FULL PRESCRIBING INFORMATION
WARNING: DERMATOLOGIC TOXICITY
Dermatologic Toxicity: Dermatologic toxicities occurred in 90% of patients and were severe (NCI-CTC grade 3 and higher) in 15% of patients receiving Vectibix monotherapy [see Dosage and Administration (2.3), Warnings and Precautions (5.1), and Adverse Reactions (6.1)].
1 INDICATIONS AND USAGE
1.1 Metastatic Colorectal Cancer
Vectibix is indicated for the treatment of patients with wild-type RAS (defined as wild-type in both KRAS and NRAS as determined by an FDA-approved test for this use) metastatic colorectal cancer (mCRC) [see Dosage and Administration (2.1)]:
• As first-line therapy in combination with FOLFOX [see Clinical Studies (14.2)].
• As monotherapy following disease progression after prior treatment with fluoropyrimidine-, oxaliplatin-, and irinotecan-containing chemotherapy [see Clinical Studies (14.1)].
Limitation of Use: Vectibix is not indicated for the treatment of patients with RAS-mutant mCRC or for whom RAS mutation status is unknown [see Dosage and Administration (2.1), Warnings and Precautions (5.2), and Clinical Pharmacology (12.1)].
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
Prior to initiation of treatment with Vectibix, assess RAS mutational status in colorectal tumors and confirm the absence of a RAS mutation in exon 2 (codons 12 and 13), exon 3 (codons 59 and 61), and exon 4 (codons 117 and 146) of both KRAS and NRAS. Information on FDA-approved tests for the detection of RAS mutations in patients with metastatic colorectal cancer is available at: http://www.fda.gov/CompanionDiagnostics.
2.2 Recommended Dose
The recommended dose of Vectibix is 6 mg/kg, administered as an intravenous infusion over 60 minutes, every 14 days. If the first infusion is tolerated, administer subsequent infusions over 30 to 60 minutes. Administer doses higher than 1000 mg over 90 minutes [see Dosage and Administration (2.4)].
Appropriate medical resources for the treatment of severe infusion reactions should be available during Vectibix infusions [see Warnings and Precautions (5.4)].
2.3 Dose Modifications
Dose Modifications for Infusion Reactions [see Warnings and Precautions (5.4) and Adverse Reactions (6.1, 6.3)]
• Reduce infusion rate by 50% in patients experiencing a mild or moderate (grade 1 or 2) infusion reaction for the duration of that infusion.
• Terminate the infusion in patients experiencing severe infusion reactions. Depending on the severity and/or persistence of the reaction, permanently discontinue Vectibix.
Dose Modifications for Dermatologic Toxicity [see Boxed Warning, Warnings and Precautions (5.1), and Adverse Reactions (6.1, 6.3)]
• Upon first occurrence of a grade 3 (NCI-CTC/CTCAE) dermatologic reaction, withhold 1 to 2 doses of Vectibix. If the reaction improves to < grade 3, reinitiate Vectibix at the original dose.
• Upon the second occurrence of a grade 3 (NCI-CTC/CTCAE) dermatologic reaction, withhold 1 to 2 doses of Vectibix. If the reaction improves to < grade 3, reinitiate Vectibix at 80% of the original dose.
• Upon the third occurrence of a grade 3 (NCI-CTC/CTCAE) dermatologic reaction, withhold 1 to 2 doses of Vectibix. If the reaction improves to < grade 3, reinitiate Vectibix at 60% of the original dose.
• Upon the fourth occurrence of a grade 3 (NCI-CTC/CTCAE) dermatologic reaction, permanently discontinue Vectibix.
Permanently discontinue Vectibix following the occurrence of a grade 4 dermatologic reaction or for a grade 3 (NCI-CTC/CTCAE) dermatologic reaction that does not recover after withholding 1 or 2 doses.
2.4 Preparation and Administration
For intravenous infusion only. Do not administer Vectibix as an intravenous push or bolus.
Preparation
Visually inspect parenteral drug products for particulate matter and discoloration prior to administration. Vectibix solution is colorless and may contain a small amount of visible translucent-to-white, amorphous, proteinaceous particles. Do not use if the solution is discolored or cloudy, or if foreign matter is present.
Prepare the solution for infusion, using aseptic technique, as follows:
• Do not shake the vial.
• Use a 21-gauge or larger gauge (smaller bore) hypodermic needle to withdraw the necessary amount of Vectibix for a dose of 6 mg/kg. Do not use needle-free devices (e.g., vial adapters) to withdraw vial contents.
• Dilute to a total volume of 100 mL with 0.9% sodium chloride injection, USP. Doses higher than 1000 mg should be diluted to 150 mL with 0.9% sodium chloride injection, USP. Do not exceed a final concentration of 10 mg/mL.
• Mix diluted solution by gentle inversion.
• Discard any unused portion of the vial.
Administration
• Administer using a low-protein-binding 0.2 μm or 0.22 μm in-line filter.
• Vectibix must be administered via infusion pump.
○ Flush line before and after Vectibix administration with 0.9% sodium chloride injection, USP, to avoid mixing with other drug products or intravenous solutions. Do not mix Vectibix with, or administer as an infusion with, other medicinal products. Do not add other medications to solutions containing panitumumab.
○ Infuse doses of 1000 mg or lower over 60 minutes through a peripheral intravenous line or indwelling intravenous catheter. If the first infusion is tolerated, administer subsequent infusions over 30 to 60 minutes. Administer doses higher than 1000 mg over 90 minutes.
• Use the diluted infusion solution of Vectibix within 6 hours of preparation if stored at room temperature, or within 24 hours of dilution if stored at 2° to 8°C (36° to 46°F). DO NOT FREEZE.
3 DOSAGE FORMS AND STRENGTHS
Injection: 100 mg/5 mL (20 mg/mL) colorless solution in single-dose vial.
Injection: 400 mg/20 mL (20 mg/mL) colorless solution in single-dose vial.
5 WARNINGS AND PRECAUTIONS
5.1 Dermatologic and Soft Tissue Toxicity
In Study 20020408, dermatologic toxicities occurred in 90% of patients and were severe (NCI-CTC grade 3 and higher) in 15% of patients with mCRC receiving Vectibix. The clinical manifestations included, but were not limited to, acneiform dermatitis, pruritus, erythema, rash, skin exfoliation, paronychia, dry skin, and skin fissures.
Monitor patients who develop dermatologic or soft tissue toxicities while receiving Vectibix for the development of inflammatory or infectious sequelae. Life-threatening and fatal infectious complications including necrotizing fasciitis, abscesses, and sepsis have been observed in patients treated with Vectibix. Life-threatening and fatal bullous mucocutaneous disease with blisters, erosions, and skin sloughing has also been observed in patients treated with Vectibix. It could not be determined whether these mucocutaneous adverse reactions were directly related to EGFR inhibition or to idiosyncratic immune-related effects (e.g., Stevens-Johnson syndrome or toxic epidermal necrolysis). Withhold or discontinue Vectibix for dermatologic or soft tissue toxicity associated with severe or life-threatening inflammatory or infectious complications [see Boxed Warning and Adverse Reactions (6.1, 6.3)]. Dose modifications for Vectibix concerning dermatologic toxicity are provided [see Dosage and Administration (2.3)].
5.2 Increased Tumor Progression, Increased Mortality, or Lack of Benefit in Patients with RAS-Mutant mCRC
Vectibix is not indicated for the treatment of patients with colorectal cancer that harbor somatic RAS mutations in exon 2 (codons 12 and 13), exon 3 (codons 59 and 61), and exon 4 (codons 117 and 146) of either KRAS or NRAS and hereafter is referred to as “RAS” [see Indications and Usage (1.1), Dosage and Administration (2.1), Clinical Pharmacology (12.1) and Clinical Studies (14)].
Retrospective subset analyses across several randomized clinical trials were conducted to investigate the role of RAS mutations on the clinical effects of anti-EGFR-directed monoclonal antibodies (panitumumab or cetuximab). Anti-EGFR antibodies in patients with tumors containing RAS mutations resulted in exposing those patients to anti-EGFR related adverse reactions without clinical benefit from these agents [see Indications and Usage (1.1), and Clinical Pharmacology (12.1)].
Additionally, in Study 20050203, 272 patients with RAS-mutant mCRC tumors received Vectibix in combination with FOLFOX and 276 patients received FOLFOX alone. In an exploratory subgroup analysis, OS was shorter (HR = 1.21, 95% CI: 1.01-1.45) in patients with RAS-mutant mCRC who received Vectibix and FOLFOX versus FOLFOX alone [see Indications and Usage (1.1)].
5.3 Electrolyte Depletion/Monitoring
Progressively decreasing serum magnesium levels leading to severe (grade 3-4) hypomagnesemia occurred in up to 7% (in Study 20080763) of patients across clinical trials. Monitor patients for hypomagnesemia and hypocalcemia prior to initiating Vectibix treatment, periodically during Vectibix treatment, and for up to 8 weeks after the completion of treatment. Other electrolyte disturbances, including hypokalemia, have also been observed. Replete magnesium and other electrolytes as appropriate.
5.4 Infusion Reactions
In Study 20020408, 4% of patients experienced infusion reactions and 1% of patients experienced severe infusion reactions (NCI-CTC grade 3-4).
Infusion reactions, manifesting as fever, chills, dyspnea, bronchospasm, and hypotension, can occur following Vectibix administration [see Adverse Reactions (6.1, 6.3)]. Fatal infusion reactions occurred in postmarketing experience. Terminate the infusion for severe infusion reactions [see Dosage and Administration (2.3)].
5.5 Acute Renal Failure in Combination with Chemotherapy
Severe diarrhea and dehydration, leading to acute renal failure and other complications, have been observed in patients treated with Vectibix in combination with chemotherapy.
5.6 Pulmonary Fibrosis/Interstitial Lung Disease (ILD)
Fatal and nonfatal cases of interstitial lung disease (ILD) (1%) and pulmonary fibrosis have been observed in patients treated with Vectibix. Pulmonary fibrosis occurred in less than 1% (2/1467) of patients enrolled in clinical studies of Vectibix. In the event of acute onset or worsening of pulmonary symptoms, interrupt Vectibix therapy. Discontinue Vectibix therapy if ILD is confirmed.
In patients with a history of interstitial pneumonitis or pulmonary fibrosis, or evidence of interstitial pneumonitis or pulmonary fibrosis, the benefits of therapy with Vectibix versus the risk of pulmonary complications must be carefully considered.
5.7 Photosensitivity
Exposure to sunlight can exacerbate dermatologic toxicity. Advise patients to wear sunscreen and hats and limit sun exposure while receiving Vectibix.
5.8 Ocular Toxicities
Serious cases of keratitis, ulcerative keratitis, and corneal perforation have occurred with Vectibix use. Monitor for evidence of keratitis, ulcerative keratitis, or corneal perforation. Interrupt or discontinue Vectibix therapy for acute or worsening keratitis, ulcerative keratitis, or corneal perforation.
5.9 Increased Mortality and Toxicity with Vectibix in Combination with Bevacizumab and Chemotherapy
In an interim analysis of an open-label, multicenter, randomized clinical trial in the first-line setting in patients with mCRC, the addition of Vectibix to the combination of bevacizumab and chemotherapy resulted in decreased OS and increased incidence of NCI-CTC grade 3-5 (87% vs 72%) adverse reactions. NCI-CTC grade 3-4 adverse reactions occurring at a higher rate in Vectibix-treated patients included rash/acneiform dermatitis (26% vs 1%), diarrhea (23% vs 12%), dehydration (16% vs 5%), primarily occurring in patients with diarrhea, hypokalemia (10% vs 4%), stomatitis/mucositis (4% vs < 1%), and hypomagnesemia (4% vs 0).
NCI-CTC grade 3-5 pulmonary embolism occurred at a higher rate in Vectibix-treated patients (7% vs 3%) and included fatal events in three (< 1%) Vectibix-treated patients.
As a result of the toxicities experienced, patients randomized to Vectibix, bevacizumab, and chemotherapy received a lower mean relative dose intensity of each chemotherapeutic agent (oxaliplatin, irinotecan, bolus 5-FU, and/or infusional 5-FU) over the first 24 weeks on study compared with those randomized to bevacizumab and chemotherapy.
5.10 Embryo-fetal Toxicity
Based on data from animal studies and its mechanism of action, Vectibix can cause fetal harm when administered to a pregnant woman. When given during organogenesis, panitumumab administration resulted in embryolethality in cynomolgus monkeys at exposures approximately 1.25 to 5-times the recommended human dose. Advise pregnant women and females of reproductive potential of the potential risk to the fetus. Advise females of reproductive potential to use effective contraception during treatment, and for at least 2 months after the last dose of Vectibix [see Use in Specific Populations (8.1, 8.3), Clinical Pharmacology (12.1)].
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the label:
- Dermatologic and Soft Tissue Toxicity [see Boxed Warning, Dosage and Administration (2.3), and Warnings and Precautions (5.1)]
- Increased Tumor Progression, Increased Mortality, or Lack of Benefit in RAS-Mutant mCRC [see Indications and Usage (1.1) and Warnings and Precautions (5.2)]
- Electrolyte Depletion/Monitoring [see Warnings and Precautions (5.3)]
- Infusion Reactions [see Dosage and Administration (2.3), and Warnings and Precautions (5.4)]
- Acute Renal Failure in Combination with Chemotherapy [see Warnings and Precautions (5.5)]
- Pulmonary Fibrosis/Interstitial Lung Disease (ILD) [see Warnings and Precautions (5.6)]
- Photosensitivity [see Warnings and Precautions (5.7)]
- Ocular Toxicities [see Warnings and Precautions (5.8)]
- Increased Mortality and Toxicity with Vectibix in combination with Bevacizumab and Chemotherapy [see Warnings and Precautions (5.9)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates in the clinical trials of a drug cannot be directly compared to rates in clinical trials of another drug and may not reflect the rates observed in practice. The adverse reaction information from clinical studies does, however, provide a basis for identifying the adverse events that appear to be related to drug use and for approximating rates.
Safety data are presented from two clinical trials in which patients received Vectibix: Study 20020408, an open-label, multinational, randomized, controlled, monotherapy clinical trial (N = 463) evaluating Vectibix with best supportive care (BSC) versus BSC alone in patients with EGFR-expressing mCRC and Study 20050203, a randomized, controlled trial (N = 1183) in patients with mCRC that evaluated Vectibix in combination with FOLFOX chemotherapy versus FOLFOX chemotherapy alone. Safety data for Study 20050203 are limited to 656 patients with wild-type KRAS mCRC. The safety profile of Vectibix in patients with wild-type RAS mCRC is similar with that seen in patients with wild-type KRAS mCRC.
Vectibix Monotherapy
In Study 20020408, the most common adverse reactions (≥ 20%) with Vectibix were skin rash with variable presentations, paronychia, fatigue, nausea, and diarrhea.
The most common (> 5%) serious adverse reactions in the Vectibix arm were general physical health deterioration and intestinal obstruction. The most frequently reported adverse reactions for Vectibix leading to withdrawal were general physical health deterioration (n = 2) and intestinal obstruction (n = 2).
For Study 20020408, the data described in Table 1 and in other sections below, except where noted, reflect exposure to Vectibix administered to patients with mCRC as a single agent at the recommended dose and schedule (6 mg/kg every 2 weeks).
| Study 20020408 | ||||
| Vectibix Plus
Best Supportive Care (N = 229) | Best Supportive Care
(N = 234) |
|||
| System Organ Class
Preferred Term | Any
Grade n (%) | Grade
3-4 n (%) | Any
Grade n (%) | Grade
3-4 n (%) |
| Eye Disorders | ||||
| Growth of eyelashes | 13 (6) | |||
| Gastrointestinal Disorders | ||||
| Nausea | 52 (23) | 2 (< 1) | 37 (16) | 1 (< 1) |
| Diarrhea | 49 (21) | 4 (2) | 26 (11) | |
| Vomiting | 43 (19) | 6 (3) | 28 (12) | 2 (< 1) |
| Stomatitis | 15 (7) | 2 (< 1) | ||
| General Disorders and Administration Site Conditions | ||||
| Fatigue | 60 (26) | 10 (4) | 34 (15) | 7 (3) |
| Mucosal inflammation | 15 (7) | 1 (< 1) | 2 (< 1) | |
| Infections and Infestations | ||||
| Paronychia | 57 (25) | 4 (2) | ||
| Respiratory, Thoracic, and Mediastinal Disorders | ||||
| Dyspnea | 41 (18) | 12 (5) | 30 (13) | 8 (3) |
| Cough | 34 (15) | 1 (< 1) | 17 (7) | |
| Skin and Subcutaneous Tissue Disorders | ||||
| Erythema | 150 (66) | 13 (6) | 2 (< 1) | |
| Pruritus | 132 (58) | 6 (3) | 4 (2) | |
| Acneiform dermatitis | 131 (57) | 17 (7) | 2 (< 1) | |
| Rash | 51 (22) | 3 (1) | 2 (< 1) | |
| Skin fissures | 45 (20) | 3 (1) | 1 (< 1) | |
| Exfoliative rash | 41 (18) | 4 (2) | ||
| Acne | 31 (14) | 3 (1) | ||
| Dry skin | 23 (10) | |||
| Nail disorder | 22 (10) | |||
| Skin exfoliation | 21 (9) | 2 (< 1) | ||
| Skin ulcer | 13 (6) | 1 (< 1) | ||
Adverse reactions in Study 20020408 that did not meet the threshold criteria for inclusion in Table 1 were conjunctivitis (4.8% vs < 1%), dry mouth (4.8% vs 0%), pyrexia (16.6% vs 13.2%), chills (3.1% vs < 1%), pustular rash (4.4% vs 0%), papular rash (1.7% vs 0%), dehydration (2.6% vs 1.7%), epistaxis (3.9% vs 0%), and pulmonary embolism (1.3% vs 0%).
In Study 20020408, dermatologic toxicities occurred in 90% of patients receiving Vectibix. Skin toxicity was severe (NCI-CTC grade 3 and higher) in 15% of patients. Ocular toxicities occurred in 16% of patients and included, but were not limited to, conjunctivitis (5%). One patient experienced an NCI-CTC grade 3 event of mucosal inflammation. The incidence of paronychia was 25% and was severe in 2% of patients [see Warnings and Precautions (5.1)].
In Study 20020408 (N = 229), median time to the development of dermatologic, nail, or ocular toxicity was 12 days after the first dose of Vectibix; the median time to most severe skin/ocular toxicity was 15 days after the first dose of Vectibix; and the median time to resolution after the last dose of Vectibix was 98 days. Severe toxicity necessitated dose interruption in 11% of Vectibix-treated patients [see Dosage and Administration (2.3)].
Subsequent to the development of severe dermatologic toxicities, infectious complications, including sepsis, septic death, necrotizing fasciitis, and abscesses requiring incisions and drainage were reported.
Vectibix in Combination with FOLFOX Chemotherapy
The most commonly reported adverse reactions (≥ 20%) in patients with wild-type KRAS mCRC receiving Vectibix (6 mg/kg every 2 weeks) and FOLFOX therapy (N = 322) in Study 20050203 were diarrhea, stomatitis, mucosal inflammation, asthenia, paronychia, anorexia, hypomagnesemia, hypokalemia, rash, acneiform dermatitis, pruritus, and dry skin (Table 2). Serious adverse reactions (≥ 2% difference between treatment arms) in Vectibix-treated patients with wild-type KRAS mCRC were diarrhea and dehydration. The commonly reported adverse reactions (≥ 1%) leading to discontinuation in patients with wild-type KRAS mCRC receiving Vectibix were rash, paresthesia, fatigue, diarrhea, acneiform dermatitis, and hypersensitivity. One grade 5 adverse reaction, hypokalemia, occurred in a patient who received Vectibix.
Table 2: Adverse Reactions (≥ 5% Difference) Observed in Patients with Wild-type KRAS Tumors Treated with Vectibix and FOLFOX Chemotherapy Compared to FOLFOX Chemotherapy Alone (Study 20050203)
| Vectibix Plus FOLFOX
(n = 322) | FOLFOX Alone
(n = 327) |
|||
| System Organ Class
Preferred Term | Any Grade
n (%) | Grade 3-4
n (%) | Any Grade
n (%) | Grade 3-4
n (%) |
| Eye Disorders | ||||
| Conjunctivitis | 58 (18) | 5 (2) | 10 (3) | |
| Gastrointestinal Disorders | ||||
| Diarrhea | 201 (62) | 59 (18) | 169 (52) | 29 (9) |
| Stomatitis | 87 (27) | 15 (5) | 42 (13) | 1 (< 1) |
| General Disorders and Administration Site Conditions | ||||
| Mucosal inflammation | 82 (25) | 14 (4) | 53 (16) | 1 (< 1) |
| Asthenia | 79 (25) | 16 (5) | 62 (19) | 11 (3) |
| Infections and Infestations | ||||
| Paronychia | 68 (21) | 11 (3) | ||
| Investigations | ||||
| Weight decreased | 58 (18) | 3 (< 1) | 22 (7) | |
| Metabolism and Nutrition Disorders | ||||
| Anorexia | 116 (36) | 14 (4) | 85 (26) | 6 (2) |
| Hypomagnesemia | 96 (30) | 21 (7) | 26 (8) | 1 (< 1) |
| Hypokalemia | 68 (21) | 32 (10) | 42 (13) | 15 (5) |
| Dehydration | 26 (8) | 8 (2) | 10 (3) | 5 (2) |
| Respiratory, Thoracic, and Mediastinal Disorders | ||||
| Epistaxis | 46 (14) | 30 (9) | ||
| Skin and Subcutaneous Tissue Disorders | ||||
| Rash | 179 (56) | 55 (17) | 24 (7) | 1 (< 1) |
| Acneiform dermatitis | 104 (32) | 33 (10) | ||
| Pruritus | 75 (23) | 3 (< 1) | 14 (4) | |
| Dry skin | 68 (21) | 5 (2) | 13 (4) | |
| Erythema | 50 (16) | 7 (2) | 14 (4) | |
| Skin fissures | 50 (16) | 1 (< 1) | 1 (< 1) | |
| Alopecia | 47 (15) | 30 (9) | ||
| Acne | 44 (14) | 10 (3) | 1 (< 1) | |
| Nail disorder | 32 (10) | 4 (1) | 4 (1) | |
| Palmar-plantar erythrodysesthesia syndrome | 30 (9) | 4 (1) | 9 (3) | 2 (< 1) |
Adverse reactions that did not meet the threshold criteria for inclusion in Table 2 were flushing (3% vs < 1%), abdominal pain (28% vs 23%), localized infection (3.7% vs < 1%), cellulitis (2.5% vs 0%), hypocalcemia (5.6% vs 2.1%), and deep vein thrombosis (5.3% vs 3.1%).
Infusion Reactions
Infusional toxicity manifesting as fever, chills, dyspnea, bronchospasm or hypotension was assessed within 24 hours of an infusion during the clinical study. Vital signs and temperature were measured within 30 minutes prior to initiation and upon completion of the Vectibix infusion. The use of premedication was not standardized in the clinical trials. Thus, the utility of premedication in preventing the first or subsequent episodes of infusional toxicity is unknown. Across clinical trials of Vectibix monotherapy, 3% (24/725) experienced infusion reactions of which < 1% (3/725) were severe (NCI-CTC grade 3-4). In one patient, Vectibix was permanently discontinued for a serious infusion reaction [see Dosage and Administration (2.2, 2.3)].
6.2 Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors, including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to panitumumab in the studies described below with the incidence of antibodies to other products may be misleading.
The immunogenicity of Vectibix has been evaluated using two different screening immunoassays for the detection of binding anti-panitumumab antibodies: an acid dissociation bridging enzyme-linked immunosorbent assay (ELISA) detecting high-affinity antibodies and a Biacore® biosensor immunoassay detecting both high- and low-affinity antibodies. For patients whose sera tested positive in screening immunoassays, an in vitro biological assay was performed to detect neutralizing antibodies.
Monotherapy: The incidence of treatment-emergent binding anti-panitumumab antibodies was 0.5% (7/1295) as detected by ELISA and 5.3% (68/1295) as detected by the Biacore® assay. The incidence of neutralizing anti-panitumumab antibodies was 0.8% (11/1295). There was no evidence of altered pharmacokinetics or safety profiles in patients who developed antibodies to panitumumab.
In combination with chemotherapy: The incidence of treatment-emergent binding anti-panitumumab antibodies was 0.9% (12/1297) as detected by the ELISA and 0.7% (9/1296) as detected by the Biacore® assay. The incidence of neutralizing anti-panitumumab antibodies was 0.2% (2/1297). No evidence of an altered safety profile was found in patients who developed antibodies to panitumumab.
6.3 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of Vectibix. Because these reactions are reported in a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
-
Skin and subcutaneous tissue disorders: Skin necrosis, angioedema, life-threatening and fatal bullous mucocutaneous disease [see Boxed Warning, Dosage and Administration (2.3), and Warnings and Precautions (5.1)]
-
Immune system disorders: Infusion reaction [see Dosage and Administration (2.3) and Warnings and Precautions (5.4)]
- Eye disorders: Keratitis/ulcerative keratitis, corneal perforation [see Warnings and Precautions (5.8)]
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on data from animal studies and its mechanism of action, Vectibix can cause fetal harm when administered to pregnant women [see Clinical Pharmacology (12.1)]. Limited available data on the use of Vectibix in pregnant women are not sufficient to inform a risk of adverse pregnancy-related outcomes. Vectibix is a human IgG monoclonal antibody and may be transferred across the placenta during pregnancy. Reproduction studies in cynomolgus monkeys treated with 1.25 to 5 times the recommended human dose of panitumumab resulted in significant embryolethality and abortions; however, no other evidence of teratogenesis was noted in offspring [see Data]. Advise pregnant women of the potential risk to the fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively. The estimated background risk of major birth defects and miscarriage for the indicated population is unknown.
Data
Animal Data
Based on animal models, EGFR is involved in prenatal development and may be essential for normal organogenesis, proliferation, and differentiation in the developing embryo. Pregnant cynomolgus monkeys were treated weekly with panitumumab during the period of organogenesis (gestation day [GD] 20-50). While no panitumumab was detected in serum of neonates from panitumumab-treated dams, anti-panitumumab antibody titers were present in 14 of 27 offspring delivered at GD 100. There were no fetal malformations or other evidence of teratogenesis noted in the offspring; however, significant increases in embryolethality and abortions occurred at doses of approximately 1.25 to 5 times the recommended human dose (based on body weight).
8.2 Lactation
Risk Summary
There are no data on the presence of panitumumab in human milk or the effects of panitumumab on the breastfed infant or on milk production. Human IgG is present in human milk, but published data suggest that breast milk antibodies do not enter the neonatal and infant circulation in substantial amounts. Because of the potential for serious adverse reactions in breastfed infants from Vectibix, advise women not to breastfeed during treatment with Vectibix and for 2 months after the final dose.
8.3 Females and Males of Reproductive Potential
Contraception
Females
Vectibix can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception during treatment with Vectibix and for 2 months after the last dose of Vectibix.
Infertility
Females
Based on results from animal fertility studies conducted in female cynomolgus monkeys, Vectibix may reduce fertility in females of reproductive potential. The effects in animal studies were reversible [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of Vectibix have not been established in pediatric patients.
The pharmacokinetics of panitumumab at doses ranging from 2.5 mg/kg intravenous weekly, 6 mg/kg intravenous every 2 weeks, or 9 mg/kg intravenous every 3 weeks were evaluated in 28 pediatric patients. Panitumumab exposures were comparable in adult and adolescent patients of 12 to 17 years of age. Limited data suggested that pediatric patients of 2 to < 12 years of age had lower panitumumab exposure and higher clearance than that in adolescent patients following 6 mg/kg intravenous administration of Vectibix. There was no evidence of an anti-tumor treatment effect in these patients.
8.5 Geriatric Use
Of the 737 patients who received Vectibix monotherapy in Study 20020408 and 20080763, 36% were 65 and over while 8% were 75 and over. No overall differences in safety or efficacy were observed in elderly patients (≥ 65 years of age) treated with Vectibix monotherapy.
Of the 322 patients in Study 20050203 who received Vectibix plus FOLFOX, 128 (40%) were 65 and over while 8% were 75 and over. Patients older than 65 years of age experienced an increased incidence of serious adverse events (52% vs 36%) and an increased incidence of serious diarrhea (15% vs 5%) as compared to younger patients.
10 OVERDOSAGE
Doses up to approximately twice the recommended therapeutic dose (12 mg/kg) resulted in adverse reactions of skin toxicity, diarrhea, dehydration, and fatigue.
11 DESCRIPTION
Panitumumab is an epidermal growth factor receptor (EGFR) antagonist for intravenous use. Panitumumab is a human IgG2 kappa monoclonal antibody with an approximate molecular weight of 147 kDa that is produced in genetically engineered mammalian (Chinese hamster ovary) cells.
Vectibix (panitumumab) Injection for intravenous use is a sterile, colorless solution with a pH range of 5.6 to 6.0, which may contain a small amount of visible translucent-to-white, amorphous, proteinaceous particles. Each single-dose 5 mL vial contains 100 mg of panitumumab, 34 mg sodium acetate, 29 mg sodium chloride, and Water for Injection, USP. Each single-dose 20 mL vial contains 400 mg of panitumumab, 136 mg sodium acetate, 117 mg sodium chloride, and Water for Injection, USP.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The EGFR is a transmembrane glycoprotein that is a member of a subfamily of type I receptor tyrosine kinases, including EGFR, HER2, HER3, and HER4. EGFR is constitutively expressed in normal epithelial tissues, including the skin and hair follicle. EGFR is overexpressed in certain human cancers, including colon and rectum cancers. Interaction of EGFR with its normal ligands (e.g., EGF, transforming growth factor-alpha) leads to phosphorylation and activation of a series of intracellular proteins, which in turn regulate transcription of genes involved with cellular growth and survival, motility, and proliferation. KRAS (Kirsten rat sarcoma 2 viral oncogene homologue) and NRAS (Neuroblastoma RAS viral oncogene homologue) are highly related members of the RAS oncogene family. Signal transduction through the EGFR can result in activation of the wild-type KRAS and NRAS proteins; however, in cells with activating RAS somatic mutations, the RAS-mutant proteins are continuously active and appear independent of EGFR regulation.
Panitumumab binds specifically to EGFR on both normal and tumor cells, and competitively inhibits the binding of ligands for EGFR. Nonclinical studies show that binding of panitumumab to the EGFR prevents ligand-induced receptor autophosphorylation and activation of receptor-associated kinases, resulting in inhibition of cell growth, induction of apoptosis, decreased proinflammatory cytokine and vascular growth factor production, and internalization of the EGFR. In vitro assays and in vivo animal studies demonstrate that panitumumab inhibits the growth and survival of selected human tumor cell lines expressing EGFR.
12.3 Pharmacokinetics
Panitumumab administered as a single agent exhibits nonlinear pharmacokinetics.
Following single-dose administrations of panitumumab as 1-hour infusions, the area under the concentration-time curve (AUC) increased in a greater than dose-proportional manner, and clearance (CL) of panitumumab decreased from 30.6 to 4.6 mL/day/kg as the dose increased from 0.75 to 9 mg/kg. However, at doses above 2 mg/kg, the AUC of panitumumab increased in an approximately dose-proportional manner.
Following the recommended dose regimen (6 mg/kg given once every 2 weeks as a 1-hour infusion), panitumumab concentrations reached steady-state levels by the third infusion with mean (± SD) peak and trough concentrations of 213 ± 59 and 39 ± 14 mcg/mL, respectively. The mean (± SD) AUC0-tau and CL were 1306 ± 374 mcg•day/mL and 4.9 ± 1.4 mL/kg/day, respectively. The elimination half-life was approximately 7.5 days (range: 3.6 to 10.9 days).
A population pharmacokinetic analysis was performed to explore the potential effects of selected covariates on panitumumab pharmacokinetics. Results suggest that age (21-88 years), gender, race (15% nonwhite), mild-to-moderate renal dysfunction, mild-to-moderate hepatic dysfunction, and EGFR membrane-staining intensity (1+, 2+, and 3+) in tumor cells had no apparent impact on the pharmacokinetics of panitumumab.
No formal pharmacokinetic studies of panitumumab have been conducted in patients with renal or hepatic impairment.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No carcinogenicity or mutagenicity studies of panitumumab have been conducted. It is not known if panitumumab can impair fertility in humans. Prolonged menstrual cycles and/or amenorrhea occurred in normally cycling, female cynomolgus monkeys treated weekly with 1.25 to 5 times the recommended human dose of panitumumab (based on body weight). Menstrual cycle irregularities in panitumumab-treated female monkeys were accompanied by both a decrease and delay in peak progesterone and 17β-estradiol levels. Normal menstrual cycling resumed in most animals after discontinuation of panitumumab treatment. A no-effect level for menstrual cycle irregularities and serum hormone levels was not identified. The effects of panitumumab on male fertility have not been studied; however, no adverse effects were observed microscopically in reproductive organs from male cynomolgus monkeys treated for 26 weeks with panitumumab at doses of up to approximately 5-fold the recommended human dose (based on body weight).
14 CLINICAL STUDIES
14.1 Recurrent or Refractory mCRC
The safety and efficacy of Vectibix was demonstrated in Study 20020408, an open-label, multinational, randomized, controlled trial of 463 patients with EGFR-expressing, metastatic carcinoma of the colon or rectum, in Study 20080763, an open-label, multicenter, multinational, randomized trial of 1010 patients with wild-type KRAS mCRC, and in Study 20100007, an open-label, multicenter, multinational, randomized trial of 377 patients with wild-type KRAS mCRC.
Study 20020408 (NCT00113763)
Patients in Study 20020408 were required to have progressed on or following treatment with a regimen(s) containing a fluoropyrimidine, oxaliplatin, and irinotecan; progression was confirmed by an independent review committee (IRC) masked to treatment assignment for 76% of the patients. Patients were randomized (1:1) to receive panitumumab at a dose of 6 mg/kg given once every 2 weeks plus BSC (N = 231) or BSC alone (N = 232) until investigator-determined disease progression. Randomization was stratified based on Eastern Cooperative Oncology Group (ECOG) performance status (PS) (0 and 1 vs 2) and geographic region (Western Europe, Eastern/Central Europe, or other). Upon investigator-determined disease progression, patients in the BSC-alone arm were eligible to receive panitumumab and were followed until disease progression was confirmed by the IRC.
Based upon IRC determination of disease progression, a statistically significant prolongation in PFS was observed in patients receiving panitumumab compared to those receiving BSC alone. The mean PFS was 96 days in the panitumumab arm and 60 days in the BSC-alone arm.
The study results were analyzed in the wild-type KRAS subgroup where KRAS status was retrospectively determined using archived paraffin-embedded tumor tissue. KRAS mutation status was determined in 427 patients (92%); of these, 243 (57%) had no detectable KRAS mutations in either codons 12 or 13. The hazard ratio for PFS in patients with wild-type KRAS mCRC was 0.45 (95% CI: 0.34-0.59) favoring the panitumumab arm. The response rate was 17% for the panitumumab arm and 0% for BSC. There were no differences in OS; 77% of patients in the BSC arm received panitumumab at the time of disease progression.
Study 20080763 (NCT01001377)
Study 20080763 was an open-label, multicenter, multinational, randomized (1:1) clinical trial, stratified by region (North America, Western Europe, and Australia versus rest of the world) and ECOG PS (0 and 1 vs 2) in patients with wild-type KRAS mCRC. A total of 1010 patients who received prior treatment with irinotecan, oxaliplatin, and a thymidylate synthase inhibitor were randomized to receive Vectibix 6 mg/kg intravenously over 60 minutes every 14 days or cetuximab 400 mg/m2 intravenously over 120 minutes on day 1 followed by 250 mg/m2 intravenously over 60 minutes every 7 days. The trial excluded patients with clinically significant cardiac disease and interstitial lung disease. The major efficacy analysis tested whether the OS of Vectibix was noninferior to cetuximab. Data for investigator-assessed PFS and objective response rate (ORR) were also collected. The criteria for noninferiority was for Vectibix to retain at least 50% of the OS benefit of cetuximab based on an OS hazard ratio of 0.55 from the NCIC CTG CO.17 study relative to BSC.
In Study 20080763, 37% of patients were women, 52% were white, 45% were Asian, and 1.3% were Hispanic or Latino. Thirty-one percent of patients were enrolled at sites in North America, Western Europe, or Australia. ECOG performance was 0 in 32% of patients, 1 in 60% of patients, and 2 in 8% of patients. Median age was 61 years. More patients (62%) had colon cancer than rectal cancer (38%). Most patients (74%) had not received prior bevacizumab.
The key efficacy analysis for Study 20080763 demonstrated that Vectibix was statistically significantly noninferior to cetuximab for OS.
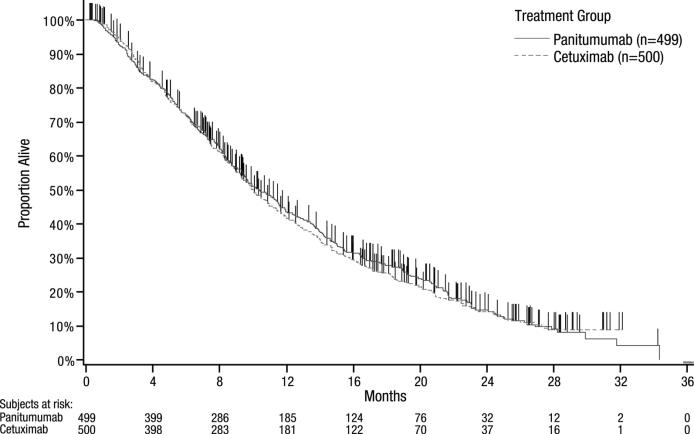
The efficacy results for Study 20080763 are presented in Table 3 and Figure 1.
Table 3: Results in Previously Treated Wild-type KRAS mCRC (Study 20080763)
| Wild-type KRAS Population | Vectibix
(n = 499)a | Cetuximab
(n = 500)a |
| OS | ||
| Number of OS events (%) | 383 (76.8) | 392 (78.4) |
| Median (months) (95% CI) | 10.4 (9.4, 11.6) | 10.0 (9.3, 11.0) |
| Hazard ratio (95% CI) | 0.97 (0.84, 1.11) | |
| PFS | ||
| Median (months) (95% CI) | 4.1 (3.2, 4.8) | 4.4 (3.2, 4.8) |
| Hazard ratio (95% CI) | 1.00 (0.88, 1.14) | |
| ORR | ||
| % (95% CI) | 22% (18%, 26%) | 19% (16%, 23%) |
a Modified intent-to-treat population that included all patients who received at least one dose of therapy
Figure 1: Kaplan-Meier Plot of Overall Survival in Patients with Wild-type KRAS mCRC (Study 20080763)
Study 20100007 (NCT01412957)
Study 20100007 was an open-label, multicenter, randomized (1:1) clinical study stratified by ECOG performance status (0 or 1 vs 2) and region (sites in Europe versus Asia versus rest of world) in patients with wild-type KRAS mCRC. Eligible patients were required to have received prior therapy with irinotecan, oxaliplatin, and a thymidylate synthase inhibitor, and have wild-type KRAS exon 2 mCRC as determined by a clinical trial assay. An assessment for RAS status (defined as KRAS exons 2, 3, and 4 and NRAS exons 2, 3, and 4) using Sanger sequencing was conducted in patients for whom tumor tissue was available.
Patients were randomized to receive Vectibix (6 mg/kg intravenously every 14 days) plus BSC or BSC alone. Patients received Vectibix and BSC or BSC until disease progression, withdrawal of consent, unacceptable toxicity, or death. Patients randomized to BSC were not offered Vectibix at the time of disease progression. The major efficacy outcome measure was OS in patients with wild-type KRAS mCRC. Secondary efficacy outcome measures included OS in the subgroup of patients with wild-type RAS mCRC; PFS and ORR in patients with wild-type KRAS; and PFS and ORR in the subgroup of patients with wild-type RAS mCRC.
A total of 377 patients were randomized, 189 to the Vectibix plus BSC arm and 188 to the BSC alone arm. Baseline demographics and disease characteristics were: median age of 61 years (range 19, 82); 57% male; 55% White, 43% Asian; 36% ECOG PS-0, 55% ECOG PS-1; 57% had a primary colon tumor and 43% had a primary rectal tumor; and 32% had prior bevacizumab exposure.
KRAS tumor mutation status was available for all patients and RAS tumor mutation status was available for 86% of the 377 patients. Among the 377 patients, 270 (72%) patients had wild-type RAS tumors, 54 (14%) had mutant RAS tumors, and 54 (14%) had unknown RAS tumor status.
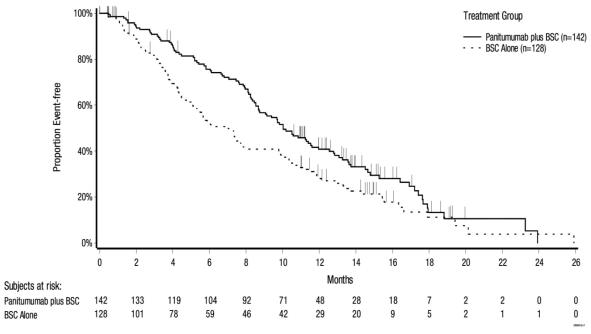
The results of the study demonstrated a statistically significant improvement in overall survival. The efficacy results for Study 20100007 are presented in Table 4 and Figure 2.
Table 4: Results in Previously Treated Wild-type KRAS and Wild-type RAS mCRC (Study 20100007)
| Wild-type KRAS
Population (n = 377) | Wild-type RAS
Population (n = 270) |
|||
| Vectibix Plus BSC
(n = 189) | BSC
(n = 188) | Vectibix Plus BSC
(n = 142) | BSC
(n = 128) |
|
| OS | ||||
| Number of deaths (%) | 136 (72) | 135 (72) | 104 (73) | 95 (74) |
| Median (months) (95% CI) | 10.0 (8.7, 11.4) | 7.4 (5.8, 9.3) | 10.0 (8.7, 11.6) | 6.9 (5.2, 7.9) |
| HR (95% CI) | 0.73 (0.57, 0.93) | 0.70 (0.53, 0.93) | ||
| p-value | 0.0096 | 0.0135 | ||
| PFS | ||||
| Number of events (%) | 182 (96) | 162 (86) | 137 (97) | 113 (88) |
| Median (months) (95% CI) | 3.6 (3.4, 5.3) | 1.7 (1.6, 1.9) | 5.2 (3.5, 5.3) | 1.7 (1.6, 2.2) |
| HR (95% CI) | 0.51 (0.41, 0.64) | 0.46 (0.35. 0.59) | ||
| p-value | < 0.0001 | < 0.0001 | ||
| ORR % (95% CI) | 27 (20.8, 33.9) | 1.6 (0.3, 4.6) | 31 (23.5, 39.3) | 2.3 (0.5, 6.7) |
Figure 2: Kaplan-Meier Plot of Overall Survival in Patients with Wild-type RAS mCRC (Study 20100007)
14.2 First-line in Combination with FOLFOX Chemotherapy
Study 20050203 (NCT00364013)
Study 20050203 was a multicenter, open-label trial that randomized (1:1) patients with mCRC who were previously untreated in the metastatic setting and who had received no prior oxaliplatin to receive Vectibix every 14 days in combination with FOLFOX or to FOLFOX alone every 14 days. Vectibix was administered at 6 mg/kg over 60 minutes prior to administration of chemotherapy. The FOLFOX regimen consisted of oxaliplatin 85 mg per m2 IV infusion over 120 minutes and leucovorin (dl-racemic) 200 mg per m2 intravenous infusion over 120 minutes at the same time on day 1 using a Y-line, followed on day 1 by 5-FU 400 mg per m2 intravenous bolus. The 5-FU bolus was followed by a continuous infusion of 5-FU 600 mg per m2 over 22 hours. On day 2, patients received leucovorin 200 mg per m2 followed by the bolus dose (400 mg per m2) and continuous infusion of 5-FU (600 mg per m2) over 22 hours. Study 20050203 excluded patients with known central nervous system metastases, clinically significant cardiac disease, interstitial lung disease, or active inflammatory bowel disease. The prespecified major efficacy measure was PFS in the subgroup of patients with wild-type KRAS mCRC as assessed by a blinded independent central review of imaging. Other key efficacy measures included OS and ORR.
In Study 20050203, in the wild-type KRAS subgroup (n = 656), 64% of patients were men, 92% White, 2% Black, and 4% Hispanic or Latino. Sixty-six percent of patients had colon cancer and 34% had rectal cancer. ECOG performance was 0 in 56% of patients, 1 in 38% of patients, and 2 in 6% of patients. Median age was 61.5 years.
The efficacy results in Study 20050203 in patients with wild-type KRAS mCRC are presented in Table 5 below.
Table 5: Results in Patients with Wild-type KRAS mCRC (Study 20050203)
| Primary Analysis | ||
| Wild-type KRAS population | Vectibix
plus FOLFOX (n = 325) | FOLFOX Alone
(n = 331) |
| PFS | ||
| Median (months) (95% CI) | 9.6 (9.2, 11.1) | 8.0 (7.5, 9.3) |
| Hazard ratio (95% CI) p-value | 0.80 (0.66, 0.97) p = 0.02 |
|
| ORR | ||
| % (95% CI) | 54% (48%, 59%) | 47% (41%, 52%) |
Exploratory Analysis of OS
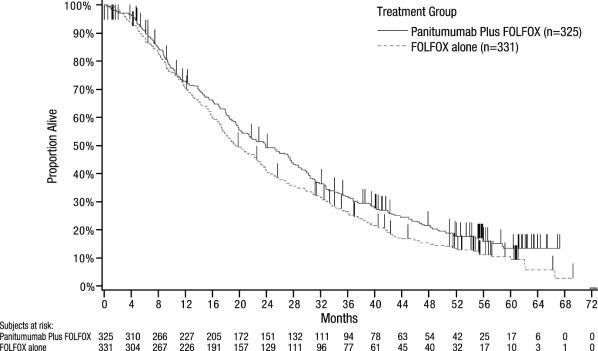
An exploratory analysis of OS with updated information based on events in 82% of patients with wild-type KRAS mCRC estimated the treatment effect of Vectibix plus FOLFOX compared with FOLFOX alone on OS (Figure 3). Median OS among 325 patients with wild-type KRAS mCRC who received Vectibix plus FOLFOX was 23.8 months (95% CI: 20.0, 27.7) vs 19.4 months (95% CI: 17.4, 22.6) among 331 patients who received FOLFOX alone (HR = 0.83, 95% CI: 0.70, 0.98).
Figure 3: Kaplan-Meier Plot of Overall Survival in Patients with Wild-type KRAS mCRC (Study 20050203)
Retrospective exploratory analyses in the RAS wild-type subgroup
Among the 656 patients with wild-type KRAS exon 2 mCRC, RAS mutation status was assessed for 620 patients using Sanger bidirectional sequencing and Surveyor®/WAVE® analysis. Of these 620 patients, approximately 17% of patients (n = 104) tumors harbored mutations in KRAS exons 3 or 4 or in NRAS exons 2, 3, and 4.
Retrospective subset analyses were then conducted among the subset of patients without RAS mutations (n = 512) as described above.
In the wild-type RAS subgroup, 65% of patients were men and 91% were White, 2% Black, and 5% Hispanic or Latino. Sixty-five percent of patients had colon cancer and 35% had rectal cancer. ECOG performance was 0 in 57% of patients, 1 in 37% of patients, and 2 in 6% of patients. Median age was 61 years.
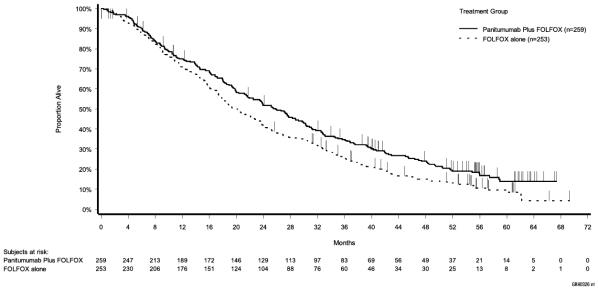
Table 6: Results in Patients with Wild-Type RAS mCRC (Study 20050203)
| Primary Analysis | ||
| Wild-type RAS population | Vectibix
plus FOLFOX (n = 259) | FOLFOX Alone
(n = 253) |
| PFS | ||
| Median (months) (95% CI) | 10.1 (9.3, 12.0) | 7.9 (7.2, 9.3) |
| Hazard ratio (95% CI) | 0.72 (0.58, 0.90) | |
| OS* | ||
| Median (months) (95% CI) | 25.8 (21.7; 29.7) | 20.2 (17.5; 23.6) |
| Hazard ratio (95% CI) | 0.77 (0.64; 0.94) | |
| ORR | ||
| % (95% CI) | 58% (51%, 64%) | 45% (39%, 51%) |
*OS with updated information based on events in 82% of patients
Figure 4: Kaplan Meier Plot of Overall Survival in Patients with Wild-Type RAS-mCRC (Study 20050203)
14.3 RAS-Mutant mCRC
Vectibix is not effective for the treatment of patients with RAS-mutant mCRC, defined as a RAS mutation in exon 2 (codons 12 and 13), exon 3 (codons 59 and 61), or exon 4 (codons 117 and 146) of KRAS and NRAS.
In Study 20050203, among patients with RAS-mutant tumors, the median PFS was 7.3 months (95% CI: 6.3, 7.9) among 272 patients receiving Vectibix plus FOLFOX and 8.7 months (95% CI: 7.6, 9.4) among patients who received FOLFOX alone (HR = 1.31, 95% CI: 1.07, 1.60). The median OS was 15.6 months (95% CI: 13.4, 17.9) among patients receiving Vectibix plus FOLFOX and 19.2 months (95% CI: 16.7, 21.8) among patients who received FOLFOX alone (HR = 1.25, 95% CI: 1.02, 1.55).
In Study 20100007, among patients with RAS-mutant tumors, no differences in OS or PFS were observed between the treatment arms [n = 54; OS HR = 0.99 (95% CI: 0.49, 2.00); PFS HR = 1.03 (95% CI: 0.56, 1.90)].
16 HOW SUPPLIED/STORAGE AND HANDLING
Vectibix is supplied as a sterile, colorless, preservative-free solution containing 20 mg/mL panitumumab in a single-dose vial. Vectibix is provided as one vial per carton.
- Each 5 mL single-dose vial contains 100 mg of panitumumab in 5 mL (20 mg/mL) (NDC 55513-954-01).
- Each 20 mL single-dose vial contains 400 mg of panitumumab in 20 mL (20 mg/mL) (NDC 55513-956-01).
Store vials in the original carton under refrigeration at 2° to 8°C (36° to 46°F) until time of use. Protect from direct sunlight. DO NOT FREEZE. Discard any unused portion remaining in the vial.
17 PATIENT COUNSELING INFORMATION
Discuss the following with patients prior to treatment with Vectibix:
Skin and eye disorders:
Advise patients to contact a healthcare professional if they experience skin or ocular/visual changes [see Boxed Warning, Dosage and Administration (2.3), Warnings and Precautions (5.1, 5.8), and Adverse Reactions (6.1, 6.3)].
Electrolyte monitoring:
Inform patients of the need for periodic monitoring of electrolytes [see Warnings and Precautions (5.3)].
Dehydration:
Advise patients of the increased risk of diarrhea and dehydration which may lead to acute renal failure and electrolyte depletion when Vectibix is administered in combination with chemotherapy [see Warnings and Precautions (5.5)].
Infusion reactions:
Advise patients of the risk of infusion reactions [see Dosage and Administration (2.3), Warnings and Precautions (5.4), and Adverse Reactions (6.1, 6.3)].
Respiratory:
Advise patients to contact a healthcare professional if they experience persistent or recurrent coughing, wheezing, dyspnea, or new-onset facial swelling [see Warnings and Precautions (5.6) and Adverse Reactions (6.1)].
Embryo-fetal Toxicity
Advise pregnant women and females of reproductive potential that Vectibix can result in fetal harm. Advise females of reproductive potential to use effective contraception during treatment with Vectibix, and for at least 2 months after the last dose and to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.10), Use in Specific Populations (8.1, 8.3)].
Lactation:
Advise women not to breastfeed during treatment with Vectibix and for 2 months after the last dose [see Use in Specific Populations (8.2)].
Infertility:
Advise females of reproductive potential of the potential for reduced fertility from Vectibix [see Use in Specific Populations (8.3)].
Sun exposure:
Advise patients to limit sun exposure (use sunscreen, wear hats) while receiving Vectibix and for 2 months after the last dose of Vectibix therapy [see Warnings and Precautions (5.7)].
Vectibix® (panitumumab)
Manufactured by:
Amgen Inc.
One Amgen Center Drive
Thousand Oaks, CA 91320-1799 USA
U.S. License No. 1080
Patent: http://pat.amgen.com/vectibix/
© 2006-2021 Amgen Inc. All rights reserved.
1xxxxxx – v25
PRINCIPAL DISPLAY PANEL
Single-Dose Vial
NDC 55513-954-01
AMGEN®
Vectibix®
(panitumumab)
Injection
100 mg
Each 5 mL single-dose vial of
Vectibix® contains 100 mg
panitumumab in a sterile,
preservative-free solution
(pH 5.8) containing 29 mg sodium
chloride and 34 mg sodium acetate
in Water for Injection, USP.
Store at 2° to 8°C.
Do not freeze or shake.
Protect from direct sunlight.
Rx Only

PRINCIPAL DISPLAY PANEL
Single-Dose Vial
NDC55513-956-01
AMGEN®
Vectibix®
(panitumumab)
Injection
400 mg
Each 20 mL single-dose vial
of Vectibix® contains 400 mg
panitumumab in a sterile,
preservative-free solution
(pH 5.8) containing 117 mg
sodium chloride and 136 mg
sodium acetate in Water for
Injection, USP.
Store at 2° to 8°C.
Do not freeze or shake.
Protect from direct sunlight.
Rx Only
