction | 150 mg taken orally once daily with food |
Discontinue ZYKADIA for patients unable to tolerate 150 mg taken orally once daily with food.
Dosage modifications for selected adverse reactions of ZYKADIA are provided in Table 2. If dose reduction is required due to an adverse reaction not listed in Table 2, then reduce the daily dose of ZYKADIA by 150 mg.
| Adverse Reaction | ZYKADIA Dose Modification |
| Gastrointestinal Adverse Reactions [see Warnings and Precautions (5.1)] | |
| Severe or intolerable nausea, vomiting, or diarrhea despite optimal antiemetic or anti-diarrheal therapy | Withhold until improved, then resume ZYKADIA at the next lower dosage. |
| Hepatotoxicity [see Warnings and Precautions (5.2)] | |
| ALT or AST elevation greater than 5 times ULN with total bilirubin elevation less than or equal to 2 times ULN | Withhold until recovery to baseline or less than or equal to 3 times ULN, then resume ZYKADIA at the next lower dosage. |
| ALT or AST elevation greater than 3 times ULN with total bilirubin elevation greater than 2 times ULN in the absence of cholestasis or hemolysis | Permanently discontinue ZYKADIA. |
| Interstitial Lung Disease/Pneumonitis [see Warnings and Precautions (5.3)] | |
| Any Grade treatment-related ILD/pneumonitis | Permanently discontinue ZYKADIA. |
| QT Interval Prolongation [see Warnings and Precautions (5.4)] | |
| QTc interval greater than 500 msec on at least 2 separate ECGs | Withhold until QTc interval is less than 481 msec or recovery to baseline if baseline QTc is greater than or equal to 481 msec, then resume ZYKADIA at the next lower dosage. |
| QTc interval prolongation in combination with torsades de pointes or polymorphic ventricular tachycardia or signs/symptoms of serious arrhythmia | Permanently discontinue ZYKADIA. |
| Hyperglycemia [see Warnings and Precautions (5.5)] | |
| Persistent hyperglycemia greater than 250 mg/dL despite optimal anti-hyperglycemic therapy | Withhold until hyperglycemia is adequately controlled, then resume ZYKADIA at the next lower dosage. If adequate hyperglycemic control cannot be achieved with optimal medical management, discontinue ZYKADIA. |
| Bradycardia [see Warnings and Precautions (5.6)] | |
| Symptomatic bradycardia that is not life-threatening | Withhold until recovery to asymptomatic bradycardia or to a heart rate of 60 bpm or above, evaluate concomitant medications known to cause bradycardia. If bradycardia cannot be attributed to another drug, resume ZYKADIA at the next lower dosage. |
| Clinically significant bradycardia requiring intervention or life-threatening bradycardia in patients taking a concomitant medication also known to cause bradycardia or a medication known to cause hypotension | Withhold until recovery to asymptomatic bradycardia or to a heart rate of 60 bpm or above. If the concomitant medication can be adjusted or discontinued, resume ZYKADIA at the next lower dosage with frequent monitoring. |
| Life-threatening bradycardia in patients who are not taking a concomitant medication also known to cause bradycardia or known to cause hypotension | Permanently discontinue ZYKADIA. |
| Pancreatitis [see Warnings and Precautions (5.7)] | |
| Lipase or amylase elevation greater than 2 times ULN | Withhold until recovery to less than 1.5 times ULN, then resume ZYKADIA at the next lower dosage. |
| Abbreviations: AST, aspartate aminotransferase; ALT, alanine aminotransferase; ULN, upper limit of normal; ILD, interstitial lung disease; ECG, electrocardiogram; bpm, beats per minute. | |
2.4 Dosage Modification for Strong CYP3A Inhibitors
Avoid concurrent use of strong CYP3A inhibitors during treatment with ZYKADIA [see Drug Interactions (7.1), Clinical Pharmacology (12.3)].
If concurrent use of a strong CYP3A inhibitor is unavoidable, reduce the ZYKADIA dose by approximately one-third, rounded to the nearest multiple of the 150 mg dosage strength. After discontinuation of a strong CYP3A inhibitor, resume the ZYKADIA dose that was taken prior to initiating the strong CYP3A inhibitor.
2.5 Dosage Modification for Patients With Severe Hepatic Impairment
For patients with severe hepatic impairment (Child-Pugh C), reduce the ZYKADIA dose by approximately one-third, rounded to the nearest multiple of the 150 mg dosage strength [see Use in Specific Populations (8.6), Clinical Pharmacology (12.3)].
3 DOSAGE FORMS AND STRENGTHS
Capsules: 150 mg hard gelatin capsule with opaque blue cap and opaque white body containing a white to off-white powder. The opaque blue cap is marked in black ink with “LDK 150MG” and the opaque white body is marked in black ink with “NVR”.
Tablets: 150 mg film-coated tablet, light blue, round, biconvex with beveled edges, without score, debossed with “NVR” on one side and “ZY1” on the other side.
5 WARNINGS AND PRECAUTIONS
5.1 Gastrointestinal Adverse Reactions
Severe gastrointestinal adverse reactions occurred in patients treated with ZYKADIA 750 mg under fasted conditions [see Adverse Reactions (6.1)]. Diarrhea, nausea, vomiting, or abdominal pain occurred in 95% of 925 patients, including severe cases (Grade 3 or 4) in 14% of patients treated with ZYKADIA across clinical studies. Diarrhea, nausea, vomiting, or abdominal pain leading to dose interruptions or reductions occurred in 36% of patients and leading to treatment discontinuation occurred in 1.6% of patients.
The incidence and severity of gastrointestinal adverse reactions were reduced for patients treated with ZYKADIA 450 mg with food in a dose optimization study (ASCEND-8). Diarrhea, nausea, vomiting, or abdominal pain occurred in 79% of 108 patients treated with ZYKADIA at the recommended dose of 450 mg with food. Of these, 53% were Grade 1 events and 24% were Grade 2 events. One patient (0.9%) experienced Grade 3 diarrhea, and one patient (0.9%) experienced Grade 3 vomiting. One patient (0.9%) required dose adjustment due to vomiting. Eleven (10%) patients had diarrhea, nausea, vomiting, or abdominal pain that required at least one dose interruption.
Monitor and manage patients using standard of care, including antidiarrheals, antiemetics, or fluid replacement, as indicated. Withhold ZYKADIA if gastrointestinal adverse reaction is severe or intolerable and is not responsive to antiemetics or antidiarrheals. Upon improvement, resume ZYKADIA at a reduced dose [see Dosage and Administration (2.3)].
5.2 Hepatotoxicity
Drug-induced hepatotoxicity occurred in patients treated with ZYKADIA [see Adverse Reactions (6.1)]. Elevations in alanine aminotransferase (ALT) > 5 times the upper limit of normal (ULN) occurred in 28% and elevations in aspartate aminotransferase (AST) > 5 times ULN occurred in 16% of 925 patients across clinical studies. Concurrent elevations in ALT > 3 times the ULN and total bilirubin > 2 times the ULN, with alkaline phosphatase < 2 times the ULN occurred in 0.3% of patients across clinical studies. Approximately 1% of patients required permanent discontinuation due to hepatotoxicity.
Monitor with liver laboratory tests, including ALT, AST, and total bilirubin, once a month and as clinically indicated, with more frequent testing in patients who develop transaminase elevations. Based on the severity of the adverse reaction, withhold ZYKADIA with resumption at a reduced dose, or permanently discontinue ZYKADIA [see Dosage and Administration (2.3)].
5.3 Interstitial Lung Disease/Pneumonitis
Severe, life-threatening, or fatal interstitial lung diseases (ILD)/pneumonitis occurred in patients treated with ZYKADIA [see Adverse Reactions (6.1)]. Across clinical studies, ILD/pneumonitis was reported in 2.4% of 925 patients treated with ZYKADIA. Grade 3 or 4 ILD/pneumonitis was reported in 1.3% of patients, with fatal events reported in 0.2% of patients. Ten patients (1.1%) discontinued ZYKADIA across clinical studies due to ILD/pneumonitis.
Monitor patients for pulmonary symptoms indicative of ILD/pneumonitis. Exclude other potential causes of ILD/pneumonitis and permanently discontinue ZYKADIA in patients diagnosed with treatment-related ILD/pneumonitis [see Dosage and Administration (2.3)].
5.4 QT Interval Prolongation
QTc interval prolongation, which may lead to an increased risk for ventricular tachyarrhythmia (e.g., torsades de pointes) or sudden death, occurred in patients treated with ZYKADIA [see Adverse Reactions (6.1)]. Across clinical studies, 6% of 919 patients with at least one post-baseline electrocardiogram (ECG) assessment had an increase from baseline of QTc > 60 msec. Approximately 1.3% of patients taking ZYKADIA 750 mg under fasted conditions were found to have a QTc > 500 msec. ZYKADIA causes concentration-dependent increases in the QTc interval [see Clinical Pharmacology (12.2)]. Across clinical studies, 0.2% of patients discontinued ZYKADIA due to QTc prolongation.
When possible, avoid use of ZYKADIA in patients with congenital long QT syndrome. Conduct periodic monitoring with ECGs and electrolytes in patients with congestive heart failure, bradyarrhythmias, electrolyte abnormalities, or those who are taking medications that are known to prolong the QTc interval. Based on the severity of the adverse reaction, withhold ZYKADIA, with resumption at a reduced dose, or permanently discontinue ZYKADIA [see Dosage and Administration (2.3)].
5.5 Hyperglycemia
Hyperglycemia occurred in patients treated with ZYKADIA [see Adverse Reactions (6.1)]. Across clinical studies, Grade 3 or 4 hyperglycemia, based on laboratory values, occurred in 13% of 925 patients.
Monitor fasting serum glucose prior to the start of ZYKADIA treatment and periodically thereafter as clinically indicated. Initiate or optimize anti-hyperglycemic medications as indicated. Based on the severity of the adverse reaction, withhold ZYKADIA with resumption at a reduced dose, or permanently discontinue ZYKADIA [see Dosage and Administration (2.3)].
5.6 Bradycardia
Bradycardia occurred in patients treated with ZYKADIA [see Adverse Reactions (6.1)]. Across clinical studies, sinus bradycardia, defined as a heart rate < 50 beats per minute, was noted as a new finding in 1.1% of 925 patients. Bradycardia was reported as an adverse reaction in 1% of patients. No patient required discontinuation and 0.1% required interruption with subsequent dose reduction for bradycardia.
Avoid using ZYKADIA in combination with other products known to cause bradycardia (e.g., beta-blockers, non-dihydropyridine calcium channel blockers, clonidine, and digoxin) to the extent possible. Monitor heart rate and blood pressure regularly. Based on the severity of the adverse reaction, withhold ZYKADIA with resumption at a reduced dose upon resolution of bradycardia, or permanently discontinue ZYKADIA [see Dosage and Administration (2.3)].
5.7 Pancreatitis
Pancreatitis occurred in patients treated with ZYKADIA [see Adverse Reactions (6.1)]. Pancreatitis, including one fatality, occurred in less than 1% of patients receiving ZYKADIA in clinical studies. Grade 3 or 4 elevations of amylase occurred in 7% of patients receiving ZYKADIA across clinical studies, while Grade 3 or 4 elevations of lipase occurred in 14% of patients.
Monitor lipase and amylase prior to the start of ZYKADIA treatment and periodically thereafter as clinically indicated. Based on the severity of the laboratory abnormalities, withhold ZYKADIA with resumption at a reduced dose [see Dosage and Administration (2.3)].
5.8 Embryo-Fetal Toxicity
Based on its mechanism of action and findings from animal studies, ZYKADIA can cause fetal harm when administered to a pregnant woman. In animal studies, administration of ceritinib to rats and rabbits during organogenesis at maternal plasma exposures below the recommended human dose caused increases in skeletal anomalies in rats and rabbits.
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with ZYKADIA and for 6 months following completion of therapy. Based on the potential for genotoxicity, advise males with female partners of reproductive potential to use condoms during treatment with ZYKADIA and for 3 months following completion of therapy [see Use in Specific Populations (8.1, 8.3), Nonclinical Toxicology (13.1)].
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Gastrointestinal Adverse Reactions [see Warnings and Precautions (5.1)]
- Hepatotoxicity [see Warnings and Precautions (5.2)]
- Interstitial Lung Disease/Pneumonitis [see Warnings and Precautions (5.3)]
- QT Interval Prolongation [see Warnings and Precautions (5.4)]
- Hyperglycemia [see Warnings and Precautions (5.5)]
- Bradycardia [see Warnings and Precautions (5.6)]
- Pancreatitis [see Warnings and Precautions (5.7)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data in the Warnings and Precautions section reflect exposure to ZYKADIA 750 mg once daily under fasted conditions in 925 patients with ALK-positive NSCLC across seven clinical studies, including ASCEND-4 and ASCEND-1, described below, a randomized active-controlled study, two single arm studies, and two dose-escalation studies. The majority of patients enrolled in these studies had received prior treatment with chemotherapy and/or crizotinib for NSCLC. Among these 925 patients, the most common adverse reactions (≥ 25% incidence) were diarrhea, nausea, vomiting, fatigue, abdominal pain, decreased appetite, and weight loss. Approximately 45% of patients initiating treatment with ZYKADIA 750 mg under fasted conditions had an adverse reaction that required at least one dose reduction and 66% of patients had an adverse reaction that required at least one dose interruption. The median time to first dose reduction due to any reason was 7 weeks.
Dose Optimization Study: Dosing Regimen of 450 mg Daily With Food
In ASCEND-8, a dose optimization study, ZYKADIA 450 mg daily with food (N = 108) was compared to 750 mg daily under fasted conditions (N = 110) in both previously treated and untreated patients with ALK-positive NSCLC. The overall safety profile of ZYKADIA 450 mg with food was consistent with ZYKADIA 750 mg fasted, except for a reduction in gastrointestinal adverse reactions, while achieving comparable steady-state exposure [see Clinical Pharmacology (12.3)]. The most common adverse reactions (≥ 25% incidence) in the 450 mg with food arm were diarrhea, nausea, abdominal pain, vomiting, and fatigue. The incidence and severity of gastrointestinal adverse reactions (diarrhea 59%, nausea 43%, and vomiting 38%) were reduced for patients treated with ZYKADIA 450 mg with food; Grade ≥ 3 adverse reactions were reported in two patients (1.9%): Grade 3 diarrhea and Grade 3 vomiting in one patient each [see Warnings and Precautions (5.1)].
In patients treated with ZYKADIA 450 mg with food, 24% of patients had an adverse reaction that required at least one dose reduction and 56% of patients had an adverse reaction that required at least one dose interruption. The median time to first dose reduction due to any reason was 8 weeks.
Previously Untreated ALK-Positive Metastatic NSCLC
The safety of ZYKADIA was evaluated in ASCEND-4, an open-label, randomized, active-controlled multicenter study of 376 previously untreated ALK-positive NSCLC patients [see Clinical Studies (14.1)]. Patients received ZYKADIA 750 mg daily (N = 189) under fasted conditions or chemotherapy and maintenance chemotherapy (N = 187). Chemotherapy regimens were pemetrexed (500 mg/m2) and investigator’s choice of cisplatin (75 mg/m2) or carboplatin [area under the curve (AUC) of 5 - 6 mg*min/mL] administered every 21 days. Patients who completed 4 cycles of chemotherapy without progressive disease received pemetrexed (500 mg/m2) as single-agent maintenance therapy every 21 days. The median duration of exposure to ZYKADIA was 18 months.
The demographic characteristics of the study population were 57% female, median age 54 years (range, 22 to 81 years), 22% age 65 years or older, 54% white, 42% Asian, 2% black, and 2% other races. Patients were enrolled in Europe (53%), Asia Pacific (42%), and South America (5%) regions. The majority of patients had adenocarcinoma (97%), never smoked (61%), and 32% had brain metastases at screening.
The following fatal adverse reactions occurred in 4 patients treated with ZYKADIA: Myocardial infarction, respiratory tract infection, pneumonitis, and unknown cause.
Serious adverse reactions were reported in 38% of patients treated with ZYKADIA. The most frequent serious adverse reactions were pneumonia (4%), pleural effusion (4%), vomiting (4%), nausea (3%), dyspnea (3%), hyperglycemia (3%), AST increased (2%), lung infection (2%), and pericardial effusion (2%).
Among patients treated with ZYKADIA, dose interruptions due to adverse reactions occurred in 77%, dose reductions were required in 66%, and adverse reactions that led to discontinuation of therapy occurred in 12% of patients. The most frequent adverse reactions, reported in at least 10% of patients treated with ZYKADIA, that led to dose interruptions or reductions were: Increased ALT (48%), increased AST (34%), vomiting (15%), increased blood creatinine (14%), increased gamma-glutamyl transpeptidase (GGT) (13%), diarrhea (13%), and nausea (13%). The most frequent adverse reactions that led to discontinuation of ZYKADIA in 1% or more of patients in ASCEND-4 were increased blood creatinine (2.1%), increased amylase (1.1%), and increased lipase (1.1%).
Tables 3 and 4 summarize adverse reactions and laboratory abnormalities, respectively, in ASCEND-4.
| ZYKADIA
N = 189 | Chemotherapy
N = 175a |
|||
| All Grades | Grade 3-4 | All Grades | Grade 3-4 | |
| % | % | % | % | |
| Gastrointestinal** | ||||
| Diarrhea | 85 | 4.8 | 11 | 1.1 |
| Nausea | 69 | 2.6 | 55 | 5 |
| Vomiting | 67 | 5 | 36 | 6 |
| Abdominal painb | 40 | 3.7 | 13 | 0 |
| Constipation | 20 | 0 | 22 | 0 |
| Esophageal disorderc | 15 | 0.5 | 8 | 0.6 |
| General | ||||
| Fatigued | 45 | 7 | 49 | 6 |
| Non-cardiac chest pain | 21 | 1.1 | 10 | 0.6 |
| Back pain | 19 | 1.6 | 18 | 2.3 |
| Pyrexia | 19 | 0 | 14 | 1.1 |
| Pain in extremity | 13 | 0 | 7 | 0 |
| Musculoskeletal pain | 11 | 0.5 | 6 | 0.6 |
| Pruritus | 11 | 0.5 | 5 | 0 |
| Metabolism and Nutrition | ||||
| Decreased appetite | 34 | 1.1 | 32 | 1.1 |
| Weight loss | 24 | 3.7 | 15 | 0.6 |
| Respiratory | ||||
| Cough | 25 | 0 | 17 | 0 |
| Skin | ||||
| Rashe | 21 | 1.1 | 8 | 0.6 |
| Neurologic | ||||
| Headache | 19 | 0.5 | 13 | 1.1 |
| Dizziness | 12 | 1.1 | 10 | 0.6 |
| Cardiac | ||||
| Prolonged QT interval | 12 | 2.6 | 1.1 | 0.6 |
| Pericarditisf | 4.2 | 1.6 | 2.3 | 1.1 |
| *National Cancer Institute Common Terminology Criteria for Adverse Events (version 4.03). **For frequency of gastrointestinal adverse reactions at the recommended dose of 450 mg with food [see Warnings and Precautions (5.1), Adverse Reactions (6.1)]. aTwelve patients randomized to chemotherapy did not receive study drug. bAbdominal pain (abdominal pain, abdominal pain upper, abdominal discomfort, and epigastric discomfort). cEsophageal disorder (dyspepsia, gastroesophageal reflux disease, and dysphagia). dFatigue (fatigue and asthenia). eRash (rash, dermatitis acneiform, rash maculo-papular). fPericarditis (pericardial effusion and pericarditis). |
||||
Additional clinically significant adverse reactions occurring in 2% or more of patients treated with ZYKADIA 750 mg under fasted conditions included: vision disorder (4% comprised of vision impairment, blurred vision, photopsia, accommodation disorder, presbyopia, reduced visual acuity, or vitreous floaters), bradycardia (4%), ILD/pneumonitis (2%), hepatotoxicity (2%), and renal failure (2%). In addition, the adverse reaction of photosensitivity was reported in 1.1% of patients.
| ZYKADIA
N = 189 | Chemotherapy
N = 175a |
|||
| All Grades | Grade 3-4 | All Grades | Grade 3-4 | |
| % | % | % | % | |
| Chemistry | ||||
| Increased ALT | 91 | 34 | 65 | 3.4 |
| Increased AST | 86 | 21 | 58 | 2.3 |
| Increased GGT | 84 | 49 | 67 | 10 |
| Increased alkaline phosphatase | 81 | 12 | 47 | 1.7 |
| Increased creatinine | 77 | 4.2 | 37 | 0.6 |
| Hyperglycemia | 53 | 10 | 67 | 10 |
| Hypophosphatemia | 38 | 3.7 | 27 | 4.0 |
| Increased amylase | 37 | 8 | 43 | 4.5 |
| Hyperbilirubinemia (total) | 15 | 0.5 | 6 | 0.6 |
| Increased lipaseb | 13 | 6 | 7 | 0.6 |
| Hematology | ||||
| Anemia | 67 | 4.2 | 84 | 11 |
| Neutropenia | 27 | 2.1 | 58 | 20 |
| Thrombocytopenia | 16 | 1.0 | 38 | 4.6 |
| Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; GGT, gamma-glutamyl transpeptidase. *National Cancer Institute Common Terminology Criteria for Adverse Events (version 4.03). aTwelve patients randomized to chemotherapy did not receive study drug. bIn the ZYKADIA arm, no patients had baseline lipase laboratory assessments, 112 had post-baseline assessments. In the chemotherapy arm, one patient had baseline lipase laboratory assessments but no post-baseline assessment; 49 patients had post-baseline assessments. |
||||
Previously Treated ALK-Positive Metastatic NSCLC
The safety of ZYKADIA was evaluated in ASCEND-1, a multicenter, single-arm, open-label clinical study of 255 ALK-positive patients (246 patients with NSCLC and 9 patients with other cancers who received ZYKADIA at a dose of 750 mg daily under fasted conditions) [see Clinical Studies (14.2)]. The median duration of exposure to ZYKADIA was 6 months.
The study population characteristics were: Median age 53 years, age less than 65 (84%), female (53%), white (63%), Asian (34%), NSCLC adenocarcinoma histology (90%), never or former smoker (97%), Eastern Cooperative Oncology Group Performance Status (ECOG PS) 0 or 1 (89%), brain metastases (49%), and number of prior therapies 2 or more (67%).
Fatal adverse reactions in patients treated with ZYKADIA occurred in 5% of patients, consisting of: Pneumonia (4 patients), respiratory failure, ILD/pneumonitis, pneumothorax, gastric hemorrhage, general physical health deterioration, pulmonary tuberculosis, cardiac tamponade, and sepsis (1 patient each).
Serious adverse reactions reported in 2% or more of patients in ASCEND-1 were convulsion, pneumonia, ILD/pneumonitis, dyspnea, dehydration, hyperglycemia, and nausea.
Dose reductions due to adverse reactions occurred in 59% of patients treated with ZYKADIA. The most frequent adverse reactions, reported in at least 10% of patients, that led to dose reductions or interruptions were: Increased ALT (29%), nausea (20%), increased AST (16%), diarrhea (16%), and vomiting (16%). Discontinuation of therapy due to adverse reactions occurred in 10% of patients treated with ZYKADIA. The most frequent adverse reactions that led to discontinuation in 1% or more of patients in ASCEND-1 were pneumonia, ILD/pneumonitis, and decreased appetite.
Tables 5 and 6 summarize adverse reactions and laboratory abnormalities, respectively, in ASCEND-1.
| ZYKADIA
N = 255 |
||
| All Grades | Grade 3-4 | |
| % | % | |
| Gastrointestinal** | ||
| Diarrhea | 86 | 6 |
| Nausea | 80 | 4 |
| Vomiting | 60 | 4 |
| Abdominal paina | 54 | 2 |
| Constipation | 29 | 0 |
| Esophageal disorderb | 16 | 1 |
| General | ||
| Fatiguec | 52 | 5 |
| Metabolism and Nutrition | ||
| Decreased appetite | 34 | 1 |
| Skin | ||
| Rashd | 16 | 0 |
| Respiratory | ||
| Interstitial lung disease/pneumonitis | 4 | 3 |
| Abbreviation: ALK, anaplastic lymphoma kinase. *National Cancer Institute Common Terminology Criteria for Adverse Events (version 4.03). **For frequency of gastrointestinal adverse reactions at the recommended dose of 450 mg with food [see Warnings and Precautions (5.1), Adverse Reactions (6.1)]. aAbdominal pain (abdominal pain, upper abdominal pain, abdominal discomfort, and epigastric discomfort). bEsophageal disorder (dyspepsia, gastroesophageal reflux disease, and dysphagia). cFatigue (fatigue and asthenia). dRash (rash, maculopapular rash, and acneiform dermatitis). |
||
Additional clinically significant adverse reactions occurring in 2% or more of patients treated with ZYKADIA 750 mg under fasted conditions included neuropathy (17% comprised of paresthesia, muscular weakness, gait disturbance, peripheral neuropathy, hypoesthesia, peripheral sensory neuropathy, dysesthesia, neuralgia, peripheral motor neuropathy, hypotonia, or polyneuropathy), vision disorder (9% comprised of vision impairment, blurred vision, photopsia, accommodation disorder, presbyopia, or reduced visual acuity), prolonged QT interval (4%), and bradycardia (3%). In addition, the adverse reaction of photosensitivity was reported in 1.2% of patients.
| ZYKADIA
N = 255 |
||
| All Grades | Grade 3-4 | |
| % | % | |
| Hematology | ||
| Anemia | 84 | 5 |
| Chemistry | ||
| Increased ALT | 80 | 27 |
| Increased AST | 75 | 13 |
| Increased creatinine | 58 | 2 |
| Hyperglycemia | 49 | 13 |
| Hypophosphatemia | 36 | 7 |
| Increased lipase | 28 | 10 |
| Hyperbilirubinemia (total) | 15 | 1 |
| Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; ALK, anaplastic lymphoma kinase. *National Cancer Institute Common Terminology Criteria for Adverse Events (version 4.03). |
||
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on ZYKADIA
Strong CYP3A Inhibitors
A strong CYP3A4/P-gp inhibitor (ketoconazole) increased the systemic exposure of ceritinib [see Clinical Pharmacology (12.3)], which may increase the incidence and severity of adverse reactions of ZYKADIA. Avoid concurrent use of strong CYP3A inhibitors during treatment with ZYKADIA. If concurrent use of strong CYP3A inhibitors is unavoidable, reduce the ZYKADIA dose [see Dosage and Administration (2.4)].
Do not consume grapefruit and grapefruit juice as they may inhibit CYP3A.
Strong CYP3A Inducers
A strong CYP3A4/P-gp inducer (rifampin) decreased the systemic exposure of ceritinib [see Clinical Pharmacology (12.3)], which may decrease the efficacy of ZYKADIA. Avoid concurrent use of strong CYP3A inducers during treatment with ZYKADIA.
7.2 Effect of ZYKADIA on Other Drugs
CYP3A Substrates
Ceritinib increased the systemic exposure of a sensitive CYP3A substrate (midazolam) [see Clinical Pharmacology (12.3)]. Avoid coadministration of ZYKADIA with sensitive CYP3A substrates. If concomitant use is unavoidable, consider dose reduction of the sensitive CYP3A substrate(s). If ZYKADIA is coadministered with other CYP3A substrates, refer to the CYP3A substrate labeling for dosage recommendation with strong CYP3A inhibitors.
CYP2C9 Substrates
Ceritinib increased the systemic exposure of a CYP2C9 substrate (warfarin) [see Clinical Pharmacology (12.3)]. Increase the frequency of INR monitoring if coadministration with warfarin is unavoidable as the anti-coagulant effect of warfarin may be enhanced.
Avoid coadministration of ZYKADIA with CYP2C9 substrates for which minimal concentration changes may lead to serious toxicities. If concomitant use of such CYP2C9 substrates is unavoidable, consider dose reduction for the coadministered CYP2C9 substrates.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on animal studies and its mechanism of action [see Clinical Pharmacology (12.1)], ZYKADIA can cause fetal harm when administered to a pregnant woman. The limited available data on the use of ZYKADIA in pregnant women are insufficient to inform a risk. Administration of ceritinib to rats and rabbits during the period of organogenesis at maternal plasma exposures below the recommended human dose caused increases in skeletal anomalies in rats and rabbits (see Data). Advise a pregnant woman of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies are 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In an embryo-fetal development study in which pregnant rats were administered daily doses of ceritinib during organogenesis, dose-related skeletal anomalies were observed at doses as low as 50 mg/kg (less than 0.5-fold the human exposure by AUC at the recommended dose). Findings included delayed ossifications and skeletal variations.
In pregnant rabbits administered ceritinib daily during organogenesis, dose-related skeletal anomalies, including incomplete ossification, were observed at doses equal to or > 2 mg/kg/day (approximately 0.015-fold the human exposure by AUC at the recommended dose). A low incidence of visceral anomalies, including absent or malpositioned gallbladder and retroesophageal subclavian cardiac artery, was observed at doses equal to or > 10 mg/kg/day (approximately 0.13-fold the human exposure by AUC at the recommended dose). Maternal toxicity and abortion occurred in rabbits at doses of 35 mg/kg or greater. In addition, embryolethality was observed in rabbits at a dose of 50 mg/kg.
8.2 Lactation
Risk Summary
There are no data regarding the presence of ceritinib or its metabolites in human milk, the effects of ceritinib on the breastfed child or its effects on milk production. Because of the potential for serious adverse reactions in breastfed children, advise women not to breastfeed during treatment with ZYKADIA and for 2 weeks following completion of therapy.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Verify pregnancy status in females of reproductive potential prior to initiating ZYKADIA [see Use in Specific Populations (8.1)].
Contraception
ZYKADIA can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Females
Advise females of reproductive potential to use effective contraception during treatment with ZYKADIA and for 6 months following completion of therapy.
Males
Based on the potential for genotoxicity, advise males with female partners of reproductive potential to use condoms during treatment with ZYKADIA and for 3 months following completion of therapy [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of ZYKADIA in pediatric patients have not been established.
11 DESCRIPTION
Ceritinib is a kinase inhibitor for oral administration. The molecular formula for ceritinib is C28H36N5O3ClS. The molecular weight is 558.14 g/mol. Ceritinib is described chemically as 5-Chloro-N4-[2-[(1-methylethyl)sulfonyl]phenyl]-N2-[5-methyl-2-(1-methylethoxy)-4-(4-piperidinyl)phenyl]-2,4-pyrimidinediamine.
The chemical structure of ceritinib is shown below:
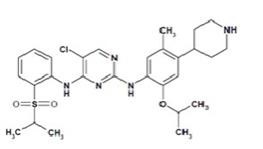
Ceritinib is a white to almost white or light yellow powder.
ZYKADIA is supplied as printed hard-gelatin capsules containing 150 mg of ceritinib and the following inactive ingredients: colloidal silicon dioxide, hard gelatin capsule shells, low substituted hydroxypropyl cellulose, magnesium stearate, microcrystalline cellulose, and sodium starch glycolate. The capsule shell is composed of FD&C Blue # 2, gelatin, and titanium dioxide.
ZYKADIA is supplied as film-coated tablets containing 150 mg of ceritinib and the following inactive ingredients: colloidal silicon dioxide, croscarmellose sodium, low-substituted hydroxypropyl cellulose, magnesium stearate, microcrystalline cellulose and povidone. The tablet coating contains FD&C Blue # 2 aluminum lake, hypromellose, polyethylene glycol 4000, talc, and titanium dioxide.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Ceritinib is a kinase inhibitor. Targets of ceritinib inhibition identified in either biochemical or cellular assays at clinically relevant concentrations include ALK, insulin-like growth factor 1 receptor (IGF-1R), insulin receptor (InsR), and ROS1. Among these, ceritinib is most active against ALK. Ceritinib inhibited autophosphorylation of ALK, ALK-mediated phosphorylation of the downstream signaling protein STAT3, and proliferation of ALK-dependent cancer cells in in vitro and in vivo assays.
Ceritinib inhibited the in vitro proliferation of cell lines expressing EML4-ALK and NPM-ALK fusion proteins and demonstrated dose-dependent inhibition of EML4-ALK-positive NSCLC xenograft growth in mice and rats. Ceritinib exhibited dose-dependent anti-tumor activity in mice bearing EML4-ALK-positive NSCLC xenografts with demonstrated resistance to crizotinib, at concentrations within a clinically relevant range.
12.2 Pharmacodynamics
Cardiac Electrophysiology
Twelve of 919 patients (1.3%) treated with ZYKADIA 750 mg once daily under fasted conditions with at least one post-baseline ECG assessment were found to have a QTc > 500 msec and 58 patients (6%) had an increase from baseline QTc > 60 msec. In ASCEND-4, a central tendency analysis of the QTc data at average steady-state concentrations demonstrated that the upper bound of the 2-sided 90% CI for QTc was 15.3 msec at ZYKADIA 750 mg once daily under fasted conditions. A pharmacokinetic/pharmacodynamic analysis suggested concentration-dependent QTc interval prolongation [see Warnings and Precautions (5.4)].
Ten of 925 patients (1.1%) had bradycardia defined as < 50 beats per minute [see Warnings and Precautions (5.6)].
12.3 Pharmacokinetics
After a single oral administration of ZYKADIA in patients, AUC and Cmax increased dose proportionally over 50 mg to 750 mg under fasted conditions.
Following ZYKADIA 750 mg once daily under fasted conditions, steady state was reached by 15 days with a geometric mean accumulation ratio of 6.2 after 3 weeks. Systemic exposure increased in a greater than dose proportional manner after repeat doses of 50 mg to 750 mg once daily under fasted conditions.
Absorption
After a single oral administration of ZYKADIA in patients, peak plasma levels (Cmax) of ceritinib were achieved around 4 to 6 hours.
Food Effect
A food effect study conducted in healthy subjects with a single 500 mg dose of ZYKADIA capsules showed that a high-fat meal (containing approximately 1000 calories and 58 grams of fat) increased ceritinib AUC by 73% and Cmax by 41% and a low-fat meal (containing approximately 330 calories and 9 grams of fat) increased ceritinib AUC by 58% and Cmax by 43% as compared with the fasted conditions.
A food effect study conducted in healthy subjects with a single 750 mg dose of ZYKADIA tablets showed that a high-fat meal (containing approximately 1000 calories and 58 grams of fat) increased ceritinib AUC by 64% and Cmax by 58% and a low-fat meal (containing approximately 330 calories and 9 grams of fat) increased ceritinib AUC by 39% and Cmax by 42% as compared with the fasted conditions.
Dose Optimization Study: Dosing Regimen of 450 mg Daily With Food
In a dose optimization study (ASCEND-8) in patients receiving 450 mg dose of ZYKADIA capsules daily with food (approximately 100 to 500 calories and 1.5 to 15 grams of fat) or 750 mg daily under fasted conditions, there was no clinically meaningful difference in the systemic steady-state exposure of ceritinib (AUC) between the 450 mg with food arm and the 750 mg fasted arm.
Distribution
Ceritinib is 97% bound to human plasma proteins, independent of drug concentration. The geometric mean apparent volume of distribution (Vd/F) is 4230 L following a single 750 mg ZYKADIA dose under fasted conditions in patients. Ceritinib also has a slight preferential distribution to red blood cells, relative to plasma, with a mean in vitro blood-to-plasma ratio of 1.35.
Elimination
Following a single 750 mg ZYKADIA dose under fasted conditions, the geometric mean apparent plasma terminal half-life (t1/2) of ceritinib was 41 hours in patients. Ceritinib demonstrates nonlinear PK over time. The geometric mean apparent clearance (CL/F) of ceritinib was lower at steady state (33.2 L/h) after 750 mg daily dosing than after a single 750 mg dose (88.5 L/h).
Metabolism: In vitro studies demonstrated that CYP3A was the major enzyme involved in the metabolic clearance of ceritinib. Following oral administration of a single 750 mg radiolabeled dose under fasted conditions, ceritinib was the main circulating component (82%) in human plasma.
Excretion: Following oral administration of a single 750 mg radiolabeled dose under fasted conditions, 92% of the administered dose was recovered in the feces (with 68% as unchanged parent compound) while 1.3% of the administered dose was recovered in the urine.
Specific Populations
Age, sex, race, body weight, and mild-to-moderate renal impairment (CLcr 30 to < 90 mL/min estimated with Cockcroft-Gault) has no clinically important effect on the systemic exposure of ceritinib based on population pharmacokinetic analyses. Patients with severe renal impairment (CLcr < 30 mL/min) were not included in the clinical trial.
Patients with Hepatic Impairment: Following a single 750 mg ZYKADIA dose under fasted conditions, the geometric mean systemic exposure (AUC0-INF) of ceritinib was increased by 66% and unbound ceritinib AUC0-INF was increased by 108% in subjects with severe (Child-Pugh C) hepatic impairment compared to subjects with normal hepatic function [see Dosage and Administration (2.5)]. Total and unbound systemic exposure of ceritinib were similar in subjects with mild (Child-Pugh A) to moderate (Child-Pugh B) hepatic impairment compared to subjects with normal hepatic function.
Drug Interaction Studies
Effect of Strong CYP3A/P-gp Inhibitors on Ceritinib: Coadministration of a single 450 mg ZYKADIA dose under fasted conditions with ketoconazole (a strong CYP3A/P-gp inhibitor) for 14 days increased ceritinib AUC by 2.9-fold and Cmax by 22%. The steady-state AUC of ceritinib at a dose of 450 mg once daily under fasted conditions with ketoconazole for 14 days was predicted to be similar to the steady-state AUC of ceritinib at a dose of 750 mg alone under fasted conditions.
Effect of Strong CYP3A/P-gp Inducers on Ceritinib: Coadministration of a single 750 mg ZYKADIA dose under fasted conditions with rifampin (a strong CYP3A/P-gp inducer) for 14 days decreased ceritinib AUC by 70% and Cmax by 44%.
Effect of Ceritinib on CYP3A Substrates: Coadministration of a single dose of midazolam (a sensitive CYP3A substrate) following 3 weeks of ZYKADIA (750 mg daily under fasted conditions) increased the midazolam AUC by 5.4-fold and Cmax by 1.8-fold compared to midazolam administered alone [see Drug Interactions (7.2)].
Effect of Ceritinib on CYP2C9 Substrates: Coadministration of a single dose of warfarin (a CYP2C9 substrate) following 3 weeks of ZYKADIA (750 mg daily under fasted conditions) increased the S-warfarin AUC by 54% with no change in Cmax compared to warfarin administered alone [see Drug Interactions (7.2)].
Effect of Acid Reducing Agents on Ceritinib: Coadministration of a single 750 mg ZYKADIA dose under fasted conditions with a proton pump inhibitor (esomeprazole) for 6 days in healthy subjects decreased ceritinib AUC by 76% and Cmax by 79%; however, coadministration of a single 750 mg ZYKADIA dose under fasted conditions with proton pump inhibitors for 6 days in a subgroup of patients from ASCEND-1 suggested less effect on ceritinib exposure than that observed in healthy subjects as AUC decreased by 30% and Cmax decreased by 25% and no clinically meaningful effect on ceritinib exposure was observed at steady-state.
Effect of Transporters on Ceritinib Disposition: Ceritinib is a substrate of efflux transporter P-gp, but is not a substrate of BCRP, MRP2, OCT1, OAT2, or OATP1B1 in vitro.
Effect of Ceritinib on Transporters: Based on in vitro data, ceritinib is unlikely to inhibit P-gp, BCRP, MRP2, OATP1B1, OATP1B3, OAT1, OAT3, OCT1, or OCT2 at clinical concentrations.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been performed with ceritinib.
Ceritinib was not mutagenic in vitro in the bacterial reverse mutation (Ames) assay but induced numerical aberrations (aneugenic) in the in vitro cytogenetic assay using human lymphocytes, and micronuclei in the in vitro micronucleus test using TK6 cells. Ceritinib was not clastogenic in the in vivo rat micronucleus assay.
There are no data on the effect of ceritinib on human fertility. Fertility/early embryonic development studies were not conducted with ceritinib. There were no adverse effects on male or female reproductive organs in general toxicology studies conducted in monkeys and rats at exposures ≥ 0.5- and 1.5-fold, respectively, of the human exposure by AUC at the recommended dose.
13.2 Animal Toxicology and/or Pharmacology
Target organs in nonclinical animal models included, but were not limited to, the pancreas, biliopancreatic/bile ducts, gastrointestinal tract, and liver. Pancreatic focal acinar cell atrophy was observed in rats at 1.5-fold the human exposure by AUC at the recommended dose. Biliopancreatic duct and bile duct necrosis was observed in rats at exposures ≥ 5% of the human exposure by AUC at the recommended dose. Bile duct inflammation and vacuolation were also noted in monkeys at exposures ≥ 0.5-fold the human exposure by AUC at the recommended dose. Frequent minimal necrosis and hemorrhage of the duodenum was exhibited in monkeys at 0.5-fold the human exposure by AUC, and in rats at an exposure similar to that observed clinically.
Ceritinib crossed the blood brain barrier in rats with a brain-to-blood exposure (AUCinf) ratio of approximately 15%.
14 CLINICAL STUDIES
14.1 Previously Untreated ALK-Positive Metastatic NSCLC
The efficacy of ZYKADIA for the treatment of patients with ALK-positive NSCLC who had not received prior systemic therapy for metastatic disease was evaluated in an open-label, randomized, active-controlled, multicenter study (ASCEND-4, NCT01828099). Patients were required to have World Health Organization (WHO) performance status 0-2 and ALK-positive NSCLC as identified by the VENTANA ALK (D5F3) CDx Assay. Neurologically stable patients with central nervous system (CNS) metastases that did not require increasing doses of steroids to manage CNS symptoms were permitted to enroll. Patients with uncontrolled diabetes mellitus; a history of ILD or interstitial pneumonitis; or a history of pancreatitis, or increased amylase, or lipase that was due to pancreatic disease were not eligible.
The major efficacy outcome measure was progression-free survival (PFS) as determined by Blinded Independent Review Committee (BIRC) according to RECIST v1.1. Additional efficacy outcome measures were overall survival (OS), overall response rate (ORR) and duration of response (DOR) determined by BIRC, overall intracranial response rate (OIRR), duration of intracranial response (DOIR) determined by BIRC neuro-radiologist, and patient reported outcomes.
Patients were randomized 1:1 to receive ZYKADIA 750 mg orally daily under fasted conditions or chemotherapy and maintenance chemotherapy. Randomization was stratified by WHO performance status, prior adjuvant/neoadjuvant chemotherapy and presence or absence of brain metastases. Patients randomized to chemotherapy received pemetrexed (500 mg/m2) and investigator’s choice of cisplatin (75 mg/m2) or carboplatin (AUC of 5 - 6 mg*min/mL) administered on Day 1 of each 21-day cycle for a maximum of 4 cycles followed by pemetrexed (500 mg/m2) every 21 days. Treatment in both arms was continued until disease progression or unacceptable toxicity.
The study population characteristics were: 57% female, median age 54 years (range, 22 to 81 years), 22% age 65 years or older, 54% white, 42% Asian, 2% black, and 2% other races. The majority of patients had adenocarcinoma (97%) and never smoked (61%). CNS metastases were present in 32% (n = 121) of patients. Approximately half (n = 55) had measurable CNS metastases as determined by BIRC neuro-radiologist and 71% (n = 39) of these patients received no prior intracranial radiotherapy. Of those randomized to chemotherapy, 43% received ZYKADIA as the next antineoplastic therapy after platinum-based chemotherapy.
Efficacy results from ASCEND-4 are summarized in Table 7 and Figure 1.
| Efficacy Parameter | ZYKADIA
(N = 189) | Chemotherapy
(N = 187) |
| Progression-Free Survival | ||
| Number of events, n (%) | 89 (47%) | 113 (60%) |
| Progressive disease, n (%) | 79 (42%) | 105 (56%) |
| Death, n (%) | 10 (5%) | 8 (4%) |
| Median PFS in months (95% CI) | 16.6 (12.6, 27.2) | 8.1 (5.8, 11.1) |
| Hazard ratio (95% CI)a | 0.55 (0.42, 0.73) | |
| P-valueb | < 0.0001 | |
| Overall Response Rate | ||
| Overall response rate, % (95% CI)c | 73 (66, 79) | 27 (21, 34) |
| Complete response, % | 1 | 0 |
| Partial response, % | 72 | 27 |
| Duration of Response | ||
| Number of responders | n = 137 | n = 50 |
| Median in months (95% CI) | 23.9 (16.6, NE) | 11.1 (7.8, 16.4) |
| Abbreviations: BIRC, Blinded Independent Review Committee; CI, confidence interval; NE, not estimable. aCox proportional hazards model stratified by brain metastases (absence vs. presence), WHO performance status (0 vs. ≥ 1), and prior adjuvant chemotherapy (absence vs. presence). bLog-rank test stratified by brain metastases (absence vs. presence), WHO performance status (0 vs. ≥ 1), and prior adjuvant chemotherapy (absence vs. presence). cClopper and Pearson exact binomial 95% CI. |
||
There was no significant difference in OS in a pre-specified interim analysis conducted at 42% of the events required for the final analysis.
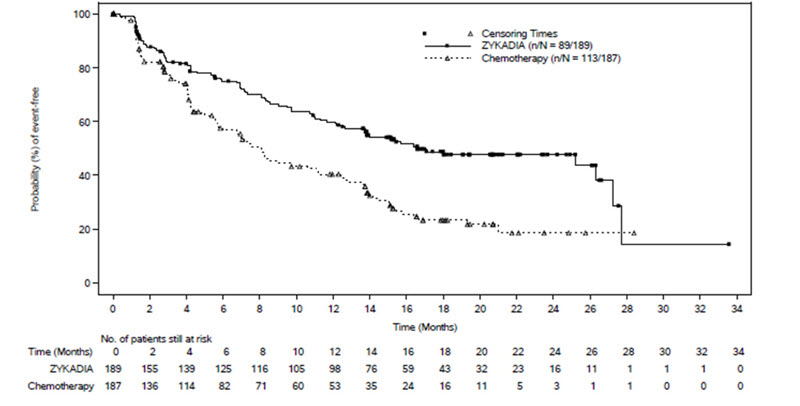
Figure 1: Kaplan-Meier Plot of Progression-Free Survival as Assessed by BIRC by Treatment Arm in ASCEND-4
Antitumor activity of ZYKADIA in the brain was assessed in patients with measurable disease as determined by the BIRC neuro-radiologist at baseline (N = 55) according to RECIST 1.1.
| Intracranial Tumor Response Assessment | ZYKADIA (N = 28) | Chemotherapy (N = 27) |
| Overall intracranial response rate,
% (95% CI)a |
57% (37, 76) |
22% (9, 42) |
| Complete response, % | 7% | 7% |
| Partial response, % | 50% | 15% |
| Duration of Intracranial Response | ||
| Number of responders | n = 16 | n = 6 |
| Median in months (95% CI) | 16.6 (8.1, NE) | NE (1.5, NE) |
| Abbreviations: BIRC, Blinded Independent Review Committee; CI, confidence interval; CNS, central nervous system; NE, not estimable. aClopper and Pearson exact binomial 95% CI. |
||
Exploratory analyses of patient-reported outcome measures suggested a delay in time to development of or worsening of shortness of breath in patients treated with ZYKADIA as compared to chemotherapy. The patient-reported delay in onset or worsening of shortness of breath may be an overestimation, because patients were not blinded to treatment assignment.
14.2 Previously Treated ALK-Positive Metastatic NSCLC
The efficacy of ZYKADIA was evaluated in a multicenter, single-arm, open-label clinical trial (ASCEND-1, NCT01283516). A total of 163 patients with metastatic ALK-positive NSCLC who progressed while receiving or were intolerant to crizotinib were enrolled. The major efficacy outcome measure was objective response rate (ORR) according to RECIST v1.0 as evaluated by both investigators and BIRC. Duration of response was an additional outcome measure. All patients received ZYKADIA at a dose of 750 mg once daily under fasted conditions.
The study population characteristics were: median age 52 years, age less than 65 (87%), female (54%), White (66%), Asian (29%), never or former smoker (97%), ECOG PS 0 or 1 (87%), progression on previous crizotinib (91%), number of prior therapies 2 or more (84%), and adenocarcinoma histology (93%). Sites of extra-thoracic metastases included brain (60%), liver (42%), and bone (42%). ALK-positivity was verified retrospectively by review of local test results for 99% of patients.
Efficacy results from ASCEND-1 are summarized in Table 9.
| Efficacy Parameter | Investigator
Assessment (N = 163) | BIRC
Assessment (N = 163) |
| Overall response rate, % (95% CI) | 55% (47, 62) | 44% (36, 52) |
| Complete response, % | 1.2% | 2.5% |
| Partial response, % | 53% | 41% |
| Duration of response, median (months) (95% CI) | 7.4 (5.4, 10.1) | 7.1 (5.6, NE) |
| Abbreviations: BIRC, Blinded Independent Review Committee; CI, confidence interval; NE, not estimable. 1Overall response rate and duration of response determined by RECIST v1.0. |
||
The analysis by the BIRC assessment was similar to the analysis by the investigator assessment.
16 HOW SUPPLIED/STORAGE AND HANDLING
ZYKADIA 150 mg capsules
Hard gelatin capsule with opaque blue cap and opaque white body; opaque blue cap marked in black ink with “LDK 150MG”, opaque white body marked in black ink with “NVR”. Available in:
Bottles of 70 capsules…………………………………………………………………………………….NDC 0078-0640-70
Store at 20°C to 25°C (68°F to 77°F); excursions permitted between 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
ZYKADIA 150 mg tablets
Film-coated tablet, light blue, round, biconvex with beveled edges, without score, debossed with “NVR” on one side and “ZY1” on the other side. Available in:
Bottles of 84 tablets…………………………………………………………………………………NDC 0078-0694-84
Store at 20°C to 25°C (68°F to 77°F); excursions permitted between 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Gastrointestinal Adverse Reactions
Inform patients that diarrhea, nausea, vomiting, and abdominal pain are the most commonly reported gastrointestinal adverse reactions. Inform patients of supportive care options, such as antiemetic and anti-diarrheal medications. Advise patients to contact their healthcare provider for severe or intolerable gastrointestinal symptoms. Inform patients that if vomiting occurs during the course of treatment, they should not take an additional dose, but should continue with the next scheduled dose of ZYKADIA [see Warnings and Precautions (5.1)].
Hepatotoxicity
Inform patients of the signs and symptoms of hepatotoxicity. Advise patients to contact their healthcare provider immediately for signs or symptoms of hepatotoxicity [see Warnings and Precautions (5.2)].
Interstitial Lung Disease/Pneumonitis
Inform patients of the risks of severe or fatal ILD/pneumonitis. Advise patients to contact their healthcare provider immediately to report new or worsening respiratory symptoms [see Warnings and Precautions (5.3)].
Arrhythmias
Inform patients of the risks of QTc interval prolongation and bradycardia. Advise patients to contact their healthcare provider immediately to report new chest pain or discomfort, changes in heartbeat, palpitations, dizziness, lightheadedness, fainting, and changes in or new use of heart or blood pressure medications [see Warnings and Precautions (5.4, 5.6)].
Hyperglycemia
Inform patients of the signs and symptoms of hyperglycemia. Advise patients to contact their healthcare provider immediately for signs or symptoms of hyperglycemia [see Warnings and Precautions (5.5)].
Pancreatitis
Inform patients of the signs and symptoms of pancreatitis and the need to monitor lipase and amylase levels prior to the start of treatment and periodically thereafter as clinically indicated [see Warnings and Precautions (5.7)].
Photosensitivity
Inform patients of the signs and symptoms of photosensitivity. Advise patients to avoid prolonged sun exposure and to use sunscreen or protective clothing during treatment with ZYKADIA [see Adverse Reactions (6.1)].
Embryo-Fetal Toxicity
- Advise pregnant women and females of reproductive potential of the potential risk to a fetus and to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.8), Use in Specific Populations (8.3)].
- Advise females of reproductive potential to use effective contraception during treatment with ZYKADIA and for 6 months following completion of therapy [see Use in Specific Populations (8.3)].
- Advise males with female partners of reproductive potential to use condoms during treatment with ZYKADIA and for 3 months following completion of therapy [see Use in Specific Populations (8.3), Nonclinical Toxicology (13.1)].
Lactation
Advise women not to breastfeed during treatment with ZYKADIA and for 2 weeks following completion of therapy [see Use in Specific Populations (8.2)].
Drug Interactions
Inform patients not to consume grapefruit and grapefruit juice during treatment with ZYKADIA [see Drug Interactions (7.1)].
Dosing Instructions
Advise patients to take ZYKADIA with food [see Dosage and Administration (2.2)]. Advise patients to make up a missed dose of ZYKADIA unless the next dose is due within 12 hours [see Dosage and Administration (2.2)].
Distributed by:
Novartis Pharmaceuticals Corporation
East Hanover, New Jersey 07936
© Novartis
T2021-132
| This Patient Information has been approved by the U.S. Food and Drug Administration. | Revised: October 2021 | ||||
| PATIENT INFORMATION
ZYKADIA® (zye kaye' dee ah) (ceritinib) capsules (ceritinib) tablets |
|||||
| Read this Patient Information leaflet that comes with ZYKADIA before you start taking it and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or treatment. | |||||
| What is the most important information I should know about ZYKADIA?
ZYKADIA may cause serious side effects, including: Stomach and intestinal (gastrointestinal) problems. ZYKADIA may cause stomach and intestinal problems, including diarrhea, nausea, vomiting, and stomach-area pain. Follow your healthcare provider’s instructions about taking medicines to help with these symptoms. Call your healthcare provider for advice if your symptoms are severe or cannot be tolerated. Liver problems. ZYKADIA may cause liver injury. Your healthcare provider should do blood tests at least every month to check your liver during treatment with ZYKADIA. Tell your healthcare provider right away if you get any of the following: |
|||||
| • you feel tired | • you have itchy skin | ||||
| • your skin or the whites of your eyes turn yellow | • you have nausea or vomiting | ||||
| • you have a decreased appetite | • you have pain on the right side of your stomach-area | ||||
| • your urine turns dark or brown (tea color) | • you bleed or bruise more easily than normal | ||||
| Lung problems (pneumonitis). ZYKADIA may cause severe or life-threatening inflammation of the lungs during treatment that can lead to death. Symptoms may be similar to those symptoms from lung cancer. Tell your healthcare provider right away if you have any new or worsening symptoms, including: | |||||
| • trouble breathing or shortness of breath | • cough with or without mucus | ||||
| • fever | • chest pain | ||||
| Heart problems. ZYKADIA may cause very slow, very fast, or abnormal heartbeats. Your healthcare provider may check your heart during treatment with ZYKADIA. Tell your healthcare provider right away if you feel new chest pain or discomfort, dizziness or lightheadedness, if you faint, or have abnormal heartbeats. Tell your healthcare provider if you start to take or have any changes in heart or blood pressure medicines. See "What are the possible side effects of ZYKADIA?" for more information about side effects. |
|||||
| What is ZYKADIA?
ZYKADIA is a prescription medicine that is used to treat people with non-small cell lung cancer (NSCLC) that:
|
|||||
Before you take ZYKADIA, tell your healthcare provider about all of your medical conditions, including if you:
|
|||||
How should I take ZYKADIA?
|
|||||
What should I avoid while taking ZYKADIA?
|
|||||
| What are the possible side effects of ZYKADIA?
ZYKADIA may cause serious side effects, including:
|
|||||
| • increased thirst | • increased hunger | • headaches | • trouble thinking or concentrating | ||
| • urinating often | • blurred vision | • tiredness | • your breath smells like fruit | ||
|
|||||
How should I store ZYKADIA?
|
|||||
| General information about the safe and effective use of ZYKADIA
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use ZYKADIA for a condition for which it was not prescribed. Do not give it to other people, even if they have the same symptoms you have. It may harm them. You can ask your healthcare provider or pharmacist for more information about ZYKADIA that is written for health professionals. |
|||||
| What are the ingredients in ZYKADIA?
Active ingredient: ceritinib Inactive ingredients capsules: colloidal silicon dioxide, hard gelatin capsule shells, low-substituted hydroxypropyl cellulose, magnesium stearate, microcrystalline cellulose, and sodium starch glycolate. Capsule shell contains FD&C Blue # 2, gelatin, and titanium dioxide. Inactive ingredients tablets: Tablet core: colloidal silicon dioxide, croscarmellose sodium, low-substituted hydroxypropyl cellulose, magnesium stearate, microcrystalline cellulose and povidone. Tablet coating: FD&C Blue # 2 aluminum lake, hypromellose, polyethylene glycol 4000, talc, and titanium dioxide. Distributed by: Novartis Pharmaceuticals Corporation, East Hanover, New Jersey 07936. For more information, go to www.US.ZYKADIA.com or call 1-888-669-6682. |
|||||
T2021-133
