FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
SUSTOL is indicated in combination with other antiemetics in adults for the prevention of acute and delayed nausea and vomiting associated with initial and repeat courses of moderately emetogenic chemotherapy (MEC) or anthracycline and cyclophosphamide (AC) combination chemotherapy regimens.
2 DOSAGE AND ADMINISTRATION
2.1 Important Administration Instructions
- For subcutaneous injection only.
- SUSTOL is intended for administration by a health care provider.
- SUSTOL is supplied as a refrigerated kit consisting of a single-dose, pre-filled, sterile syringe, a special thin walled 18 Ga 5/8" administration needle, two syringe warming pouches, and a Point Lok® needle protection device. See the SUSTOL Instructions for Use included in the kit for complete administration instructions with illustrations.
- Do not substitute non-kit components for any of the components from the kit for administration.
Preparation
- At least 60 minutes prior to administration, remove the SUSTOL kit from refrigeration.
- Unpack the kit to allow the SUSTOL syringe and all other contents to warm to room temperature.
- Activate one of the syringe warming pouches, and wrap the SUSTOL syringe in the warming pouch for 5 to 6 minutes to warm SUSTOL to body temperature.
- Prior to administration, inspect the SUSTOL syringe visually for particulate matter and discoloration. Note that the syringe is amber colored glass. SUSTOL should not be administered if particulate matter or discoloration is observed, the tip cap is missing or has been tampered with, or if the Luer fitting is missing or dislodged.
Administration
- Use standard aseptic technique when performing the injection.
- Administer SUSTOL as a single subcutaneous injection in the skin of the back of the upper arm or in the skin of the abdomen at least one inch away from the umbilicus. Avoid injecting SUSTOL anywhere the skin is burned, hardened, inflamed, swollen, or otherwise compromised [see Warnings and Precautions (5.1)]. Topical anesthetic may be used at the injection site prior to administration of SUSTOL.
- Due to the viscosity of SUSTOL, the time required for injection is greater than most medications administered subcutaneously. SUSTOL requires a slow, sustained injection which may take up to 20 to 30 seconds. Pressing the plunger harder will NOT expel SUSTOL faster.
2.2 Recommended Dosage
The recommended dosage of SUSTOL is 10 mg administered subcutaneously. Administer SUSTOL in combination with dexamethasone at least 30 minutes before the initiation of MEC or AC combination chemotherapy. Administer SUSTOL on Day 1 of chemotherapy and not more frequently than once every 7 days because of the extended-release properties of the formulation.
For patients receiving MEC, the recommended dexamethasone dosage is 8 mg intravenously on Day 1. For patients receiving AC combination chemotherapy regimens, the recommended dexamethasone dosage is 20 mg intravenously on Day 1, followed by 8 mg orally, twice a day, on Days 2, 3 and 4.
If SUSTOL is administered with an NK1 receptor antagonist, see the prescribing information of the NK1 receptor antagonist for the recommended dexamethasone dosage.
2.3 Dosage Adjustment in Renal Impairment
In patients with moderate renal impairment (creatinine clearance of 30 to 59 mL/min), administer SUSTOL on Day 1 of chemotherapy and not more frequently than once every 14 days. Avoid SUSTOL in patients with severe renal impairment (creatinine clearance of less than 30 mL/min) [see Use in Specific Populations (8.6)].
3 DOSAGE FORMS AND STRENGTHS
SUSTOL is supplied as a clear, colorless to slightly yellow, viscous liquid and is available as an:
- Extended-Release Injection: 10 mg/0.4 mL in a single-dose pre-filled syringe.
4 CONTRAINDICATIONS
SUSTOL is contraindicated in patients with hypersensitivity to granisetron, any of the components of SUSTOL, or to any of the other 5-HT3 receptor antagonists [see Warnings and Precautions (5.3), Description (11)].
5 WARNINGS AND PRECAUTIONS
5.1 Injection Site Reactions (ISRs)
Infections
Infections at the injection site occurred in 0.4% (7 of 1814) of patients with cancer and 0.2% (1 of 412) of healthy subjects in clinical trials. Infections had a median onset of 9 days (range 7 to 16 days) following SUSTOL administration. One patient who was neutropenic at the time of the infection was hospitalized. All patients with infection were treated with antibiotics and had complete resolution.
Bruising and/or hematomas
Bruising and/or hematomas at the injection site occurred in 426 of 1131 (38%) patients treated with SUSTOL 10 mg with a median time to onset of 2 days. Bruising and/or hematomas with a delayed onset (onset 5 or more days following SUSTOL administration) were reported in 175 (15%) patients. Severe bruising or hematoma (e.g., greater than 4 cm bruise or hematoma) occurred in 3% of patients. Patients receiving concomitant anticoagulant and antiplatelet medications were at greater risk for severe injection site bruising and hematomas.
Bleeding
Bleeding at the injection site occurred in 70 of 1814 (4%) patients treated with SUSTOL. One patient required emergency management. Bleeding for longer than 5 days was reported in 23 (1%) patients.
Pain and Tenderness
In a clinical trial that collected information about injection site pain and tenderness from patient diaries, pain with or without tenderness at the injection site was reported by 91 of 456 (20%) of patients treated with SUSTOL 10 mg, and an additional 50 of 456 (11%) of patients reported tenderness without pain. Pain and/or tenderness severe enough to require taking pain medication, interfere with patient activity level, or cause significant discomfort at rest was reported in 2% of patients. Among all patients who reported pain and/or tenderness with SUSTOL 10 mg in clinical trials, the median duration was 5 days, and pain lasting longer than 7 days occurred in 6% of patients.
Nodules
Nodules at the injection site occurred in 203 of 1131 (18%) of patients treated with SUSTOL 10 mg. Nodules persisted for a median of 15 days and 73 patients (6%) had nodules with durations longer than 21 days.
Management of ISRs
- Monitor patients for ISRs following SUSTOL injection. Some ISRs (infections, bruising, and hematoma) may occur up to 2 weeks or more after SUSTOL administration.
- In patients receiving antiplatelet agents or anticoagulants, consider the increased risk of bruising or severe hematoma prior to the use of SUSTOL.
- In patients with ongoing or unresolved ISRs, administer SUSTOL at a site away from areas affected by ISRs [see Dosage and Administration (2.1)].
5.2 Gastrointestinal Disorders
Constipation
In clinical trials, 224 of 1131 (20%) of patients treated with SUSTOL 10 mg reported constipation compared to 13% to 15% in the 5-HT3 receptor antagonist control arms. Hospitalization due to constipation or fecal impaction was reported in 5 SUSTOL-treated patients (0.3%). Monitor patients for the development of constipation while receiving treatment with SUSTOL taking into consideration the extended-release properties of the SUSTOL polymer formulation over at least 5 to 7 days, particularly in patients receiving opioid medications. Consider optimizing bowel regimens in patients using SUSTOL.
Progressive Ileus and Gastric Distention
SUSTOL may mask a progressive ileus and/or gastric distention. This should be particularly considered before use of SUSTOL in patients who have had recent abdominal surgery. Monitor for decreased bowel activity, particularly in patients with risk factors for gastrointestinal obstruction.
5.3 Hypersensitivity Reactions
Hypersensitivity reactions, including anaphylaxis, have been reported in granisetron-treated patients who have exhibited hypersensitivity to other 5-HT3 receptor antagonists [see Contraindications (4)]. Avoid SUSTOL in patients who have had hypersensitivity reactions to other 5-HT3 receptor antagonists [see Contraindications (4)].
Due to the extended-release properties of the SUSTOL polymer formulation, exposure to granisetron may continue for 5 to 7 days following administration. Hypersensitivity reactions may occur up to 7 days or longer following SUSTOL administration and may have an extended course. Inform patients of the signs and symptoms of anaphylaxis, and instruct them to seek immediate medical care should signs and symptoms occur. If hypersensitivity reactions occur, administer appropriate treatment and monitor patients until signs and symptoms resolve.
5.4 Serotonin Syndrome
The development of serotonin syndrome has been reported with 5-HT3 receptor antagonists. Most reports have been associated with concomitant use of serotonergic drugs (e.g., selective serotonin reuptake inhibitors (SSRIs), serotonin and norepinephrine reuptake inhibitors (SNRIs), monoamine oxidase inhibitors, mirtazapine, fentanyl, lithium, tramadol, and intravenous methylene blue). Some of the reported cases were fatal. Serotonin syndrome occurring with overdose of another 5-HT3 receptor antagonist alone has also been reported. The majority of reports of serotonin syndrome related to 5-HT3 receptor antagonist use occurred in a post-anesthesia care unit or an infusion center.
Symptoms associated with serotonin syndrome may include the following combination of signs and symptoms: mental status changes (e.g., agitation, hallucinations, delirium, and coma), autonomic instability (e.g., tachycardia, labile blood pressure, dizziness, diaphoresis, flushing, hyperthermia), neuromuscular symptoms (e.g., tremor, rigidity, myoclonus, hyperreflexia, incoordination), seizures, with or without gastrointestinal symptoms (e.g., nausea, vomiting, diarrhea). Patients should be monitored for the emergence of serotonin syndrome, especially with concomitant use of SUSTOL and other serotonergic drugs. If symptoms of serotonin syndrome occur, discontinue SUSTOL and initiate supportive treatment. Patients should be informed of the increased risk of serotonin syndrome, especially if SUSTOL is used concomitantly with other serotonergic drugs [see Drug Interactions (7.1)].
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Injection Site Reactions [see Warnings and Precautions (5.1)]
- Gastrointestinal Disorders [see Warnings and Precautions (5.2)]
- Hypersensitivity Reactions [see Warnings and Precautions (5.3)]
- Serotonin Syndrome [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
SUSTOL
The safety of a 10 mg subcutaneous dose of SUSTOL was evaluated in two double-blind, randomized, active-controlled studies, in which 210 patients (23%) received MEC and 467 patients (51%) received AC combination chemotherapy. The data described below reflect exposure to a single 10 mg dose of SUSTOL in 924 patients whose mean age was 56 years (range 19 to 91 years); 76% of patients were female; 70% of patients were Caucasian, 16% Asian, 10% Black, and 4% other races. Dexamethasone was co-administered with SUSTOL in Study 1 and Study 2 and an NK1 receptor antagonist was co-administered with SUSTOL in Study 2.
Table 1 lists the most common adverse reactions reported in at least 3% of patients following a single-dose of SUSTOL 10 mg in Study 1 and/or Study 2. Overall, injection site reactions (ISRs) were the most common group of adverse reactions in SUSTOL-treated patients. Specific types of ISRs reported by SUSTOL-treated patients are shown in Table 2.
| Study 1 | Study 2 | |||
|---|---|---|---|---|
| Adverse Reaction | SUSTOL 10 mg subcutaneous (N=468) % | Palonosetron hydrochloride 0.25 mg intravenous (N=463) % | SUSTOL 10 mg subcutaneous (N=456) % | Ondansetron 0.15 mg/kg intravenous (N=459) % |
| Injection Site Reactions, any* | 37 | 15† | 62 | See footnote† |
| Constipation | 14 | 11 | 22 | 15 |
| Fatigue | 11 | 10 | 21 | 24 |
| Headache | 9 | 9 | 13 | 19 |
| Diarrhea | 8 | 7 | 9 | 8 |
| Abdominal Pain | 7 | 7 | 7 | 4 |
| Insomnia | 4 | 2 | 5 | 6 |
| Dyspepsia | 3 | 3 | 6 | 7 |
| Dizziness | 3 | 2 | 5 | 5 |
| Asthenia | 4 | 6 | 2 | 2 |
| Gastroesophageal Reflux | 1 | 1 | 5 | 4 |
Injection Site Reactions (ISRs)
Injection site reactions occurred in 37% (175/468) in Study 1, Cycle 1 only, and 62% (281/456) in Study 2 of SUSTOL-treated patients. The ISR manifestations included pain, erythema, mass/nodule, swelling/induration, and bleeding. The incidence of individual ISRs is shown in Table 2. Patients may have experienced one or more types of injection site reactions; a total of 213 of 924 patients had three or more.
ISR reporting procedures included both investigator- and patient-reported outcomes in Study 2, while Study 1 used only investigator reporting.
| Injection Site Reaction | Study 1 Treatment Arm (Subcutaneous Injection) | Study 2*,†
SUSTOL (N=456) % |
|
|---|---|---|---|
| SUSTOL (N=468) % | Saline Control (N=463) % |
||
|
|||
| Total Subjects with at least 1 ISR | 37 | 15 | 62 |
| Pain | 3 | 1 | 20 |
| Tenderness | 4 | 1 | 27 |
| Bruising/Hematoma | 22 | 10 | 45 |
| Bleeding | 2 | 1 | 4 |
| Erythema/Redness | 11 | 3 | 17 |
| Swelling/Induration | 1 | 0 | 10 |
| Mass/Nodule | 11 | 1 | 18 |
| Infection at injection site | <1 | 0 | 1 |
| Other‡ | 2 | 1 | 1 |
Less common adverse reactions reported in less than 3% of SUSTOL-treated patients in clinical trials are syncope, elevation of serum transaminase levels, pancreatitis, atrial fibrillation, somnolence, flushing, and hypersensitivity reactions (e.g., anaphylaxis, urticaria).
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of other formulations of granisetron. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
| System Organ Class | Adverse Reactions |
|---|---|
| Cardiovascular | bradycardia, chest pain, palpitations, sick sinus syndrome |
7 DRUG INTERACTIONS
7.1 Serotonergic Drugs
Serotonin syndrome (including altered mental status, autonomic instability, and neuromuscular symptoms) has been described following the concomitant use of 5-HT3 receptor antagonists and other serotonergic drugs, including selective serotonin reuptake inhibitors (SSRIs) and serotonin and noradrenaline reuptake inhibitors (SNRIs). Monitor for the emergence of serotonin syndrome. If symptoms occur, discontinue SUSTOL and initiate supportive treatment [see Warnings and Precautions (5.4)].
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no available data on the use of SUSTOL in pregnant women. Limited published data on granisetron use during pregnancy are not sufficient to inform a drug-associated risk. In animal reproduction studies, no adverse developmental effects were observed in pregnant rats and rabbits administered granisetron hydrochloride during organogenesis at intravenous doses up to 61 times and 41 times respectively the maximum recommended human dose (MRHD) of SUSTOL 10 mg/week [see Data].
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Animal Data
Reproduction studies with granisetron hydrochloride have been performed in pregnant rats following administration during the period of organogenesis at intravenous doses up to 9 mg/kg/day (approximately 61 times the maximum recommended human dose (MRHD) of SUSTOL 10 mg/week, based on body surface area) and oral doses up to 125 mg/kg/day (approximately 851 times the MRHD of SUSTOL 10 mg/week, based on body surface area). Reproduction studies have been performed in pregnant rabbits in which granisetron hydrochloride was administered during the period of organogenesis at intravenous doses up to 3 mg/kg/day (approximately 41 times the MRHD of SUSTOL 10 mg/week, based on body surface area) and at oral doses up to 32 mg/kg/day (approximately 436 times the MRHD of SUSTOL 10 mg/week, based on body surface area). These studies did not reveal any evidence of impaired fertility or harm to the fetus due to granisetron hydrochloride.
Reproduction studies with the polymer vehicle for SUSTOL have been performed in pregnant rats and rabbits following administration of the polymer vehicle during the period of organogenesis at subcutaneous doses up to 0.295 and 1.18 g per day, respectively, (approximately 45 and 36 times, respectively the amount of polymer vehicle present in the maximum recommended /weekly single human dose of SUSTOL, based on body surface area). These studies did not reveal any evidence of impaired fertility or harm to the fetus due to the polymer vehicle. A pre and postnatal development study with the polymer vehicle for SUSTOL in rats showed no evidence of any adverse effects on pre and postnatal development at subcutaneous doses (administered on gestation days 7 through lactation day 20) up to 0.295 g per day (approximately 45 times the amount of polymer vehicle present in the maximum recommended /weekly single human dose of SUSTOL, based on body surface area).
8.2 Lactation
Risk Summary
There are no data on the presence of SUSTOL in human milk, the effects of SUSTOL on the breastfed infant, or the effects of SUSTOL on milk production. The lack of clinical data during lactation precludes a clear determination of the risk of SUSTOL to an infant during lactation; therefore, the developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for SUSTOL and any potential adverse effects on the breastfed infant from SUSTOL or from the underlying maternal condition.
8.4 Pediatric Use
The safety and effectiveness of SUSTOL in pediatric patients under 18 years of age have not been established.
SUSTOL is not recommended for use in pediatric patients less than 12 years of age because the product administration requires a large gauge needle and an extended administration time.
8.5 Geriatric Use
Of the 738 patients administered 10 mg of SUSTOL in the comparator controlled studies, 177 (24%) were 65 and over while 39 (5%) were 75 and over. No overall differences in safety or effectiveness were observed between these patients and younger patients; and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
8.6 Renal Impairment
Breakdown products of the polymer vehicle in SUSTOL can be detected in urine of healthy subjects [see Clinical Pharmacology (12.3)]. There are no pharmacokinetic data regarding elimination of the polymer vehicle of SUSTOL in patients with renal impairment and the clinical significance of potential prolonged elimination is not known. Avoid SUSTOL in patients with severe renal impairment. In patients with moderate renal impairment, administer SUSTOL not more frequently than once every 14 days [see Dosage and Administration (2.3)].
10 OVERDOSAGE
There is no specific antidote for granisetron overdosage. In the case of overdosage, symptomatic treatment should be given. Overdosage of up to 38.5 mg of granisetron hydrochloride, as a single intravenous injection, has been reported without symptoms or with only the occurrence of headache.
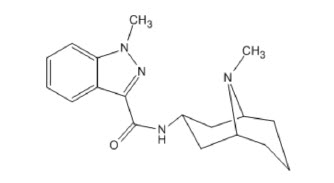
11 DESCRIPTION
SUSTOL (granisetron) extended-release injection, contains granisetron, a serotonin-3 (5-HT3) receptor antagonist. Granisetron is 1-methyl-N-[(1R,3r,5S)-9-methyl-9-azabicyclo[3.3.1]non-3-yl]-1H-indazole-3-carboxamide with a molecular weight of 312.4. Its empirical formula is C18H24N4O, with the following chemical structure:
 |
Granisetron is a white to off-white crystalline solid that is insoluble in water.
SUSTOL is a sterile, clear, colorless to slightly yellow, viscous liquid supplied in a single-dose, pre-filled syringe. Each syringe contains 10 mg granisetron incorporated in an extended-release polymer formulation; the inactive ingredients are triethylene glycol poly(orthoester) polymer, 392 mg and polyethylene glycol monomethyl ether, NF, 98 mg.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Granisetron is a selective 5-hydroxytryptamine3 (5-HT3) receptor antagonist with little or no affinity for other serotonin receptors, including 5-HT1, 5-HT1A, 5-HT1B/C, 5-HT2; for alpha1-, alpha2-, or beta-adrenoreceptors; for dopamine-D2; or for histamine-H1; benzodiazepine; picrotoxin or opioid receptors.
Serotonin receptors of the 5-HT3 type are located peripherally on vagal nerve terminals and centrally in the chemoreceptor trigger zone of the area postrema. During chemotherapy that induces vomiting, mucosal enterochromaffin cells release serotonin, which stimulates 5-HT3 receptors. This evokes vagal afferent discharge, inducing vomiting. Animal studies demonstrate that, in binding to 5-HT3 receptors, granisetron blocks serotonin stimulation and subsequent vomiting after emetogenic stimuli such as cisplatin. In the ferret animal model, a single granisetron injection prevented vomiting due to high-dose cisplatin or arrested vomiting within 5 to 30 seconds.
12.2 Pharmacodynamics
Cardiac Electrophysiology
The effect of SUSTOL on QTc prolongation was evaluated in a double-blind randomized, four-way crossover, placebo and positive (moxifloxacin) controlled study in 51 adult male and female healthy subjects. At 2-fold the recommended dosage of SUSTOL, there was no significant effect on the QTcF interval.
In 142 cancer patients, 24-hour Holter monitoring and 12-lead ECGs were evaluated. QTcF greater than 450 msec were seen in a total of 20 (19%) patients administered SUSTOL and 9 (31%) patients administered intravenous palonosetron hydrochloride. In the SUSTOL group, one patient had a QTcF interval greater than 500 msec and 4 patients had a change from baseline QTcF greater than 60 msec.
12.3 Pharmacokinetics
Absorption
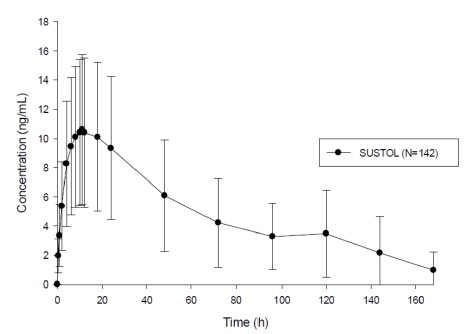
SUSTOL is an extended-release injection formulation of granisetron using a polymer-based drug delivery system. Following a single-dose administration in healthy subjects, granisetron is released from the polymer over an extended period of time and remains detectable in plasma for 7 days post-dose (Figure 1). A mean concentration of 3.5 ng/mL (range 0 to 14 ng/mL) was observed at 5 days post-dose [see Warnings and Precautions (5.3)].
 |
The granisetron pharmacokinetic parameters following injection of SUSTOL were similar between the abdomen and upper arm injection sites as shown in Table 3.
| Parameter* | Injection Site Location | |
|---|---|---|
| Abdomen N=113 | Upper Arm N=113 |
|
|
||
| Cmax (ng/mL) | 9.8 ± 4.8 | 10.8 ± 4.6 |
| Tmax (hours) [range] | 12 [1 to 144] | 11 [1 to 120] |
| AUCinf, ng.h/mL | 680 ± 362 | 720 ± 366 |
In patients, peak plasma granisetron concentrations were delayed compared to healthy subjects with a median Tmax of approximately 24 hours.
Distribution
Plasma protein binding of granisetron is approximately 65% and granisetron distributes freely between plasma and red blood cells.
Metabolism
Published data suggests that granisetron is metabolized by CYP1A1 and CYP3A4. Granisetron metabolism involves N-demethylation and aromatic ring oxidation followed by conjugation.
Elimination
Metabolism: Following a single 10 mg subcutaneous injection of SUSTOL, the terminal elimination half-life of granisetron was approximately 24 hours and was comparable between healthy subjects and patients.
Granisetron clearance is predominantly by hepatic metabolism.
Excretion: Approximately 12% of a granisetron dose, following intravenous administration of granisetron hydrochloride, is eliminated unchanged in the urine in 48 hours. The remainder of the dose is excreted as metabolites, 49% in the urine and 34% in the feces.
Specific Populations
Age: Geriatric Population: In a pooled analysis of samples collected from 56 cancer patients in 3 clinical studies, the mean Cmax and mean AUC0-inf for granisetron following subcutaneous administration of a 10 mg dose of SUSTOL was 39% and 76% higher in patients 65 years of age and older than in patients less than 65 years of age. These pharmacokinetic differences are not considered clinically meaningful taking into consideration the small number of patients 65 years of age and older in the analysis (n = 16) and the high inter-patient variability.
Sex: In a pooled analysis of samples collected from 56 cancer patients in 3 clinical studies, the mean Cmax and mean AUC0-inf for granisetron following subcutaneous administration of a 10 mg dose of SUSTOL was 34% and 132% higher in males than in females. These pharmacokinetic differences are not considered meaningful taking into consideration the small number of males in the analysis (n = 13) and the high inter-patient variability.
Renal Impairment: The total clearance of granisetron was similar in patients with severe renal failure compared to patients with normal renal function following a single 40 mcg/kg intravenous dose of granisetron hydrochloride.
Breakdown products of the polymer vehicle in SUSTOL can be detected in urine of healthy subjects [see Clinical Pharmacology (12.3)]. There are no pharmacokinetic data regarding elimination of the polymer vehicle of SUSTOL in patients with renal impairment [see Dosage and Administration (2.3), Use in Specific Populations (8.6)].
Hepatic Impairment: The total clearance of granisetron was approximately 50% lower in patients with hepatic impairment (neoplastic liver disease) compared to patients with normal hepatic function following a single 40 mcg/kg intravenous dose of granisetron hydrochloride. The high inter-subject variability in the pharmacokinetic parameters of granisetron in patients with hepatic impairment limits the interpretation of these findings.
Drug Interaction Studies
Effect of Other Drugs on Granisetron
Granisetron is metabolized by the hepatic cytochrome P-450 drug-metabolizing enzymes CYP1A1 and CYP3A4. Inducers or inhibitors of CYP1A1 and CYP3A4 enzymes may affect the clearance and half-life of granisetron. In in vitro human microsomal studies, ketoconazole inhibited ring oxidation of granisetron. However, the potential for an in vivo pharmacokinetic interaction with ketoconazole is not known.
Phenobarbital: Administration of intravenous granisetron hydrochloride with phenobarbital, an enzyme inducer, resulted in a 25% increase in total plasma clearance of granisetron. The clinical significance of this interaction is not known.
Effect of Granisetron on Other Drugs
Granisetron does not induce or inhibit the cytochrome P-450 drug-metabolizing enzyme system in vitro. In addition, the in vitro activity of the cytochrome P-450 subfamily 3A4, which is involved in the metabolism of some narcotic analgesics, is not modified by granisetron.
Polymer Breakdown Products
In a metabolic fate study conducted in healthy subjects, breakdown products of the triethylene glycol poly(orthoester) polymer vehicle of the SUSTOL formulation including triethylene glycol (TEG), pentaerythritol (PE), and the oxidative metabolite of TEG, triethylene glycol monocarboxylic acid (TEG acid) were detected in urine with incomplete recovery by the end of study period (10 days). Accumulation of these metabolites in plasma was not noted. The recovery of the polymer load was incomplete in this study and could be due to insufficient sampling and assay sensitivity issues preventing detection of additional metabolites [see Dosage and Administration (2.3), Use in Specific Populations (8.6)].
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
In a 24-month carcinogenicity study of granisetron hydrochloride, rats were treated orally with 1, 5 or 50 mg/kg/day. The 50 mg/kg/day dose was reduced to 25 mg/kg/day during week 59 due to toxicity. For a 60 kg person, the 1, 5, and 25 mg/kg/day doses represent approximately 7, 34 and 169 times, respectively, the maximum recommended human dose (MRHD) (10 mg granisetron/week, approximately 6.2 mg/m2/week) of SUSTOL 10 mg/week, based on body surface area. There was a statistically significant increase in the incidence of hepatocellular carcinomas and adenomas in male rats treated with 5 mg/kg/day (approximately 34 times the MRHD of SUSTOL 10 mg/week, based on body surface area) and above, and in female rats treated with 25 mg/kg/day (approximately 169 times the MRHD of SUSTOL 10 mg/week, based on body surface area). No increase in liver tumors was observed at a dose of 1 mg/kg/day (approximately 7 times the MRHD of SUSTOL 10 mg/week, based on body surface area) in male rats and 5 mg/kg/day (approximately 34 times the MRHD of SUSTOL 10 mg/week, based on body surface area) in female rats.
A 24-month subcutaneous carcinogenicity study in rats evaluated the carcinogenic potenial of the polymer vehicle in SUSTOL. There were no drug-related neoplasms up to 0.295 g/week (approximately 9 to 30 times the amounnt of polymer vehicle present in the MRHD of SUSTOL 10 mg/week, based on body surface area).
In a 12-month oral toxicity study with granisetron hydrochloride, rats were treated at 100 mg/kg/day (approximately 677 times the MRHD of SUSTOL 10 mg/week, based on body surface area). This dose produced hepatocellular adenomas in male and female rats while no such tumors were found in the control rats. A 24-month mouse carcinogenicity study of granisetron did not show a statistically significant increase in tumor incidence, but the study was not conclusive. Because of the tumor findings in rat studies, SUSTOL should be prescribed only at the dose and for the indication recommended [see Indications and Usage (1), Dosage and Administration (2.2)].
Granisetron hydrochloride was not mutagenic in an in vitro Ames test and mouse lymphoma cell forward mutation assay, and in vivo mouse micronucleus test and in vitro and ex vivo rat hepatocyte unscheduled DNA synthesis (UDS) assays. However, granisetron hydrochloride was positive in a mouse lymphoma assay with metabolic activation and produced a significant increase in UDS in HeLa cells in vitro and a significant increased incidence of cells with polyploidy in an in vitro human lymphocyte chromosomal aberration test.
Neither SUSTOL nor the polymer vehicle for SUSTOL was mutagenic in the Ames test, mouse lymphoma assay, and the in vivo rat bone marrow micronucleus test.
Granisetron hydrochloride at subcutaneous doses up to 6 mg/kg/day (approximately 40 times the MRHD of SUSTOL 10 mg/week, based on body surface area), and oral doses up to 100 mg/kg/day (approximately 677 times the MRHD of SUSTOL 10 mg/week, based on body surface area) was found to have no effect on fertility and reproductive performance of male and female rats.
The polymer vehicle for SUSTOL at subcutaneous doses up to 0.295 g per day (approximately 137 times the amount of polymer vehicle present in the maximum recommended /weekly single human dose of SUSTOL, based on body surface area) was found to have no adverse effect on fertility and reproductive performance of male and female rats.
14 CLINICAL STUDIES
In a randomized, multicenter, double-blind, parallel group study, a single 10 mg subcutaneous dose of SUSTOL was compared to a single 0.25 mg intravenous dose of palonosetron hydrochloride in cancer patients administered moderately emetogenic (MEC) or anthracycline plus cyclophosphamide (AC) combination chemotherapy. SUSTOL or palonosetron hydrochloride was administered 30 minutes prior to chemotherapy on Day 1. Patients also received either 8 or 20 mg intravenous dexamethasone on Day 1 depending on chemotherapy regimen. Patients who received 20 mg of intravenous dexamethasone also received oral dexamethasone 8 mg twice daily on Days 2, 3, and 4.
In this study of 733 patients (371 in the SUSTOL 10 mg arm and 362 in the palonosetron arm), 79% of patients were female and 63% were Caucasian. The mean age was 57 years (range 22 to 91 years), 55% received MEC and 45% received AC combination chemotherapy regimens. The most common MEC regimens were carboplatin/paclitaxel (31%).
The primary endpoints were proportion of patients with complete response (CR) [defined as no emetic episodes (vomiting or retching) and no use of rescue medication] during the acute phase (0 to 24 hours) and the delayed phase (>24 to 120 hours) following the administration of chemotherapy in Cycle 1. The study design allowed for assessment of non-inferiority of SUSTOL to palonosetron hydrochloride in the acute and delayed phase of MEC and of AC combination chemotherapy.
Non-inferiority of SUSTOL to palonosetron hydrochloride was demonstrated in the acute and delayed phases of MEC and of AC combination chemotherapy [Table 4].
| Moderately Emetogenic Chemotherapy | |||
| Complete Response | SUSTOL 10 mg subcutaneous N=200 n (%) | Palonosetron hydrochloride, 0.25 mg intravenous N=206 n (%) | Difference (95% CI †): SUSTOL minus Palonosetron |
|---|---|---|---|
|
|||
| Acute Phase (0 – 24 hours) | 166 (83) | 183 (89) | -6 (-13, 1) |
| Delayed Phase (>24 – 120 hours) | 137 (69) | 144 (70) | -1 (-10, 8) |
| AC Combination Chemotherapy | |||
| Complete Response | SUSTOL 10 mg subcutaneous N=171 n (%) | Palonosetron hydrochloride, 0.25 mg intravenous N=156 n (%) | Difference (95% CI ‡): SUSTOL minus Palonosetron |
| Acute Phase (0 – 24 hours) | 120 (70) | 99 (64) | 6 (-3, 17) |
| Delayed Phase (>24 – 120 hours) | 85 (50) | 74 (47) | 2 (-9, 13) |
16 HOW SUPPLIED/STORAGE AND HANDLING
SUSTOL extended-release injection is supplied in cartons of six kits (NDC 47426-101-06) each kit contains:
- One sterile single-dose amber colored glass syringe which contains 10 mg granisetron/0.4 mL,
- One sterile 18 Ga x 5/8" special thin walled administration needle,
- Two sodium acetate syringe warming pouches,
- One Point Lok needle protection device.
Storage
Store SUSTOL in the refrigerator at 2°C to 8°C (36°F to 46°F).
SUSTOL can be placed back in the refrigerator after being kept at room temperature. SUSTOL can remain at room temperature for up to a maximum of 7 days.
Protect from light. Do not freeze.
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Administration
- SUSTOL is intended for subcutaneous injection by a health care provider.
Injection Site Reactions [see Warnings and Precautions (5.1)]
- Inform the patient that ISRs may occur and may include infections, bruising and/or hematomas, bleeding, pain and tenderness, and nodules.
- Inform the patient that some ISRs (infections, bruising, and hematoma) may occur up to 2 weeks or more after SUSTOL administration.
- Instruct the patient to seek immediate medical care for the following ISRs:
ο signs of infection at the injection site.
ο injection site bleeding that is severe or lasts for longer than one day. - Advise the patient to tell their healthcare provider if they experience:
ο pain or tenderness severe enough to require treatment with pain medication or interfere with daily activity.
ο bruising and/or hematoma or a persistent nodule at the injection site.
Gastrointestinal
Advise the patient to report new or worsening constipation to their healthcare provider and seek immediate medical care if signs and symptoms of an ileus occur [see Warnings and Precautions (5.2)].
Hypersensitivity reactions
Advise the patient that hypersensitivity reactions may occur up to 7 days or longer following SUSTOL administration. Inform the patient of the signs and symptoms of hypersensitivity reactions, and have them seek immediate medical care should signs and symptoms occur [see Warnings and Precautions (5.1, 5.3)].
Serotonin Syndrome
Advise the patient of the possibility of serotonin syndrome, especially with concomitant use of SUSTOL and another serotonergic agent such as medications to treat depression and migraines. Advise the patient to seek immediate medical attention if the following symptoms occur: changes in mental status, autonomic instability, neuromuscular symptoms with or without gastrointestinal symptoms [see Warnings and Precautions (5.4)].
Manufactured for: Heron Therapeutics, Inc., San Diego, CA 92121, USA
Patent: https://www.herontx.com/patents/
SUSTOL® is a registered trademark of Heron Therapeutics, Inc.
Copyright © 2017-2023 Heron Therapeutics, Inc.
All rights reserved.
MEDICATION GUIDE
SUSTOL® (sus' tol)
(granisetron)
extended-release injection, for subcutaneous use
What is the most important information I should know about SUSTOL?
SUSTOL can cause serious side effects, including:
-
Injection site reactions. Some injection site reactions may be serious and require medical care. Injection site reactions may include infections, bruising, swelling that is caused by blood that collects under the skin (hematoma), bleeding, pain and tenderness, and small bumps (nodules) at the injection site. Infections, bruising, and hematomas can happen up to 2 weeks or more after receiving SUSTOL.
Tell your healthcare provider if you have:
ο pain or tenderness that you need to take pain medicine for or if you have pain that interferes with your daily activity.
ο bruising, hematoma, or a nodule at the injection site that does not go away. Your risk of severe bruising and hematomas at the injection site is increased if you take a blood thinner medicine (anticoagulant or antiplatelet medicine).
Get medical care right away if you have:
ο signs of an infection at the injection site, including continued redness or warmth, or if you have a fever.
ο bleeding at the injection site that is severe or lasts for longer than 24 hours. - Stomach and intestinal problems. Problems having a bowel movement (constipation) that may be serious, can happen up to 7 days after treatment with SUSTOL. These problems may be more likely in people taking opioid pain medicines. SUSTOL can make it harder to identify certain stomach and bowel problems you may have. Tell your healthcare provider if you have constipation or your constipation worsens after you receive SUSTOL. Get medical care right away if you have pain or swelling in your stomach-area (abdomen).
-
Serious allergic reactions. Serious allergic reactions have happened in people who receive SUSTOL and who have had allergic reactions to other medicines used to help prevent nausea and vomiting called 5-HT3 receptor antagonists. Serious allergic reactions can happen up to 7 days or longer after treatment. Get emergency medical help right away if you have any signs or symptoms of a serious allergic reaction, including:
ο hives
ο breathing trouble
ο swollen face
ο chest pain
What is SUSTOL?
- SUSTOL is a prescription medicine called an "antiemetic."
- SUSTOL is used in adults to help prevent the nausea and vomiting that happens right away or later with certain anti-cancer medicines (chemotherapy).
It is not known if SUSTOL is safe and effective in children under 18 years of age.
Who should not receive SUSTOL?
Do not receive SUSTOL if you are allergic to:
- granisetron or any of the ingredients in SUSTOL. See the end of this Medication Guide for a complete list of ingredients in SUSTOL.
- any other 5-HT3 receptor antagonist medicine used to help prevent nausea and vomiting.
What should I tell my healthcare provider before receiving SUSTOL?
Before receiving SUSTOL, tell your healthcare provider about all of your medical conditions, including if you:
- have constipation
- have had recent stomach-area (abdominal) surgery
- have kidney problems
- are pregnant or plan to become pregnant. It is not known if SUSTOL will harm your unborn baby.
- are breastfeeding or plan to breastfeed. It is not known if SUSTOL passes into your breast milk. Talk to your healthcare provider about the best way to feed your baby if you will receive SUSTOL.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Using SUSTOL with certain other medicines can cause serious side effects.
Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine.
How will I receive SUSTOL?
- SUSTOL will be given to you by an injection under your skin (subcutaneously) in the back of your upper arm or in your stomach-area (abdomen) on Day 1 of your chemotherapy cycle.
- SUSTOL will be given to you by a healthcare provider.
- SUSTOL is usually given about 30 minutes before you receive your anti-cancer medicine (chemotherapy).
- You should not receive SUSTOL more often than 1 time every 7 days.
What are the possible side effects of SUSTOL?
SUSTOL may cause serious side effects, including:
- See "What is the most important information I should know about SUSTOL?"
-
Serotonin Syndrome. A possible life-threatening problem called serotonin syndrome can happen with medicines called 5-HT3 receptor antagonists, including SUSTOL, especially when used with medicines used to treat depression and migraine headaches called serotonin reuptake inhibitors (SSRIs), serotonin and norepinephrine reuptake inhibitors (SNRIs), monoamine oxidase inhibitors (MAOIs), and certain other medicines. Tell your healthcare provider or nurse right away if you have any of the following symptoms of serotonin syndrome:
ο agitation, seeing things that are not there (hallucinations), confusion, or coma
ο fast heartbeat or unusual and frequent changes in your blood pressure
ο dizziness, sweating, flushing, or fever
ο tremors, stiff muscles, muscle twitching, overactive reflexes, or loss of coordination
ο seizures
ο nausea, vomiting, or diarrhea
The most common side effects of SUSTOL include: injection site reactions, constipation, fatigue, headache, diarrhea, stomach-area (abdominal) pain, trouble sleeping or falling asleep, indigestion, dizziness, weakness, and heartburn.
These are not all the possible side effects of SUSTOL. Tell your healthcare provider if you have any side effect that bothers you or does not go away.
Call your doctor for medical advice about side effects.You may report side effects to FDA at 1-800-FDA-1088.
General Information about the safe and effective use of SUSTOL
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use SUSTOL for a condition for which it was not prescribed. Do not give SUSTOL to other people, even if they have the same symptoms that you have. It may harm them.You can ask your healthcare provider or pharmacist for information about SUSTOL that is written for health professionals.
What are the ingredients in SUSTOL?
Active ingredient: granisetron
Inactive ingredients: triethylene glycol poly(orthoester) polymer and polyethylene glycol monomethyl ether
Manufactured for: Heron Therapeutics, Inc., San Diego, CA 92121
Patent: https://www.herontx.com/patents/
SUSTOL® is a registered trademark of Heron Therapeutics, Inc.
Copyright © 2017-2023 Heron Therapeutics, Inc.
All rights reserved.
For more information, go to www.SUSTOL.com or call 1-844-437-6611.
This Medication Guide has been approved by the U.S. Food and Drug Administration
Revised: 06/2023
| SUSTOL®
(granisetron) extended-release injection 10 mg/0.4 mL Instructions for Use | You must read these complete instructions before you administer SUSTOL for the first time. SUSTOL must be administered by a healthcare provider. |
|
|---|---|---|
| What is SUSTOL? | SUSTOL is a subcutaneous injection that provides long-acting antiemetic medication to patients receiving chemotherapy. SUSTOL contains a highly viscous polymer that provides sustained release of granisetron. SUSTOL is supplied as part of an administration kit. |
|
| When should you administer SUSTOL? | Administer SUSTOL at least 30 minutes before chemotherapy on Day 1 of chemotherapy treatment and not more frequently than once every 7 days, because of the extended-release properties of the formulation. | |
| Before you begin to prepare SUSTOL Injection | Read these critical instructions.
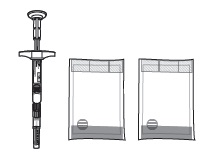
Do not substitute any of the components from the kit for administration. Check to make sure the SUSTOL kit contains: |
|
|
||
| Bring SUSTOL to room temperature | STEP | To bring SUSTOL to room temperature: |
 | 1 | Remove SUSTOL kit from refrigeration. Open carton and remove tray. Open tray and unpack warming pouches. Wait a minimum of 60 minutes prior to use to allow SUSTOL and the warming pouches to warm to room temperature. SUSTOL may be left out at room temperature overnight, and for up to 7 days, before it must be discarded. SUSTOL may be re-refrigerated if not used within the 7 day period. Warming SUSTOL prior to injection decreases the viscosity and makes administration easier. Bringing SUSTOL to room temperature prior to warming with the sodium acetate warming pouch will facilitate warming of SUSTOL to body temperature. |
| Prepare the syringe | STEP | Follow these steps to prepare SUSTOL for use: |
| 1 |  Examine all contents of the package. Do not use if any component is damaged. Examine all contents of the package. Do not use if any component is damaged. |
|
| 2 | Check the expiration date on the syringe. Do not use if the expiration date has passed. | |
| 3 |  Remove the perforated flag label from the plunger rod. Remove the perforated flag label from the plunger rod. |
|
 | 4 | To ensure that the syringe contains the full dose, check that the gray stopper is close to or below the black line on the syringe barrel label as shown in the illustration. Do NOT expel air bubbles. For a subcutaneous injection, air bubbles in the syringe will not harm the patient. |
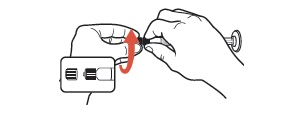 | 5 | Unscrew the tip cap from the end of the pre-filled glass syringe. Do NOT use if the tip cap is missing or has been tampered with. Do NOT use if the Luer fitting is missing or dislodged. |
| 6 | Remove the hub cover from the 18G x 5/8" needle supplied with the syringe. Attach the needle to the syringe and seat the needle securely. Do NOT remove the blue needle cover. Do not substitute with any other needle
Use ONLY the 18G x 5/8" needle provided with the syringe because of the highly viscous nature of the medication. |
|
| Warm SUSTOL to body temperature | STEP | Just prior to administering the injection, follow these steps: |
 | SUSTOL must be warmed to body temperature immediately prior to injection to reduce viscosity and facilitate administration. SUSTOL is packaged with two warming pouches. The warming pouches use a non- toxic solution of sodium acetate to create an exothermic (warming) reaction. The second warming pouch can be used if SUSTOL is not immediately administered within the 15 minutes provided by the first warming pouch and needs to be re-warmed. Before activating the warming pouch, make sure that the SUSTOL kit was removed from the refrigerator and that the warming pouches were unpacked from the kit at least 60 minutes prior to use to allow SUSTOL and the warming pouches to warm to room temperature. |
|
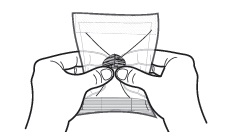 | 1 | To activate the warming pouch, locate the metal disc and grasp with thumb and forefingers of both hands. Flex (bend) the disc rapidly until crystals begin to form (fluid will start to turn white). Massage warming pouch for a few seconds to soften and to increase the activation rate. |
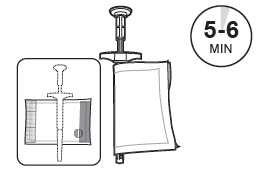 | 2 | Wrap the SUSTOL syringe and needle in the syringe warming pouch. Use the outline on the warming pouch label as a placement guide for the syringe and needle. Fold the warming pouch over the syringe and press fastening strips together. Allow SUSTOL to warm for 5 - 6 minutes in the warming pouch. SUSTOL must be warmed to body temperature immediately prior to injection to reduce viscosity and facilitate administration. |
 | 3 | Keep the SUSTOL syringe and needle in the warming pouch until you are ready to administer SUSTOL. The warming pouch will keep the SUSTOL syringe at body temperature for 10 - 15 minutes. If more than 15 minutes has elapsed since warming the SUSTOL syringe, then re-warm the syringe using the second warming pouch prior to injecting SUSTOL.Sodium acetate is a non-toxic salt. In case of a puncture to a warming pouch and the spilling of the solution or crystals, clean up with soap and water. In case of eye contact, flush with water for 15 minutes. In case of skin contact, wash with soap and water. Seek medical attention if irritation develops or persists. Used warming pouches may be safely disposed of in the trash. |
| Select an injection site | STEP | To select an injection site, use these guidelines: |
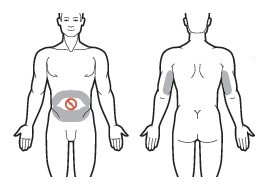 | 1 | Use the skin of the abdomen at least one inch from the umbilicus or the skin of the back of the upper arms as sites for subcutaneous injections. For SUBCUTANEOUS injection only. For SUBCUTANEOUS injection only.Do NOT inject within 1 inch of the umbilicus or anywhere the skin is: • Burned • Hardened • Inflamed • Swollen • Compromised For subsequent injections, use the same general area of the body. However, be sure not to inject in the exact same place. Administer new injections at least 1 inch from the previous site. |
| Inject SUSTOL | STEP | Follow these steps to inject SUSTOL: |
| 1 | Remove the syringe from the warming pouch.The white finger flange on the syringe provides better control when administering SUSTOL. Do NOT remove white finger flange. |
|
| 2 | Remove the blue needle cover. Use standard sharps safety techniques to avoid needle sticks. | |
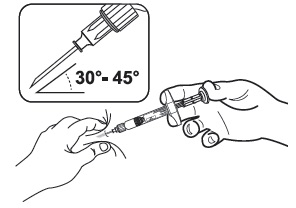 | 3 | Use standard aseptic technique when performing the injection. Pinch the skin and underlying subcutaneous tissue with your non-dominant hand to create a "tent." Insert the needle into the subcutaneous tissue at a 30° - 45° angle with your dominant hand. Administer SUSTOL ONLY as a subcutaneous injection. NEVER administer intravenously, intramuscularly or intraperitoneally. |
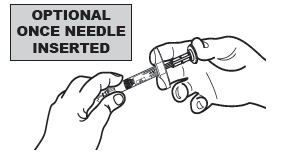 | 4 | For additional stability, once the needle is inserted into the subcutaneous tissue, release the "tent" and hold the syringe barrel with your non-dominant hand. |
| 5 | Press the plunger using a slow, firm, and steady push until you have delivered the entire dose. Do not use excessive force to inject the material more quickly, as this will only result in greater difficulty with the injection.Due to the high viscosity of SUSTOL, you will experience resistance when injecting SUSTOL. Apply firm and steady pressure to the plunger for about 20-30 seconds. Pressing harder will NOT expel SUSTOL faster. Do NOT advance or reposition the syringe in the subcutaneous tissue during the injection. |
|
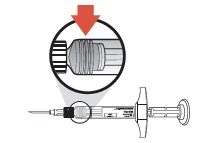 | 6 | Because SUSTOL is highly viscous, before withdrawing the needle, make sure that the medication is completely expelled by checking the position of the stopper at the base of the syringe barrel. |
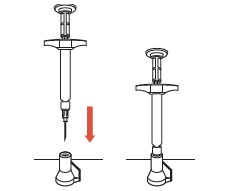 | 7 | Withdraw the needle. Do not remove the needle from the syringe. Use the enclosed Point-Lok® device to prevent needle sticks. Place the Point-Lok needle protection device, supplied within the SUSTOL kit, on a secured flat surface. Do not hold the Point-Lok device in your hand. Carefully insert the exposed needle into the opening at the top of the Point-Lok device and push down until the needle is fully seated in the Point-Lok device. This action will seal the needle tip and lock the needle firmly into the Point-Lok device. Discard the protected needle and attached syringe using your organization's standard sharps disposal procedures. |
| 8 | Dispense enclosed Medication Guide to each Patient. | |
| Marketed by: Heron Therapeutics, Inc. 4242 Campus Point Court, Suite 200 San Diego, CA 92121 © 2017 Heron Therapeutics, Inc. Revision Date: May 2017 | Questions about how to use SUSTOL?
Call the support line toll-free at 844-HERON11 (844-437-6611) or visit www.SUSTOL.com LAM-0000363 |
|
Principal Panel - Carton of Six Kits
SUSTOL®
(granisetron) extended-release injection 10 mg/0.4 mL
FOR SUBCUTANEOUS USE ONLY.
REMOVE CARTON FROM REFRIGERATOR AND UNPACK ALL CONTENTS AT LEAST 60 MINUTES PRIOR TO USE.
Each single-dose unit contains:
One sterile single-dose glass syringe which contains 10 mg granisetron
One sterile 18Gx5/8" administration needle
Two sodium acetate warming pouches
One Point-Lok®,needle protection device
Instructions for Use and Package Insert
Must be refrigerated
Store at 2°C to 8°C (36°F to 46°F); DO NOT FREEZE.
Protect from Light.
SUSTOL is to be administered once weekly.
NDC: 47426-101-06
Contains:
6 single-dose
PRE-FILLED syringe units
Rx only
HERON
THERAPEUTICS
 |