| HIGHLIGHTS OF EMERGENCY USE AUTHORIZATION (EUA)
These highlights of the EUA do not include all the information needed to use BEBTELOVIMAB under the EUA. See the FULL FACT SHEET FOR HEALTHCARE PROVIDERS for BEBTELOVIMAB. BEBTELOVIMAB injection for intravenous use Original EUA Authorized Date: 02/2022 Revised EUA Authorized Date: 11/2022 ----------------------------RECENT MAJOR CHANGES-------------------------- Dosage and Administration, Dose Preparation and 03/2022 Administration (2.3): updated administration materials Use in Specific Populations, Pregnancy (8.1): added hypersensitivity reactions in pregnant women 05/2022 Clinical Pharmacology, Microbiology (12.4): updated 11/2022 neutralizing data -------------------EMERGENCY USE AUTHORIZATION-------------------- The U.S. Food and Drug Administration (FDA) has issued an Emergency Use Authorization (EUA) for the emergency use of bebtelovimab for the treatment of mild-to-moderate coronavirus disease 2019 (COVID-19) in adults and pediatric patients (12 years of age and older weighing at least 40 kg):
Bebtelovimab is authorized only for the duration of the declaration that circumstances exist justifying the authorization of the emergency use of bebtelovimab under section 564(b)(1) of the Act, 21 U.S.C. § 360bbb-3(b)(1), unless the authorization is terminated or revoked sooner. See Full Fact Sheet for Healthcare Providers for the justification for emergency use of drugs during the COVID-19 pandemic, information on available alternatives, and additional information on COVID-19. | ------------------------DOSAGE AND ADMINISTRATION-----------------------
The dosage in adults (18 years and older) and pediatric patients (≥12 years of age and weighing at least 40 kg) is bebtelovimab 175 mg administered as a single intravenous injection over at least 30 seconds. Administer bebtelovimab as soon as possible after positive results of direct SARS-CoV-2 viral testing and within 7 days of symptom onset. (2.1) ---------------------DOSAGE FORMS AND STRENGTHS---------------------- Injection: 175 mg/2 mL (87.5 mg/mL) in a single-dose vial. (3) -------------------------------CONTRAINDICATIONS------------------------------ No contraindications have been identified based on the limited available data for the emergency use of bebtelovimab authorized under this EUA. (4) ------------------------WARNINGS AND PRECAUTIONS-----------------------
|
| TABLE OF CONTENTS*
1 EMERGENCY USE AUTHORIZATION 2 DOSAGE AND ADMINISTRATION 2.1 Dosage 2.2 Dosage Adjustment in Specific Populations 2.3 Dose Preparation and Administration 3 DOSAGE FORMS AND STRENGTHS 4 CONTRAINDICATIONS 5 WARNINGS AND PRECAUTIONS 5.1 Hypersensitivity Including Anaphylaxis and Infusion-Related Reactions 5.2 Clinical Worsening After SARS-CoV-2 Monoclonal Antibody Administration 5.3 Limitations of Benefit and Potential for Risk in Patients with Severe COVID-19 6 ADVERSE REACTIONS 6.1 Adverse Reactions from Clinical Studies 6.4 Required Reporting for Serious Adverse Events and Medication Errors 7 DRUG INTERACTIONS 8 USE IN SPECIFIC POPULATIONS 8.1 Pregnancy 8.2 Lactation 8.4 Pediatric Use 8.5 Geriatric Use | 10 OVERDOSAGE
11 DESCRIPTION 12 CLINICAL PHARMACOLOGY 12.1 Mechanism of Action 12.2 Pharmacodynamics 12.3 Pharmacokinetics 12.4 Microbiology 13 NONCLINICAL TOXICOLOGY 13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility 13.2 Animal Toxicology and/or Pharmacology 14 CLINICAL STUDIES 14.1 Phase 2 Data from the Placebo-Controlled Portion of BLAZE-4 (Low Risk Subjects; Treatment Arms 9-11) 14.2 Phase 2 Data from the Randomized, Open-Label Portion of BLAZE-4 (High Risk Subjects; Treatment Arms 12-13) 14.3 Phase 2 Data from the Non-Randomized, Open-Label Portion of BLAZE-4 (High Risk Subjects; Treatment Arm 14) 14.4 Overall Benefit-Risk Assessment and Limitations of Data Supporting the Benefits of the Product 16 HOW SUPPLIED/STORAGE AND HANDLING 17 PATIENT COUNSELING INFORMATION 18 MANUFACTURER INFORMATION * Sections or subsections omitted from the EUA are not listed |
1 EMERGENCY USE AUTHORIZATION
The U.S. Food and Drug Administration (FDA) has issued an Emergency Use Authorization (EUA) for the emergency use of bebtelovimab for the treatment of mild-to-moderate coronavirus disease 2019 (COVID-19) in adults and pediatric patients (12 years of age and older weighing at least 40 kg):
- with positive results of direct SARS-CoV-2 viral testing, and
- who are at high risk1 for progression to severe COVID-19, including hospitalization or death, and
- for whom alternative COVID-19 treatment options approved or authorized by FDA are not accessible or clinically appropriate [see Clinical Studies (14.4)].
LIMITATIONS OF AUTHORIZED USE
- Bebtelovimab is not authorized for treatment of mild-to-moderate COVID-19 in geographic regions where infection is likely to have been caused by a non-susceptible SARS-CoV-2 variant based on available information including variant susceptibility to this drug and regional variant frequency.
- FDA's determination and any updates will be available at: https://www.fda.gov/emergency-preparedness-and-response/mcm-legal-regulatory-and-policy-framework/emergency-use-authorization#coviddrugs.2
- Bebtelovimab is not authorized for use in patients, who:
- are hospitalized due to COVID-19, OR
- require oxygen therapy and/or respiratory support due to COVID-19, OR
- require an increase in baseline oxygen flow rate and/or respiratory support due to COVID-19 and are on chronic oxygen therapy and/or respiratory support due to underlying non-COVID-19 related comorbidity.
Treatment with bebtelovimab has not been studied in patients hospitalized due to COVID-19. Monoclonal antibodies, such as bebtelovimab, may be associated with worse clinical outcomes when administered to hospitalized patients with COVID-19 requiring high flow oxygen or mechanical ventilation [see Warnings and Precautions (5.3)].
Bebtelovimab is not FDA-approved for any use, including for use as treatment of COVID-19 [see Emergency Use Authorization (1)].
Bebtelovimab is authorized only for the duration of the declaration that circumstances exist justifying the authorization of the emergency use of bebtelovimab under section 564(b)(1) of the Act, 21 U.S.C. § 360bbb-3(b)(1), unless the authorization is terminated or revoked sooner.
- 1
- For information on medical conditions and factors associated with increased risk for progression to severe COVID-19, see the Centers for Disease Control and Prevention (CDC) website: https://www.cdc.gov/coronavirus/2019-ncov/hcp/clinical-care/underlyingconditions.html. Healthcare providers should consider the benefit-risk for an individual patient.
- 2
- FDA will monitor conditions to determine whether use in a geographic region is consistent with this scope of authorization, referring to available information, including information on variant susceptibility [see Microbiology (12.4)], and CDC regional variant frequency data available at: https://covid.cdc.gov/covid-data-tracker/#variant-proportions.
Justification for Emergency Use of Drugs During the COVID-19 Pandemic
There is currently an outbreak of Coronavirus Disease 2019 (COVID-19) caused by SARS-CoV-2, a novel coronavirus. The Secretary of HHS has declared that:
- A public health emergency related to COVID-19 has existed since January 27, 2020.
- Circumstances exist justifying the authorization of emergency use of drugs and biological products during the COVID-19 pandemic (March 27, 2020 declaration).
An EUA is a FDA authorization for the emergency use of an unapproved product or unapproved use of an approved product (i.e., drug, biological product, or device) in the United States under certain circumstances including, but not limited to, when the Secretary of HHS declares that there is a public health emergency that affects the national security or the health and security of United States citizens living abroad, and that involves biological agent(s) or a disease or condition that may be attributable to such agent(s). Criteria for issuing an EUA include:
- The biological agent(s) can cause a serious or life-threatening disease or condition;
- Based on the totality of the available scientific evidence (including data from adequate and well-controlled clinical trials, if available), it is reasonable to believe that
- the product may be effective in diagnosing, treating, or preventing the serious or life-threatening disease or condition; and
- The known and potential benefits of the product - when used to diagnose, prevent, or treat such disease or condition - outweigh the known and potential risks of the product, taking into consideration the material threat posed by the biological agent(s);
- There is no adequate, approved, and available alternative to the product for diagnosing, preventing, or treating the serious or life-threatening disease or condition.
Information Regarding Available Alternatives for the EUA Authorized Use
Veklury (remdesivir) is FDA-approved for the treatment of COVID-19 in adults and pediatric patients (12 years of age and older weighing at least 40 kg) with positive results of direct SARS-CoV-2 viral testing, who are not hospitalized and have mild-to-moderate COVID-19, and who are at high risk for progression to severe COVID-19, including hospitalization or death. Veklury is administered via intravenous infusion for a total treatment duration of 3 days.
Although Veklury is an approved alternative treatment of mild-to-moderate COVID-19 in adults and pediatric patients (12 years of age and older weighing at least 40 kg) with positive results of direct SARS-CoV-2 viral testing, and who are at high risk for progression to severe COVID-19, including hospitalization or death, FDA does not consider Veklury to be an adequate alternative to bebtelovimab for this authorized use because it may not be feasible or practical for certain patients (e.g., it requires a 3-day treatment duration).
Other therapeutics are currently authorized for the same use as bebtelovimab. For additional information on all products authorized for treatment or prevention of COVID-19, please see https://www.fda.gov/emergency-preparedness-and-response/mcm-legal-regulatory-and-policy-framework/emergency-use-authorization.
For information on clinical studies of bebtelovimab and other therapies for the treatment of COVID-19, see www.clinicaltrials.gov.
2 DOSAGE AND ADMINISTRATION
2.1 Dosage
The dosage in adults (18 years and older) and pediatric patients (≥12 years of age and weighing at least 40 kg) is bebtelovimab 175 mg.
Administer bebtelovimab as soon as possible after positive results of direct SARS-CoV-2 viral testing and within 7 days of symptom onset.
Bebtelovimab must be administered as a single intravenous injection over at least 30 seconds.
2.2 Dosage Adjustment in Specific Populations
No dosage adjustment is recommended in pregnant or lactating individuals, in geriatrics, in individuals with renal impairment, or in individuals with mild hepatic impairment [see Clinical Pharmacology (12.3)].
2.3 Dose Preparation and Administration
General Information
- Bebtelovimab should be prepared by a qualified healthcare professional using aseptic technique.
- Inspect bebtelovimab vial visually for particulate matter and discoloration. Bebtelovimab is clear to opalescent and colorless to slightly yellow to slightly brown solution. Discard the vial if the solution is cloudy, discolored or visible particles are observed.
- Bebtelovimab may only be administered in settings in which healthcare providers have immediate access to medications to treat a severe infusion reaction, such as anaphylaxis, and the ability to activate the emergency medical system (EMS), as necessary.
- Clinically monitor patients for possible infusion-related reactions during administration and observe patients for at least 1 hour after injection is complete.
Materials Needed for Administration
- 1 bebtelovimab vial (175 mg/2 mL)
- 1 disposable polypropylene dosing syringe capable of holding 2 mL
- 0.9% Sodium Chloride Injection for flushing
- Optional: 1 syringe extension set made of polyethylene or polyvinylchloride with or without di-ethylhexylphthalate (DEHP)
Preparation
- Remove bebtelovimab vial from refrigerated storage and allow to equilibrate to room temperature for approximately 20 minutes before preparation. Do not expose to direct heat. Do not shake vial. Inspect the vial.
- Withdraw 2 mL from the vial into the disposable syringe.
- Discard any product remaining in the vial.
- This product is preservative-free and therefore, should be administered immediately.
- If immediate administration is not possible, store the syringe for up to 24 hours at refrigerated temperature (2°C to 8°C [36°F to 46°F]) and up to 7 hours at room temperature (20°C to 25°C [68°F to 77°F]). If refrigerated, allow the prepared syringe to equilibrate to room temperature for approximately 20 minutes prior to administration.
- If used, attach and prime the syringe extension set.
- Administer the entire contents of the syringe via IV injection over at least 30 seconds.
- After the entire contents of the syringe have been administered, flush the injection line with 0.9% Sodium Chloride to ensure delivery of the required dose.
3 DOSAGE FORMS AND STRENGTHS
Bebtelovimab is a sterile, preservative-free, clear to opalescent and colorless to slightly yellow to slightly brown solution available as:
- Injection: 175 mg/2 mL (87.5 mg/mL) in a single-dose vial
4 CONTRAINDICATIONS
No contraindications have been identified based on the limited available data for the emergency use of bebtelovimab authorized under this EUA.
5 WARNINGS AND PRECAUTIONS
There are limited clinical data available for bebtelovimab. Serious and unexpected adverse events may occur that have not been previously reported with bebtelovimab use.
5.1 Hypersensitivity Including Anaphylaxis and Infusion-Related Reactions
Serious hypersensitivity reactions, including anaphylaxis, have been observed with administration of other SARS-CoV-2 monoclonal antibodies and could occur with administration of bebtelovimab. If signs and symptoms of a clinically significant hypersensitivity reaction or anaphylaxis occur, immediately discontinue administration and initiate appropriate medications and/or supportive care.
Infusion-related reactions, which may occur up to 24 hours after the injection, have been observed in clinical trials of bebtelovimab when administered with other monoclonal antibodies and may occur with use of bebtelovimab alone. These reactions may be severe or life threatening.
Signs and symptoms of infusion-related reactions may include:
- fever, difficulty breathing, reduced oxygen saturation, chills, fatigue, arrhythmia (e.g., atrial fibrillation, sinus tachycardia, bradycardia), chest pain or discomfort, weakness, altered mental status, nausea, headache, bronchospasm, hypotension, hypertension, angioedema, throat irritation, rash including urticaria, pruritus, myalgia, vasovagal reactions (e.g., pre-syncope, syncope), dizziness and diaphoresis.
Administer appropriate medications and/or supportive care if an infusion-related reaction occurs.
Hypersensitivity reactions occurring more than 24 hours after the injection have also been reported with the use of SARS-CoV-2 monoclonal antibodies under Emergency Use Authorization.
5.2 Clinical Worsening After SARS-CoV-2 Monoclonal Antibody Administration
Clinical worsening of COVID-19 after administration of SARS-CoV-2 monoclonal antibody treatment has been reported and may include signs or symptoms of fever, hypoxia or increased respiratory difficulty, arrhythmia (e.g., atrial fibrillation, sinus tachycardia, bradycardia), fatigue, and altered mental status. Some of these events required hospitalization. It is not known if these events were related to SARS-CoV-2 monoclonal antibody use or were due to progression of COVID-19.
5.3 Limitations of Benefit and Potential for Risk in Patients with Severe COVID 19
Treatment with bebtelovimab has not been studied in patients hospitalized due to COVID-19. Monoclonal antibodies, such as bebtelovimab, may be associated with worse clinical outcomes when administered to hospitalized patients with COVID-19 requiring high flow oxygen or mechanical ventilation. Therefore, bebtelovimab is not authorized for use in patients, regardless of age, who:
- are hospitalized due to COVID-19, OR
- require oxygen therapy and/or respiratory support due to COVID-19, OR
- require an increase in baseline oxygen flow rate and/or respiratory support due to COVID-19 and are on chronic oxygen therapy and/or respiratory support due to underlying non-COVID-19 related comorbidity.
6 ADVERSE REACTIONS
6.1 Adverse Reactions from Clinical Studies
The following adverse reactions have been observed in the clinical studies of bebtelovimab that supported the EUA. The adverse reaction rates observed in these clinical studies cannot be directly compared to rates in the clinical studies of other products and may not reflect the rates observed in clinical practice. Additional adverse events associated with bebtelovimab may become apparent with more widespread use.
The safety of bebtelovimab is primarily based on exposure of 602 ambulatory (non-hospitalized) subjects who received doses of bebtelovimab, alone or in combination with bamlanivimab and etesevimab, in the phase 1 and phase 2 portions of BLAZE-4, a randomized, single-dose clinical trial.
The following adverse reactions (i.e., adverse events assessed as causally related) have been observed in those who have received bebtelovimab, alone or in combination with bamlanivimab and etesevimab, at the authorized dose or higher:
- Infusion-related reactions (n=2, 0.3%)
- Pruritus (n=2, 0.3%)
- Rash (n=5, 0.8%)
The most common treatment-emergent adverse events observed in subjects treated with bebtelovimab, alone or in combination with bamlanivimab and etesevimab, at the authorized dose or higher, included nausea (0.8%) and vomiting (0.7%).
6.4 Required Reporting for Serious Adverse Events and Medication Errors
The prescribing healthcare provider and/or the provider's designee is/are responsible for mandatory reporting of all serious adverse events* and medication errors potentially related to bebtelovimab within 7 calendar days from the healthcare provider's awareness of the event, using FDA Form 3500 (for information on how to access this form, see below). The FDA requires that such reports, using FDA Form 3500, include the following:
- Patient demographics and baseline characteristics (e.g., patient identifier, age or date of birth, gender, weight, ethnicity, and race)
- A statement "Bebtelovimab use for COVID-19 under Emergency Use Authorization (EUA)” under the “Describe Event, Problem, or Product Use/Medication Error” heading
- Information about the serious adverse event or medication error (e.g., signs and symptoms, test/laboratory data, complications, timing of drug initiation in relation to the occurrence of the event, duration of the event, treatments required to mitigate the event, evidence of event improvement/disappearance after stopping or reducing the dosage, evidence of event reappearance after reintroduction, clinical outcomes).
- Patient's preexisting medical conditions and use of concomitant products
- Information about the product (e.g., dosage, route of administration, NDC #).
Submit adverse event and medication error reports, using Form 3500, to FDA MedWatch using one of the following methods:
- Complete and submit the report online: www.fda.gov/medwatch/report.htm
- Complete and submit a postage-paid FDA Form 3500 (https://www.fda.gov/media/76299/download) and return by:
- Mail to MedWatch, 5600 Fishers Lane, Rockville, MD 20852-9787, or
- Fax to 1-800-FDA-0178, or
- Call 1-800-FDA-1088 to request a reporting form
In addition, please provide a copy of all FDA MedWatch forms to:
- Eli Lilly and Company, Global Patient Safety
- Fax: 1-317-277-0853
- E-mail: mailindata_gsmtindy@lilly.com
- Or call Eli Lilly and Company at 1-855-LillyC19 (1-855-545-5921) to report adverse events.
The prescribing health care provider and/or the provider's designee is/are responsible for mandatory responses to requests from FDA for information about adverse events and medication errors following receipt of bebtelovimab.
*Serious adverse events are defined as:
- Death;
- A life-threatening adverse event;
- Inpatient hospitalization or prolongation of existing hospitalization;
- A persistent or significant incapacity or substantial disruption of the ability to conduct normal life functions;
- A congenital anomaly/birth defect;
- Other important medical event, which may require a medical or surgical intervention to prevent death, a life-threatening event, hospitalization, disability, or congenital anomaly.
7 DRUG INTERACTIONS
Bebtelovimab is not renally excreted or metabolized by cytochrome P450 enzymes; therefore, interactions with concomitant medications that are renally excreted or that are substrates, inducers, or inhibitors of cytochrome P450 enzymes are unlikely.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Severe hypersensitivity reactions and infusion-related reactions, have been observed with administration of bebtelovimab, including in pregnant patients [see Warnings and Precautions (5.1)]. There are risks to the mother and fetus associated with untreated COVID-19 in pregnancy as well as potential risks to the fetus associated with severe maternal hypersensitivity and infusion-related reactions (see Clinical Considerations).
There are insufficient data to evaluate a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. Bebtelovimab should only be used during pregnancy if the potential benefit outweighs the potential risk for the mother and the fetus. There are maternal and fetal risks associated with untreated COVID-19 in pregnancy (see Clinical Considerations).
Data
Nonclinical reproductive toxicity studies have not been performed with bebtelovimab. In tissue cross reactivity studies using human fetal tissues, no binding of clinical concern was detected for bebtelovimab. Human immunoglobulin G1 (IgG1) antibodies are known to cross the placental barrier; therefore, bebtelovimab has the potential to be transferred from the mother to the developing fetus. It is unknown whether the potential transfer of bebtelovimab provides any treatment benefit or risk to the developing fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Clinical Considerations
Disease-associated maternal and/or embryo-fetal risk
COVID-19 in pregnancy is associated with adverse maternal and fetal outcomes, including preeclampsia, eclampsia, preterm birth, premature rupture of membranes, venous thromboembolic disease, and fetal death.
Maternal Adverse Reactions
Pregnant patients who develop severe hypersensitivity and infusion-related reactions should be managed appropriately, including obstetrical care [see Warnings and Precautions (5.1)].
8.2 Lactation
Risk Summary
There are no available data on the presence of bebtelovimab in human or animal milk, the effects on the breastfed infant, or the effects on milk production. Maternal IgG is known to be present in human milk. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for bebtelovimab and any potential adverse effects on the breastfed child from bebtelovimab or from the underlying maternal condition. Breastfeeding individuals with COVID-19 should follow practices according to clinical guidelines to avoid exposing the infant to COVID-19.
8.4 Pediatric Use
Bebtelovimab is not authorized for use in pediatric individuals under 12 years of age or weighing less than 40 kg. The safety and effectiveness of bebtelovimab have not been assessed in pediatric patients. The recommended dosing regimen in patients 12 years to less than 18 years of age, weighing at least 40 kg, is expected to result in comparable serum exposures of bebtelovimab as those observed in adults.
8.5 Geriatric Use
Of the 602 patients receiving bebtelovimab in BLAZE-4, 10.5% were 65 years of age and older and 3.3% were 75 years of age and older. Based on population PK analyses of samples from 573 patients over an age range of 14 to 89 years, there was no impact of age on PK. Therefore, there is no difference in the PK of bebtelovimab in geriatric patients compared to younger patients.
10 OVERDOSAGE
Doses up to 1750 mg of bebtelovimab (10 times the authorized dose of bebtelovimab) have been administered in clinical trials without dose-limiting toxicity. Treatment of overdose with bebtelovimab should consist of general supportive measures including monitoring of vital signs and observation of the clinical status of the patient. There is no specific antidote for overdose with bebtelovimab.
11 DESCRIPTION
Bebtelovimab is a human immunoglobulin G-1 (IgG1 variant) monoclonal antibody consisting of 2 identical light chain polypeptides composed of 215 amino acids each and 2 identical heavy chain polypeptides composed of 449 amino acids produced by a Chinese Hamster Ovary (CHO) stable bulk culture or cell line with a molecular weight of 144 kDa.
Bebtelovimab injection is a sterile, preservative-free, clear to opalescent and colorless to slightly yellow to slightly brown solution in a single-dose vial for intravenous injection.
Each mL contains 87.5 mg of bebtelovimab, L-histidine (0.4 mg), L-histidine hydrochloride monohydrate (0.6 mg), sodium chloride (2.9 mg), sucrose (60 mg), polysorbate 80 (0.5 mg), and Water for Injection. The bebtelovimab solution has a pH range of 5.5-6.5.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Bebtelovimab is a recombinant neutralizing human IgG1λ monoclonal antibody (mAb) to the spike protein of SARS-CoV-2 and is unmodified in the Fc region. Bebtelovimab binds the spike protein with a dissociation constant KD = 0.046 to 0.075 nM and blocks spike protein attachment to the human ACE2 receptor with an IC50 value of 0.39 nM (0.056 mcg/mL).
12.2 Pharmacodynamics
The exposure-response relationships of bebtelovimab for viral loads and clinical outcomes are unknown.
12.3 Pharmacokinetics
A summary of PK parameters of bebtelovimab following administration of a single dose of 175 mg bebtelovimab is provided in Table 1.
|
Abbreviations: CV = coefficient of variation; Cmax = maximum concentration; Cday,29 = drug concentration on day 29; AUCinf = area under the concentration versus time curve from zero to infinity; Vss = steady-state volume of distribution. |
|
| Bebtelovimab (175 mg)
N=585 |
|
| Systemic Exposure | |
| Geometric Mean (%CV) Cmax, mcg/mL | 59.9 (31.9) |
| Geometric Mean (%CV) Cday 29, mcg/mL | 4.55 (70.9) |
| Geometric Mean (%CV) AUCinf, mcg day/mL | 539 (41.5) |
| Distribution | |
| Geometric Mean (%CV) Vss (L) | 4.55 (25.8) |
| Elimination | |
| Geometric Mean (%CV) Elimination Half-Life (day) | 11.5 (27.0) |
| Geometric Mean (%CV) Clearance (L/day) | 0.325 (41.5) |
Specific Populations:
The PK profile of bebtelovimab was not affected by age, sex, race, or baseline viral load based on a population PK analysis. Body weight had no clinically relevant effect on the PK of bebtelovimab in adults with COVID-19 over the body weight range of 45 kg to 194 kg.
Patients with renal impairment
Renal impairment is not expected to impact the PK of bebtelovimab, since mAbs with molecular weight >69 kDa are known not to undergo renal elimination. Similarly, dialysis is not expected to impact the PK of bebtelovimab.
Patients with hepatic impairment
Bebtelovimab is expected to be degraded into small peptides and amino acids via catabolic pathways in the same manner as other IgG monoclonal antibodies and human endogenous IgG antibodies.
Based on population PK analysis, there is no significant difference in PK of bebtelovimab in patients with mild hepatic impairment compared to patients with normal hepatic function. Bebtelovimab has not been studied in patients with moderate or severe hepatic impairment.
12.4 Microbiology
Antiviral Activity
The cell culture neutralization activity of bebtelovimab against SARS-CoV-2 was measured in a dose-response model quantifying plaque reduction using cultured Vero E6 cells. Bebtelovimab neutralized the USA/WA/1/2020 isolate of SARS-CoV-2 with an estimated EC50 value = 0.044 nM (6.4 ng/mL).
Bebtelovimab demonstrated antibody-dependent cell-mediated cytotoxicity on Jurkat reporter cells expressing FcγRIIIa following engagement with target cells expressing spike protein. Bebtelovimab did not elicit complement-dependent cytotoxicity activity in cell-based assays.
Antibody Dependent Enhancement (ADE) of Infection
The risk that bebtelovimab could mediate viral uptake and replication by immune cells was studied in THP-1 and Raji cell lines and primary human macrophages. In general, experiments with bebtelovimab did not demonstrate productive viral infection in immune cells exposed to SARS-CoV-2 at concentrations of mAb down to 60,000-fold below the approximate EC50 value for neutralization.
Antiviral Resistance
There is a potential risk of treatment failure due to the development of viral variants that are resistant to bebtelovimab.
Nonclinical selection studies using a directed evolution of a yeast displayed Spike RBD identified that substitutions at residues K444, V445, G446, and P499 interfered with bebtelovimab's ability to block the Spike RBD:ACE-2 interaction. Pseudotyped virus-like particle (VLP) neutralization assays confirmed a 5-fold or greater reduction in susceptibility to bebtelovimab of viral variants with the following substitutions: K444E (>862), K444N (>1,901-fold), K444Q (208-fold), K444T (>1,814-fold), V445A (111-fold), V445F (369-fold), V445G (>730-fold), G446D (69-fold), G446R (7-fold), G446V (8-fold), P499H (>1,606-fold), P499R (>1,870-fold), and P499S (25-fold). In the context of Delta spike protein, G446V substitution had reduced susceptibility of 16.4-fold.
Pseudotyped VLP assessment using the full-length spike genes from different variant lineages indicate that bebtelovimab retains activity (<5-fold reduction) against the Alpha (B.1.1.7, UK origin), Beta (B.1.351, South Africa origin), Gamma (P.1, Brazil origin), Delta (B.1.617.2, India origin), Delta [+K417N] (AY.1/AY.2, India origin), Epsilon (B.1.427/B.1.429, California origin), Iota (B.1.526, New York origin), Kappa (B.1.617.1, India origin), Lambda (C.37, Peru origin), Omicron (B.1.1.529/BA.1, South Africa origin), Omicron [+R346K] (BA.1.1), Omicron BA.2, Omicron BA.2 [+L452Q] (BA.2.12.1), Omicron BA.2 [+D339H, G446S, N460K, R493Q (reversion)] (BA.2.75), Omicron BA.2 [BA.2.75+R346T+F486S] (BA.2.75.2), Omicron BA.4/BA.5, and Omicron BA.4 [+R346T] (BA.4.6/BF.7) variant lineages (Table 2). The Mu (B.1.621, Colombia origin) variant showed a reduction in susceptibility to bebtelovimab of 5.3-fold. The Omicron BA.5 [+N444T, N460K] (BQ.1), and Omicron BA.5 [+R346T, N444T, N460K] (BQ.1.1) variants showed a large reduction in susceptibility to bebtelovimab of >672-fold.
|
a Key substitutions occurring in the receptor binding domain of spike protein are listed. Pseudotyped VLP contained the full-length spike protein reflective of the consensus sequence for each of the variant lineages with the exception of BA.2.75.2 which is a full-length spike of BA.2.75+R346T+F486S substitutions. |
||||
|
b No change: <5-fold reduction in susceptibility. |
||||
|
c Isolates of the B.1.526 lineage harbor several spike protein amino acid substitutions, and not all isolates contain the E484K substitution (as of February 2021). |
||||
|
d Bebtelovimab is unlikely to be active against this variant. |
||||
| Lineage with Spike Protein Substitution | Country First Identified | WHO Nomenclature | Key Substitutions Testeda | Fold Reduction in Susceptibility |
| B.1.1.7 | UK | Alpha | N501Y | No changeb |
| B.1.351 | South Africa | Beta | K417N + E484K + N501Y | No changeb |
| P.1 | Brazil | Gamma | K417T + E484K + N501Y | No changeb |
| B.1.617.2/AY.3 | India | Delta | L452R + T478K | No changeb |
| AY.1/AY.2 (B.1.617.2 sublineages) | India | Delta [+K417N] | L452R + T478K + K417N | No changeb |
| B.1.427/B.1.429 | USA (California) | Epsilon | L452R | No changeb |
| B.1.526c | USA (New York) | Iota | E484K | No changeb |
| B.1.617.1 | India | Kappa | L452R + E484Q | No changeb |
| C.37 | Peru | Lambda | L452Q + F490S | No changeb |
| B.1.621 | Colombia | Mu | R346K + E484K + N501Y | 5.3 |
| B.1.1.529/BA.1 | South Africa | Omicron [BA.1] | G339D + S371L + S373P + S375F + K417N + N440K + G446S + S477N + T478K + E484A + Q493R + G496S + Q498R + N501Y + Y505H | No changeb |
| BA.1.1 | South Africa | Omicron [+R346K] | BA.1 + R346K | No changeb |
| BA.2 | South Africa | Omicron [BA.2] | G339D + S371F + S373P + S375F + T376A + D405N + R408S + K417N + N440K + S477N + T478K + E484A + Q493R + Q498R + N501Y + Y505H | No changeb |
| BA.2.12.1 | USA | Omicron [BA.2+L452Q] | BA.2 + L452Q | No changeb |
| BA.2.75 | India | Omicron [BA.2+D339H, G446S, N460K, R493Q (reversion)] | BA.2 + D339H + G446S + N460K + R493Q (reversion) | No changeb |
| BA.2.75.2 | India | Omicron [BA.2.75+R346T+F486S] | BA.2.75 + R346T + F486S | No changeb |
| BA.4/BA.5 | South Africa | Omicron [BA.4/BA.5] | G339D + S371F + S373P + S375F + T376A + D405N + R408S + K417N + N440K + L452R + S477N + T478K + E484A + F486V + Q498R + N501Y + Y505H | No changeb |
| BA.4.6/BF.7 | USA/Belgium | Omicron [BA.4+R346T] | BA.4 + R346T | No changeb |
| BQ.1 | Nigeria | Omicron [BA.5+K444T+N460K] | BA.5 + K444T + N460K | >672d |
| BQ.1.1 | Multiple | Omicron [BA.5+R346T+K444T+N460K] | BA.5 + R346T + K444T + N460K | >672d |
In authentic SARS-CoV-2 assays, bebtelovimab retained activity (<5-fold reduction) against variant virus isolates from the Alpha (B.1.1.7), Beta (B.1.351), Gamma (P.1), Delta (B.1.617.2/AY.3), Omicron (B.1.1.529/BA.1), Omicron [+R346K] (BA.1.1), Omicron BA.2, Omicron BA.2 [+L452Q] (BA.2.12.1), Omicron BA.2 [+D339H, G446S, N460K, R493Q (reversion)] (BA.2.75), Omicron BA.4, Omicron BA.4 [+R346T] (BA.4.6), and Omicron BA.5 lineages, as well as SARS-CoV-2 (USA/WA/1/2020 isolate) engineered to express the L452R substitution present in the Epsilon (B.1.427/B.1.429) lineage or the E484K substitution present in the Iota (B.1.526) lineage (Table 3).
|
a The B.1.1.7, B.1.351, B.1.617.2, B.1.1.529/BA.1, and BA.2 variants were assessed using cell culture-expanded virus isolates and tested using a plaque reduction assay; the B.1.351, P.1, B.1.617.2, B.1.1.529/BA.1, BA.1.1, BA.2, BA.2.12.1, BA.2.75, BA.4, BA.4.6, and BA.5 variants were assessed using cell culture-expanded isolates and tested using a microneutralization assay with a CPE-based endpoint titer to determine the IC>99; the B.1.526/E484K, B.1.427/B.1.429/L452R, and BA.2.75 spike substitutions were assessed using recombinant SARS-CoV-2 (USA/WA/1/2020 isolate with E484K, L452R, or full spike of BA.2.75) and tested using a plaque reduction assay. |
||||
|
b Key substitutions occurring in receptor binding domain of spike protein which are associated with each lineage. |
||||
|
c No change: <5-fold reduction in susceptibility when compared to ancestral control isolate using the same methodology. |
||||
|
d These viral variants have been tested with two different neutralization methodologies, both yielding <5-fold reductions in susceptibility. |
||||
|
e Isolates of the B.1.526 lineage harbor several spike protein amino acid substitutions, and not all isolates contain the E484K substitution (as of February 2021). |
||||
| Lineage with Spike Protein Substitution | Country First Identified | WHO Nomenclature | Key Substitutions Testedb | Fold Reduction in Susceptibility |
| B.1.1.7 | UK | Alpha | N501Y | No changec |
| B.1.351 | South Africa | Beta | K417N, E484K, N501Y | No changec,d |
| P.1 | Brazil | Gamma | K417T, E484K, N501Y | No changec |
| B.1.617.2/AY.3 | India | Delta | L452R, T478K | No changec,d |
| B.1.427/B.1.429 | USA (California) | Epsilon | L452R | No changec |
| B.1.526e | USA (New York) | Iota | E484K | No changec |
| B.1.1.529/BA.1 | South Africa | Omicron | G339D + S371L + S373P + S375F + K417N + N440K + G446S + S477N + T478K + E484A + Q493R + G496S + Q498R + N501Y + Y505H | No changec,d |
| BA.1.1 | South Africa | Omicron [+R346K] | BA.1 + R346K | No changec |
| BA.2 | South Africa | Omicron [BA.2] | G339D + S371F + S373P + S375F + T376A + D405N + R408S + K417N + N440K + S477N + T478K + E484A + Q493R + Q498R + N501Y + Y505H | No changec, d |
| BA.2.12.1 | USA | Omicron [BA.2+L452Q] | BA.2 + L452Q | No changec |
| BA.2.75 | India | Omicron [BA.2+D339H, G446S, N460K, R493Q (reversion)] | BA.2 + D339H + G446S + N460K + R493Q (reversion) | No changec, d |
| BA.4 | South Africa | Omicron [BA.4] | G339D + S371F + S373P + S375F + T376A + D405N + R408S + K417N + N440K + L452R + S477N + T478K + E484A + F486V + Q498R + N501Y + Y505H | No changec |
| BA.4.6 | USA | Omicron [BA.4+R346T] | BA.4 + R346T | No changec |
| BA.5 | South Africa | Omicron [BA.4/BA.5] | G339D + S371F + S373P + S375F + T376A + D405N + R408S + K417N + N440K + L452R + S477N + T478K + E484A + F486V + Q498R + N501Y + Y505H | No changec |
Genotypic analysis and phenotypic testing are ongoing to monitor for potential bebtelovimab-resistance-associated spike variations in clinical trials. Baseline sequencing data are available for 611 of the subjects in the BLAZE-4 (Arms 9-14) Study. Of these, 552 (90.3%) were infected with a variant of interest or concern, as designated by the WHO. No subject was infected with virus of the Omicron lineage or sub-lineages. The majority of subjects in the trial were infected with Delta (49.9%) and Alpha (28.6%). These were distributed across the treatment groups with Delta and Alpha infection rates of 60.2% and 23.1% in placebo, 31.3% and 41.8% in bebtelovimab alone arms, and 58.3% and 21.9% in the bebtelovimab with bamlanivimab and etesevimab arms, respectively. Gamma and Mu infections comprised 5.6% and 3.8% of the total infections respectively. Subjects infected with Beta, Delta [+K417N], Iota, and Lambda variants were the minority with 0.5%, 0.8%, 0.7%, and 0.5% total infections, respectively. All other subjects in the trial had SARS-CoV-2 infections from either non-WHO classified viruses (3.3%), or the lineage was not able to be determined based on the baseline sequence data (6.4%). Detection of viral variants with a 5-fold or greater reduction in susceptibility to bebtelovimab at baseline has been rare, with only one G446V substitution (8-fold shift) observed transiently out of 611 subjects in the BLAZE-4 (Arms 9-14) study that had baseline sequencing available (0.2%, 1/611).
Analysis of treatment-emergent variants focused on changes at amino acid positions with known phenotypically confirmed bebtelovimab-associated variations (i.e., K444, V445, G446, and P499) in serial viral samples obtained in the BLAZE-4 (Arms 9-14) bebtelovimab Phase 2 Study. Treatment-emergent substitutions detected at ≥15% or ≥50% allele fractions at these positions included K444E/N, V445G, G446V, and P499H/R. These substitutions resulted in a 5-fold or greater reduction in susceptibility to bebtelovimab in pseudotyped VLP assays: K444E (>862), K444N (>1,901-fold), V445G (>730-fold), G446V (8-fold), P499H (>1,606-fold), and P499R (>1,870-fold). Additional treatment-emergent substitutions detected at ≥15% or >50% allele fractions outside the epitope in at least 2 subjects included C379F (n=2) and G404C (n=2), seen in bebtelovimab in combination with bamlanivimab and etesevimab arms.
Considering all substitutions detected at ≥15% allele fraction at positions K444, V445, G446, and P499, 5.5% (11/199) of subjects treated with bebtelovimab alone harbored a variant that was treatment-emergent. This was more frequent than observed in the placebo arm (0%, 0/112), or when bebtelovimab was administered together with bamlanivimab and etesevimab (0.3%, 1/312). The appearance of these treatment-emergent bebtelovimab resistance-associated substitutions was associated with higher viral loads in the subjects in whom they were detected, but none of these subjects were hospitalized. The majority of the variants were first detected on Day 5 (n=3) and Day 7 (n=6) following treatment initiation.
It is possible that bebtelovimab resistance-associated variants could have cross-resistance to other mAbs targeting the receptor binding domain of SARS-CoV-2. The clinical impact is not known.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis, mutagenesis, and reproductive toxicology studies with bebtelovimab have not been conducted.
13.2 Animal Toxicology and/or Pharmacology
In toxicology studies, bebtelovimab had no adverse effects when administered intravenously to rats.
In tissue cross reactivity studies using human adult and fetal tissues, no binding of clinical concern was detected for bebtelovimab.
Antiviral Activity In Vivo
Prophylactic administration of bebtelovimab to male Syrian golden hamsters (n=5 to 8 per group) resulted in 2 to 4 log10 decreases in viral genomic RNA and viral replication (subgenomic RNA) from lung tissue, as well as decreases in lung weight and improvements in body weight compared to controls.
The applicability of these findings to a treatment setting is not known.
14 CLINICAL STUDIES
The data supporting this EUA for treatment of mild-to-moderate COVID-19 are primarily based on analyses of data from the Phase 2 portion of the BLAZE-4 trial (NCT04634409) that enrolled both low risk and high risk subjects (treatment arms 9-14). This trial evaluated the clinical efficacy data from subjects receiving 175 mg bebtelovimab alone and together with 700 mg bamlanivimab and 1,400 mg of etesevimab.
BLAZE-4 is a Phase 1/2, randomized, single-dose clinical trial evaluating treatment of subjects with mild-to-moderate COVID-19 (subjects with COVID-19 symptoms who are not hospitalized). Efficacy of bebtelovimab, alone and together with bamlanivimab and etesevimab, was evaluated in low risk adults (i.e., those not at high-risk to progress to severe COVID-19) in a randomized part of the trial which included a placebo control arm (treatment arms 9-11). Low risk adults were randomized with a 1:1:1 ratio. High-risk adults and pediatric subjects (12 years of age and older weighing at least 40 kg) received open-label active treatments. One cohort of high risk subjects was randomized with 2:1 ratio (treatment arms 12 and 13). Another cohort of high risk subject was enrolled with no randomization (treatment arm 14). The trial enrolled subjects who were not hospitalized and had 1 or more COVID-19 symptoms that were at least mild in severity. Treatment was initiated within 3 days of obtaining the clinical sample for the first positive SARS-CoV-2 viral infection determination.
BLAZE-4 was conducted prior to the emergence of the Omicron variant. No subject in BLAZE-4 was infected with virus of the Omicron lineage or sub-lineages. The majority of participants in the trial were infected with Delta (49.8%) and Alpha (28.6%).
14.1 Phase 2 Data from the Placebo-Controlled Portion of BLAZE-4 (Low Risk Subjects; Treatment Arms 9-11)
In this portion of the trial, adult subjects were treated with a single infusion of bamlanivimab 700 mg, etesevimab 1,400 mg, and bebtelovimab 175 mg (N=127), 175 mg bebtelovimab alone (N=125), or placebo (N=128). The majority (96.8%) of the subjects enrolled in these treatment arms did not meet the criteria for high-risk.
At baseline, median age was 35 years (with 1 placebo subject aged 65 or older); 56% of subjects were female, 79% were White, 36% were Hispanic or Latino, and 19% were Black or African American. Subjects had mild (74%) to moderate (26%) COVID-19; the mean duration of symptoms was 3.6 days; mean viral load by cycle threshold (CT) was 24.63 at baseline. The baseline demographics and disease characteristics were well balanced across treatment arms with the exception of baseline serology status. A higher percentage of subjects in the placebo arm were positive for baseline serology (15% vs. 9% for bamlanivimab, etesevimab, and bebtelovimab together, and 7% for bebtelovimab alone). Participants enrolled in these treatment arms had not received SARS-CoV-2 vaccine at baseline.
The primary endpoint was the proportion of subjects with persistently high viral load (PHVL) by Day 7. PHVL occurred in 26 subjects treated with placebo (21%) as compared to 16 (13%) subjects treated with bamlanivimab 700 mg, etesevimab 1,400 mg, and bebtelovimab 175 mg together [p=0.098], and 17 (14%) subjects treated with bebtelovimab 175 mg alone [p=0.147], a 38% (95% CI: -9%, 65%) and 34% (95% CI: -15%, 62%) relative reduction, respectively.
Secondary endpoints included mean change in viral load from baseline to Day 3, 5, 7, and 11 (Figure 1).
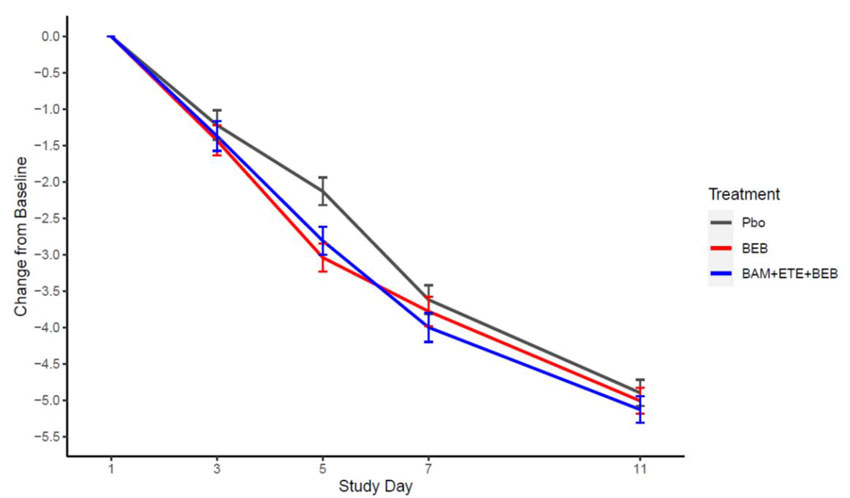
Figure 1: SARS-CoV-2 Viral Load Change from Baseline (Mean ± SE) by Visit from the Placebo-Controlled Portion of BLAZE-4 in Low Risk Adults (700 mg bamlanivimab, 1,400 mg etesevimab, 175 mg bebtelovimab together and 175 mg bebtelovimab alone).
For the secondary endpoint of COVID-19 related hospitalization (defined as ≥24 hours of acute care) or death by any cause by Day 29, these events occurred in 2 (1.6%) subjects treated with placebo as compared with 3 (2.4%) events in subjects treated with bamlanivimab 700 mg, etesevimab 1,400 mg, and bebtelovimab 175 mg together and 2 (1.6%) events in subjects treated with bebtelovimab 175 mg alone. There was 1 subject treated with bamlanivimab 700 mg, etesevimab 1,400 mg, and bebtelovimab 175 mg together who died on Day 5. Conclusions are limited as COVID-19 related hospitalization and death rates are expected to be low in a low risk population.
The median time to sustained symptom resolution as recorded in a trial specific daily symptom diary was 7 days (95%CI: 6, 8 days) for subjects treated with bamlanivimab 700 mg, etesevimab 1,400 mg, and bebtelovimab 175 mg together [p=0.289] and 6 days (95% CI: 5, 7 days) for subjects treated with bebtelovimab 175 mg alone [p=0.003] as compared with 8 days (95% CI: 7, 9 days) for subjects treated with placebo. Symptoms assessed were cough, shortness of breath, feeling feverish, fatigue, body aches and pains, sore throat, chills, and headache. Sustained symptom resolution was defined as absence of any of these symptoms, except for allowance of mild fatigue and cough, in two consecutive assessments.
14.2 Phase 2 Data from the Randomized, Open-Label Portion of BLAZE-4 (High Risk Subjects; Treatment Arms 12-13)
In this portion of the trial, subjects were treated with a single infusion of bamlanivimab 700 mg, etesevimab 1,400 mg, and bebtelovimab 175 mg (N=50) or 175 mg bebtelovimab alone (N=100). The majority (91.3%) of the subjects enrolled in these dose arms meet the criteria for high-risk.
At baseline, median age was 50 years (with 28 subjects aged 65 or older); 52% of subjects were female, 75% were White, 18% were Hispanic or Latino, and 18% were Black or African American. Subjects had mild (75%) to moderate (25%) COVID-19; the mean duration of symptoms was 4.7 days; mean viral load by cycle threshold (CT) was 26.66 at baseline; and 20.7% of subjects had at least one dose of a COVID-19 vaccine. There were 2 pediatric patients enrolled (ages 14 and 17), one in each treatment arm. The baseline demographics and disease characteristics were well balanced across treatment groups.
The primary objective for these treatment arms was to characterize the safety profile of bebtelovimab 175 mg by evaluating adverse events and serious adverse events. Efficacy endpoints included the proportion of subjects with COVID-19 related hospitalization or death by any cause by Day 29, mean change in viral load from baseline to Days 3, 5, 7, and 11 and time to sustained symptom resolution.
The proportion of subjects with COVID-19 related hospitalization (defined as ≥24 hours of acute care) or death by any cause was assessed by Day 29. Events occurred in 2 (4%) subjects treated with bamlanivimab 700 mg, etesevimab 1,400 mg, and bebtelovimab 175 mg together and 3 (3%) subjects treated with bebtelovimab 175 mg alone. There was 1 subject treated with bebtelovimab 175 mg alone who died on Day 34.
Mean changes in viral load from baseline to Day 3, 5, 7, and 11 are shown in Figure 2.
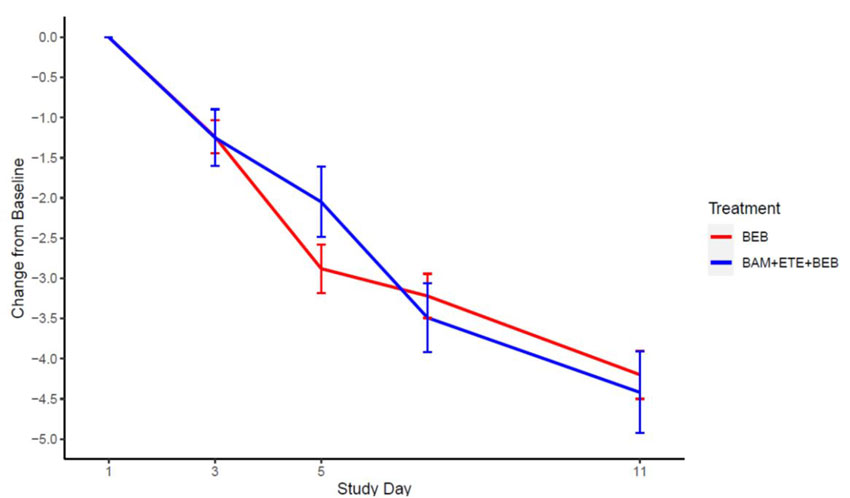
Figure 2: SARS-CoV-2 Viral Load Change from Baseline (Mean ± SE) by Visit from the Open-Label Portion of BLAZE-4 (700 mg bamlanivimab, 1,400 mg etesevimab, 175 mg bebtelovimab together and 175 mg bebtelovimab alone).
The median time to sustained symptom resolution as recorded in a trial specific daily symptom diary was 7 days for subjects treated with bebtelovimab 175 mg alone.
14.3 Phase 2 Data from the Non-Randomized, Open-Label Portion of BLAZE-4 (High Risk Subjects; Treatment Arm 14)
In this portion of the trial, subjects were treated with a single infusion of bamlanivimab 700 mg, etesevimab 1,400 mg, and bebtelovimab 175 mg (N=176). The majority (97.7%) of the subjects enrolled meet the criteria for high-risk.
At baseline, median age was 51 years (with 35 subjects aged 65 or older); 56% of subjects were female, 80% were White, 28% were Hispanic or Latino, and 16% were Black or African American. Subjects had mild (73%) to moderate (27%) COVID-19; the mean duration of symptoms was 4 days; mean viral load by cycle threshold (CT) was 23.45 at baseline; and 31% of subjects had at least one dose of a COVID-19 vaccine. There were 2 pediatric patients enrolled (ages 14 and 15).
The primary objective for this treatment arm was to characterize the safety profile of bamlanivimab 700 mg, etesevimab 1,400 mg, and bebtelovimab 175 mg by evaluating adverse events and serious adverse events. Efficacy endpoints included the proportion of subjects with COVID-19 related hospitalization or death by any cause by Day 29, mean change in viral load from baseline to Days 3, 5, 7, and 11, and time to sustained symptom resolution.
The proportion of subjects with COVID-19 related hospitalization (defined as ≥24 hours of acute care) or death by any cause was assessed by Day 29. Events occurred in 3 subjects (1.7%), and no subjects died.
Mean changes in viral load from baseline to Day 3, 5, 7, and 11 were -1.4, -3.1, -4.0, and -5.4, respectively.
The median time to sustained symptom resolution as recorded in a trial specific daily symptom diary was 8 days.
14.4. Overall Benefit-Risk Assessment and Limitations of Data Supporting the Benefits of the Product
Based on the data from BLAZE-4, bebtelovimab has been shown to improve symptoms in patients with mild-to-moderate COVID-19. Additionally, a reduction in SARS-CoV-2 viral load on Day 5 was observed relative to placebo, though the clinical significance of this is unclear. The placebo-controlled phase 2 data are limited by enrollment of only subjects without risk factors for progression to severe COVID-19, and the trial was not powered or designed to determine a difference in the clinical outcomes of hospitalization or death between the placebo and bebtelovimab treatment arms [see Clinical Studies (14.1)]. Bebtelovimab has been studied in individuals who have risk factors for progression to severe COVID-19, but the efficacy analyses are limited due to the lack of a concurrent placebo control arm for this population [see Clinical Studies (14.2, 14.3)].
However, based on the totality of scientific evidence available, including the available Phase 2 and pharmacokinetic data, along with the nonclinical viral neutralization data for Omicron and other variants of concern, it is reasonable to believe that bebtelovimab may be effective for the treatment of patients with mild-to-moderate COVID-19 to reduce the risk of progression to hospitalization or death. In addition, the mechanism of action for bebtelovimab is similar to other neutralizing SARS-CoV-2 monoclonal antibodies, including bamlanivimab and etesevimab, that have data from Phase 3 clinical trials showing a reduction in hospitalization or death in high risk patients infected with other SARS-CoV-2 variants. The safety profile of bebtelovimab is acceptable with monitorable risks and is comparable to other SARS-CoV-2 monoclonal antibodies, including bamlanivimab and etesevimab. Considered together, these data support that the known and potential benefits of treatment with bebtelovimab outweigh the known and potential risks in adults and pediatric patients (12 years of age and older weighing at least 40 kg) with positive results of direct SARS-CoV-2 viral testing and who are at high risk for progression to severe COVID-19, including hospitalization or death, and for whom alternative COVID-19 treatment options approved or authorized by FDA are not accessible or clinically appropriate.
Clinical data summarized above were similar for bebtelovimab alone as compared to the combination of bamlanivimab, etesevimab and bebtelovimab administered together. Bebtelovimab retains activity against currently circulating variants [see Microbiology (12.4)].
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
As a healthcare practitioner, you must communicate to the patient and/or caregiver information consistent with the “FACT SHEET FOR PATIENTS, PARENTS AND CAREGIVERS” and provide them with a copy of this Fact Sheet prior to administration of bebtelovimab. However, if providing this information will delay the administration of bebtelovimab to a degree that would endanger the life of a patient, the information must be provided to the parent and/or caregiver as soon as feasible after bebtelovimab administration.
Remind patients treated with bebtelovimab that they should continue to self-isolate and use infection control measures (e.g., wear mask, isolate, social distance, avoid sharing personal items, clean and disinfect “high touch” surfaces, and frequent handwashing) according to CDC guidelines.
For additional information visit: www.LillyAntibody.com/bebtelovimab
If you have questions, please contact: 1-855-LillyC19 (1-855-545-5921)
18 MANUFACTURER INFORMATION
Eli Lilly and Company, Indianapolis, IN 46285, USA
Copyright © 2022, Eli Lilly and Company. All rights reserved.
Literature revised November 4, 2022
BEB-0009-EUA HCP-20221104
Fact Sheet for Patients, Parents, and Caregivers
Emergency Use Authorization (EUA) of Bebtelovimab for Coronavirus Disease 2019
(COVID-19)
You are being given this Fact Sheet because your healthcare provider believes it is necessary to provide you or your child with bebtelovimab for the treatment of mild-to-moderate coronavirus disease 2019 (COVID-19) in adults and children (12 years of age and older weighing at least 88 pounds [40 kg]) with positive results of direct severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) viral testing, and who are at high risk for progression to severe COVID-19, including hospitalization or death, and for whom other COVID-19 treatment options approved or authorized by FDA are not available or clinically appropriate. This Fact Sheet contains information to help you understand the potential risks and potential benefits of receiving bebtelovimab, which you or your child have received or may receive.
The U.S. Food and Drug Administration (FDA) has issued an Emergency Use Authorization (EUA) to make bebtelovimab available during the COVID-19 pandemic (for more details about an EUA please see “What is an Emergency Use Authorization?” at the end of this document). Bebtelovimab is not an FDA-approved medicine in the United States. Read this Fact Sheet for information about bebtelovimab. Talk to your healthcare provider about your options or if you have any questions. It is your choice for you or your child to receive bebtelovimab or stop it at any time.
What is COVID-19?
COVID-19 is caused by a virus called a coronavirus (SARS-CoV-2). You can get COVID-19 through contact with another person who has the virus.
COVID-19 illnesses have ranged from very mild (including some with no reported symptoms) to severe, including illness resulting in death. While information so far suggests that most COVID-19 illness is mild, serious illness can happen and may cause some of your or your child's other medical conditions to become worse. Older people and people of all ages with severe, or long lasting (chronic) medical conditions like heart disease, lung disease, diabetes, and obesity, for example, seem to be at higher risk of being hospitalized for COVID-19. Older age, with or without other conditions, also places people at higher risk of being hospitalized for COVID-19.
What is bebtelovimab?
Bebtelovimab is an investigational medicine used for the treatment of mild-to-moderate coronavirus disease 2019 (COVID-19) in adults and children (12 years of age and older weighing at least 88 pounds [40 kg]):
- with positive results of direct SARS-CoV-2 viral testing, and
- who are at high risk3 for progression to severe COVID-19, including hospitalization or death, and
- for whom other COVID-19 treatment options approved or authorized by FDA are not available or clinically appropriate.
There is limited information known about the safety and effectiveness of using bebtelovimab for the treatment of mild-to-moderate COVID-19.
For more information on EUA, see the “What is an Emergency Use Authorization (EUA)?” section at the end of this Fact Sheet.
Bebtelovimab is not authorized for use in people who:
- are likely to be infected with a SARS-CoV-2 variant that is not able to be treated by bebtelovimab based on the circulating variants in your area (ask your health care provider about FDA and CDC's latest information on circulating variants by geographic area), or
- are hospitalized due to COVID-19, or
- require oxygen therapy and/or respiratory support due to COVID-19, or
- require an increase in baseline oxygen flow rate and/or respiratory support due to COVID-19 and are on chronic oxygen therapy and/or respiratory support due to underlying non-COVID-19 related comorbidity.
What should I tell my healthcare provider before I or my child receive bebtelovimab?
Tell your healthcare provider about all your or your child's medical conditions including if you or your child:
- Have any allergies
- Are pregnant or plan to become pregnant
- Are breastfeeding or plan to breastfeed
- Have any serious illnesses
- Are taking any medicines (prescription, and over-the-counter, vitamins, or herbal products)
How will I or my child receive bebtelovimab?
Bebtelovimab will be given as an injection through a vein (intravenously or IV) over at least 30 seconds. You will be observed by your healthcare provider for at least 1 hour after you receive bebtelovimab.
What are the important possible side effects of bebtelovimab?
- Allergic reactions. Allergic reactions can happen during and after injection with bebtelovimab. Tell your healthcare provider right away if you or your child develop any of the following signs and symptoms of allergic reaction: fever, difficulty breathing, low oxygen level in your blood, chills, tiredness, fast or slow heart rate, chest discomfort or pain, weakness, confusion, nausea, headache, shortness of breath, low or high blood pressure, wheezing, swelling of your lips, face, or throat, rash including hives, itching, muscle aches, dizziness, feeling faint, and sweating. These reactions may be severe or life threatening.
The side effects of receiving any medicine by vein may include brief pain, bleeding, bruising of the skin, soreness, swelling, and possible infection at the injection site.
These are not all the possible side effects of bebtelovimab. Not many people have received bebtelovimab. Serious and unexpected side effects may happen. All of the risks are not known at this time.
It is possible that bebtelovimab could interfere with your body's own ability to fight off a future infection of SARS-CoV-2. Similarly, bebtelovimab may reduce the body's immune response to a vaccine for SARS-CoV-2. Talk to your healthcare provider if you have any questions.
What other treatment choices are there?
Veklury (remdesivir) is FDA-approved for the treatment of mild-to-moderate COVID-19 in certain adults and pediatric patients. Talk with your doctor to see if Veklury is appropriate for you.
Like bebtelovimab, FDA may allow for the emergency use of other medicines to treat people with COVID-19. Go to https://www.fda.gov/emergency-preparedness-and-response/mcm-legal-regulatory-and-policy-framework/emergency-use-authorization for information on the emergency use of other medicines that are authorized by FDA to treat people with COVID-19. Your healthcare provider may talk with you about clinical trials for which you may be eligible.
It is your choice for you or your child to be treated or not to be treated with bebtelovimab. Should you decide not to receive it or for your child to not receive it, it will not change your or your child's standard medical care.
What if I am pregnant or breastfeeding?
There is limited experience treating pregnant women or breastfeeding mothers with bebtelovimab. Severe allergic reactions have been observed with administration of bebtelovimab, including in pregnant patients.
For a mother and unborn baby, the benefit of receiving bebtelovimab may be greater than the risk from the treatment. If pregnant or breastfeeding, discuss your options and specific situation with your healthcare provider.
How do I report side effects with bebtelovimab?
Contact your healthcare provider if you have any side effects that bother you or do not go away.
Report side effects to FDA MedWatch at www.fda.gov/medwatch, or call 1-800-FDA-1088 or to Eli Lilly and Company, Inc. as shown below.
| Fax Number | Telephone Number |
| 1-317-277-0853 | 1-855-LillyC19 (1-855-545-5921) |
How can I learn more about COVID-19?
- Ask your healthcare provider
- Visit https://www.cdc.gov/COVID19
- Contact your local or state public health department
What is an Emergency Use Authorization?
The United States FDA has made bebtelovimab available under an emergency access mechanism called an Emergency Use Authorization (EUA). The EUA is supported by a Secretary of Health and Human Service (HHS) declaration that circumstances exist to justify the emergency use of drugs and biological products during the COVID-19 pandemic.
Bebtelovimab for the treatment of mild-to-moderate COVID-19 in adults and children (12 years of age and older weighing at least 88 pounds [40 kg]) and who are at high risk of developing severe COVID-19, including hospitalization or death, and for whom other COVID-19 treatment options approved or authorized by FDA are not available or clinically appropriate has not undergone the same type of review as an FDA-approved product. In issuing an EUA under the COVID-19 public health emergency, the FDA has determined, among other things, that based on the total amount of scientific evidence available, including data from adequate and well-controlled clinical trials, it is reasonable to believe that the product may be effective for diagnosing, treating, or preventing COVID-19, or a serious or life-threatening disease or condition caused by COVID-19; that the known and potential benefits of the product, when used to diagnose, treat, or prevent such disease or condition, outweigh the known and potential risks of such product; and that there are no adequate, approved and available alternatives.
All of these criteria must be met to allow for the product to be used in the treatment of patients during the COVID-19 pandemic. The EUA for bebtelovimab is in effect for the duration of the COVID-19 declaration justifying emergency use of bebtelovimab, unless terminated or revoked (after which bebtelovimab may no longer be used under the EUA).
Additional Information
For general questions, visit the website or call the telephone number provided below.
| Website | Telephone Number |
| www.LillyAntibody.com/bebtelovimab | 1-855-LillyC19 (1-855-545-5921) |
Literature revised November 4, 2022
Eli Lilly and Company, Indianapolis, IN 46285, USA
Copyright © 2022, Eli Lilly and Company. All rights reserved.
BEB-0003-EUA PAT-20221104
- 3
- For information on medical conditions and factors associated with increased risk for progression to severe COVID-19, see the Centers for Disease Control and Prevention (CDC) website: https://www.cdc.gov/coronavirus/2019-ncov/need-extra-precautions/people-with-medical-conditions.html. Healthcare providers should consider the benefit-risk for an individual patient.
