Rx only
CONTRAINDICATIONS AND WARNINGS
Claravis™ must not be used by female patients who are or may become pregnant. There is an extremely high risk that severe birth defects will result if pregnancy occurs while taking Claravis in any amount, even for short periods of time. Potentially any fetus exposed during pregnancy can be affected. There are no accurate means of determining whether an exposed fetus has been affected.
Birth defects which have been documented following isotretinoin exposure include abnormalities of the face, eyes, ears, skull, central nervous system, cardiovascular system, and thymus and parathyroid glands. Cases of IQ scores less than 85 with or without other abnormalities have been reported. There is an increased risk of spontaneous abortion, and premature births have been reported.
Documented external abnormalities include: skull abnormality; ear abnormalities (including anotia, micropinna, small or absent external auditory canals); eye abnormalities (including microphthalmia); facial dysmorphia; cleft palate. Documented internal abnormalities include: CNS abnormalities (including cerebral abnormalities, cerebellar malformation, hydrocephalus, microcephaly, cranial nerve deficit); cardiovascular abnormalities; thymus gland abnormality; parathyroid hormone deficiency. In some cases death has occurred with certain of the abnormalities previously noted.
If pregnancy does occur during treatment of a female patient who is taking Claravis, Claravis must be discontinued immediately and she should be referred to an Obstetrician-Gynecologist experienced in reproductive toxicity for further evaluation and counseling.
Special Prescribing Requirements
Because of isotretinoin's teratogenicity and to minimize fetal exposure, Claravis is approved for marketing only under a special restricted distribution program approved by the Food and Drug Administration. This program is called iPLEDGE™. Claravis must only be prescribed by prescribers who are registered and activated with the iPLEDGE program. Claravis must only be dispensed by a pharmacy registered and activated with iPLEDGE, and must only be dispensed to patients who are registered and meet all the requirements of iPLEDGE (see PRECAUTIONS).
| Female Patients of Childbearing Potential | Male Patients, And Female Patients Not of Childbearing Potential | |
| PRESCRIBER | ||
| Confirms patient counseling | X | X |
| Enters the two contraception methods chosen by the patient | X | |
| Enters pregnancy test results | X | |
| PATIENT | ||
| Answers educational questions before every prescription | X | |
| Enters two forms of contraception | X | |
| PHARMACIST | ||
| Contacts system to get an authorization | X | X |
DESCRIPTION
Isotretinoin, USP, a retinoid, is available as Claravis™ (isotretinoin capsules USP) in 10 mg, 20 mg, 30 mg and 40 mg hard gelatin capsules for oral administration. Chemically, isotretinoin is 13-cis-retinoic acid and is related to both retinoic acid and retinol (vitamin A). It is a yellow to orange crystalline powder. The structural formula is:
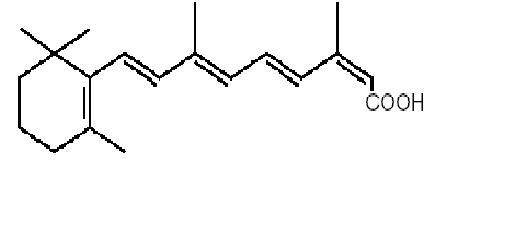
C20H28O2 Molecular Weight: 300.44
Each capsule contains the following inactive ingredients: butylated hydroxyanisole, edetate disodium, gelatin, hydrogenated vegetable oil, polysorbate 80, soybean oil, titanium dioxide, white wax (beeswax), and vitamin E.
In addition, the 10 mg capsule contains black iron oxide and FD&C yellow no. 6. The 20 mg capsule contains black iron oxide, red iron oxide and yellow iron oxide. The 30 mg capsule contains red iron oxide and yellow iron oxide. The 40 mg capsule contains colloidal silicon dioxide, FD&C yellow no. 6, and sodium lauryl sulfate.
The edible imprinting ink contains: 10 mg strength, D&C red no. 7 calcium lake, ethylene glycol monoethyl ether, FD&C yellow no. 6 aluminum lake, shellac glaze and titanium dioxide; 20 mg strength, ammonium hydroxide, propylene glycol, shellac glaze, simethicone and titanium dioxide; the 30 mg strength, D&C yellow no. 10 aluminum lake, FD&C blue no.1 aluminum lake, FD&C blue no. 2 aluminum lake, FD&C red no. 40 aluminum lake, iron oxide black, propylene glycol, and shellac glaze; 40 mg strength, ammonium hydroxide, FD&C blue no. 2 aluminum lake, iron oxide black, propylene glycol, and shellac glaze.
Meets dissolution test 2.
CLINICAL PHARMACOLOGY
Isotretinoin is a retinoid, which when administered in pharmacologic dosages of 0.5 to 1 mg/kg/day (see DOSAGE AND ADMINISTRATION), inhibits sebaceous gland function and keratinization. The exact mechanism of action of isotretinoin is unknown.
Nodular Acne
Clinical improvement in nodular acne patients occurs in association with a reduction in sebum secretion. The decrease in sebum secretion is temporary and is related to the dose and duration of treatment with Claravis, and reflects a reduction in sebaceous gland size and an inhibition of sebaceous gland differentiation.1
Pharmacokinetics
Absorption
Due to its high lipophilicity, oral absorption of isotretinoin is enhanced when given with a high-fat meal. In a crossover study, 74 healthy adult subjects received a single 80 mg oral dose (2 x 40 mg capsules) of Claravis under fasted and fed conditions. Both peak plasma concentration (Cmax) and the total exposure (AUC) of isotretinoin were more than doubled following a standardized high-fat meal when compared with Claravis given under fasted conditions (see Table 2 ). The observed elimination half-life was unchanged. This lack of change in half-life suggests that food increases the bioavailability of isotretinoin without altering its disposition. The time to peak concentration (Tmax) was also increased with food and may be related to a longer absorption phase. Therefore, Claravis capsules should always be taken with food (see DOSAGE AND ADMINISTRATION). Clinical studies have shown that there is no difference in the pharmacokinetics of isotretinoin between patients with nodular acne and healthy subjects with normal skin.
| Claravis 2 x 40 mg Capsules |
AUCo-∞ (ng • hr/mL) |
Cmax (ng/mL) |
Tmax (hr) |
t½ (hr) |
| Fed | 10,004 (22%) | 862 (22%) | 5.3 (77%) | 21 (39%) |
| Fasted | 3,703 (46%) | 301 (63%) | 3.2 (56%) | 21 (30%) |
Metabolism
Following oral administration of isotretinoin, at least three metabolites have been identified in human plasma: 4-oxo-isotretinoin, retinoic acid (tretinoin), and 4-oxo-retinoic acid (4-oxo-tretinoin). Retinoic acid and 13-cis-retinoic acid are geometric isomers and show reversible interconversion. The administration of one isomer will give rise to the other. Isotretinoin is also irreversibly oxidized to 4-oxo-isotretinoin, which forms its geometric isomer 4-oxo-tretinoin.
After a single 80 mg oral dose of isotretinoin to 74 healthy adult subjects, concurrent administration of food increased the extent of formation of all metabolites in plasma when compared to the extent of formation under fasted conditions.
All of these metabolites possess retinoid activity that is in some in vitro models more than that of the parent isotretinoin. However, the clinical significance of these models is unknown. After multiple oral dose administration of isotretinoin to adult cystic acne patients (≥18 years), the exposure of patients to 4-oxo-isotretinoin at steady-state under fasted and fed conditions was approximately 3.4 times higher than that of isotretinoin.
In vitro studies indicate that the primary P450 isoforms involved in isotretinoin metabolism are 2C8, 2C9, 3A4, and 2B6. Isotretinoin and its metabolites are further metabolized into conjugates, which are then excreted in urine and feces.
Elimination
Following oral administration of an 80 mg dose of 14C-isotretinoin as a liquid suspension, 14C-activity in blood declined with a half-life of 90 hours. The metabolites of isotretinoin and any conjugates are ultimately excreted in the feces and urine in relatively equal amounts (total of 65% to 83%). After a single 80 mg oral dose of isotretinoin to 74 healthy adult subjects under fed conditions, the mean + SD elimination half-lives (t½) of isotretinoin and 4-oxo-isotretinoin were 21 + 8.2 hours and 24 + 5.3 hours, respectively. After both single and multiple doses, the observed accumulation ratios of isotretinoin ranged from 0.90 to 5.43 in patients with cystic acne.
Special Patient Populations
Pediatric Patients
The pharmacokinetics of isotretinoin were evaluated after single and multiple doses in 38 pediatric patients (12 to 15 years) and 19 adult patients (≥ 18 years) who received isotretinoin for the treatment of severe recalcitrant nodular acne. In both age groups, 4-oxo-isotretinoin was the major metabolite; tretinoin and 4-oxo-tretinoin were also observed. The dose-normalized pharmacokinetic parameters for isotretinoin following single and multiple doses are summarized in Table 3 for pediatric patients. There were no statistically significant differences in the pharmacokinetics of isotretinoin between pediatric and adult patients.
| Parameter | Isotretinoin (Single Dose) | Isotretinoin (Steady-State) |
| Cmax (ng/mL) | 573.25 (278.79) | 731.98 (361.86) |
| AUC(0 to 12) (ng·hr/mL) | 3033.37 (1394.17) | 5082 (2184.23) |
| AUC(0 to 24) (ng·hr/mL) | 6003.81 (2885.67) | -- |
| Tmax (hr) | 6 (1 to 24.6) | 4 (0 to 12) |
| CSSmin (ng/mL) | -- | 352.32 (184.44) |
| T½ (hr) | -- | 15.69 (5.12) |
| CL/F (L/hr) | -- | 17.96 (6.27) |
In pediatric patients (12 to 15 years), the mean ± SD elimination half-lives (t½) of isotretinoin and 4-oxo-isotretinoin were 15.7 ± 5.1 hours and 23.1 ± 5.7 hours, respectively. The accumulation ratios of isotretinoin ranged from 0.46 to 3.65 for pediatric patients.
INDICATIONS AND USAGE
Severe Recalcitrant Nodular Acne
Claravis (isotretinoin capsules, USP) is indicated for the treatment of severe recalcitrant nodular acne. Nodules are inflammatory lesions with a diameter of 5 mm or greater. The nodules may become suppurative or hemorrhagic. “Severe,” by definition,2 means “many” as opposed to “few or several” nodules. Because of significant adverse effects associated with its use, Claravis should be reserved for patients with severe nodular acne who are unresponsive to conventional therapy, including systemic antibiotics. In addition, Claravis is indicated only for those female patients who are not pregnant, because Claravis can cause severe birth defects (see Boxed CONTRAINDICATIONS AND WARNINGS ).
A single course of therapy for 15 to 20 weeks has been shown to result in complete and prolonged remission of disease in many patients.1,3,4 If a second course of therapy is needed, it should not be initiated until at least 8 weeks after completion of the first course, because experience has shown that patients may continue to improve while off Claravis. The optimal interval before retreatment has not been defined for patients who have not completed skeletal growth (see WARNINGS, Skeletal, Bone Mineral Density, Hyperostosis, and Premature Epiphyseal Closure).
CONTRAINDICATIONS
Allergic Reactions
Claravis is contraindicated in patients who are hypersensitive to this medication or to any of its components (see PRECAUTIONS, Hypersensitivity).
WARNINGS
Psychiatric Disorders
Claravis may cause depression, psychosis and, rarely, suicidal ideation, suicide attempts, suicide, and aggressive and/or violent behaviors. No mechanism of action has been established for these events (see ADVERSE REACTIONS, Psychiatric). Prescribers should read the brochure, Recognizing Psychiatric Disorders in Adolescents and Young Adults: A Guide for Prescribers of Isotretinoin. Prescribers should be alert to the warning signs of psychiatric disorders to guide patients to receive the help they need. Therefore,prior to initiation of Claravis therapy, patients and family members should be asked about any history of psychiatric disorder, and at each visit during therapy patients should be assessed for symptoms of depression, mood disturbance, psychosis, or aggression to determine if further evaluation may be necessary. Signs and symptoms of depression, as described in the brochure (“Recognizing Psychiatric Disorders in Adolescents and Young Adults”), include sad mood, hopelessness, feelings of guilt, worthlessness or helplessness, loss of pleasure or interest in activities, fatigue, difficulty concentrating, change in sleep pattern, change in weight or appetite, suicidal thoughts or attempts, restlessness, irritability, acting on dangerous impulses, and persistent physical symptoms unresponsive to treatment. Patients should stop Claravis and the patient or a family member should promptly contact their prescriber if the patient develops depression, mood disturbance, psychosis, or aggression, without waiting until the next visit. Discontinuation of Claravis therapy may be insufficient; further evaluation may be necessary. While such monitoring may be helpful, it may not detect all patients at risk. Patients may report mental health problems or family history of psychiatric disorders. These reports should be discussed with the patient and/or the patient’s family. A referral to a mental health professional may be necessary. The physician should consider whether Claravis therapy is appropriate in this setting; for some patients the risks may outweigh the benefits of Claravis therapy.
Pseudotumor Cerebri
Claravis use has been associated with a number of cases of pseudotumor cerebri (benign intracranial hypertension), some of which involved concomitant use of tetracyclines. Concomitant treatment with tetracyclines should therefore be avoided. Early signs and symptoms of pseudotumor cerebri include papilledema, headache, nausea and vomiting, and visual disturbances. Patients with these symptoms should be screened for papilledema and, if present, they should be told to discontinue Claravis immediately and be referred to a neurologist for further diagnosis and care (see ADVERSE REACTIONS, Neurological).
Serious Skin Reactions
There have been post-marketing reports of erythema multiforme and severe skin reactions [e.g., Stevens-Johnson Syndrome (SJS), toxic epidermal necrolysis (TEN)] associated with isotretinoin use. These events may be serious and result in death, life-threatening events, hospitalization, or disability. Patients should be monitored closely for severe skin reactions, and discontinuation of Claravis should be considered if warranted.
Pancreatitis
Acute pancreatitis has been reported in patients with either elevated or normal serum triglyceride levels. In rare instances, fatal hemorrhagic pancreatitis has been reported. Claravis should be stopped if hypertriglyceridemia cannot be controlled at an acceptable level or if symptoms of pancreatitis occur.
Lipids
Elevations of serum triglycerides in excess of 800 mg/dL have been reported in patients treated with Claravis. Marked elevations of serum triglycerides were reported in approximately 25% of patients receiving Claravis in clinical trials. In addition, approximately 15% developed a decrease in high-density lipoproteins and about 7% showed an increase in cholesterol levels. In clinical trials, the effects on triglycerides, HDL, and cholesterol were reversible upon cessation of Claravis therapy. Some patients have been able to reverse triglyceride elevation by reduction in weight, restriction of dietary fat and alcohol, and reduction in dose while continuing Claravis.5
Blood lipid determinations should be performed before Claravis is given and then at intervals until the lipid response to Claravis is established, which usually occurs within 4 weeks. Especially careful consideration must be given to risk/benefit for patients who may be at high risk during Claravis therapy (patients with diabetes, obesity, increased alcohol intake, lipid metabolism disorder or familial history of lipid metabolism disorder). If Claravis therapy is instituted, more frequent checks of serum values for lipids and/or blood sugar are recommended (see PRECAUTIONS, Laboratory Tests).
The cardiovascular consequences of hypertriglyceridemia associated with Claravis are unknown.
Animal Studies
In rats given 8 or 32 mg/kg/day of isotretinoin (1.3 to 5.3 times the recommended clinical dose of 1 mg/kg/day after normalization for total body surface area) for 18 months or longer, the incidences of focal calcification, fibrosis and inflammation of the myocardium, calcification of coronary, pulmonary and mesenteric arteries, and metastatic calcification of the gastric mucosa were greater than in control rats of similar age. Focal endocardial and myocardial calcifications associated with calcification of the coronary arteries were observed in two dogs after approximately 6 to 7 months of treatment with isotretinoin at a dosage of 60 to 120 mg/kg/day (30 to 60 times the recommended clinical dose of 1 mg/kg/day, respectively, after normalization for total body surface area).
Hearing Impairment
Impaired hearing has been reported in patients taking Claravis; in some cases, the hearing impairment has been reported to persist after therapy has been discontinued. Mechanism(s) and causality for this event have not been established. Patients who experience tinnitus or hearing impairment should discontinue Claravis treatment and be referred for specialized care for further evaluation (see ADVERSE REACTIONS, Special Senses).
Hepatotoxicity
Clinical hepatitis considered to be possibly or probably related to Claravis therapy has been reported. Additionally, mild to moderate elevations of liver enzymes have been observed in approximately 15% of individuals treated during clinical trials, some of which normalized with dosage reduction or continued administration of the drug. If normalization does not readily occur or if hepatitis is suspected during treatment with Claravis, the drug should be discontinued and the etiology further investigated.
Inflammatory Bowel Disease
Claravis has been associated with inflammatory bowel disease (including regional ileitis) in patients without a prior history of intestinal disorders. In some instances, symptoms have been reported to persist after Claravis treatment has been stopped. Patients experiencing abdominal pain, rectal bleeding or severe diarrhea should discontinue Claravis immediately (see ADVERSE REACTIONS, Gastrointestinal).
Skeletal
Bone Mineral Density
Effects of multiple courses of Claravis on the developing musculoskeletal system are unknown. There is some evidence that long-term, high dose, or multiple courses of therapy with isotretinoin have more of an effect than a single course of therapy on the musculoskeletal system. In an open-label clinical trial (N = 217) of a single course of therapy with Claravis for severe recalcitrant nodular acne, bone density measurements at several skeletal sites were not significantly decreased (lumbar spine change > -4% and total hip change > -5%) or were increased in the majority of patients. One patient had a decrease in lumbar spine bone mineral density >4% based on unadjusted data. Sixteen (7.9%) patients had decreases in lumbar spine bone mineral density > 4%, and all the other patients (92%) did not have significant decreases or had increases (adjusted for body mass index). Nine patients (4.5%) had a decrease in total hip bone mineral density > 5% based on unadjusted data. Twenty one (10.6%) patients had decreases in total hip bone mineral density > 5%, and all the other patients (89%) did not have significant decreases or had increases (adjusted for body mass index). Follow-up studies performed in eight of the patients with decreased bone mineral density for up to 11 months thereafter demonstrated increasing bone density in five patients at the lumbar spine, while the other three patients had lumbar spine bone density measurements below baseline values. Total hip bone mineral densities remained below baseline (range –1.6% to –7.6%) in five of eight patients (62.5%).
In a separate open-label extension study of ten patients, ages 13 to 18 years, who started a second course of Claravis 4 months after the first course, two patients showed a decrease in mean lumbar spine bone mineral density up to 3.25% (see PRECAUTIONS, Pediatric Use).
Spontaneous reports of osteoporosis, osteopenia, bone fractures, and delayed healing of bone fractures have been seen in the Claravis population. While causality to Claravis has not been established, an effect cannot be ruled out. Longer term effects have not been studied. It is important that Claravis be given at the recommended doses for no longer than the recommended duration.
Hyperostosis
A high prevalence of skeletal hyperostosis was noted in clinical trials for disorders of keratinization with a mean dose of 2.24 mg/kg/day. Additionally, skeletal hyperostosis was noted in six of eight patients in a prospective study of disorders of keratinization.6 Minimal skeletal hyperostosis and calcification of ligaments and tendons have also been observed by x-ray in prospective studies of nodular acne patients treated with a single course of therapy at recommended doses. The skeletal effects of multiple Claravis treatment courses for acne are unknown.
In a clinical study of 217 pediatric patients (12 to 17 years) with severe recalcitrant nodular acne, hyperostosis was not observed after 16 to 20 weeks of treatment with approximately 1 mg/kg/day of Claravis given in two divided doses. Hyperostosis may require a longer time frame to appear. The clinical course and significance remain unknown.
Vision Impairment
Visual problems should be carefully monitored. All Claravis patients experiencing visual difficulties should discontinue Claravis treatment and have an ophthalmological examination (see ADVERSE REACTIONS, Special Senses).
Corneal Opacities
Corneal opacities have occurred in patients receiving Claravis for acne and more frequently when higher drug dosages were used in patients with disorders of keratinization. The corneal opacities that have been observed in clinical trial patients treated with Claravis have either completely resolved or were resolving at follow-up 6 to 7 weeks after discontinuation of the drug (see ADVERSE REACTIONS, Special Senses).
Decreased Night Vision
Decreased night vision has been reported during Claravis therapy and in some instances the event has persisted after therapy was discontinued. Because the onset in some patients was sudden, patients should be advised of this potential problem and warned to be cautious when driving or operating any vehicle at night.
PRECAUTIONS
Claravis must only be prescribed by prescribers who are registered and activated with the iPLEDGE program. Claravis must only be dispensed by a pharmacy registered and activated with iPLEDGE, and must only be dispensed to patients who are registered and meet all the requirements of iPLEDGE. Registered and activated pharmacies must receive isotretinoin only from wholesalers registered with iPLEDGE.
iPLEDGE program requirements for wholesalers, prescribers, and pharmacists are described below:
Wholesalers
For the purpose of the iPLEDGE program, the term wholesaler refers to wholesaler, distributor, and/or chain pharmacy distributor. To distribute Claravis, wholesalers must be registered with iPLEDGE, and agree to meet all iPLEDGE requirements for wholesale distribution of isotretinoin products. Wholesalers must register with iPLEDGE by signing and returning the iPLEDGE wholesaler agreement that affirms they will comply with all iPLEDGE requirements for distribution of isotretinoin. These include:
- Registering prior to distributing isotretinoin and re-registering annually thereafter
- Distributing only FDA approved isotretinoin product
- Only shipping isotretinoin to
-wholesalers registered in the iPLEDGE program with prior written consent from the manufacturer or
-pharmacies licensed in the U.S. and registered and activated in the iPLEDGE program - Notifying the isotretinoin manufacturer (or delegate) of any non-registered and/or non-activated pharmacy or unregistered wholesaler that attempts to order isotretinoin
- Complying with inspection of wholesaler records for verification of compliance with the iPLEDGE program by the isotretinoin manufacturer (or delegate)
- Returning to the manufacturer (or delegate) any undistributed product if registration is revoked by the manufacturer or if the wholesaler chooses to not re-register annually
Prescribers
To prescribe isotretinoin, the prescriber must be registered and activated with the pregnancy risk management program iPLEDGE. Prescribers can register by signing and returning the completed registration form. Prescribers can only activate their registration by affirming that they meet requirements and will comply with all iPLEDGE requirements by attesting to the following points:
- I know the risk and severity of fetal injury/birth defects from isotretinoin.
- I know the risk factors for unplanned pregnancy and the effective measures for avoidance of unplanned pregnancy.
- I have the expertise to provide the patient with detailed pregnancy prevention counseling or I will refer her to an expert for such counseling, reimbursed by the manufacturer.
- I will comply with the iPLEDGE program requirements described in the booklets entitled The Guide to Best Practices for the iPLEDGE Program and The iPLEDGE Program Prescriber Contraception Counseling Guide.
- Before beginning treatment of female patients of childbearing potential with isotretinoin and on a monthly basis, the patient will be counseled to avoid pregnancy by using two forms of contraception simultaneously and continuously one month before, during, and one month after isotretinoin therapy, unless the patient commits to continuous abstinence.
- I will not prescribe isotretinoin to any female patient of childbearing potential until verifying she has a negative screening pregnancy test and monthly negative CLIA-certified (Clinical Laboratory Improvement Amendment) pregnancy tests. Patients should have a pregnancy test at the completion of the entire course of isotretinoin and another pregnancy test one month later.
- I will report any pregnancy case that I become aware of while the female patient is on isotretinoin or one month after the last dose to the pregnancy registry.
To prescribe isotretinoin, the prescriber must access the iPLEDGE system via the internet (www.ipledgeprogram.com) or telephone (1-866-495-0654) to:
- Register each patient in the iPLEDGE program.
- Confirm monthly that each patient has received counseling and education.
- For female patients of childbearing potential:
-
- Enter patient’s two chosen forms of contraception each month.
- Enter monthly result from CLIA-certified laboratory conducted pregnancy test.
Isotretinoin must only be prescribed to female patients who are known not to be pregnant as confirmed by a negative CLIA-certified laboratory conducted pregnancy test.
Isotretinoin must only be dispensed by a pharmacy registered and activated with the pregnancy risk management program iPLEDGE and only when the registered patient meets all the requirements of the iPLEDGE program. Meeting the requirements for a female patient of childbearing potential signifies that she:
- Has been counseled and has signed a Patient Information/Informed Consent About Birth Defects (for female patients who can get pregnant) form that contains warnings about the risk of potential birth defects if the fetus is exposed to isotretinoin. The patient must sign the informed consent form before starting treatment and patient counseling must also be done at that time and on a monthly basis thereafter.
-
Has had two negative urine or serum pregnancy tests with a sensitivity of at least 25 mIU/mL before receiving the initial isotretinoin prescription. The first test (a screening test) is obtained by the prescriber when the decision is made to pursue qualification of the patient for isotretinoin. The second pregnancy test (a confirmation test) must be done in a CLIA-certified laboratory. The interval between the two tests should be at least 19 days.
- For patients with regular menstrual cycles, the second pregnancy test should be done during the first 5 days of the menstrual period immediately preceding the beginning of isotretinoin therapy and after the patient has used two forms of contraception for one month.
-For patients with amenorrhea, irregular cycles, or using a contraceptive method that precludes withdrawal bleeding, the second pregnancy test must be done immediately preceding the beginning of isotretinoin therapy and after the patient has used two forms of contraception for one month. - Has had a negative result from a urine or serum pregnancy test in a CLIA-certified laboratory before receiving each subsequent course of isotretinoin. A pregnancy test must be repeated every month, in a CLIA-certified laboratory, prior to the female patient receiving each prescription.
- Has selected and has committed to use two forms of effective contraception simultaneously, at least one of which must be a primary form, unless the patient commits to continuous abstinence from heterosexual contact, or the patient has undergone a hysterectomy or bilateral oophorectomy, or has been medically confirmed to be post-menopausal. Patients must use two forms of effective contraception for at least one month prior to initiation of isotretinoin therapy, during isotretinoin therapy, and for one month after discontinuing isotretinoin therapy. Counseling about contraception and behaviors associated with an increased risk of pregnancy must be repeated on a monthly basis.
If the patient has unprotected heterosexual intercourse at any time one month before, during,
or one month after therapy, she must:
- Stop taking isotretinoin immediately, if on therapy
- Have a pregnancy test at least 19 days after the last act of unprotected heterosexual intercourse
- Start using two forms of effective contraception simultaneously again for one month before resuming isotretinoin therapy
- Have a second pregnancy test after using two forms of effective contraception for one month as described above depending on whether she has regular menses or not.
Effective forms of contraception include both primary and secondary forms of contraception:
Primary forms
- tubal sterilization
- partner's vasectomy
- intrauterine device
- hormonal (combination oral contraceptives, transdermal patch, injectables, implantables, or vaginal ring)
Secondary forms
Barrier forms:
- male latex condom with or without spermicide
- diaphragm with spermicide
- cervical cap with spermicide
Other:
- vaginal sponge (contains spermicide)
Any birth control method can fail. There have been reports of pregnancy from female patients who have used oral contraceptives, as well as transdermal patch/injectable/implantable/vaginal ring hormonal birth control products; these pregnancies occurred while these patients were taking Claravis. These reports are more frequent for female patients who use only a single method of contraception. Therefore, it is critically important that female patients of childbearing potential use two effective forms of contraception simultaneously. Patients must receive written warnings about the rates of possible contraception failure (included in patient education kits).
Using two forms of contraception simultaneously substantially reduces the chances that a female will become pregnant over the risk of pregnancy with either form alone. A drug interaction that decreases effectiveness of hormonal contraceptives has not been entirely ruled out for Claravis (see PRECAUTIONS, Drug Interactions). Although hormonal contraceptives are highly effective, prescribers are advised to consult the package insert of any medication administered concomitantly with hormonal contraceptives, since some medications may decrease the effectiveness of these birth control products.
Patients should be prospectively cautioned not to self-medicate with the herbal supplement St. John’s Wort because a possible interaction has been suggested with hormonal contraceptives based on reports of breakthrough bleeding on oral contraceptives shortly after starting St. John's Wort. Pregnancies have been reported by users of combined hormonal contraceptives who also used some form of St. John's Wort.
If a pregnancy does occur during isotretinoin treatment, isotretinoin must be discontinued immediately. The patient should be referred to an Obstetrician-Gynecologist experienced in reproductive toxicity for further evaluation and counseling. Any suspected fetal exposure during or one month after isotretinoin therapy must be reported immediately to the FDA via the MedWatch number 1-800-FDA-1088 and also the iPLEDGE pregnancy registry at 1-866-495-0654 or via the internet (www.ipledgeprogram.com).
All Patients
Isotretinoin is contraindicated in female patients who are pregnant. To receive isotretinoin all patients must meet all of the following conditions:
- Must be registered with the iPLEDGE program by the prescriber
- Must understand that severe birth defects can occur with the use of isotretinoin by female patients
- Must be reliable in understanding and carrying out instructions
- Must sign a Patient Information/Informed Consent (for all patients) form that contains warnings about the potential risks associated with isotretinoin
- Must fill and pick up the prescription within 7 days of the date of specimen collection for the pregnancy test for female patients of childbearing potential
- Must fill and pick up the prescription within 30 days of the office visit for male patients and female patients not of childbearing potential
- Must not donate blood while on isotretinoin and for one month after treatment has ended
- Must not share isotretinoin with anyone, even someone who has similar symptoms
Female Patients of Childbearing Potential
Isotretinoin is contraindicated in female patients who are pregnant. In addition to the requirements for all patients described above, female patients of childbearing potential must meet the following conditions:
- Must NOT be pregnant or breast-feeding
- Must comply with the required pregnancy testing at a CLIA-certified laboratory
- Must fill and pick up the prescription within 7 days of the date of specimen collection for the pregnancy test
- Must be capable of complying with the mandatory contraceptive measures required for isotretinoin therapy, or commit to continuous abstinence from heterosexual intercourse, and understand behaviors associated with an increased risk of pregnancy
- Must understand that it is her responsibility to avoid pregnancy one month before, during and one month after isotretinoin therapy
- Must have signed an additional Patient Information/Informed Consent About Birth Defects (for female patients who can get pregnant) form, before starting isotretinoin, that contains warnings about the risk of potential birth defects if the fetus is exposed to isotretinoin
- Must access the iPLEDGE system via the internet (www.ipledgeprogram.com) or telephone (1-866-495-0654), before starting isotretinoin, on a monthly basis during therapy, and one month after the last dose to answer questions on the program requirements and to enter the patient’s two chosen forms of contraception
- Must have been informed of the purpose and importance of providing information to the iPLEDGE program should she become pregnant while taking isotretinoin or within one month of the last dose
Pharmacists
To dispense isotretinoin, pharmacies must be registered and activated with the pregnancy risk management program iPLEDGE.
The Responsible Site Pharmacist must register the pharmacy by signing and returning the completed registration form. After registration, the Responsible Site Pharmacist can only activate the pharmacy registration by affirming that they meet requirements and will comply with all iPLEDGE requirements by attesting to the following points:
- I know the risk and severity of fetal injury/birth defects from isotretinoin.
- I will train all pharmacists, who participate in the filling and dispensing of isotretinoin prescriptions, on the iPLEDGE program requirements.
- I will comply and seek to ensure all pharmacists who participate in the filling and dispensing of isotretinoin prescriptions comply with the iPLEDGE program requirements described in the booklet entitled Pharmacist Guide for the iPLEDGE Program.
- I will obtain Claravis product only from iPLEDGE registered wholesalers.
- I will not sell, buy, borrow, loan or otherwise transfer isotretinoin in any manner to or from another pharmacy.
- I will return to the manufacturer (or delegate) any unused product if registration is revoked by the manufacturer or if the pharmacy chooses to not reactivate annually.
- I will not fill isotretinoin for any party other than a qualified patient.
To dispense isotretinoin, the pharmacist must:
- be trained by the Responsible Site Pharmacist concerning the iPLEDGE program requirements.
- obtain authorization from the iPLEDGE program via the internet (www.ipledgeprogram.com) or telephone (1-866-495-0654) for every isotretinoin prescription. Authorization signifies that the patient has met all program requirements and is qualified to receive isotretinoin.
- write the Risk Management Authorization (RMA) number on the prescription.
Claravis must only be dispensed:
- in no more than a 30 day supply
- with a Claravis Medication Guide
- after authorization from the iPLEDGE program
- prior to the “do not dispense to patient after” date provided by the iPLEDGE system (within 30 days of the office visit for male patients and female patients not of childbearing potential and within 7 days of the date of specimen collection for female patients of childbearing potential)
- with a new prescription for refills and another authorization from the iPLEDGE program (No automatic refills are allowed)
A Claravis Medication Guide must be given to the patient each time Claravis is dispensed, as required by law. This Claravis Medication Guide is an important part of the risk management program for the patients.
Claravis must not be prescribed, dispensed or otherwise obtained through the internet or any other means outside of the iPLEDGE program. Only FDA-approved isotretinoin products must be distributed, prescribed, dispensed, and used. Patients must fill isotretinoin prescriptions only at US licensed pharmacies.
A description of the iPLEDGE program educational materials available with iPLEDGE is provided below. The main goal of these educational materials is to explain the iPLEDGE program requirements and to reinforce the educational messages.
- TheGuide to Best Practices for the iPLEDGE Program includes: isotretinoin teratogenic potential, information on pregnancy testing, and the method to complete a qualified isotretinoin prescription.
- TheiPLEDGE Program Prescriber Contraception Counseling Guide includes: specific information about effective contraception, the limitations of contraceptive methods, behaviors associated with an increased risk of contraceptive failure and pregnancy and the methods to evaluate pregnancy risk.
- The Pharmacist Guide for the iPLEDGE Program includes: isotretinoin teratogenic potential and the method to obtain authorization to dispense an isotretinoin prescription.
- The iPLEDGE program is a systematic approach to comprehensive patient education about their responsibilities and includes education for contraception compliance and reinforcement of educational messages. The iPLEDGE program includes information on the risks and benefits of isotretinoin which is linked to the Medication Guide dispensed by pharmacists with each isotretinoin prescription.
- Female patients not of childbearing potential and male patients, and female patients of childbearing potential are provided with separate booklets. Each booklet contains information on isotretinoin therapy including precautions and warnings, a Patient Information/Informed Consent (for all patients) form, and a toll-free line which provides isotretinoin information in two languages.
- The booklet for female patients not of childbearing potential and male patients, TheiPLEDGEProgram Guide to Isotretinoin for Male Patients and Female Patients Who Cannot Get Pregnant, also includes information about male reproduction and a warning not to share isotretinoin with others or to donate blood during isotretinoin therapy and for one month following discontinuation of isotretinoin
- The booklet for female patients of childbearing potential, TheiPLEDGEProgram Guide to Isotretinoin for Female Patients Who Can Get Pregnant, includes a referral program that offers female patients free contraception counseling, reimbursed by the manufacturer, by a reproductive specialist; and a second Patient Information/Informed Consent About Birth Defects (for female patients who can get pregnant) form concerning birth defects
- The booklet, The iPLEDGE Program Birth Control Workbook includes information on the types of contraceptive methods, the selection and use of appropriate, effective contraception, the rates of possible contraceptive failure and a toll-free contraception counseling line.
- In addition, there is a patient educational DVD with the following videos — “Be Prepared, Be Protected” and “Be Aware: The Risk of Pregnancy While on Isotretinoin” (see Information for Patients).
General
Although an effect of Claravis on bone loss is not established, physicians should use caution when prescribing Claravis to patients with a genetic predisposition for age-related osteoporosis, a history of childhood osteoporosis conditions, osteomalacia, or other disorders of bone metabolism. This would include patients diagnosed with anorexia nervosa and those who are on chronic drug therapy that causes drug-induced osteoporosis/osteomalacia and/or affects vitamin D metabolism, such as systemic corticosteroids and any anticonvulsant.
Patients may be at increased risk when participating in sports with repetitive impact where the risks of spondylolisthesis with and without pars fractures and hip growth plate injuries in early and late adolescence are known. There are spontaneous reports of fractures and/or delayed healing in patients while on therapy with Claravis or following cessation of therapy with Claravis while involved in these activities. While causality to Claravis has not been established, an effect must not be ruled out.
Information for Patients
See PRECAUTIONS and Boxed CONTRAINDICATIONS AND WARNINGS .
- Patients must be instructed to read the Medication Guide supplied as required by law when Claravis is dispensed. The complete text of the Medication Guide is reprinted at the end of this document. For additional information, patients must also be instructed to read the iPLEDGE program patient educational materials. All patients must sign the Patient Information/Informed Consent (for all patients) form.
- Female patients of childbearing potential must be instructed that they must not be pregnant when Claravis therapy is initiated, and that they should use two forms of effective contraception simultaneously for one month before starting Claravis, while taking Claravis, and for one month after Claravis has been stopped, unless they commit to continuous abstinence from heterosexual intercourse. They should also sign a second Patient Information/Informed Consent About Birth Defects (for female patients who can get pregnant) form prior to beginning Claravis therapy. They should be given an opportunity to view the patient DVD provided by the manufacturer to the prescriber. The DVD includes information about contraception, the most common reasons that contraception fails, and the importance of using two forms of effective contraception when taking teratogenic drugs and comprehensive information about types of potential birth defects, which could occur if a female patient who is pregnant takes Claravis at any time during pregnancy. Female patients should be seen by their prescribers monthly and have a urine or serum pregnancy test, in a CLIA-certified laboratory, performed each month during treatment to confirm negative pregnancy status before another Claravis prescription is written (see Boxed CONTRAINDICATIONS AND WARNINGS and PRECAUTIONS).
- Claravis is found in the semen of male patients taking Claravis, but the amount delivered to a female partner would be about one million times lower than an oral dose of 40 mg. While the no-effect limit for isotretinoin induced embryopathy is unknown, 20 years of postmarketing reports include four with isolated defects compatible with features of retinoid exposed fetuses; however two of these reports were incomplete, and two had other possible explanations for the defects observed.
- Prescribers should be alert to the warning signs of psychiatric disorders to guide patients to receive the help they need. Therefore, prior to initiation of isotretinoin treatment, patients and family members should be asked about any history of psychiatric disorder, and at each visit during treatment patients should be assessed for symptoms of depression, mood disturbance, psychosis, or aggression to determine if further evaluation may be necessary. Signs and symptoms of depression include sad mood, hopelessness, feelings of guilt, worthlessness or helplessness, loss of pleasure or interest in activities, fatigue, difficulty concentrating, change in sleep pattern, change in weight or appetite, suicidal thoughts or attempts, restlessness, irritability, acting on dangerous impulses, and persistent physical symptoms unresponsive to treatment. Patients should stop isotretinoin and the patient or a family member should promptly contact their prescriber if the patient develops depression, mood disturbance, psychosis, or aggression, without waiting until the next visit. Discontinuation of isotretinoin treatment may be insufficient; further evaluation may be necessary. While such monitoring may be helpful, it may not detect all patients at risk. Patients may report mental health problems or family history of psychiatric disorders. These reports should be discussed with the patient and/or the patient’s family. A referral to a mental health professional may be necessary. The physician should consider whether isotretinoin therapy is appropriate in this setting; for some patients the risks may outweigh the benefits of isotretinoin therapy.
- Patients must be informed that some patients, while taking isotretinoin or soon after stopping isotretinoin, have become depressed or developed other serious mental problems. Symptoms of depression include sad, “anxious” or empty mood, irritability, acting on dangerous impulses, anger, loss of pleasure or interest in social or sports activities, sleeping too much or too little, changes in weight or appetite, school or work performance going down, or trouble concentrating. Some patients taking isotretinoin have had thoughts about hurting themselves or putting an end to their own lives (suicidal thoughts). Some people tried to end their own lives. And some people have ended their own lives. There were reports that some of these people did not appear depressed. There have been reports of patients on isotretinoin becoming aggressive or violent. No one knows if isotretinoin caused these behaviors or if they would have happened even if the person did not take isotretinoin. Some people have had other signs of depression while taking isotretinoin.
- Patients must be informed that they must not share Claravis with anyone else because of the risk of birth defects and other serious adverse events.
- Patients must be informed not to donate blood during therapy and for one month following discontinuation of the drug because the blood might be given to a pregnant female patient whose fetus must not be exposed to Claravis.
- Patients should be reminded to take Claravis with a meal (see DOSAGE AND ADMINISTRATION). To decrease the risk of esophageal irritation, patients should swallow the capsules with a full glass of liquid.
- Patients should be informed that transient exacerbation (flare) of acne has been seen, generally during the initial period of therapy.
- Wax epilation and skin resurfacing procedures (such as dermabrasion, laser) should be avoided during Claravis therapy and for at least 6 months thereafter due to the possibility of scarring (see ADVERSE REACTIONS, Skin and Appendages).
- Patients should be advised to avoid prolonged exposure to UV rays or sunlight.
- Patients should be informed that they may experience decreased tolerance to contact lenses during and after therapy.
- Patients should be informed that approximately 16% of patients treated with Claravis in a clinical trial developed musculoskeletal symptoms (including arthralgia) during treatment. In general, these symptoms were mild to moderate, but occasionally required discontinuation of the drug. Transient pain in the chest has been reported less frequently. In the clinical trial, these symptoms generally cleared rapidly after discontinuation of Claravis, but in some cases persisted (see ADVERSE REACTIONS, Musculoskeletal). There have been rare postmarketing reports of rhabdomyolysis, some associated with strenuous physical activity (see Laboratory Tests, CPK).
- Pediatric patients and their caregivers should be informed that approximately 29% (104/358) of pediatric patients treated with Claravis developed back pain. Back pain was severe in 13.5% (14/104) of the cases and occurred at a higher frequency in female patients than male patients. Arthralgias were experienced in 22% (79/358) of pediatric patients. Arthralgias were severe in 7.6% (6/79) of patients. Appropriate evaluation of the musculoskeletal system should be done in patients who present with these symptoms during or after a course of Claravis. Consideration should be given to discontinuation of Claravis if any significant abnormality is found.
- Neutropenia and rare cases of agranulocytosis have been reported. Claravis should be discontinued if clinically significant decreases in white cell counts occur.
- Patients should be advised that severe skin reactions (Stevens-Johnson Syndrome and toxic epidermal necrolysis) have been reported in post-marketing data. Claravis should be discontinued if clinically significant skin reactions occur.
Hypersensitivity
Anaphylactic reactions and other allergic reactions have been reported. Cutaneous allergic reactions and serious cases of allergic vasculitis, often with purpura (bruises and red patches) of the extremities and extracutaneous involvement (including renal) have been reported. Severe allergic reaction necessitates discontinuation of therapy and appropriate medical management.
Drug Interactions
- Vitamin A: Because of the relationship of Claravis to vitamin A, patients should be advised against taking vitamin supplements containing vitamin A to avoid additive toxic effects.
- Tetracyclines: Concomitant treatment with Claravis and tetracyclines should be avoided because Claravis use has been associated with a number of cases of pseudotumor cerebri (benign intracranial hypertension), some of which involved concomitant use of tetracyclines.
- Micro-dosed Progesterone Preparations: Micro-dosed progesterone preparations (“minipills” that do not contain an estrogen) may be an inadequate method of contraception during Claravis therapy. Although other hormonal contraceptives are highly effective, there have been reports of pregnancy from female patients who have used combined oral contraceptives, as well as transdermal patch/injectable/implantable/vaginal ring hormonal birth control products. These reports are more frequent for female patients who use only a single method of contraception. It is not known if hormonal contraceptives differ in their effectiveness when used with Claravis. Therefore, it is critically important for female patients of childbearing potential to select and commit to use two forms of effective contraception simultaneously, at least one of which must be a primary form (see PRECAUTIONS).
- Norethindrone/ethinyl estradiol: In a study of 31 premenopausal female patients with severe recalcitrant nodular acne receiving OrthoNovum® 7/7/7 Tablets as an oral contraceptive agent, Claravis at the recommended dose of 1 mg/kg/day, did not induce clinically relevant changes in the pharmacokinetics of ethinyl estradiol and norethindrone and in the serum levels of progesterone, follicle-stimulating hormone (FSH) and luteinizing hormone (LH). Prescribers are advised to consult the package insert of medication administered concomitantly with hormonal contraceptives, since some medications may decrease the effectiveness of these birth control products.
- St. John’s Wort: Claravis use is associated with depression in some patients (see WARNINGS, Psychiatric Disorders and ADVERSE REACTIONS, Psychiatric). Patients should be prospectively cautioned not to self-medicate with the herbal supplement St. John’s Wort because a possible interaction has been suggested with hormonal contraceptives based on reports of breakthrough bleeding on oral contraceptives shortly after starting St. John’s Wort. Pregnancies have been reported by users of combined hormonal contraceptives who also used some form of St. John’s Wort.
- Phenytoin: Claravis has not been shown to alter the pharmacokinetics of phenytoin in a study in seven healthy volunteers. These results are consistent with the in vitro finding that neither isotretinoin nor its metabolites induce or inhibit the activity of the CYP 2C9 human hepatic P450 enzyme. Phenytoin is known to cause osteomalacia. No formal clinical studies have been conducted to assess if there is an interactive effect on bone loss between phenytoin and Claravis. Therefore, caution should be exercised when using these drugs together.
- Systemic Corticosteroids: Systemic corticosteroids are known to cause osteoporosis. No formal clinical studies have been conducted to assess if there is an interactive effect on bone loss between systemic corticosteroids and Claravis. Therefore, caution should be exercised when using these drugs together.
Laboratory Tests
• Pregnancy Test
- Female patients of childbearing potential must have had two negative urine or serum pregnancy tests with a sensitivity of at least 25 mIU/mL before receiving the initial Claravis prescription. The first test (a screening test) is obtained by the prescriber when the decision is made to pursue qualification of the patient for Claravis. The second pregnancy test (a confirmation test) must be done in a CLIA-certified laboratory. The interval between the two tests must be at least 19 days.
- For patients with regular menstrual cycles, the second pregnancy test must be done during the first 5 days of the menstrual period immediately preceding the beginning of Claravis therapy and after the patient has used 2 forms of contraception for 1 month.
- For patients with amenorrhea, irregular cycles, or using a contraceptive method that precludes withdrawal bleeding, the second pregnancy test must be done immediately preceding the beginning of Claravis therapy and after the patient has used 2 forms of contraception for 1 month.
- Each month of therapy, patients must have a negative result from a urine or serum pregnancy test. A pregnancy test must be repeated each month, in a CLIA-certified laboratory, prior to the female patient receiving each prescription.
- Lipids: Pretreatment and follow-up blood lipids should be obtained under fasting conditions. After consumption of alcohol, at least 36 hours should elapse before these determinations are made. It is recommended that these tests be performed at weekly or biweekly intervals until the lipid response to Claravis is established. The incidence of hypertriglyceridemia is one1 patient in four on Claravis therapy (see WARNINGS, Lipids).
- Liver Function Tests: Since elevations of liver enzymes have been observed during clinical trials, and hepatitis has been reported, pretreatment and follow-up liver function tests should be performed at weekly or biweekly intervals until the response to Claravis has been established (see WARNINGS, Hepatotoxicity).
- Glucose: Some patients receiving Claravis have experienced problems in the control of their blood sugar. In addition, new cases of diabetes have been diagnosed during Claravis therapy, although no causal relationship has been established.
- CPK: Some patients undergoing vigorous physical activity while on Claravis therapy have experienced elevated CPK levels; however, the clinical significance is unknown. There have been rare postmarketing reports of rhabdomyolysis, some associated with strenuous physical activity. In a clinical trial of 217 pediatric patients (12 to 17 years) with severe recalcitrant nodular acne, transient elevations in CPK were observed in 12% of patients, including those undergoing strenuous physical activity in association with reported musculoskeletal adverse events such as back pain, arthralgia, limb injury, or muscle sprain. In these patients, approximately half of the CPK elevations returned to normal within 2 weeks and half returned to normal within 4 weeks. No cases of rhabdomyolysis were reported in this trial.
Carcinogenesis, Mutagenesis and Impairment of Fertility
In male and female Fischer 344 rats given oral isotretinoin at dosages of 8 or 32 mg/kg/day (1.3 to 5.3 times the recommended clinical dose of 1 mg/kg/day, respectively, after normalization for total body surface area) for greater than 18 months, there was a dose-related increased incidence of pheochromocytoma relative to controls. The incidence of adrenal medullary hyperplasia was also increased at the higher dosage in both sexes. The relatively high level of spontaneous pheochromocytomas occurring in the male Fischer 344 rat makes it an equivocal model for study of this tumor; therefore, the relevance of this tumor to the human population is uncertain.
The Ames test was conducted with isotretinoin in two laboratories. The results of the tests in one laboratory were negative while in the second laboratory a weakly positive response (less than 1.6 x background) was noted in S. typhimurium TA100 when the assay was conducted with metabolic activation. No dose-response effect was seen and all other strains were negative. Additionally, other tests designed to assess genotoxicity (Chinese hamster cell assay, mouse micronucleus test, S. cerevisiae D7 assay, in vitro clastogenesis assay with human-derived lymphocytes, and unscheduled DNA synthesis assay) were all negative.
In rats, no adverse effects on gonadal function, fertility, conception rate, gestation or parturition were observed at oral dosages of isotretinoin of 2, 8, or 32 mg/kg/day (0.3, 1.3, or 5.3 times the recommended clinical dose of 1 mg/kg/day, respectively, after normalization for total body surface area).
In dogs, testicular atrophy was noted after treatment with oral isotretinoin for approximately 30 weeks at dosages of 20 or 60 mg/kg/day (10 or 30 times the recommended clinical dose of 1 mg/kg/day, respectively, after normalization for total body surface area). In general, there was microscopic evidence for appreciable depression of spermatogenesis but some sperm were observed in all testes examined and in no instance were completely atrophic tubules seen. In studies of 66 men, 30 of whom were patients with nodular acne under treatment with oral isotretinoin, no significant changes were noted in the count or motility of spermatozoa in the ejaculate. In a study of 50 men (ages 17 to 32 years) receiving Claravis therapy for nodular acne, no significant effects were seen on ejaculate volume, sperm count, total sperm motility, morphology or seminal plasma fructose.
Nursing Mothers
It is not known whether this drug is excreted in human milk. Because of the potential for adverse effects, nursing mothers should not receive Claravis.
Pediatric Use
The use of isotretinoin in pediatric patients less than 12 years of age has not been studied. The use of isotretinoin for the treatment of severe recalcitrant nodular acne in pediatric patients ages 12 to 17 years should be given careful consideration, especially for those patients where a known metabolic or structural bone disease exists (see PRECAUTIONS, General). Use of isotretinoin in this age group for severe recalcitrant nodular acne is supported by evidence from a clinical study comparing 103 pediatric patients (13 to 17 years) to 197 adult patients (≥18 years). Results from this study demonstrated that isotretinoin, at a dose of 1 mg/kg/day given in two divided doses, was equally effective in treating severe recalcitrant nodular acne in both pediatric and adult patients.
In studies with isotretinoin, adverse reactions reported in pediatric patients were similar to those described in adults except for the increased incidence of back pain and arthralgia (both of which were sometimes severe) and myalgia in pediatric patients (see ADVERSE REACTIONS).
In an open-label clinical trial (N = 217) of a single course of therapy with Claravis for severe recalcitrant nodular acne, bone density measurements at several skeletal sites were not significantly decreased (lumbar spine change > -4% and total hip change > -5%) or were increased in the majority of patients. One patient had a decrease in lumbar spine bone mineral density > 4% based on unadjusted data. Sixteen (7.9%) patients had decreases in lumbar spine bone mineral density > 4%, and all the other patients (92%) did not have significant decreases or had increases (adjusted for body mass index). Nine patients (4.5%) had a decrease in total hip bone mineral density > 5% based on unadjusted data. Twenty-one (10.6%) patients had decreases in total hip bone mineral density > 5%, and all the other patients (89%) did not have significant decreases or had increases (adjusted for body mass index). Follow-up studies performed in eight of the patients with decreased bone mineral density for up to 11 months thereafter demonstrated increasing bone density in five patients at the lumbar spine, while the other three patients had lumbar spine bone density measurements below baseline values. Total hip bone mineral densities remained below baseline (range -1.6% to -7.6%) in five of eight patients (62.5%).
In a separate open-label extension study of ten patients, ages 13 to 18 years, who started a second course of isotretinoin 4 months after the first course, two patients showed a decrease in mean lumbar spine bone mineral density up to 3.25% (see WARNINGS, Skeletal, Bone Mineral Density).
Geriatric Use
Clinical studies of isotretinoin did not include sufficient numbers of subjects aged 65 years and over to determine whether they respond differently from younger subjects. Although reported clinical experience has not identified differences in responses between elderly and younger patients, effects of aging might be expected to increase some risks associated with isotretinoin therapy (see WARNINGS and PRECAUTIONS).
ADVERSE REACTIONS
Clinical Trials and Postmarketing Surveillance
The adverse reactions listed below reflect the experience from investigational studies of Claravis, and the postmarketing experience. The relationship of some of these events to Claravis therapy is unknown. Many of the side effects and adverse reactions seen in patients receiving Claravis are similar to those described in patients taking very high doses of vitamin A (dryness of the skin and mucous membranes, e.g., of the lips, nasal passage, and eyes).
Dose Relationship
Cheilitis and hypertriglyceridemia are usually dose related. Most adverse reactions reported in clinical trials were reversible when therapy was discontinued; however, some persisted after cessation of therapy (see WARNINGS and ADVERSE REACTIONS:).
Body as a Whole
Allergic reactions, including vasculitis, systemic hypersensitivity (see PRECAUTIONS, Hypersensitivity), edema, fatigue, lymphadenopathy, weight loss.
Cardiovascular
Palpitation, tachycardia, vascular thrombotic disease, stroke.
Endocrine/Metabolic
Hypertriglyceridemia (see WARNINGS, Lipids), alterations in blood sugar levels (see PRECAUTIONS, Laboratory Tests).
Gastrointestinal
Inflammatory bowel disease (see WARNINGS, Inflammatory Bowel Disease), hepatitis (see WARNINGS, Hepatotoxicity), pancreatitis (see WARNINGS, Lipids), bleeding and inflammation of the gums, colitis, esophagitis/esophageal ulceration, ileitis, nausea, other nonspecific gastrointestinal symptoms.
Hematologic
Allergic reactions (see PRECAUTIONS, Hypersensitivity), anemia, thrombocytopenia, neutropenia, rare reports of agranulocytosis (see PRECAUTIONS, Information for Patients). See PRECAUTIONS, Laboratory Tests for other hematological parameters.
Musculoskeletal
Skeletal hyperostosis, calcification of tendons and ligaments, premature epiphyseal closure, decreases in bone mineral density (see WARNINGS, Skeletal), musculoskeletal symptoms (sometimes severe) including back pain, myalgia, and arthralgia (see PRECAUTIONS, Information for Patients), transient pain in the chest (see PRECAUTIONS, Information for Patients), arthritis, tendonitis, other types of bone abnormalities, elevations of CPK/rare reports of rhabdomyolysis (see PRECAUTIONS, Laboratory Tests).
Neurological
Pseudotumor cerebri (see WARNINGS, Pseudotumor Cerebri), dizziness, drowsiness, headache, insomnia, lethargy, malaise, nervousness, paresthesias, seizures, stroke, syncope, weakness.
Psychiatric
Suicidal ideation, suicide attempts, suicide, depression, psychosis, aggression, violent behaviors (see WARNINGS, Psychiatric Disorders), emotional instability.
Of the patients reporting depression, some reported that the depression subsided with discontinuation of therapy and recurred with reinstitution of therapy.
Reproductive System
Abnormal menses.
Respiratory
Bronchospasms (with or without a history of asthma), respiratory infection, voice alteration.
Skin and Appendages
Acne fulminans, alopecia (which in some cases persists), bruising, cheilitis (dry lips), dry mouth, dry nose, dry skin, epistaxis, eruptive xanthomas,7 erythema multiforme, flushing, fragility of skin, hair abnormalities, hirsutism, hyperpigmentation and hypopigmentation, infections (including disseminated herpes simplex), nail dystrophy, paronychia, peeling of palms and soles, photoallergic/photosensitizing reactions, pruritus, pyogenic granuloma, rash (including facial erythema, seborrhea, and eczema), Stevens-Johnson syndrome, sunburn susceptibility increased, sweating, toxic epidermal necrolysis, urticaria, vasculitis (including Wegener's granulomatosis; see PRECAUTIONS, Hypersensitivity), abnormal wound healing (delayed healing or exuberant granulation tissue with crusting; see PRECAUTIONS, Information for Patients).
Special Senses: Hearing: hearing impairment (see WARNINGS, Hearing Impairment), tinnitus.
Vision: corneal opacities (see WARNINGS, Corneal Opacities), decreased night vision which may persist (see WARNINGS, Decreased Night Vision), cataracts, color vision disorder, conjunctivitis, dry eyes, eyelid inflammation, keratitis, optic neuritis, photophobia, visual disturbances.
Urinary System: glomerulonephritis (see PRECAUTIONS, Hypersensitivity), nonspecific urogenital findings (see PRECAUTIONS, Laboratory Tests for other urological parameters).
Laboratory
Elevation of plasma triglycerides (see WARNINGS, Lipids), decrease in serum high-density lipoprotein (HDL) levels, elevations of serum cholesterol during treatment.
Increased alkaline phosphatase, SGOT (AST), SGPT (ALT), GGTP or LDH (see WARNINGS, Hepatotoxicity).
Elevation of fasting blood sugar, elevations of CPK (see PRECAUTIONS, Laboratory Tests), hyperuricemia.
Decreases in red blood cell parameters, decreases in white blood cell counts (including severe neutropenia and rare reports of agranulocytosis; (see PRECAUTIONS, Information for Patients), elevated sedimentation rates, elevated platelet counts, thrombocytopenia.
White cells in the urine, proteinuria, microscopic or gross hematuria.
OVERDOSAGE
The oral LD50 of isotretinoin is greater than 4000 mg/kg in rats and mice (>600 times the recommended clinical dose of 1 mg/kg/day after normalization of the rat dose for total body surface area and >300 times the recommended clinical dose of 1 mg/kg/day after normalization of the mouse dose for total body surface area) and is approximately 1960 mg/kg in rabbits (653 times the recommended clinical dose of 1 mg/kg/day after normalization for total body surface area). In humans, overdosage has been associated with vomiting, facial flushing, cheilosis, abdominal pain, headache, dizziness, and ataxia. These symptoms quickly resolve without apparent residual effects.
Claravis causes serious birth defects at any dosage (see Boxed CONTRAINDICATIONS AND WARNINGS ). Female patients of childbearing potential who present with isotretinoin overdose must be evaluated for pregnancy. Patients who are pregnant should receive counseling about the risks to the fetus, as described in the Boxed CONTRAINDICATIONS AND WARNINGS . Non-pregnant patients must be warned to avoid pregnancy for at least one month and receive contraceptive counseling as described in PRECAUTIONS. Educational materials for such patients can be obtained by calling the manufacturer. Because an overdose would be expected to result in higher levels of isotretinoin in semen than found during a normal treatment course, male patients should use a condom, or avoid reproductive sexual activity with a female patient who is or might become pregnant, for one month after the overdose. All patients with isotretinoin overdose should not donate blood for at least one month.
DOSAGE AND ADMINISTRATION
Claravis should be administered with a meal (see PRECAUTIONS, Information for Patients).
The recommended dosage range for Claravis is 0.5 to 1 mg/kg/day given in two divided doses with food for 15 to 20 weeks. In studies comparing 0.1, 0.5, and 1 mg/kg/day,8 it was found that all dosages provided initial clearing of disease, but there was a greater need for retreatment with the lower dosages. During treatment, the dose may be adjusted according to response of the disease and/or the appearance of clinical side effects – some of which may be dose related. Adult patients whose disease is very severe with scarring or is primarily manifested on the trunk may require dose adjustments up to 2 mg/kg/day, as tolerated. Failure to take Claravis with food will significantly decrease absorption. Before upward dose adjustments are made, the patients should be questioned about their compliance with food instructions.
The safety of once daily dosing with Claravis has not been established. Once daily dosing is not recommended.
If the total nodule count has been reduced by more than 70% prior to completing 15 to 20 weeks of treatment, the drug may be discontinued. After a period of 2 months or more off therapy, and if warranted by persistent or recurring severe nodular acne, a second course of therapy may be initiated. The optimal interval before retreatment has not been defined for patients who have not completed skeletal growth. Long-term use of Claravis, even in low doses, has not been studied, and is not recommended. It is important that Claravis be given at the recommended doses for no longer than the recommended duration. The effect of long-term use of Claravis on bone loss is unknown (see WARNINGS, Skeletal, Bone Mineral Density, Hyperostosis and Premature Epiphyseal Closure).
Contraceptive measures must be followed for any subsequent course of therapy (see PRECAUTIONS).
| Body Weight | Total mg/day | |||
| kilograms | pounds | 0.5 mg/kg | 1 mg/kg | 2 mg/kg |
| 40 | 88 | 20 | 40 | 80 |
| 50 | 110 | 25 | 50 | 100 |
| 60 | 132 | 30 | 60 | 120 |
| 70 | 154 | 35 | 70 | 140 |
| 80 | 176 | 40 | 80 | 160 |
| 90 | 198 | 45 | 90 | 180 |
| 100 | 220 | 50 | 100 | 200 |
INFORMATION FOR PHARMACISTS
Access the iPLEDGE system via the internet (www.ipledgeprogram.com) or telephone (1-866-495-0654) to obtain an authorization and the “do not dispense to patient after” date. Claravis must only be dispensed in no more than a 30-day supply.
REFILLS REQUIRE A NEW PRESCRIPTION AND A NEW AUTHORIZATION FROM THE iPLEDGE SYSTEM.
A Claravis Medication Guide must be given to the patient each time Claravis is dispensed, as required by law. This Claravis Medication Guide is an important part of the risk management program for the patient.
HOW SUPPLIED
Claravis™ (isotretinoin capsules, USP) is available as:
| 20 mg: | Two-piece hard gelatin capsule with brown opaque cap and brown opaque body filled with yellow oily dispersion. Imprinted in white ink barr on one piece and 935 on the other piece. |
|
| Available in cartons of 30 capsules containing 3 prescription blister packs of 10 capsules NDC 54868-6307-0. |
| 30 mg: | Two-piece hard gelatin capsule with orange opaque cap and orange opaque body filled with yellow oily dispersion. Imprinted in black ink barr on one piece and 454 on the other piece. |
|
| Available in cartons of 30 capsules containing 3 prescription blister packs of 10 capsules NDC 54868-6185-0. |
| 40 mg: | Two-piece hard gelatin capsule with light orange opaque cap and light orange opaque body filled with yellow oily dispersion. Imprinted in black ink barr on one piece and 936 on the other piece. |
|
| Available in cartons of 30 capsules containing 3 prescription blister packs of 10 capsules NDC 54868-6308-0. |
Store at 20° to 25°C (68° to 77°F) [See USP Controlled Room Temperature].
Protect from light.
KEEP THIS AND ALL MEDICATIONS OUT OF THE REACH OF CHILDREN
REFERENCES
1. Peck GL, Olsen TG, Yoder FW, et al. Prolonged remissions of cystic and conglobate acne with 13-cis-retinoic acid. N Engl J Med 300:329-333, 1979. 2. Pochi PE, Shalita AR, Strauss JS, Webster SB. Report of the consensus conference on acne classification. J Am Acad Dermatol 24:495-500, 1991. 3. Farrell LN, Strauss JS, Stranieri AM. The treatment of severe cystic acne with 13-cis-retinoic acid: evaluation of sebum production and the clinical response in a multiple-dose trial. J Am Acad Dermatol 3:602-611, 1980. 4. Jones H, Blanc D, Cunliffe WJ. 13-cis-retinoic acid and acne. Lancet 2:1048-1049, 1980. 5. Katz RA, Jorgensen H, Nigra TP. Elevation of serum triglyceride levels from oral isotretinoin in disorders of keratinization. Arch Dermatol 116:1369-1372, 1980. 6. Ellis CN, Madison KC, Pennes DR, Martel W, Voorhees JJ. Isotretinoin therapy is associated with early skeletal radiographic changes. J Am Acad Dermatol 10:1024-1029, 1984. 7. Dicken CH, Connolly SM. Eruptive xanthomas associated with isotretinoin (13-cis-retinoic acid). Arch Dermatol 116:951-952, 1980. 8. Strauss JS, Rapini RP, Shalita AR, et al. Isotretinoin therapy for acne: results of a multicenter dose-response study. J Am Acad Dermatol 10:490-496, 1984.
OrthoNovum 7/7/7 is a registered trademark of Ortho-McNeil Pharmaceutical, Inc.
---------------------------------------------------------------------------------------------------------------------
Patient Information/Informed Consent About Birth Defects (for femalepatients who canget pregnant)
To be completed by the patient (and her parent or guardian* if patient is under age 18) and signed by her doctor.
Read each item below and initial in the space provided to show that you understand each item and agree to follow your doctor’s instructions. Do not sign this consent and do not take isotretinoin if there is anything that you do not understand.
*A parent or guardian of a minor patient (under age 18) must also read and initial each item before signing the consent.
______________________________________________________________________________
(Patient’s Name)
- I understand that there is a very high chance that my unborn baby could have severe birth defects if I am pregnant or become pregnant while taking isotretinoin. This can happen with any amount and even if taken for short periods of time. This is why I must not be pregnant while taking isotretinoin.
Initial: __________ - I understand that I must not get pregnant one month before, during the entire time of my treatment, and for one month after the end of my treatment with isotretinoin.
Initial: __________ - I understand that I must avoid sexual intercourse completely, or I must use two separate, effective forms of birth control (contraception) at the same time. The only exceptions are if I have had surgery to remove the uterus (a hysterectomy) or both of my ovaries (bilateral oophorectomy), or my doctor has medically confirmed that I am post-menopausal.
Initial: __________ - I understand that hormonal birth control products are among the most effective forms of birth control. Combination birth control pills and other hormonal products include skin patches, shots, under-the-skin implants, vaginal rings, and intrauterine devices (IUDs). Any form of birth control can fail. That is why I must use two different birth control methods at the same time, starting one month before, during, and for one month after stopping therapy every time I have sexual intercourse, even if one of the methods I choose is hormonal birth control.
Initial: __________ - I understand that the following are effective forms of birth control:
Primary form
- tubal sterilization (tying my tubes)
- partner's vasectomy
- intrauterine device
- hormonal (combination birth control pills, skin patches, shots, under-the skin implants, or vaginal ring.
Secondary forms
- Barrier forms:
- male latex condom with or without spermicide
- diaphragm with spermicide
- cervical cap with spermicide
Other:
- vaginal sponge (contains spermicide)
-
A diaphragm, and cervical cap must each be used with spermicide, a special cream that kills sperm.
I understand that at least one of my two forms of birth control must be a primary method.
Initial: __________ - I will talk with my doctor about any medicines including herbal products I plan to take during my isotretinoin treatment because hormonal birth control methods may not work if I am taking certain medicines or herbal products.
Initial: __________ - I may receive a free birth control counseling session from a doctor or other family planning expert. My isotretinoin doctor can give me an isotretinoin Patient Referral Form for this free consultation.
Initial: __________ - I must begin using the birth control methods I have chosen as described above at least one month before I start taking isotretinoin.
Initial: __________ - I cannot get my first prescription for isotretinoin unless my doctor has told me that I have two negative pregnancy test results. The first pregnancy test should be done when my doctor decides to prescribe isotretinoin. The second pregnancy test must be done in a lab during the first 5 days of my menstrual period right before starting isotretinoin therapy treatment, or as instructed by my doctor. I will then have one pregnancy test; in a lab.
- every month during treatment
- at the end of treatment
- and 1 month after stopping treatment
-
I must not start taking isotretinoin until I am sure that I am not pregnant, have negative results from two pregnancy tests, and the second test has been done in a lab.
Initial:__________
- I have read and understand the materials my doctor has given to me, including TheiPLEDGEProgram Guide for Isotretinoin for Female Patients Who Can Get Pregnant, TheiPLEDGEBirth Control Workbook and The iPLEDGE Program Patient Introductory Brochure.
My doctor gave me and asked me to watch the DVD containing a video about birth control and a video about birth defects and isotretinoin.
I was told about a private counseling line that I may call for more information about birth control. I have received information on emergency birth control.
Initial: __________
- I must stop taking isotretinoin right away and call my doctor if I get pregnant, miss my expected menstrual period, stop using birth control, or have sexual intercourse without using my two birth control methods at any time.
Initial: __________ - My doctor gave me information about the purpose and importance of providing information to the iPLEDGE program should I become pregnant while taking isotretinoin or within one month of the last dose. I also understand that if I become pregnant, information about my pregnancy, my health, and my baby’s health may be shared with the makers of isotretinoin, authorized parties who maintain the iPLEDGE program for the makers of isotretinoin, and government health regulatory authorities.
Initial: __________ - I understand that being qualified to receive isotretinoin in the iPLEDGE program means
that I:
- have had two negative urine or blood pregnancy tests before receiving the first isotretinoin prescription. The second test must be done in a lab. I must have a negative result from a urine or blood pregnancy test done in a lab repeated each month before I receive another isotretinoin prescription.
- have chosen and agreed to use two forms of effective birth control at the same time. At least one method must be a primary form of birth control, unless I have chosen never to have sexual contact with a male (abstinence), or I have undergone a hysterectomy. I must use two forms of birth control for at least one month before I start isotretinoin therapy, during therapy, and for one month after stopping therapy. I must receive counseling, repeated on a monthly basis, about birth control and behaviors associated with an increased risk of pregnancy.
- have signed a Patient Information/Informed Consent About Birth Defects (for female patients who can get pregnant) that contains warnings about the chance of possible birth defects if I am pregnant or become pregnant and my unborn baby is exposed to isotretinoin.
- have interacted with the iPLEDGE program before starting isotretinoin and on a monthly basis to answer questions on the program requirements and to enter my two chosen forms of birth control.
Initial: ______
My doctor has answered all my questions about isotretinoin and I understand that it is my responsibility not to get pregnant one month before, during isotretinoin treatment, or for one month after I stop taking isotretinoin.
Initial: ______
I now authorize my doctor ________________ to begin my treatment with isotretinoin.
Patient Signature:_____________________________________ Date: ______
Parent/Guardian Signature (if under age 18):________________ Date:______
Please print: Patient Name and Address_______________________________
______________________________ Telephone _______________________
I have fully explained to the patient, __________________, the nature and purpose of the treatment described above and the risks to female patients of childbearing potential. I have asked the patient if she has any questions regarding her treatment with isotretinoin and have answered those questions to the best of my ability.
Doctor Signature: __________________________________ Date: ______
PLACE THE ORIGINAL SIGNED DOCUMENTS IN THE PATIENT’S MEDICAL RECORD. PLEASE PROVIDE A COPY TO THE PATIENT.
--------------------------------------------------------------------------------------------------------------------
Patient Information/Informed Consent (for all patients)
To be completed by patient (and parent or guardian if patient is under age 18) and signed by the doctor.
Read each item below and initial in the space provided if you understand each item and agree to follow your doctor’s instructions. A parent or guardian of a patient under age 18 must also read and understand each item before signing the agreement.
Do not sign this agreement and do not take isotretinoin if there is anything that you do not understand about all the information you have received about using isotretinoin.
- I,__________________________________________________________________________
(Patient's Name) understand that isotretinoin is a medicine used to treat severe nodular acne that cannot be cleared up by any other acne treatments, including antibiotics. In severe nodular acne, many red, swollen, tender lumps form in the skin. If untreated, severe nodular acne can lead to permanent scars.
Initials: __________ - My doctor has told me about my choices for treating my acne.
Initials: __________ - I understand that there are serious side effects that may happen while I am taking isotretinoin. These have been explained to me. These side effects include serious birth defects in babies of pregnant patients. [Note: There is a second Patient Information/Informed Consent About Birth Defects (for female patients who can get pregnant)].
Initials: __________ - I understand that some patients, while taking isotretinoin or soon after stopping isotretinoin, have become depressed or developed other serious mental problems. Symptoms of depression include sad, “anxious” or empty mood, irritability, acting on dangerous impulses, anger, loss of pleasure or interest in social or sports activities, sleeping too much or too little, changes in weight or appetite, school or work performance going down, or trouble concentrating. Some patients taking isotretinoin have had thoughts about hurting themselves or putting an end to their own lives (suicidal thoughts). Some people tried to end their own lives. And some people have ended their own lives. There were reports that some of these people did not appear depressed. There have been reports of patients on isotretinoin becoming aggressive or violent. No one knows if isotretinoin caused these behaviors or if they would have happened even if the person did not take isotretinoin. Some people have had other signs of depression while taking isotretinoin (see #7 below).
Initials: __________ - Before I start taking isotretinoin, I agree to tell my doctor if I have ever had symptoms of depression (see #7 below), been psychotic, attempted suicide, had any other mental problems, or take medicine for any of these problems. Being psychotic means having a loss of contact with reality, such as hearing voices or seeing things that are not there.
Initials: __________ - Before I start taking isotretinoin, I agree to tell my doctor if, to the best of my knowledge, anyone in my family has ever had symptoms of depression, been psychotic, attempted suicide, or had any other serious mental problems.
Initials: __________ - Once I start taking isotretinoin, I agree to stop using isotretinoin and tell my doctor right away if any of the following signs and symptoms of depression or psychosis happen. I:
· Start to feel sad or have crying spells
· Lose interest in activities I once enjoyed
· Sleep too much or have trouble sleeping
· Become more irritable, angry, or aggressive than usual (for example, temper outbursts, thoughts of violence)
· Have a change in my appetite or body weight
· Have trouble concentrating
· Withdraw from my friends or family
· Feel like I have no energy
· Have feelings of worthlessness or guilt
· Start having thoughts about hurting myself or taking my own life (suicidal thoughts)
· Start acting on dangerous impulses
· Start seeing or hearing things that are not real
Initials: __________ -
I agree to return to see my doctor every month I take isotretinoin to get a new prescription for isotretinoin, to check my progress, and to check for signs of side effects.
Initials: __________ - Is otretinoin will be prescribed just for me – I will not share isotretinoin with other people because it may cause serious side effects, including birth defects.
Initials: __________ - I will not give blood while taking isotretinoin or for one month after I stop taking isotretinoin. I understand that if someone who is pregnant gets my donated blood, her baby may be exposed to isotretinoin and may be born with serious birth defects.
Initials: __________ - I have read the The iPLEDGEProgram Patient Introductory Brochure, and other materials my provider gave me containing important safety information about isotretinoin. I understand all the information I received.
Initials: __________ - My doctor and I have decided I should take isotretinoin. I understand that I must be qualified in the iPLEDGE program to have my prescription filled each month. I understand that I can stop taking isotretinoin at any time. I agree to tell my doctor if I stop taking isotretinoin.
Initials: __________
I now allow my doctor _______________________ to begin my treatment with isotretinoin.
Patient Signature:_________________________________Date:____________________
Parent/Guardian Signature (if under age 18):____________________Date: ___________
Patient Name (print)__________________________________
Patient Address___________________________________ Telephone (____-____-____)
I have:
• fully explained to the patient, ______________________________, the nature and purpose of isotretinoin treatment, including its benefits and risks.
• given the patient the appropriate educational materials, The iPLEDGEProgram Patient Introductory Brochure and asked the patient if he/she has any questions regarding his/her treatment with isotretinoin.
• answered those questions to the best of my ability.
Doctor Signature:_______________________________________Date:___________
PLACE THE ORIGINAL SIGNED DOCUMENTS IN THE PATIENT'S MEDICAL RECORD. PLEASE PROVIDE A COPY TO THE PATIENT.
------------------------------------------------------------------------------------------------------------------
MEDICATION GUIDE
Claravis™
(isotretinoin capsules, USP)
Read the Medication Guide that comes with Claravis before you start taking it and each time you get a prescription. There may be new information. This information does not take the place of talking with your doctor about your medical condition or your treatment.
What is the most important information I should know about Claravis?
- Claravis is used to treat a type of severe acne (nodular acne) that has not been helped by other treatments, including antibiotics.
- Because Claravis can cause birth defects, Claravis is only for patients who can understand and agree to carry out all of the instructions in the iPLEDGE program.
- Claravis may cause serious mental health problems.
- Birth defects (deformed babies), loss of a baby before birth (miscarriage), death of the baby, and early (premature) births. Female patients who are pregnant or who plan to become pregnant must not take Claravis. Female patients must not get pregnant:
- for one month before starting Claravis
- while taking Claravis
- for one month after stopping Claravis.
If you get pregnant while taking Claravis, stop taking it right away and call your doctor. Doctors and patients should report all cases of pregnancy to:
- FDA MedWatch at 1-800-FDA-1088, and
- the iPLEDGE pregnancy registry at 1-866-495-0654
2. Serious mental health problems. Claravis may cause:
- depression
- psychosis (seeing or hearing things that are not real)
- suicide. Some patients taking Claravis have had thoughts about hurting themselves or putting an end to their own lives (suicidal thoughts). Some people tried to end their own lives. And some people have ended their own lives.
Stop Claravis and call your doctor right away if you or a family member notices that you have any of the following signs and symptoms of depression or psychosis:
- start to feel sad or have crying spells
- lose interest in activities you once enjoyed
- sleep too much or have trouble sleeping
- become more irritable, angry, or aggressive than usual (for example, temper outbursts, thoughts of violence)
- have a change in your appetite or body weight
- have trouble concentrating
- withdraw from your friends or family
- feel like you have no energy
- have feelings of worthlessness or guilt
- start having thoughts about hurting yourself or taking your own life (suicidal thoughts)
- start acting on dangerous impulses
- start seeing or hearing things that are not real
After stopping Claravis, you may also need follow-up mental health care if you had any of these symptoms.
What is Claravis?
Claravis is a medicine taken by mouth to treat the most severe form of acne (nodular acne) that cannot be cleared up by any other acne treatments, including antibiotics. Claravis can cause serious side effects (see “What is the most important information I should know about Claravis?”). Claravis can only be:
- prescribed by doctors that are registered in the iPLEDGE program
- dispensed by a pharmacy that is registered with the iPLEDGE program
- given to patients who are registered in the iPLEDGE program and agree to do everything required in the program
What is severe nodular acne?
Severe nodular acne is when many red, swollen, tender lumps form in the skin. These can be the size of pencil erasers or larger. If untreated, nodular acne can lead to permanent scars.
Who should not take Claravis?
- Do not take Claravis if you are pregnant, plan to become pregnant, or become pregnant during Claravis treatment. Claravis causes severe birth defects. See “What is the most important information I should know about Claravis?”
- Do not take Claravis ifyou are allergic to anything in it. See the end of this Medication Guide for a complete list of ingredients in Claravis.
What should I tell my doctor before taking Claravis?
Tell your doctor if you or a family member has any of the following health conditions:
- mental problems
- asthma
- liver disease
- diabetes
- heart disease
- bone loss (osteoporosis) or weak bones
- an eating problem called anorexia nervosa (where people eat too little)
- food or medicine allergies
Tell your doctor if you are pregnant or breastfeeding. Claravis must not be used by women who are pregnant or breastfeeding.
Tell your doctor about all of the medicines you take including prescription and non-prescription medicines, vitamins and herbal supplements. Claravis and certain other medicines can interact with each other, sometimes causing serious side effects. Especially tell your doctor if you take:
- Vitamin A supplements. Vitamin A in high doses has many of the same side effects as Claravis. Taking both together may increase your chance of getting side effects.
- Tetracycline antibiotics. Tetracycline antibiotics taken with Claravis can increase the chances of getting increased pressure in the brain.
- Progestin-only birth control pills (mini-pills). They may not work while you take Claravis. Ask your doctor or pharmacist if you are not sure what type you are using.
- Dilantin (phenytoin). This medicine taken with Claravis may weaken your bones.
- Corticosteroid medicines. These medicines taken with Claravis may weaken your bones.
- St. John’s Wort. This herbal supplement may make birth control pills work less effectively.
These medicines should not be used with Claravis unless your doctor tells you it is okay.
Know the medicines you take. Keep a list of them to show to your doctor and pharmacist. Do not take any new medicine without talking with your doctor.
How should I take Claravis?
- You must take Claravis exactly as prescribed. You must also follow all the instructions of the iPLEDGE program. Before prescribing Claravis, your doctor will:
- explain the iPLEDGE program to you
- have you sign the Patient Information/Informed Consent form (for all patients). Female patients who can get pregnant must also sign another consent form.
You will not be prescribed Claravis if you cannot agree to or follow all the instructions of the iPLEDGE program.
- You will get no more than a 30-day supply of Claravis at a time. This is to make sure you are following the Claravis iPLEDGE program. You should talk with your doctor each month about side effects.
- The amount of Claravis you take has been specially chosen for you. It is based on your body weight, and may change during treatment.
- Take Claravis 2 times a day with a meal, unless your doctor tells you otherwise. Swallow your Claravis capsules whole with a full glass of liquid.Do not chew or suck on the capsule. Claravis can hurt the tube that connects your mouth to your stomach (esophagus) if it is not swallowed whole.
- If you miss a dose, just skip that dose. Do not take 2 doses at the same time.
- If you take too much Claravis or overdose, call your doctor or poison control center right away.
- Your acne may get worse when you first start taking Claravis. This should last only a short while. Talk with your doctor if this is a problem for you.
- You must return to your doctor as directed to make sure you don’t have signs of serious side effects. Your doctor may do blood tests to check for serious side effects from Claravis. Female patients who can get pregnant will get a pregnancy test each month.
- Female patients who can get pregnant must agree to use two separate forms of effective birth control at the same time one month before, while taking, and for one month after taking Claravis. You must access the iPLEDGE system to answer questions about the program requirements and to enter your two chosen forms of birth control. To access the iPLEDGE system, go to www.ipledgeprogram.com or call 1-866-495-0654.
You must talk about effective birth control methods with your doctor or go for a free visit to talk about birth control with another doctor or family planning expert. Your doctor can arrange this free visit, which will be paid for by the company that makes Claravis.
If you have sex at any time without using two forms of effective birth control, get pregnant, or miss your expected period, stop using Claravis and call your doctor right away.
What should I avoid while taking Claravis?
- Do not get pregnant while taking Claravis and for one month after stopping Claravis. See "What is the most important information I should know about Claravis?”
- Do not breast feed while taking Claravis and for one month after stopping Claravis. We do not know if Claravis can pass through your milk and harm the baby.
- Do not give blood while you take Claravis and for one month after stopping Claravis. If someone who is pregnant gets your donated blood, her baby may be exposed to Claravis and may be born with birth defects.
- Do not take other medicines or herbal products with Claravis unless you talk to your doctor. See “What should I tell my doctor before taking Claravis?”
- Do not drive at night until you know if Claravis has affected your vision. Claravis may decrease your ability to see in the dark.
- Do not have cosmetic procedures to smooth your skin, including waxing, dermabrasion, or laser procedures, while you are using Claravis and for at least 6 months after you stop. Claravis can increase your chance of scarring from these procedures. Check with your doctor for advice about when you can have cosmetic procedures.
- Avoid sunlight and ultraviolet lights as much as possible. Tanning machines use ultraviolet lights. Claravis may make your skin more sensitive to light.
- Do not share Claravis with other people. It can cause birth defects and other serious health problems.
What are the possible side effects of Claravis?
- Claravis can cause birth defects (deformed babies), loss of a baby before birth (miscarriage), death of the baby, and early (premature) births. See “What is the most important information I should know about Claravis?”
- Claravis may cause serious mental health problems. See “What is the most important information I should know about Claravis?”
- serious brain problems. Claravis can increase the pressure in your brain. This can lead to permanent loss of eyesight and, in rare cases, death. Stop taking Claravis and call your doctor right away if you get any of these signs of increased brain pressure:
- bad headache
- blurred vision
- dizziness
- nausea or vomiting
- seizures (convulsions)
- stroke
- skin problems. Skin rash can occur in patients taking Claravis. In some patients a rash can be serious. Stop using Claravis and call your doctor right away if you develop conjunctivitis (red or inflamed eyes, like “pink eye”), a rash with a fever, blisters on legs, arms or face and/or sores in your mouth, throat, nose, eyes, or if your skin begins to peel.
- stomach area (abdomen)problems. Certain symptoms may mean that your internal organs are being damaged. These organs include the liver, pancreas, bowel (intestines), and esophagus (connection between mouth and stomach). If your organs are damaged, they may not get better even after you stop taking Claravis. Stop taking Claravis and call your doctor if you get:
- severe stomach, chest or bowel pain
- trouble swallowing or painful swallowing
- new or worsening heartburn
- diarrhea
- rectal bleeding
- yellowing of your skin or eyes
- dark urine
- bone and muscle problems. Claravis may affect bones, muscles, and ligaments and cause pain in your joints or muscles. Tell your doctor if you plan hard physical activity during treatment with Claravis. Tell your doctor if you get:
- back pain
- joint pain
- broken bone. Tell all healthcare providers that you take Claravis if you break a bone.
Stop Claravis and call your doctor right away if you have muscle weakness. Muscle weakness with or without pain can be a sign of serious muscle damage.
Claravis may stop long bone growth in teenagers who are still growing.
- hearing problems. Stop using Claravis and call your doctor if your hearing gets worse or if you have ringing in your ears. Your hearing loss may be permanent.
- vision problems. Claravis may affect your ability to see in the dark. This condition usually clears up after you stop taking Claravis, but it may be permanent. Other serious eye effects can occur. Stop taking Claravis and call your doctor right away if you have any problems with your vision or dryness of the eyes that is painful or constant. If you wear contact lenses, you may have trouble wearing them while taking Claravis and after treatment.
- lipid (fats and cholesterol in blood) problems. Claravis can raise the level of fats and cholesterol in your blood. This can be a serious problem. Return to your doctor for blood tests to check your lipids and to get any needed treatment. These problems usually go away when Claravis treatment is finished.
- serious allergic reactions. Stop taking Claravis and get emergency care right away if you develop hives, a swollen face or mouth, or have trouble breathing. Stop taking Claravis and call your doctor if you get a fever, rash, or red patches or bruises on your legs.
- blood sugar problems. Claravis may cause blood sugar problems including diabetes. Tell your doctor if you are very thirsty or urinate a lot.
- decreased red and white blood cells. Call your doctor if you have trouble breathing, faint, or feel weak.
- The common, less serious side effects of Claravis are dry skin, chapped lips, dry eyes, and dry nose that may lead to nosebleeds. Call your doctor if you get any side effect that bothers you or that does not go away.
These are not all of the possible side effects with Claravis. Your doctor or pharmacist can give you more detailed information. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store Claravis?
- Store at 20° to 25°C (68° to 77°F) [See USP Controlled Room Temperature]. Protect from light.
- Keep Claravis and all medicines out of the reach of children.
General Information about Claravis.
Medicines are sometimes prescribed for conditions that are not mentioned in Medication Guides. Do not use Claravis for a condition for which it was not prescribed. Do not give Claravis to other people, even if they have the same symptoms that you have. It may harm them.
This Medication Guide summarizes the most important information about Claravis. If you would like more information, talk with your doctor. You can ask your doctor or pharmacist for information about Claravis that is written for healthcare professionals. You can also call iPLEDGE program at 1-866-495-0654 or visit www.ipledgeprogram.com.
What are the ingredients in Claravis?
Active Ingredient:Isotretinoin
Inactive Ingredients: Each capsule contains the following inactive ingredients: butylated hydroxyanisole, edetate disodium, gelatin, hydrogenated vegetable oil, polysorbate 80, soybean oil, titanium dioxide, white wax (beeswax), and vitamin E.
In addition, the 10 mg capsule contains black iron oxide and FD&C yellow no. 6. The 20 mg capsule contains black iron oxide, red iron oxide and yellow iron oxide. The 30 mg capsule contains red iron oxide and yellow iron oxide. The 40 mg capsule contains colloidal silicon, sodium lauryl sulfate, and FD&C yellow no. 6.
The edible imprinting ink contains: 10 mg strength, D&C red no. 7 calcium lake, ethylene glycol monoethyl ether, FD&C yellow no. 6 aluminum lake, shellac glaze and titanium dioxide; 20 mg strength, ammonium hydroxide, propylene glycol, shellac glaze, simethicone and titanium dioxide; the 30 mg strength, D&C yellow no. 10 aluminum lake, FD&C blue no.1 aluminum lake, FD&C blue no. 2 aluminum lake, FD&C red no. 40 aluminum lake, iron oxide black, propylene glycol, and shellac glaze; the 40 mg strength, ammonium hydroxide, FD&C blue no. 2 aluminum lake, iron oxide black, propylene glycol, and shellac glaze.
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Dilantin is a registered trademark of Warner-Lambert Company LLC.
BARR LABORATORIES, INC.
Pomona, NY 10970
Rev. B 5/2012

PRINCIPAL DISPLAY PANEL, part 4 of 4
Claravis 20 mg Capsules Prescription Blister 10s Pack Text
Barr
Laboratories Inc.
Claravis TM
(isotretinoin capsules, USP)
Each capsule contains 20 mg isotretinoin
Prescription Blister Pack
10 Capsules
Rx only
PATIENT:
READ INFORMATION CAREFULLY.
CAUSES BIRTH DEFECTS
DO NOT GET PREGNANT
20 mg PHARMACISTS: DISPENSE INTACT.
THIS AREA FOR
PRESCRIPTION LABEL

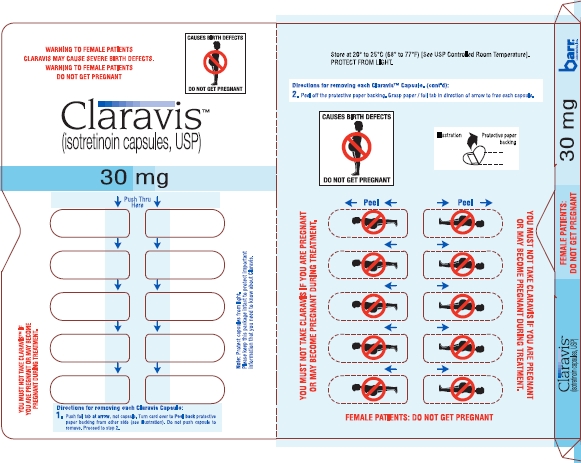
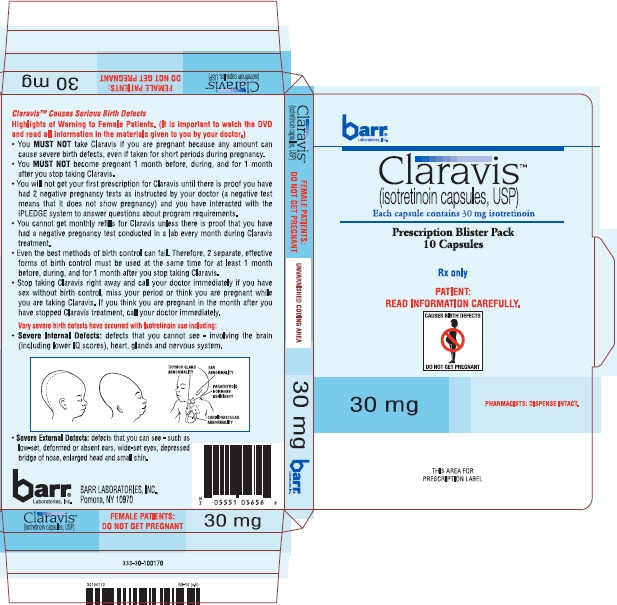

PRINCIPAL DISPLAY PANEL, part 4 of 4
Claravis 30 mg Capsules Prescription Blister 10s Pack Text
Barr
Laboratories Inc.
Claravis TM
(isotretinoin capsules, USP)
Each capsule contains 30 mg isotretinoin
Prescription Blister Pack
10 Capsules
Rx only
PATIENT:
READ INFORMATION CAREFULLY.
CAUSES BIRTH DEFECTS
DO NOT GET PREGNANT
30 mg PHARMACISTS: DISPENSE INTACT.
THIS AREA FOR
PRESCRIPTION LABEL

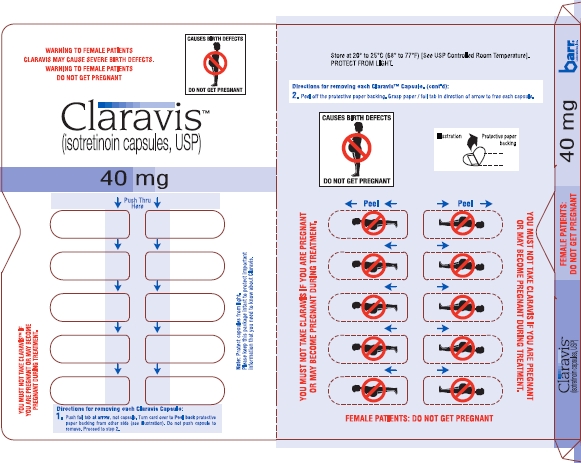
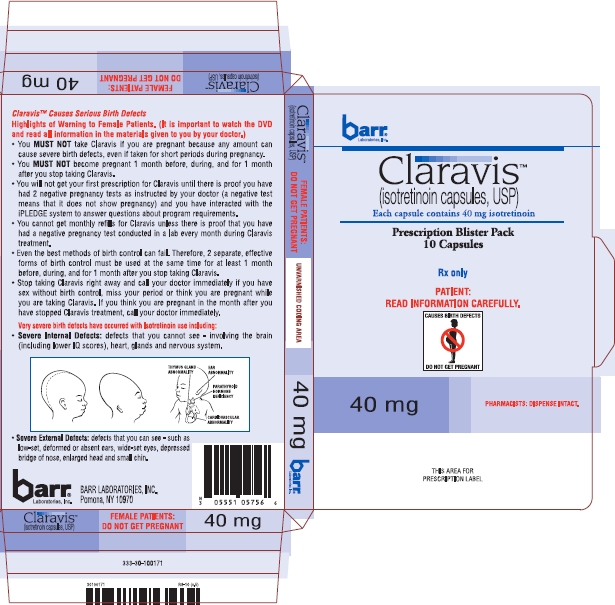

PRINCIPAL DISPLAY PANEL, part 4 of 4
Claravis 40 mg Capsules Prescription Blister 10s Pack Text
Barr
Laboratories Inc.
Claravis TM
(isotretinoin capsules, USP)
Each capsule contains 40 mg isotretinoin
Prescription Blister Pack
10 Capsules
Rx only
PATIENT:
READ INFORMATION CAREFULLY.
CAUSES BIRTH DEFECTS
DO NOT GET PREGNANT
40 mg PHARMACISTS: DISPENSE INTACT.
THIS AREA FOR
PRESCRIPTION LABEL
