CYTOGAM- human cytomegalovirus immune globulin liquid
CSL Behring AG
----------
CYTOGAM®
Cytomegalovirus Immune Globulin Intravenous (Human)
DESCRIPTION
CYTOGAM, Cytomegalovirus Immune Globulin Intravenous (Human) (CMV-IGIV), is an immunoglobulin G (IgG) containing a standardized amount of antibody to Cytomegalovirus (CMV). CMV-IGIV is formulated in final vial as a sterile liquid. The globulin is stabilized with 5% sucrose and 1% Albumin (Human). CYTOGAM contains no preservative. The purified immunoglobulin is derived from pooled adult human plasma selected for high titers of antibody for Cytomegalovirus (CMV).1 Source material for fractionation may be obtained from another U.S. licensed manufacturer. Pooled plasma was fractionated by ethanol precipitation of the proteins according to Cohn Methods 6 and 9, modified to yield a product suitable for intravenous administration. A widely utilized solvent-detergent viral inactivation process is also used.2 Certain manufacturing operations may be performed by other firms. Each milliliter contains: 50 ± 10 mg of immunoglobulin, primarily IgG, and trace amounts of IgA and IgM; 50 mg of sucrose; 10 mg of Albumin (Human). The sodium content is 20-30 mEq per liter, i.e., 0.4-0.6 mEq per 20 mL or 1.0-1.5 mEq per 50 mL. The solution should appear colorless and translucent.
CLINICAL PHARMACOLOGY
CYTOGAM contains IgG antibodies representative of the large number of normal persons who contributed to the plasma pools from which the product was derived. The globulin contains a relatively high concentration of antibodies directed against Cytomegalovirus (CMV). In the case of persons who may be exposed to CMV, CYTOGAM can raise the relevant antibodies to levels sufficient to attenuate or reduce the incidence of serious CMV disease.
INDICATIONS AND USAGE
Cytomegalovirus Immune Globulin Intravenous (Human) is indicated for the prophylaxis of cytomegalovirus disease associated with transplantation of kidney, lung, liver, pancreas and heart. In transplants of these organs other than kidney from CMV seropositive donors into seronegative recipients, prophylactic CMV-IGIV should be considered in combination with ganciclovir.
CLINICAL STUDIES
Clinical studies have shown a 50% reduction in primary CMV disease in renal transplant patients given CMV-IGIV3 and a 56% reduction in serious CMV disease4 in liver transplant patients given CMV-IGIV. CMV-IGIV prophylaxis was associated with increased survival in liver transplant recipients.5
In two separate clinical trials, CYTOGAM was shown to provide effective prophylaxis in renal transplant recipients at risk for primary CMV disease. In the first randomized trial,3 the incidence of virologically confirmed CMV-associated syndromes was reduced from 60% in controls (n=35) to 21% in recipients of CMV immune globulin (n=24) (P <0.01); marked leukopenia was reduced from 37% in controls to 4% in globulin recipients (P <0.01); and fungal or parasitic superinfections were not seen in globulin recipients but occurred in 20% of controls (P=0.05). Serious CMV disease was reduced from 46% to 13%. There was a concomitant but not statistically significant reduction in the incidence of CMV pneumonia (17% of controls as compared with 4% of globulin recipients). There was no effect on rates of viral isolation or seroconversion although the rate of viremia was less in CYTOGAM recipients. In a subsequent non-randomized trial in renal transplant recipients (n=36),6 the incidence of virologically confirmed CMV-associated syndrome was reduced to 36% in the globulin recipients in comparison to a 60% incidence in control patients (n=35) in the randomized trial. The rates of serious CMV disease, and concomitant fungal and parasitic superinfection were similar to patients receiving CMV-IGIV in the first trial.
In a randomized, double-blind, placebo-controlled trial in liver transplant recipients,4 the incidence of serious CMV-associated disease was reduced from 26% in the 72 control patients to 12% in the 69 CMV-IGIV recipients (p=0.02); serious CMV-associated disease included CMV disease in 2 or more organs, CMV pneumonia, or CMV-associated invasive fungal infection, the incidence of which was 18% in controls and 7% in CMV-IGIV recipients (p=0.04). In follow-up5 of the liver transplant patients studied in this randomized controlled trial and a subsequent open-label trial,7 the one year survival of the 72 control patients was 72% versus 86% in the 90 recipients of CMV-IGIV (p=0.03). In the randomized control trial, the reduction in serious CMV-associated disease in CMV seronegative recipients of livers from a CMV seropositive donor (7/19 in the CMV-IGIV group vs. 9/19 in control) was less than in transplants with other donor and recipient serologic status (1/50 in the CMV-IGIV group vs. 10/53 in the control group). This finding was similar to that of Merigan et al.8 in a study of ganciclovir prophylaxis after heart transplantation. In this study, patients received ganciclovir IV at 5 mg/kg twice a day for the initial 14 days post-transplant, then at 6 mg/kg each day for 5 days per week through day 28.
Recent studies of combined prophylaxis with CMV-IGIV and ganciclovir have shown reductions in the incidence of serious CMV associated disease in CMV seronegative recipients of CMV seropositive organs below that expected from one drug alone.9-12
Ham et al.9 used CMV-IGIV with a dosage schedule of 150 mg/kg CMV-IGIV within 72 hours of transplant; 100 mg/kg at two, four, six and eight weeks following liver transplant and then 50 mg/kg at 12 and 16 weeks post-transplant in combination with ganciclovir (10 mg/kg/day for 14 days). The incidence of CMV disease was reduced from an expected 60-80% rate to 7% in 15 seronegative recipients of a seropositive organ.
Snydman10 using the CMV-IGIV dosage schedule listed under DOSAGE AND ADMINISTRATION section in combination with ganciclovir (10 mg/kg/day for 14 days) reduced the incidence of serious CMV disease in D+R- liver transplant recipients receiving placebo or one drug from 16/47 (34%) to 3/41 (7%) in patients receiving both drugs for prophylaxis.
Martin11 using CMV-IGIV 100 mg/kg every two weeks for six weeks followed by 50 mg/kg every two weeks with a final dose at week 16, in combination with ganciclovir 10 mg/kg/day for 14 days after transplantation, observed severe CMV disease in 1/74 (1%) of CMV seronegative recipients of a kidney from a CMV seropositive donor, in 0/14 (0%) of CMV seronegative recipients of a kidney-pancreas transplant from a CMV seropositive donor and in 1/12 (8%) of CMV seronegative recipients of a liver from a CMV seropositive donor. The incidence of serious CMV disease with combined CMV-IGIV and ganciclovir prophylaxis was lower than previous experience with single drug prophylaxis.
Valantine and Luikart12 compared prophylaxis with CMV-IGIV (biweekly for three months) in combination with ganciclovir prophylaxis (IV at 5 mg/kg twice a day for the initial 14 days post-transplant, then at 6 mg/kg through day 28) in 16 CMV seronegative recipients of hearts from CMV seropositive donors with 16 matched controls receiving ganciclovir alone. The actuarial incidence of CMV disease was reduced from 55% in the ganciclovir group to 46% in the combined group (p ≤0.06) and survival was increased from 61% to 94% (p ≤0.001). In heart-lung or lung transplant patients in whom either the donor or recipient was CMV seropositive, the actuarial incidence of CMV disease in patients receiving ganciclovir alone (n=25) was 85% as compared to 36% of the 33 patients receiving both CMV-IGIV and ganciclovir (p ≤0.05). Survival was 60% in the ganciclovir group and 80% in patients receiving CMV-IGIV and ganciclovir (p ≤0.01).
CONTRAINDICATIONS
CYTOGAM should not be used in individuals with a history of a prior severe reaction associated with the administration of this or other human immunoglobulin preparations. Persons with selective immunoglobulin A deficiency have the potential for developing antibodies to immunoglobulin A and could have anaphylactic reactions to subsequent administration of blood products that contain immunoglobulin A, including CYTOGAM.
WARNINGS
Because CMV-IGIV is made from human blood, it may carry a risk of transmitting infectious agents, e.g., viruses, the variant Creutzfeldt-Jakob disease (vCJD) agent and, theoretically, the Creutzfeldt-Jakob disease (CJD) agent. The risk of transmission of recognized blood-borne viruses is considered to be low because of the viral inactivation and removal properties in the Cohn-Oncley cold ethanol precipitation procedure used for purification of immune globulin products.13-15 Until 1993, cold ethanol manufactured immune globulins licensed in the United States had not been documented to transmit any viral agent. However, during a brief period in late 1993 to early 1994, intravenous immune globulin made by one U.S. manufacturer was associated with transmission of Hepatitis C virus.16 To further guard against possible transmission of blood-borne viruses, including Hepatitis C, CMV-IGIV is treated with a solvent detergent viral inactivation procedure2 known to inactivate a wide spectrum of lipid enveloped viruses, including HIV-1, HIV-2, Hepatitis B, and Hepatitis C.17 However, because new blood-borne viruses may yet emerge, some of which may not be inactivated by the manufacturing process or by solvent detergent treatment, CMV-IGIV, like any other blood product, should be given only if a benefit is expected.
Immune Globulin Intravenous (Human) products have been reported to be associated with renal dysfunction, acute renal failure, osmotic nephrosis and death.18-25 Patients predisposed to acute renal failure include patients with any degree of pre-existing renal insufficiency, diabetes mellitus, age greater than 65, volume depletion, sepsis, paraproteinemia or patients receiving known nephrotoxic drugs. Especially in such patients, IGIV products should be administered at the minimum concentrations available and the minimum rate of infusion practicable. While these reports of renal dysfunction and acute renal failure have been associated with the use of many IGIV products, those containing sucrose as a stabilizer (and given at daily doses of 350 mg/kg or greater) account for a disproportionate share of the total number.18 CYTOGAM contains sucrose as a stabilizer. See PRECAUTIONS and DOSAGE AND ADMINISTRATION sections for important information intended to reduce the risk of acute renal failure.
During administration, the patient's vital signs should be monitored continuously and careful observation made for any symptoms throughout the infusion. Epinephrine should be available for the treatment of an acute anaphylactic reaction (see PRECAUTIONS section).
PRECAUTIONS
General
CYTOGAM does not contain a preservative. The vial should be entered only once for administration purposes and the infusion should begin within 6 hours. The infusion schedule should be adhered to closely (see DOSAGE AND ADMINISTRATION: Infusion section). Do not use if the solution is turbid.
Although systemic allergic reactions are rare (see ADVERSE REACTIONS section), epinephrine and diphenhydramine should be available for treatment of acute allergic symptoms. If hypotension or anaphylaxis occur, the administration of the immunoglobulin should be discontinued immediately and an antidote should be given as noted above.
Renal Function
Assure that patients are not volume depleted prior to the initiation of IGIV. Periodic monitoring of renal function tests and urine output is particularly important in patients judged to have a potential increased risk for developing acute renal failure. Renal function, including the measurement of blood urea nitrogen (BUN) and serum creatinine should be assessed prior to the initial infusion of CYTOGAM and again at appropriate intervals thereafter. If renal function deteriorates, discontinuation of the product should be considered. The recommended rate of CYTOGAM infusion for prophylaxis of CMV disease in solid organ transplant patients is 60 mg Ig/kg/hr (see DOSAGE AND ADMINISTRATION).
Aseptic Meningitis Syndrome
An aseptic meningitis syndrome (AMS) has been reported to occur infrequently in association with Immune Globulin Intravenous (Human) (IGIV) treatment.26-29 The syndrome usually begins within several hours to two days following IGIV treatment. It is characterized by symptoms and signs including severe headache, nuchal rigidity, drowsiness, fever, photophobia, painful eye movements, and nausea and vomiting. Cerebrospinal fluid (CSF) studies are frequently positive with pleocytosis up to several thousand cells per cu.mm., predominantly from the granulocytic series, and elevated protein levels up to several hundred mg/dL. Patients exhibiting such symptoms and signs should receive a thorough neurological examination, including CSF studies, to rule out other causes of meningitis. AMS may occur more frequently in association with high dose (2 g/kg) IGIV treatment. Discontinuation of IGIV treatment has resulted in remission of AMS within several days without sequelae.
Hemolysis
Immune Globulin Intravenous (Human) (IGIV) products can contain blood group antibodies which may act as hemolysins and induce in vivo coating of red blood cells with immunoglobulin, causing a positive direct antiglobulin reaction and, rarely, hemolysis.30-32 Hemolytic anemia can develop subsequent to IGIV therapy due to enhanced RBC sequestration33 (see ADVERSE REACTIONS). IGIV recipients should be monitored for clinical signs and symptoms of hemolysis (see PRECAUTIONS: Laboratory Tests).
Transfusion-Related Acute Lung Injury (TRALI)
There have been reports of noncardiogenic pulmonary edema [Transfusion-Related Acute Lung Injury (TRALI)] in patients administered IGIV.34 TRALI is characterized by severe respiratory distress, pulmonary edema, hypoxemia, normal left ventricular function, and fever and typically occurs within 1-6 hours after transfusion. Patients with TRALI may be managed using oxygen therapy with adequate ventilatory support.
IGIV recipients should be monitored for pulmonary adverse reactions. If TRALI is suspected, appropriate tests should be performed for the presence of anti-neutrophil antibodies in both the product and patient serum (see PRECAUTIONS: Laboratory Tests).
Thrombotic Events
Thrombotic events have been reported in association with IGIV35-37 (see ADVERSE REACTIONS). Patients at risk may include those with a history of atherosclerosis, multiple cardiovascular risk factors, advanced age, impaired cardiac output, and/or known or suspected hyperviscosity. The potential risks and benefits of IGIV should be weighed against those of alternative therapies for all patients for whom IGIV administration is being considered. Baseline assessment of blood viscosity should be considered in patients at risk for hyperviscosity, including those with cryoglobulins, fasting chylomicronemia/markedly high triacylglycerols (triglycerides), or monoclonal gammopathies (see PRECAUTIONS: Laboratory Tests).
Laboratory Tests
If signs and/or symptoms of hemolysis are present after IGIV infusion, appropriate confirmatory laboratory testing should be done (see PRECAUTIONS).
If TRALI is suspected, appropriate tests should be performed for the presence of anti-neutrophil antibodies in both the product and the patient serum (see PRECAUTIONS).
Because of the potentially increased risk of thrombosis, baseline assessment of blood viscosity should be considered in patients at risk for hyperviscosity, including those with cryoglobulins, fasting chylomicronemia/markedly high triacylglycerols (triglycerides), or monoclonal gammopathies (see PRECAUTIONS).
Drug Interactions
Antibodies present in immune globulin preparations may interfere with the immune response to live virus vaccines such as measles, mumps, and rubella; therefore, vaccination with live virus vaccines should be deferred until approximately three months after administration of CYTOGAM. If such vaccinations were given shortly after CYTOGAM, a revaccination may be necessary. Admixtures of CYTOGAM with other drugs have not been evaluated. It is recommended that CYTOGAM be administered separately from other drugs or medications which the patient may be receiving (see DOSAGE AND ADMINISTRATION section).
Pregnancy
Animal reproduction studies have not been conducted with Cytomegalovirus Immune Globulin Intravenous (Human). It is also not known whether Cytomegalovirus Immune Globulin Intravenous (Human) can cause fetal harm when administered to a pregnant woman or can affect reproduction capacity. Cytomegalovirus Immune Globulin Intravenous (Human) should be given to a pregnant woman only if clearly needed.
Information for Patients
Patients should be instructed to report all infections directly to their physician and to CSL Behring Pharmacovigilance at 1-866-915-6958. The risks and benefits of this product should be discussed with the patient. In addition, patients should be instructed to immediately report symptoms of decreased urine output, sudden weight gain, and/or shortness of breath (which may suggest kidney damage) to their physician.
ADVERSE REACTIONS
Minor reactions such as flushing, chills, muscle cramps, back pain, fever, nausea, vomiting, arthralgia, and wheezing were the most frequent adverse reactions observed during the clinical trials of CYTOGAM, Cytomegalovirus Immune Globulin Intravenous (Human). The incidence of these reactions during the clinical trials was less than 6.0% of all infusions and such reactions were most often related to infusion rates. A decrease in blood pressure was observed in 1 of 1039 infusions in clinical trials of CYTOGAM. If a patient develops a minor side effect, slow the rate immediately or temporarily interrupt the infusion.
Increases in serum creatinine and blood urea nitrogen (BUN) have been observed as soon as one to two days following IGIV infusion. Progression to oliguria or anuria requiring dialysis has been observed. Types of severe renal adverse events that have been seen following IGIV therapy include acute renal failure, acute tubular necrosis, proximal tubular nephropathy and osmotic nephrosis.18-25
Severe reactions such as angioneurotic edema and anaphylactic shock, although not observed during clinical trials, are a possibility. Clinical anaphylaxis may occur even when the patient is not known to be sensitized to immune globulin products. A reaction may be related to the rate of infusion; therefore, carefully adhere to the infusion rates as outlined under DOSAGE AND ADMINISTRATION. If anaphylaxis or drop in blood pressure occurs, discontinue infusion and use antidote such as diphenhydramine and adrenalin.
Postmarketing
The following adverse reactions have been identified and reported during the post-approval use of IGIV products:38
Respiratory: Apnea, Acute Respiratory Distress Syndrome (ARDS), Transfusion-Related Acute Lung Injury (TRALI), cyanosis, hypoxemia, pulmonary edema, dyspnea, bronchospasm
Cardiovascular: Cardiac arrest, thromboembolism, vascular collapse, hypotension
Neurological: Coma, loss of consciousness, seizures, tremor
Integumentary: Stevens-Johnson syndrome, epidermolysis, erythema multiforme, bullous dermatitis
Hematologic: Pancytopenia, leukopenia, hemolysis, positive direct antiglobulin (Coombs) test
General/Body as a Whole: Pyrexia, Rigors
Musculoskeletal: Back pain
Gastrointestinal: Hepatic dysfunction, abdominal pain
Because postmarketing reporting of these reactions is voluntary and the at-risk populations are of uncertain size, it is not always possible to reliably estimate the frequency of the reaction or establish a causal relationship to exposure to the product. Such is also the case with literature reports authored independently.
OVERDOSAGE
Although few data are available, clinical experience with other immunoglobulin preparations suggests that the major manifestations would be those related to volume overload.
DOSAGE AND ADMINISTRATION
The maximum recommended total dosage per infusion is 150 mg Ig/kg, administered according to the following schedule:
| Type of Transplant | ||
|---|---|---|
| Kidney | Liver, Pancreas, Lung, Heart | |
| Within 72 hours of transplant: | 150 mg/kg | 150 mg/kg |
| 2 weeks post transplant: | 100 mg/kg | 150 mg/kg |
| 4 weeks post transplant: | 100 mg/kg | 150 mg/kg |
| 6 weeks post transplant: | 100 mg/kg | 150 mg/kg |
| 8 weeks post transplant: | 100 mg/kg | 150 mg/kg |
| 12 weeks post transplant: | 50 mg/kg | 100 mg/kg |
| 16 weeks post transplant: | 50 mg/kg | 100 mg/kg |
Preparation for Administration
Remove the tab portion of the vial cap and clean the rubber stopper with 70% alcohol or equivalent. DO NOT SHAKE VIAL; AVOID FOAMING.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration whenever solution and container permit. Infuse the solution only if it is colorless, free of particulate matter and not turbid.
Infusion
Infusion should begin within 6 hours after entering the vial and should be complete within 12 hours of entering the vial. Vital signs should be taken preinfusion, mid-way and post-infusion as well as before any rate increase. CYTOGAM should be administered through an intravenous line using an administration set that contains an in-line filter (pore size 15µ) and a constant infusion pump (i.e., IVAC pump or equivalent). A smaller in-line filter (0.2µ) is also acceptable. Pre-dilution of CYTOGAM before infusion is not recommended. CYTOGAM should be administered through a separate intravenous line. If this is not possible, CYTOGAM may be "piggybacked" into a pre-existing line if that line contains either Sodium Chloride Injection, USP, or one of the following dextrose solutions (with or without NaCl added): 2.5% dextrose in water, 5% dextrose in water, 10% dextrose in water, 20% dextrose in water. If a pre-existing line must be used, the CYTOGAM should not be diluted more than 1:2 with any of the above-named solutions. Admixtures of CYTOGAM with any other solutions have not been evaluated.
Initial Dose
Administer intravenously at 15 mg Ig per kg body weight per hour. If no adverse reactions occur after 30 minutes, the rate may be increased to 30 mg Ig/kg/hr; if no adverse reactions occur after a subsequent 30 minutes, then the infusion may be increased to 60 mg Ig/kg/hr (volume not to exceed 75 mL/hour). DO NOT EXCEED THIS RATE OF ADMINISTRATION. The patient should be monitored closely during and after each rate change.
Subsequent Doses
Administer at 15 mg Ig/kg/hr for 15 minutes. If no adverse reactions occur, increase to 30 mg Ig/kg/hr for 15 minutes and then increase to a maximum rate of 60 mg Ig/kg/hr (volume not to exceed 75 mL/hour). DO NOT EXCEED THIS RATE OF ADMINISTRATION. The patient should be monitored closely during each rate change.
CYTOGAM should be used with caution in patients with pre-existing renal insufficiency and in patients judged to be at increased risk of developing renal insufficiency (including, but not limited to those with diabetes mellitus, age greater than 65, volume depletion, paraproteinemia, sepsis and patients receiving known nephrotoxic drugs). In these cases especially, it is important to assure that patients are not volume depleted prior to CYTOGAM infusion. While most cases of renal insufficiency have occurred in patients receiving total doses of 350 mg Ig/kg or greater, no prospective data are presently available to identify a maximum safe dose, concentration or rate of infusion in patients determined to be at increased risk of acute renal failure. In the absence of prospective data, recommended doses should not be exceeded and the concentration and infusion rate selected should be the minimum practicable.
Potential adverse reactions are: flushing, chills, muscle cramps, back pain, fever, nausea, vomiting, wheezing, drop in blood pressure. Minor adverse reactions have been infusion rate related – if the patient develops a minor side effect (i.e., nausea, back pain, flushing), slow the rate or temporarily interrupt the infusion. If anaphylaxis or drop in blood pressure occurs, discontinue infusion and use antidote such as diphenhydramine and adrenalin.
To prevent the transmission of hepatitis viruses or other infectious agents from one person to another, sterile disposable syringes and needles should be used. The syringes and needles should not be reused.
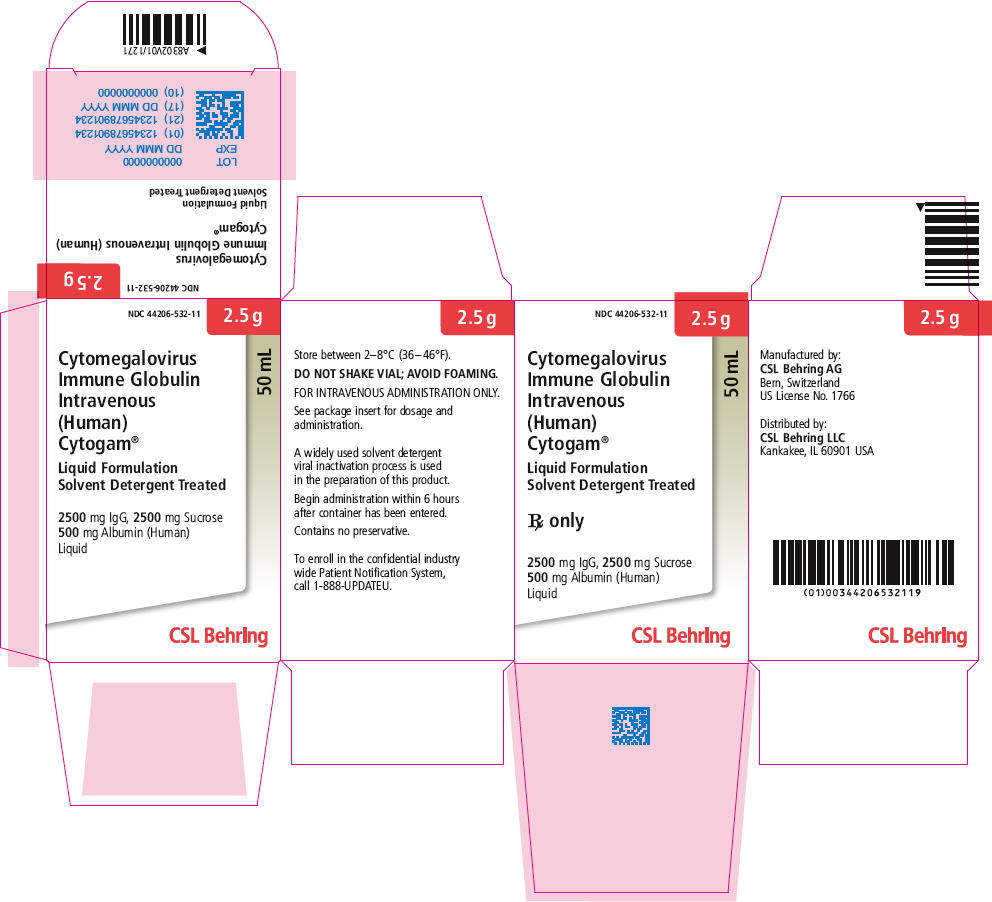
HOW SUPPLIED
CYTOGAM is supplied in a 50 mL single-dose vial (50 mg/mL).
The product presentation includes a package insert and the following component:
| Presentation | Carton NDC Number | Component |
|---|---|---|
| 2.5 g | 44206-532-11 | CYTOGAM in a single-use vial [NDC 44206-532-90] |
REFERENCES
- Snydman DR, McIver J, Leszczynski J, et al. A pilot trial of a novel cytomegalovirus immune globulin in renal transplant recipients. Transplantation 1984;38:553-557.
- Horowitz B, Wiebe ME, Lippin A, et al. Inactivation of viruses in labile blood derivatives. Transfusion 1985;25:516-522.
- Snydman DR, Werner BG, Heinze-Lacey BH, et al. Use of cytomegalovirus immune globulin to prevent cytomegalovirus disease in renal transplant recipients. N Engl J Med 1987;317:1049-1054.
- Snydman DR, Werner BG, Dougherty NN, et al. Cytomegalovirus Immune Globulin prophylaxis in liver transplantation. A randomized, double-blind, placebo-controlled trial. Ann Int Med 1993;119:984-991.
- Falagas ME, Snydman DR, Ruthazer R, et al. Cytomegalovirus Immune Globulin (CMVIG) prophylaxis is associated with increased survival after orthotopic liver transplantation. Clin Transplant 1997;11:432-437.
- Snydman DR, Werner BG, Tilney NL, et al. A final analysis of primary cytomegalovirus disease prevention in renal transplant recipients with a cytomegalovirus immune globulin: Comparison of randomized and open-label trials. Transplant Proc 1991;23:1357-1360.
- Snydman DR, Werner BG, Dougherty NN, et al. A further analysis of the use of Cytomegalovirus Immune Globulin in orthotopic liver transplant patients at risk for primary infection. Transplant Proc 1994;26 Suppl 1:23-27.
- Merigan TC, Renlund DG, Keay S, et al. A controlled trial of ganciclovir to prevent cytomegalovirus disease after heart transplantation. N Engl J Med 1992;326:1182-1186.
- Ham JM, Shelden SR, Godkin RR, et al. Cytomegalovirus prophylaxis with ganciclovir, acyclovir and CMV hyperimmune globulin in liver transplant patients receiving OKT3 induction. Transplant Proc 1995;27 (5 Suppl 1):31-33.
- Snydman DR. Combined CMV-IGIV and ganciclovir prophylaxis in CMV seronegative transplant recipients from CMV seropositive donors. Report on file.
- Martin M. CMV prophylaxis with combination ganciclovir and CMV hyperimmune globulin followed by high-dose acyclovir in solid organ transplant recipients. Report on file.
- Valantine H, Luikart H. Impact of CMV hyperimmune globulin on outcome after cardiothoracic transplantation: A comparative study of combined prophylaxis with CMVIG plus ganciclovir vs. ganciclovir alone. Report on file.
- Bossell, et al. Safety of therapeutic immune globulin preparations with respect to transmission of human T-lymphotropic virus type III / lymphadenopathy-associated virus infection. MMWR 1996;35:231-233.
- Wells MA, Wittek AE, Epstein JS, et al. Inactivation and partition of human T-cell lymphotropic virus type III, during ethanol fractionation of plasma. Transfusion 1986;26:210-213.
- McIver J, Grady G. Immunoglobulin preparations. In: Churchill WH, and Kurtz SR, editors. Transfusion Medicine. Boston: Blackwell Scientific Publications; 1988.
- Schneider L, Geha R. Outbreak of Hepatitis C associated with intravenous immunoglobulin administration – United States, October 1993 - June 1994. MMWR 1994;43:505-509.
- Edwards CA, Piet MPJ, Chin S, et al. Tri(nButyl) phosphate detergent treatment of licensed therapeutic and experimental blood derivatives. Vox Sang 1987;52:53-59.
- Cayco AV, Perazella MA, Hayslett JP. Renal insufficiency after intravenous immune globulin therapy: A report of two cases and an analysis of the literature. J Am Soc Nephrol 1997;8:1788-1794.
- Cantu TG, Hoehn-Saric EW, Burgess KM, Racusen L, Scheel PJ. Acute renal failure associated with immunoglobulin therapy. Am J Kidney Dis 1995;25:228-234.
- Hansen-Schmidt S, Silomon J, Keller F. Osmotic nephrosis due to high-dose intravenous immunoglobulin therapy containing sucrose (but not with glycine) in a patient with immunoglobulin A nephritis. Am J Kidney Dis 1996;28: 451-453.
- Tan E, Hajinazarian M, Bay W, Neff J, Mendell JR. Acute renal failure resulting from intravenous immunoglobulin therapy. Arch Neurol 1993;50:137-139.
- Winward D, Brophy MT. Acute renal failure after administration of intravenous immunoglobulin: Review of the literature and case report. Pharmacotherapy 1995;15:765-772.
- Phillips AO. Renal failure and intravenous immunoglobulin [letter; comment]. Clin Nephrol 1992;37:217.
- Lindberg HA, Wald MH, Barker MH. Renal changes following administration of hypertonic solutions. Arch Intern Med 1939;63:907-918.
- Rigdon RH, Cardwell ES. Renal lesions following the intravenous injection of a hypertonic solution of sucrose. Arch Intern Med 1942;69:670-690.
- Sekul E, Culper E, Dalakas M. Aseptic meningitis associated with high-dose intravenous immunoglobulin therapy; Frequency and risk factors. Ann Intern Med 1994;121:259-262.
- Kato E, Shindo S, Eto Y, et al. Administration of immune globulin associated with aseptic meningitis. JAMA 1988; 259:3269-3270.
- Casteels Van Daele M, Wijindaele L, Hunnick K, et al. Intravenous immunoglobulin and acute aseptic meningitis. N Engl J Med 1990;323:614-615.
- Scribner C, Kapit R, Philips E, et al. Aseptic meningitis and intravenous immunoglobulin therapy. Ann Intern Med 1994;121:305-306.
- Copelan EA, Strohm PL, Kennedy MS, Tutschka PJ. Hemolysis following intravenous immune globulin therapy. Transfusion 1986;26:410-412.
- Thomas MJ, Misbah SA, Chapel HM, Jones M, Elrington G, Newsom-Davis J. Hemolysis after high-dose intravenous Ig. Blood 1993;15:3789.
- Reinhart WH, Berchtold PE. Effect of high dose intravenous immunoglobulin therapy on blood rheology. Lancet 1992;339:662-664.
- Kessary-Shoham H, Levy Y, Shoenfeld Y, Lorber M, Gershon H. In vivo administration of intravenous immunoglobulin (IVIg) can lead to enhanced erythrocyte sequestration. J Autoimmun 1999;13:129-135.
- Rizk A, Gorson KC, Kenney L, Weinstein R. Transfusion-related acute lung injury after the infusion of IVIG. Transfusion 2001;41:264-268.
- Dalakas MC. High-dose intravenous immunoglobulin and serum viscosity: risk of precipitant thromboembolic events. Neurology 1994;44:223-226.
- Woodruff RK, Grigg AP, Firkin FC, Smith IL. Fatal thrombotic events during treatment of autoimmune thrombocytopenia with intravenous immunoglobulin in elderly patients. Lancet 1986;2:217-218.
- Wolberg AS, Kon RH, Monroe DM, Hoffman M. Coagulation factor XI is a contaminant in intravenous immunoglobulin preparations. Am J Hematol 2000;65:30-34.
- Pierce LR, Jain N. Risks associated with the use of intravenous immunoglobulin. Trans Med Rev 2003;17:241-251.
For additional information concerning Cytomegalovirus Immune Globulin Intravenous (Human) contact:
CSL Behring Medical Affairs
CSL Behring LLC
King of Prussia, PA 19406 USA
1-800-504-5434
Manufactured by:
CSL Behring AG
Bern, Switzerland
US License No. 1766
Distributed by:
CSL Behring LLC
Kankakee, IL 60901 USA
Revised May 2018
PRINCIPAL DISPLAY PANEL - 50 mL Vial Label
Cytomegalovirus
Immune Globulin Intravenous (Human)
Cytogam®
NDC 44206-532-90
2500 mg IgG Liquid
Liquid Formulation Solvent Detergent Treated
Store between 2–8°C (36–46°F). DO NOT SHAKE VIAL; AVOID FOAMING.
FOR INTRAVENOUS ADMINISTRATION ONLY. See package insert for dosage
and administration. Begin administration within 6 hours after container has
been entered. Contains no preservative. A widely used solvent detergent viral
inactivation process is used in the preparation of this product.
Rx only
A8303/129
Manufactured by:
CSL Behring AG
Bern, Switzerland
US License No. 1766
Distributed by:
CSL Behring LLC
Kankakee, IL 60901
USA
2.5 g
50 mL

| CYTOGAM
human cytomegalovirus immune globulin liquid |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - CSL Behring AG (481152762) |