FULL PRESCRIBING INFORMATION
WARNING: POTENTIAL RISK OF OSTEOSARCOMA
- In male and female rats, parathyroid hormone caused an increase in the incidence of osteosarcoma (a malignant bone tumor). The occurrence of osteosarcoma was dependent on parathyroid hormone dose and treatment duration. This effect was observed at parathyroid hormone exposure levels ranging from 3 to 71 times the exposure levels in humans receiving a 100 mcg dose of NATPARA. These data could not exclude a risk to humans [see Warnings and Precautions (5.1), Nonclinical Toxicology (13.1)].
- Because of a potential risk of osteosarcoma, use NATPARA only in patients who cannot be well-controlled on calcium and active forms of vitamin D alone and for whom the potential benefits are considered to outweigh this potential risk [see Indications and Usage (1), Warnings and Precautions (5.1)].
- Avoid use of NATPARA in patients who are at increased baseline risk for osteosarcoma, such as patients with Paget's disease of bone or unexplained elevations of alkaline phosphatase, pediatric and young adult patients with open epiphyses, patients with hereditary disorders predisposing to osteosarcoma or patients with a prior history of external beam or implant radiation therapy involving the skeleton [see Warnings and Precautions (5.1)].
- Because of the risk of osteosarcoma, NATPARA is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the NATPARA REMS Program [see Warnings and Precautions (5.2)].
1 INDICATIONS AND USAGE
NATPARA is a parathyroid hormone indicated as an adjunct to calcium and vitamin D to control hypocalcemia in patients with hypoparathyroidism.
Limitations of Use
- Because of the potential risk of osteosarcoma, NATPARA is recommended only for patients who cannot be well-controlled on calcium supplements and active forms of vitamin D alone [see Warnings and Precautions (5.1)].
- NATPARA was not studied in patients with hypoparathyroidism caused by calcium-sensing receptor mutations.
- NATPARA was not studied in patients with acute post-surgical hypoparathyroidism.
2 DOSAGE AND ADMINISTRATION
2.1 Dosing Guidelines
The dose of NATPARA should be individualized based on total serum calcium (albumin-corrected) and 24-hour urinary calcium excretion. The recommended NATPARA dose is the minimum dose required to prevent both hypocalcemia and hypercalciuria. This dose will generally be the dose that maintains total serum calcium (albumin-corrected) within the lower half of the normal range (i.e., between 8 and 9 mg/dL) without the need for active forms of vitamin D and with calcium supplementation sufficient and individualized to meet the patient's daily requirements.
Doses of active forms of vitamin D and calcium supplements will need to be adjusted when using NATPARA.
2.2 Before Initiating NATPARA and During Therapy with NATPARA
- Confirm 25-hydroxyvitamin D stores are sufficient. If insufficient, replace to sufficient levels per standard of care.
- Confirm serum calcium is above 7.5 mg/dL before starting NATPARA.
- The goal of NATPARA treatment is to achieve serum calcium within the lower half of the normal range.
2.3 Initiating NATPARA
- 1.
- Initiate NATPARA 50 mcg once daily as a subcutaneous injection in the thigh (alternate thigh every day).
- 2.
- In patients using active forms of vitamin D, decrease the dose of active vitamin D by 50%, if serum calcium is above 7.5 mg/dL.
- 3.
- In patients using calcium supplements, maintain calcium supplement dose.
- 4.
- Measure serum calcium concentration within 3 to 7 days.
- 5.
- Adjust dose of active vitamin D or calcium supplement or both based on serum calcium value and clinical assessment (i.e., signs and symptoms of hypocalcemia or hypercalcemia). Suggested adjustments to active vitamin D and calcium supplement based on serum calcium levels are provided below (see Table 1).
| Adjust First | Adjust Second | |
|---|---|---|
| Serum Calcium | Active Vitamin D Forms | Calcium Supplement |
|
||
| Above the Upper Limit of Normal (10.6 mg/dL) | Decrease or Discontinue* | Decrease |
| Greater than 9 mg/dL and below the Upper Limit of Normal (10.6 mg/dL) | Decrease or Discontinue* | No change or decrease if active vitamin D has been discontinued |
| Less than or equal to 9 mg/dL and above 8 mg/dL | No change | No change |
| Lower than 8 mg/dL | Increase | Increase |
- 6.
- Repeat steps 4 and 5 until target serum calcium levels are within the lower half of the normal range, active vitamin D has been discontinued and calcium supplementation is sufficient to meet daily requirements.
2.4 NATPARA Dose Adjustments
The dose of NATPARA may be increased in increments of 25 mcg every four weeks up to a maximum daily dose of 100 mcg if serum calcium cannot be maintained above 8 mg/dL without an active form of vitamin D and/or oral calcium supplementation.
The dose of NATPARA may be decreased to as low as 25 mcg per day if total serum calcium is repeatedly above 9 mg/dL after the active form of vitamin D has been discontinued and calcium supplement has been decreased to a dose sufficient to meet daily requirements.
After a NATPARA dose change monitor clinical response as well as serum calcium. Adjust active vitamin D and calcium supplements per steps 4-6 above if indicated [see Dosage and Administration (2.3)].
2.5 NATPARA Maintenance Dose
The maintenance dose should be the lowest dose that achieves a total serum calcium (albumin-corrected) within the lower half of the normal total serum calcium range (i.e., approximately 8 and 9 mg/dL), without the need for active forms of vitamin D and with calcium supplementation sufficient to meet daily requirements. Monitor serum calcium and 24-hour urinary calcium per standard of care once a maintenance dose is achieved.
2.6 NATPARA Dose Interruption or Discontinuation
Abrupt interruption or discontinuation of NATPARA can result in severe hypocalcemia. Resume treatment with, or increase the dose of, an active form of vitamin D and calcium supplements if indicated in patients interrupting or discontinuing NATPARA, monitor for signs and symptoms of hypocalcemia and serum calcium levels [see Warnings and Precautions (5.4)].
In the case of a missed dose, the next NATPARA dose should be administered as soon as reasonably feasible and additional exogenous calcium should be taken in the event of hypocalcemia.
2.7 Reconstitution and Administration Instructions
- Patients and caregivers who will administer NATPARA should receive appropriate training and instruction by a trained healthcare professional prior to first use of NATPARA.
- Follow the Instructions for Use to reconstitute NATPARA using the mixing device for reconstitution and to administer NATPARA using the pen delivery device (i.e., Q-Cliq® pen).
- Inspect NATPARA visually for particulate matter and discoloration prior to administration.
- Discard the needle in a puncture-resistant container following administration.
- Store the Q-Cliq pen containing the remaining doses of NATPARA in a refrigerator.
- All reconstituted NATPARA medication cartridges older than 14 days must be discarded [see How Supplied/Storage and Handling (16.2)].
3 DOSAGE FORMS AND STRENGTHS
NATPARA is supplied as a multiple-dose, dual-chamber glass cartridge containing a sterile powder and diluent in 4 dosage strengths.
| For injection: | 25 mcg per dose strength | (0.4 mg for reconstitution with 1.13 mL) |
| For injection: | 50 mcg per dose strength | (0.8 mg for reconstitution with 1.13 mL) |
| For injection: | 75 mcg per dose strength | (1.21 mg for reconstitution with 1.13 mL) |
| For injection: | 100 mcg per dose strength | (1.61 mg for reconstitution with 1.13 mL) |
4 CONTRAINDICATIONS
NATPARA is contraindicated in patients with a known hypersensitivity to any component of this product. Hypersensitivity reactions (e.g., anaphylaxis, angioedema, and urticaria) have occurred with NATPARA [see Warnings and Precautions (5.6), Adverse Reactions (6.3)].
5 WARNINGS AND PRECAUTIONS
5.1 Potential Risk of Osteosarcoma
In male and female rats, parathyroid hormone caused an increase in the incidence of osteosarcoma (a malignant bone tumor). The occurrence of osteosarcoma was observed to be dependent on parathyroid hormone dose and treatment duration. This effect was observed at parathyroid hormone exposure levels ranging from 3 to 71 times the exposure levels for humans receiving a 100 mcg dose of NATPARA. These data could not exclude a risk to humans [see Nonclinical Toxicology (13.1)].
Because of a potential risk of osteosarcoma, use NATPARA only in patients who cannot be well-controlled on calcium supplements and active forms of vitamin D alone and for whom the potential benefits are considered to outweigh this potential risk [see Limitations of Use (1)].
To further mitigate the potential risk of osteosarcoma, avoid use of NATPARA in patients who are at increased risk for osteosarcoma, such as patients with Paget's disease of bone or unexplained elevations of alkaline phosphatase, pediatric and young adult patients with open epiphyses, patients with hereditary disorders predisposing to osteosarcoma, or patients with a prior history of external beam or implant radiation therapy involving the skeleton. Instruct patients to promptly report clinical symptoms (e.g., persistent localized pain) and signs (e.g., soft tissue mass tender to palpation) that could be consistent with osteosarcoma.
NATPARA is available only through a restricted program under a REMS [see Warnings and Precautions (5.2)].
5.2 NATPARA REMS Program
Because of the potential risk of osteosarcoma associated with NATPARA therapy, NATPARA is available only through a restricted REMS program called the NATPARA REMS Program. Under the NATPARA REMS Program, only certified healthcare providers can prescribe and only certified pharmacies can dispense NATPARA. Further information is available at www.NATPARAREMS.com or by telephone at 1-855-NATPARA (1-855-628-7272).
5.3 Hypercalcemia
Severe hypercalcemia has been reported with NATPARA. In the pivotal trial, 3 patients randomized to NATPARA required administration of IV fluids to correct hypercalcemia during treatment with NATPARA. The risk is highest when starting or increasing the dose of NATPARA, but can occur at any time. Monitor serum calcium and patients for signs and symptoms of hypercalcemia. Treat hypercalcemia per standard practice and consider holding and/or lowering the dose of NATPARA if severe hypercalcemia occurs [see Dosage and Administration (2), Adverse Reactions (6.1)].
5.4 Hypocalcemia
Severe hypocalcemia has been reported in patients taking NATPARA, including cases of hypocalcemia that resulted in seizures. The risk is highest when NATPARA is withheld, missed or abruptly discontinued, but can occur at any time. Monitor serum calcium and patients for signs and symptoms of hypocalcemia. Resume treatment with, or increase the dose of, an active form of vitamin D or calcium supplements or both if indicated in patients interrupting or discontinuing NATPARA to prevent severe hypocalcemia [see Dosage and Administration (2.6), Adverse Reactions (6.1)].
5.5 Risk of Digoxin Toxicity with Concomitant Use of Digitalis Compounds
The inotropic effects of digoxin are affected by serum calcium levels. Hypercalcemia of any cause may predispose to digoxin toxicity. In patients using NATPARA concomitantly with digitalis compounds, monitor serum calcium and digoxin levels and patients for signs and symptoms of digitalis toxicity. Adjustment of digoxin and/or NATPARA may be needed. No drug-drug interaction study has been conducted with digoxin and NATPARA [see Drug Interactions (7), Adverse Reactions (6.1)].
5.6 Hypersensitivity
There have been reports of hypersensitivity reactions in patients taking NATPARA. Reactions included anaphylaxis, dyspnea, angioedema, urticaria, and rash. If signs or symptoms of a serious hypersensitivity reaction occur, discontinue treatment with NATPARA, treat hypersensitivity reaction according to the standard of care, and monitor until signs and symptoms resolve [see Contraindications (4), Adverse Reactions (6.3)]. Monitor for hypocalcemia if NATPARA is discontinued [see Dosage and Administration (2.6)].
6 ADVERSE REACTIONS
The following serious adverse reactions are described in greater detail in other sections of the label:
- Osteosarcoma [see Boxed Warning, Warnings and Precautions (5.1)]
- Hypercalcemia [see Warnings and Precautions (5.3)]
- Hypocalcemia [see Warnings and Precautions (5.4)]
- Hypersensitivity [see Contraindications (4), Warnings and Precautions (5.6)]
6.1 Adverse Reactions in Clinical Trials for Hypoparathyroidism
Because clinical trials are conducted under widely varying conditions, the adverse reaction rates observed cannot be directly compared to rates in other clinical trials and may not reflect the rates observed in clinical practice.
NATPARA was studied in a placebo-controlled trial [see Clinical Studies (14)].
The data described in Table 2 below reflect exposure to NATPARA in 84 patients, including 78 exposed for 24 weeks. The mean age of the trial population was 47 years and ranged from 19 to 74 years old. Seventy-nine percent (79%) were females. Ninety-six percent (96%) were Caucasian, 0.8% were Black, and 1.6% were Asian. Patients had had hypoparathyroidism for on average 15 years and hypoparathyroidism was caused by post-surgical complications in 71% of cases, idiopathic hypoparathyroidism in 25% of cases, DiGeorge Syndrome in 3% of cases, and auto-immune hypoparathyroidism in 1% of cases. Prior to trial enrollment, participants were receiving a median (interquartile range) daily oral calcium dose of 2000 (1250, 3000) mg and a median daily oral active vitamin D dose equivalent to 0.75 (0.5, 1) mcg of calcitriol. The mean eGFR at baseline was 97.4 mL/min/1.73 m2 and 45%, 10% and 0% had mild, moderate and severe renal impairment, respectively, at baseline. During the trial, most patients received 100 mcg and the dose range was 50 to 100 mcg administered subcutaneously once daily in the thigh.
Table 2 lists common adverse reactions associated with NATPARA use in the clinical trial. Common adverse reactions were reactions that occurred in ≥5% of subjects and occurred more commonly on NATPARA than on placebo.
| Adverse Reaction | Placebo (N=40) % | NATPARA (N=84) % |
|---|---|---|
|
||
| Paresthesia | 25 | 31 |
| Hypocalcemia* | 23 | 27 |
| Headache | 23 | 25 |
| Hypercalcemia* | 3 | 19 |
| Nausea | 18 | 18 |
| Hypoesthesia | 10 | 14 |
| Diarrhea | 3 | 12 |
| Vomiting | 0 | 12 |
| Arthralgia | 10 | 11 |
| Hypercalciuria* | 8 | 11 |
| Pain in extremity | 8 | 10 |
| Upper respiratory tract infection | 5 | 8 |
| Abdominal pain upper | 3 | 7 |
| Sinusitis | 5 | 7 |
| Blood 25-hydroxycholecalciferol decreased | 3 | 6 |
| Hypertension | 5 | 6 |
| Hypoesthesia facial | 3 | 6 |
| Neck pain | 3 | 6 |
Hypercalcemia
In the overall pivotal trial, a greater proportion of patients on NATPARA had albumin-corrected serum calcium above the normal range (8.4 to 10.6 mg/dL). During the entire trial duration 3 patients on NATPARA and 1 patient on placebo had a calcium level above 12 mg/dL. Table 3 displays the number of subjects who had albumin-corrected serum calcium levels above the normal range (8.4 to 10.6 mg/dL) by study treatment period in the placebo-controlled study based on routine monitoring at each trial visit. More patients randomized to NATPARA had hypercalcemia in both phases of the study (note: all trial participants underwent a 50% reduction in active vitamin D dose at randomization).
| Titration Period (Weeks 0-12)* | Maintenance Period (Weeks 12-24) |
|||
|---|---|---|---|---|
| Albumin-corrected serum calcium | Placebo N=40 | NATPARA N=84 | Placebo N=40 | NATPARA N=84 |
|
||||
| >10.6 to ≤12 mg/dL | 0% | 30% | 0% | 10% |
| >12 to ≤13 mg/dL | 0% | 2% | 3% | 0% |
Hypocalcemia
Table 4 displays the number of subjects who had albumin-corrected serum calcium levels below 8.4 mg/dL by treatment period in the placebo-controlled study based on routine monitoring at each trial visit. More patients randomized to placebo had hypocalcemia of less than 7 mg/dL in the titration phase (note: all trial participants underwent a 50% reduction in active vitamin D dose at randomization). More patients randomized to NATPARA had hypocalcemia of less than 7 mg/dL in the dose maintenance phase.
| Titration Period (Weeks 0-12) | Maintenance Period (Weeks 12-24) |
|||
|---|---|---|---|---|
| Albumin-corrected serum calcium | Placebo N=40 | NATPARA N=84 | Placebo N=40 | NATPARA N=84 |
| ≥7 to <8.4 mg/dL | 98% | 79% | 75% | 71% |
| <7 mg/dL | 18% | 6% | 0% | 12% |
The risk of hypocalcemia increases when NATPARA is withdrawn. At the end of the trial, NATPARA and placebo were withdrawn, calcium and active vitamin D were returned to baseline doses and subjects were followed for 4 weeks. During this withdrawal phase, more patients previously randomized to NATPARA experienced an albumin-corrected serum calcium value of less than 7 mg/dL (5.0% versus 17% for previous treatment with placebo and NATPARA, respectively). Twenty subjects (24%) previously randomized to NATPARA experienced adverse reactions of hypocalcemia in the post-treatment phase compared to three subjects (8%) previously randomized to placebo. Five subjects previously randomized to NATPARA with albumin-corrected serum calcium below 7 mg/dL required treatment with IV calcium gluconate to correct hypocalcemia.
Hypercalciuria
Treatment with NATPARA did not lower 24-hour urinary calcium excretion in the placebo-controlled trial. The proportion of subjects with hypercalciuria (defined as urine calcium levels of >300 mg/24 hours) was similar at baseline and trial end in the NATPARA and placebo groups. The median (IQR) 24-hour Urine Calcium at trial end was similar between NATPARA [231 (168-351) mg/24 hours], and placebo [232 (139-342) mg/24 hours]. At trial end, serum calcium values between NATPARA and placebo were also similar. Risk of hypercalciuria throughout the trial was related to serum calcium levels. To minimize the risk of hypercalciuria, NATPARA should be dosed to a target albumin-corrected total serum calcium within the lower half of the normal range (i.e., between 8 and 9 mg/dL) [see Dosage and Administration (2.1)].
6.2 Immunogenicity
NATPARA may trigger the development of antibodies. In the placebo-controlled study in adults with hypoparathyroidism, the incidence of anti-PTH antibodies was 8.6% (3/35) and 5.9% (1/17) in subjects who received subcutaneous administration of 50 to 100 mcg NATPARA or placebo once daily for 24 weeks, respectively.
Across all clinical studies in subjects with hypoparathyroidism following treatment with NATPARA for up to 2.6 years, the immunogenicity incidence rate was 16.1% (14/87). These 14 subjects had low titer anti-PTH antibodies and, of these, 3 subjects subsequently became antibody negative. One of these subjects had antibodies with neutralizing activity; this subject maintained a clinical response with no evidence of immune-related adverse reactions. Anti-PTH antibodies did not appear to affect efficacy or safety during the clinical trials but their longer-term impact is unknown.
Immunogenicity assay results are highly dependent on the sensitivity and specificity of the assay and may be influenced by several factors such as: assay methodology, sample handling, timing of sample collection, concomitant medication, and underlying diseases. For these reasons, comparison of the incidence of antibodies to NATPARA with the incidence of antibodies to other products may be misleading.
6.3 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of NATPARA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Hypersensitivity reactions (e.g., anaphylaxis, dyspnea, angioedema, urticaria, and rash).
- Seizures due to hypocalcemia
7 DRUG INTERACTIONS
7.1 Alendronate
Co-administration of alendronate and NATPARA leads to reduction in the calcium-sparing effect, which can interfere with the normalization of serum calcium. Concomitant use of NATPARA with alendronate is not recommended.
7.2 Digoxin
NATPARA causes transient increase in calcium and therefore, concomitant use of NATPARA and cardiac glycosides (e.g., digoxin) may predispose patients to digitalis toxicity if hypercalcemia develops. Digoxin efficacy is reduced if hypocalcemia is present. In patients using NATPARA concomitantly with digoxin, carefully monitor serum calcium and digoxin levels, and patients for signs and symptoms of digoxin toxicity. Adjustment of digoxin and/or NATPARA may be needed. No drug-drug interaction study has been conducted with digoxin and NATPARA.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Available data with NATPARA Injection use in pregnant women are insufficient to inform a drug associated risk of birth defects, miscarriage or adverse maternal or fetal outcomes. There are disease associated risks to the mother and the fetus related to hypocalcemia in pregnancy. (see Clinical Considerations). In animal reproduction studies, no adverse developmental effects were observed when pregnant rats and rabbits were administered parathyroid hormone subcutaneously during the period of organogenesis at doses resulting in 123 times and 8 times, respectively, the human exposure at the 100 mcg/day clinical dose. When female rats were administered parathyroid hormone subcutaneously prior to birth and continuing to weaning at doses resulting in 10 times the human exposure at the 100 mcg/day clinical dose, increased incidence of dehydration, broken palate and palate injuries related to incisor misalignment and mortality were observed in offspring (see Data).
All pregnancies have a background risk of major birth defects, loss, and other adverse outcomes.In the U.S. general population, the estimated major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Clinical Considerations
Disease-Associated Maternal and Embryo/Fetal Risk
Maternal hypocalcemia can result in an increased rate of spontaneous abortion, premature and dysfunctional labor, and possibly preeclampsia.
Fetal/Neonatal adverse reactions
Infants born to mothers with hypocalcemia can have associated fetal and neonatal hyperparathyroidism, which in turn can cause fetal and neonatal skeletal demineralization, subperiosteal bone resorption, osteitis fibrosa cystica and neonatal seizures. Infants born to mothers with hypocalcemia should be carefully monitored for signs of hypocalcemia or hypercalcemia, including neuromuscular irritability (ranging from myotonic jerks to seizures), apnea, cyanosis and cardiac rhythm disorders.
Data
Animal Data
In pregnant rats given subcutaneous doses up to 1000 mcg/kg/day during organogenesis there were no findings observed at 123 times the 100 mcg/day clinical dose based on AUC. In pregnant rabbits given subcutaneous doses of 5, 10 and 50 mcg/kg/day during organogenesis, various skeletal alterations including incomplete ossification in <35% of litters given 10 mcg/kg/day which were statistically significant but within historical control range at exposures 8 times the 100 mcg/kg/day clinical dose. There was a fetus with spina bifida in the 50 mcg/kg/day dose group at 72 times the 100 mcg/day clinical dose based on AUC. Given the association of folic acid deficiency and neural tube defects this finding may be related to decreased body weight and food consumption in the pregnant rabbits.
Developmental effects were observed in a peri /postnatal study in pregnant rats given subcutaneous doses of 100, 300, 1000 mcg/kg/day from organogenesis through lactation while an entire litter was stillborn in the 300 mcg/kg/day group (34 times the 100 mcg/day clinical dose based on AUC) and an entire litter from the 1000 mcg/kg/day (123 times the 100 mcg/day clinical dose based on AUC) was dead by postnatal day 4. Increased incidence of morbidity associated with dehydration, broken palate and palate injuries related to incisor misalignment and mortality were found in pups from litters given 100 mcg/kg/day (10 times the 100 mcg/day clinical dose based on AUC). At 300 mcg/kg/day there was a litter with kidney dilatation and another with an extra liver lobe. There was a single pup with a diaphragmatic hernia from a litter exposed to 1000 mcg/kg/day.
8.2 Lactation
Risk Summary
There are no data available on the presence of parathyroid hormone in the breast milk, the effects on the breastfed infant or the effects on milk production. Parathyroid hormone is present in the milk of lactating rats (see Data). When a drug is present in animal milk, it is likely that the drug will be present in human milk. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for NATPARA and any potential adverse effects on the breastfed child from NATPARA or from the underlying maternal condition.
Data
Animal Data
In a lactation transfer study where rats were given subcutaneous doses of 300 and 1000 mcg/kg/day from day 17 of gestation through day 16 of lactation, mean parathyroid hormone concentration in milk was approximately 10 ng/mL at the dose of 1000 mcg/kg/day, 42 times lower in milk than in plasma. The concentration of drug in animal milk does not necessarily predict the concentration of drug in human milk.
8.4 Pediatric Use
Safety and efficacy in patients less than 18 years of age has not been established. Avoid use of NATPARA in patients who are at increased baseline risk for osteosarcoma including pediatric and young adult patients with open epiphyses [see Boxed Warning, Warnings and Precautions (5.1)].
8.5 Geriatric Use
Clinical studies of NATPARA did not include sufficient numbers of subjects aged 65 and over to determine whether response in these subjects is different from younger subjects. In general, dose selection for elderly individuals should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy [see Clinical Pharmacology (12.3)].
8.6 Renal Impairment
Clinical studies of NATPARA did not include sufficient numbers of subjects with moderate and severe renal impairment to determine whether they respond differently from subjects with mild renal impairment or normal renal function. Some of the mechanisms of action of NATPARA (e.g., conversion of 25-OH vitamin D to 1,25-OH2 vitamin D) are dependent on renal function. NATPARA is eliminated by the kidney and maximum drug levels increased with renal impairment [see Clinical Pharmacology (12.3)].
10 OVERDOSAGE
Accidental overdose in studies in hypoparathyroidism occurred in 1 subject who received a 150 mcg dose and experienced mild palpitations. Serum calcium 24 hours later was 10.3 mg/dL. In the event of overdose, the patient should be carefully monitored for hypercalcemia by a medical professional [see Adverse Reactions (6.1)].
11 DESCRIPTION
The active ingredient in NATPARA, parathyroid hormone, is produced by recombinant DNA technology using a modified strain of Escherichia coli. Parathyroid hormone has 84 amino acids and a molecular weight of 9425 daltons; the amino acid sequence for parathyroid hormone is shown below in Figure 1.
Figure 1: Amino Acid Sequence of Parathyroid Hormone

NATPARA (parathyroid hormone) for injection for subcutaneous use is supplied as a medication cartridge, which is comprised of a multiple-dose, dual-chamber glass cartridge containing a sterile lyophilized powder and a sterile diluent, within a plastic cartridge holder. The sterile lyophilized powder contains either 0.4 mg or 0.8 mg or 1.21 mg or 1.61 mg of parathyroid hormone, depending on dosage strength, and 4.5 mg sodium chloride, 30 mg mannitol, and 1.26 mg citric acid monohydrate. The volume of the sterile diluent is 1.13 mL and the diluent contains a 3.2 mg/mL aqueous solution of m-cresol.
The disposable NATPARA medication cartridge is designed for use with a reusable mixing device for product reconstitution and a reusable Q-Cliq pen for drug delivery. The Q-Cliq pen delivers a fixed volumetric dose of 71.4 µL. Using the Q-Cliq pen, each NATPARA dual chamber cartridge delivers 14 doses of NATPARA [see Dosage Forms and Strengths (3)].
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
NATPARA is a parathyroid hormone. Parathyroid hormone raises serum calcium by increasing renal tubular calcium reabsorption, increasing intestinal calcium absorption (i.e., by converting 25-OH vitamin D to 1,25-OH2 vitamin D) and by increasing bone turnover which releases calcium into the circulation.
12.2 Pharmacodynamics
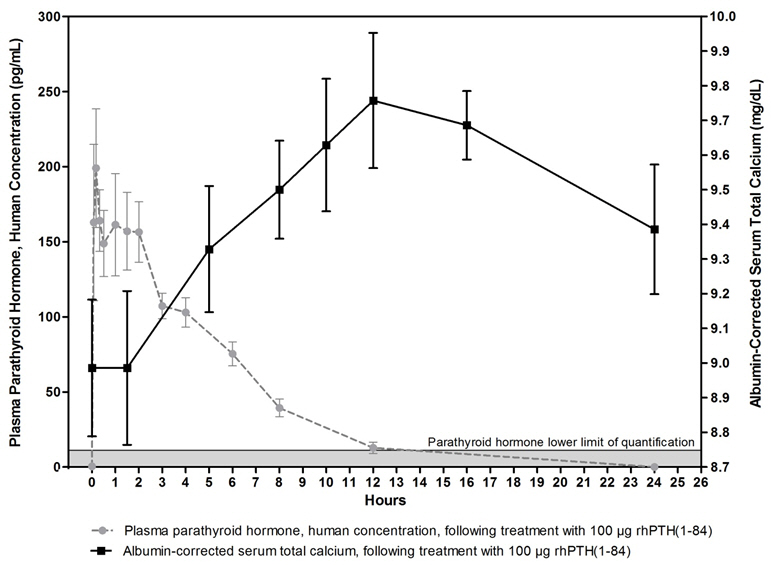
The pharmacodynamics in subjects with hypoparathyroidism after single subcutaneous administration of 50 and 100 mcg dose of NATPARA in the thigh were evaluated.
Treatment with NATPARA increases serum calcium levels (Figure 2). The increase in serum calcium levels in hypoparathyroidism subjects occurs in a dose-related manner. Mean peak serum calcium levels are reached between 10 and 12 hours following a single subcutaneous injection and the increase in serum calcium above baseline is sustained for more than 24 hours after administration. The maximum mean increases of serum calcium, which occurred at 12 hours, were approximately 0.5 mg/dL and 0.7 mg/dL from baseline with the 50 mcg and 100 mcg doses, respectively. The mean calcium intake for the 50 and 100 mcg doses was 1700 mg [see Clinical Pharmacology (12.3)].
12.3 Pharmacokinetics
Following single subcutaneous(SC) injections of NATPARA at 50 mcg and 100 mcg in subjects with hypoparathyroidism, peak plasma concentrations (mean Tmax) of NATPARA occurs within 5 to 30 minutes and a second usually smaller peak at 1 to 2 hours. The plasma AUC increased in a dose-proportional manner from 50 mcg to 100 mcg. The apparent terminal half-life (t1/2) was 3.02 and 2.83 hours for the 50 and 100 mcg dose, respectively.
Mean unadjusted concentration-time profiles of parathyroid hormone in plasma following SC administration of 100 mcg of NATPARA are presented in Figure 2. One 100 mcg dose of NATPARA provides a 24-hour calcemic response in hypoparathyroidism subjects.
Figure 2 Mean (±SE) Unadjusted Plasma Parathyroid Hormone and Albumin-Corrected Serum Calcium Concentration Following 100 mcg SC Administration in Subjects with Hypoparathyroidism
Metabolism:
In vitro and in vivo studies demonstrated that the clearance of parathyroid hormone is primarily a hepatic process with a lesser role played by the kidneys.
Excretion:
In the liver, most of the intact parathyroid hormone is cleaved by cathepsins. In the kidney, a small amount of parathyroid hormone binds to physiologic PTH-1 receptors, but most is filtered at the glomerulus. C-terminal fragments are also cleared efficiently by glomerular filtration.
Hepatic Impairment:
A pharmacokinetic study was conducted in 6 men and 6 women with moderate hepatic impairment (Child-Pugh Classification of 7-9 [Grade B]) as compared with a matched group of 12 subjects with normal hepatic function. Following a single 100-mcg subcutaneous dose, the mean Cmax and baseline-corrected Cmax values were 18% to 20% greater in the moderately impaired subjects than in those with normal function. There were no apparent differences in the serum total calcium concentration-time profiles between the 2 hepatic function groups. No dose adjustment for NATPARA is recommended in patients with mild to moderate hepatic impairment.
Renal Impairment:
Pharmacokinetics following a single NATPARA 100 mcg subcutaneous dose was evaluated in 16 subjects with normal renal function (creatinine clearance (CLcr) >90 mL/min) and 16 subjects with renal impairment. The mean maximum concentration (Cmax) of parathyroid hormone following administration of 100 mcg NATPARA in subjects with mild (CLcr 60 to 90 mL/min) and moderate (CLcr 30 to 60 mL/min) renal impairment was approximately 22% higher than that observed in subjects with normal renal function. Exposure to parathyroid hormone as measured by AUC0-last and baseline-corrected AUC0-last was approximately 3.9% and 2.5%, respectively, higher than that observed for subjects with normal renal function. No studies were conducted in patients with severe renal impairment or in renal impairment patients on dialysis.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
In a 104-week carcinogenicity study in rats, parathyroid hormone was given subcutaneously at doses of 10, 50 and 150 mcg/kg/day. These doses resulted in systemic exposures that were, respectively 3 to 71 times higher than systemic exposure observed in humans following a subcutaneous dose of 100 mcg/day based on AUC. Systemic exposure at the 10 mcg/kg/day dose of parathyroid hormone was 3-5 times greater AUC than the exposure observed in hypoparathyroidism subjects at the clinical dose of 100 mcg/day. This is the lowest dose at which a parathyroid hormone-related increase in bone tumors was observed in rats. Higher exposures resulted in a marked dose-related increase in all bone tumors including osteoma, osteoblastoma and osteosarcomas in both sexes. The bone tumors in rats occurred in association with a large increase in bone mass and focal osteoblast hyperplasia. However, since bone metabolism in the rat differs from that in humans, the relevance of these animal findings to humans is uncertain.
Parathyroid hormone is not genotoxic in any of the following test systems: the bacterial reverse mutation (Ames) assay or the in vitro mammalian cell forward-gene mutation (AS52/XPRT) assay study with and without metabolic activation.
No effect on fertility was observed in male and female rats given parathyroid hormone at doses up to 1000 mcg/kg/day (120 times systemic exposure after a clinical dose of 100 mcg/day).
13.2 Animal Toxicology and/or Pharmacology
In pregnant rats given subcutaneous doses up to 1000 mcg/kg/day during organogenesis there were no findings observed at 123 times the 100 mcg/day clinical dose based on AUC. In pregnant rabbits given subcutaneous doses of 5, 10 and 50 mcg/kg/day during organogenesis, various skeletal alterations including in complete ossification in <35% of litters given 10 mcg/kg/day which were statistically significant but within historical control range at exposures 8 times the 100 mcg/kg/day clinical dose. There was a fetus with spina bifida in the 50 mcg/kg/day dose group at 72 times the 100 mcg/day clinical dose based on AUC. Given the association of folic acid deficiency and neural tube defects this finding may be related to decreased body weight and food consumption in the pregnant rabbits.
Developmental effects were observed in a peri-/post-natal study in pregnant rats given subcutaneous doses of 100, 300, 1000 mcg/kg/day from organogenesis through lactation while an entire litter was stillborn in the 300 mcg/kg/day group (34 times the 100 mcg/day clinical dose based on AUC) and an entire litter from the 1000 mcg/kg/day (123 times the 100 mcg/day clinical dose based on AUC) was dead by postnatal day 4. Increased incidence of morbidity associated with dehydration, broken palate and palate injuries related to incisor misalignment and mortality were found in pups from litters given 100 mcg/kg/day (10 times the 100 mcg/day clinical dose based on AUC). At 300 mcg/kg/day there was a litter with kidney dilatation and another with an extra liver lobe. There was a single pup with a diaphragmatic hernia from a litter exposed to 1000 mcg/kg/day.
14 CLINICAL STUDIES
Study in Patients with Established Hypoparathyroidism
The efficacy of NATPARA was evaluated in a 24-week, randomized, double-blind, placebo-controlled, multicenter trial. In this trial, patients with established hypoparathyroidism receiving calcium and active forms of vitamin D (vitamin D metabolite or analogs) were randomized (2:1) to NATPARA (n=84) or placebo (n=40). The mean age was 47 years (range, 19 to 74 years), 79.0% were females and 96.0% were Caucasian, 0.8% were Black, and 1.6% were Asian. Patients had hypoparathyroidism for on average 15 years and hypoparathyroidism was caused by post-surgical complications in 71% of cases, idiopathic hypoparathyroidism in 25%, DiGeorge Syndrome in 3%, and auto-immune hypoparathyroidism in 1%. Patients with hypoparathyroidism due to calcium-sensing receptor mutations were excluded from the trial. The mean eGFR at baseline was 97.4 mL/min/1.73 m2 and 45%, 10% and 0% had mild, moderate and severe renal impairment, respectively, at baseline.
Before randomization, participants entered a 2-16 weeks run-in phase. In this phase calcium supplement and active vitamin D doses were adjusted to target an albumin-corrected serum calcium concentration between 8.0 and 9.0 mg/dL and 25-hydroxyvitamin D was replaced in patients with insufficient stores. At randomization, baseline serum calcium was 8.6 mg and participants were receiving a median (interquartile range) daily oral calcium dose of 2000 (1250, 3000) mg and a median daily oral active vitamin D dose equivalent to 0.75 mcg (0.5, 1) of calcitriol.
At randomization, active forms of vitamin D were reduced by 50% and patients were randomized to NATPARA 50 mcg daily or placebo. Randomization was followed by a 12-week NATPARA titration phase and a 12-week NATPARA dose maintenance phase. During the titration phase NATPARA was increased by 25 mcg increments every four weeks up to a maximum of 100 mcg. Titration was indicated for patients who could not achieve independence from active vitamin D and who could not reduce oral calcium to 500 mg or less per day. At end of treatment, 56% of subjects randomized to NATPARA were receiving 100 mcg of NATPARA per day, 26% were receiving 75 mcg of NATPARA per day, and 18% were receiving 50 mcg of NATPARA per day. Doses of co-administered active forms of vitamin D and calcium were adjusted (reduced or increased) to maintain albumin-corrected serum calcium within a desired target range throughout the trial in both arms.
For the efficacy analysis, subjects that fulfilled three components of a three-part response criterion were considered responders. A responder was defined as an individual who had: at least a 50% reduction from baseline in the dose of active vitamin D, at least a 50% reduction from baseline in the dose of oral calcium supplementation and an albumin-corrected total serum calcium concentration between 7.5 mg/dL and 10.6 mg/dL.
At the end of treatment, significantly (p-value <0.001) more subjects treated with NATPARA [46/84 (54.8%)] compared to placebo [1/40 (2.5%)] met the response criterion. Forty-two percent (35/84) of subjects randomized to NATPARA were independent of active forms of vitamin D and were on no more than 500 mg of oral calcium, compared with 2.5% (1/40) of subjects randomized to placebo (p<0.001). There were no differences in the proportion of patients with a calcium level between 7.5 mg and 10.6 mg at end of treatment between subjects randomized to NATPARA and placebo.
Table 5 shows the proportion of individuals who, at the end of treatment, fulfilled the 3-part response criterion. Table 6 provides results on individual components of the response criterion.
| Placebo (N=40) | NATPARA (N=84) |
|
|---|---|---|
| Efficacy Endpoint | ||
| Responder* at End of Treatment, based on investigator-prescribed data - n (%) | 1 (2.5) | 46 (54.8) (p <0.001) † |
| Components of the Efficacy Endpoint | |||
|---|---|---|---|
| Placebo (N=40) | NATPARA (N=84) |
||
| ACSC = albumin-corrected total serum calcium; EOT = end of treatment; N = total number of subjects; n = number of subjects meeting the specified endpoint; SD = standard deviation; ULN = upper limit of normal | |||
|
|||
| Oral Calcium | Reduction ≥50% - n (%) | 3 (7.5) | 58 (69.0) |
| Percent Change from Baseline - Mean (SD) * | 6.5 (38.5) | -51.8 (44.6) (p <0.001) |
|
| Oral Active Vitamin D | Reduction ≥50% - n (%) | 18 (45.0) | 73 (86.9) |
| ACSC maintained between ≥7.5 mg/dL to ≤ULN - n (%) | 35 (87.5) | 73 (86.9) | |
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
NATPARA (parathyroid hormone) for injection for subcutaneous use is supplied as a medication cartridge, which is comprised of a multiple-dose, dual-chamber glass cartridge containing a sterile lyophilized powder and a sterile diluent, within a plastic cartridge holder. The medication cartridge is available in 4 dosage strengths (25, 50, 75, and 100 mcg/dose). The 25 mcg/dose cartridge contains 0.4 mg parathyroid hormone; the 50 mcg/dose cartridge contains 0.8 mg parathyroid hormone; the 75 mcg/dose cartridge contains 1.21 mg parathyroid hormone; the 100 mcg/dose cartridge contains 1.61 mg parathyroid hormone.
NATPARA is supplied in the following packages:
-
- 2 cartridges of 25 mcg/dose strength (NDC 68875-0202-2)
- 2 cartridges of 50 mcg/dose strength (NDC 68875-0203-2)
- 2 cartridges of 75 mcg/dose strength (NDC 68875-0204-2)
- 2 cartridges of 100 mcg/dose strength (NDC 68875-0205-2)
The disposable NATPARA medication cartridge is designed for use with a reusable mixing device for product reconstitution and a reusable Q-Cliq pen injector for drug delivery. The Q-Cliq pen is designed to deliver a fixed volumetric dose of 71.4 µL. Using the Q-Cliq pen, each NATPARA medication cartridge delivers 14 doses; each dose contains 25, 50, 75, or 100 mcg of NATPARA depending on the product dosage strength.
Designed for use with 31G × 8 mm BD Ultra-Fine™ Pen Needles.
The mixing device, provided in a separate carton, is designed to enable reconstitution of the product before the first use of each cartridge. The mixing device can be used to reconstitute up to 6 NATPARA medication cartridges.
The Q-Cliq pen, packaged in a separate carton, can be used for up to 2 years of daily treatment by replacing the reconstituted cartridge every two weeks (14 days).
Instructions for use of the mixing device and the Q-Cliq pen are provided with the NATPARA medication cartridges.
16.2 Storage and Handling
Prior to reconstitution, the dual-chamber NATPARA medication cartridge should be stored in the package provided at refrigerated temperature, 36 to 46°F (2 to 8°C). After reconstitution, the medication cartridge should be stored in the Q-Cliq pen under refrigeration at 36 to 46°F (2 to 8°C). The reconstituted product can be used for up to 14 days under these conditions. Store away from heat and light. Avoid exposure to elevated temperatures. Discard reconstituted NATPARA medication cartridges after 14 days.
Do not freeze or shake. Do not use NATPARA if it has been frozen or shaken.
The mixing device and empty Q-Cliq pen can be stored at room temperature.
Safely discard needles.
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide and Instructions for Use).
General Counseling Information – Prior to treatment, patients should fully understand the risks and benefits of NATPARA. Ensure that all patients receive the Medication Guide and Instructions for Use document prior to initiating NATPARA therapy.
Potential Risk of Osteosarcoma
[see Warnings and Precautions (5.1)]
Advise patients that the active ingredient in NATPARA, parathyroid hormone, caused an increase in the incidence of osteosarcoma (a malignant bone tumor) in male and female rats in dedicated lifelong carcinogenicity studies and that the risk of osteosarcoma in rats was dependent on parathyroid hormone dose administered, on treatment duration and occurred at exposure levels close to the clinical exposure range. Based on these findings NATPARA may carry a potential risk to humans.
Patients should be advised that because of a potential risk of osteosarcoma, NATPARA is only recommended for patients who cannot be well-controlled on oral calcium supplementation and on active forms of vitamin D. In addition, use of NATPARA should be avoided in patients who have risk factors for osteosarcoma unless the benefits of using NATPARA in these patients are determined to outweigh this potential risk.
Instruct patients to promptly report signs and symptoms of possible osteosarcoma such as persistent localized pain or occurrence of a new soft tissue mass that is tender to palpation.
NATPARA REMS
[see Warnings and Precautions (5.1, 5.2)]
- NATPARA is available only through a restricted program called the NATPARA REMS Program, because of the potential risk of osteosarcoma.
- Counsel patients on the benefits and risks of NATPARA using the NATPARA Patient Brochure.
- Patients must sign the NATPARA REMS Patient-Prescriber Acknowledgment Form.
- Provide patient with a copy of the NATPARA Patient Brochure and NATPARA REMS Patient-Prescriber Acknowledgment Form.
- NATPARA is only available through certified pharmacies, provide information to your patients about how they will receive prescriptions:
- Submit the NATPARA prescription to the NATPARA REMS Program Coordinating Center (by fax or email).
- The REMS Program Coordinating Center will send the prescription to a certified pharmacy to fill after verifying that the prescriber is certified and a Patient-Prescriber Acknowledgment Form is on record.
- The REMS Program Coordinating Center will call the patient and provide the name and phone number of the certified pharmacy that will be dispensing NATPARA.
- The certified pharmacy will contact the patient to arrange the date to ship NATPARA once the prescription is filled.
Severe Hypercalcemia
[see Warnings and Precautions (5.3)]
Instruct patients that severe hypercalcemia can occur when starting or adjusting NATPARA dose and/or when making changes to co-administered drugs known to raise serum calcium. Instruct patients to: report symptoms of hypercalcemia promptly, report any changes to co-administered drug(s) known to influence calcium levels and follow recommended serum calcium monitoring.
Severe Hypocalcemia
[see Warnings and Precautions (5.4)]
Instruct patients that severe hypocalcemia can occur if NATPARA dosing is abruptly interrupted or discontinued. Instruct patients to report symptoms of hypocalcemia promptly, report interruption in NATPARA dosing and follow recommended serum calcium monitoring. In the event of NATPARA dose interruption, patients should contact their healthcare provider, as their doses of active vitamin D and calcium supplementation may need adjustment.
Digoxin Toxicity
[see Warnings and Precautions (5.5)]
Instruct patients to report use of digoxin containing medication, and follow recommended serum calcium monitoring.
Hypersensitivity
[see Warnings and Precautions (5.6)]
Instruct patients that serious hypersensitivity reactions (anaphylaxis, dyspnea, angioedema, urticaria, rash) can occur with NATPARA. Instruct patients to immediately report symptoms of serious hypersensitivity reactions and to seek medical attention if a reaction occurs.
Lactation
Advise women that infants exposed to parathyroid hormone through breast milk should be monitored for symptoms of hypercalcemia or hypocalcemia.
Dosing Instructions
Instruct patients to read the Instructions for Use document carefully. The patient or caregiver should be instructed by a physician or an appropriately qualified healthcare professional in the proper technique for administering subcutaneous injections using the mixing device and the Q-Cliq pen, including the use of aseptic technique. The patient and caregiver should be cautioned that needles must not be re-used and instructed in safe disposal procedures. A puncture-resistant container for disposal of used needles should be supplied to the patient along with instructions for safe disposal of the full container. Instruct patients to never share their devices with other patients. Advise patients to never transfer the contents of the delivery device to a syringe.
After reconstitution, each NATPARA medication cartridge can be used for 14 subcutaneous injections. After the use period, only the cartridge should be discarded. The Q-Cliq pen can be used for up to 2 years by replacing the reconstituted medication cartridge every two weeks (14 days).
Common Adverse Reactions
[see Adverse Reactions (6.1)]
Inform patients that the most common adverse reactions occurring in patients on NATPARA were paresthesia, hypocalcemia, headache, hypercalcemia, nausea, hypoaesthesia, diarrhea, vomiting, arthralgia, hypercalciuria and pain in extremity.
Manufactured by:
Takeda Pharmaceuticals U.S.A., Inc.
Lexington, MA 02421 USA
U.S. License Number 1898
For information about NATPARA contact:
Takeda Pharmaceuticals U.S.A., Inc.
Lexington, MA 02421
USA
1-800-828-2088
www.NATPARA.com
NATPARA® is a registered trademark of Takeda Pharmaceuticals U.S.A., Inc.
Q-Cliq® is a registered trademark of Takeda Pharmaceuticals U.S.A., Inc.
Ultra-Fine™ is a trademark of Becton, Dickinson and company.
©2023 Takeda Pharmaceuticals U.S.A., Inc. All rights reserved.
Medication Guide
NATPARA® (nat-PAH-rah)
(parathyroid hormone)
for injection
What is the most important information I should know about NATPARA?
-
possible bone cancer (osteosarcoma). During animal drug testing, NATPARA caused some rats to develop a bone cancer called osteosarcoma. It is not known if people who take NATPARA will have a higher chance of getting osteosarcoma. Tell your healthcare provider right away if you have pain in any areas of your body that does not go away, or any new or unusual lumps or swelling under your skin that is tender to touch. These are some of the signs and symptoms of osteosarcoma and your healthcare provider may need to do further tests.
- NATPARA is only available through the NATPARA Risk Evaluation and Mitigation Strategy (REMS) Program. The purpose of the NATPARA REMS program is to inform patients about the potential risk of osteosarcoma associated with the use of NATPARA. For more information about this REMS program, call 1-855-NATPARA (1-855-628-7272) or go to www.NATPARAREMS.com.
NATPARA may cause serious side effects, including:
- high blood calcium (hypercalcemia). NATPARA can cause some people to have a higher blood calcium level than normal. Your healthcare provider should check your blood calcium before you start and during your treatment with NATPARA. Tell your healthcare provider if you have nausea, vomiting, constipation, low energy, or muscle weakness. These may be signs that you have too much calcium in your blood.
- low blood calcium (hypocalcemia). People who stop using or miss a dose of NATPARA may have an increased risk of severe low blood calcium levels. Tell your healthcare provider if you have tingling of your lips, tongue, fingers and feet, twitching of face muscles, cramping of feet and hands, seizures, depression, or have problems thinking or remembering.
Tell your healthcare provider right away if you have any of these signs and symptoms of high or low blood calcium levels.
What is NATPARA?
- NATPARA is a prescription parathyroid hormone (PTH) used with calcium and vitamin D to control low blood calcium (hypocalcemia) in people with low PTH blood levels (hypoparathyroidism).
- NATPARA is only for people who do not respond well to treatment with calcium and active forms of vitamin D alone, because it may increase the possible risk of bone cancer (osteosarcoma).
- NATPARA was not studied in people with hypoparathyroidism caused by calcium-sensing receptor mutations.
- NATPARA was not studied in people who get sudden hypoparathyroidism after surgery.
It is not known if NATPARA is safe and effective for children 18 years of age and younger.
NATPARA should not be used in children and young adults whose bones are still growing.
Do not use NATPARA if you are allergic to parathyroid hormone or any of the ingredients in NATPARA. See the end of this Medication Guide for a complete list of ingredients in NATPARA.
Before you start using NATPARA, tell your healthcare provider about all of your medical conditions, including if you:
- have Paget's disease or other bone disease
- have or have had cancer in your bones
- have or have had radiation therapy
- have or had too much calcium in your blood
- have high blood levels of certain electrolytes (alkaline phosphatase)
- are pregnant or plan to become pregnant. It is not known if NATPARA will harm your unborn baby.
- are breastfeeding or plan to breastfeed. It is not known if NATPARA passes into your breast milk. You and your healthcare provider should decide if you will use NATPARA or breastfeed. You should not do both.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
NATPARA and other medicines may affect each other causing side effects.
Especially tell your healthcare provider if you are taking medicines that contain digoxin, alendronate, calcium supplements or food products that contain calcium, or active vitamin D.
Know the medicines you take. Keep a list of them to show your healthcare provider or pharmacist each time you get a new medicine.
How should I use NATPARA?
- Read the detailed Instructions for Use at the end of this Medication Guide.
- Use NATPARA exactly as your healthcare provider tells you to.
- NATPARA is given with the Q-Cliq® pen injector. Before you use NATPARA for the first time, a healthcare provider will show you how to use the Q-Cliq pen the right way and how to properly mix NATPARA using the mixing device.
- Do not stop taking or change your dose of NATPARA unless your healthcare provider tells you to. Your calcium level could become dangerously low.
- Give NATPARA 1 time each day in your thigh just under your skin (subcutaneous).
- After you mix NATPARA, each medicine cartridge can be used for 14 injections (14 doses). After you use the 14 doses throw away the cartridge.
- Do not throw away the Q-Cliq pen. It can be re-used for up to 2 years, by changing the mixed NATPARA medicine cartridges every 2 weeks (14 days).
- Look at NATPARA for any discoloration or particles in the medicine. It should be colorless. It is normal to see small particles in the liquid.
- Do not transfer the medicine from the NATPARA medicine cartridge to a syringe. This can cause you to use the wrong dose of NATPARA.
- Your healthcare provider should check your blood calcium level when you start and while you are using NATPARA. After you start NATPARA, your healthcare provider may change your doses of calcium and active vitamin D.
- If you miss a day or forget to give your daily NATPARA injection, give your injection as soon as you remember and call your healthcare provider right away. You may need to take more calcium. Take your next dose of NATPARA the next day as prescribed.
- If you use more than your daily dose of NATPARA, call your healthcare provider right away.
What are the possible side effects of NATPARA?
NATPARA may cause serious side effects, including:
- See "What is the most important information I should know about NATPARA?"
- Allergic (hypersensitivity) reaction, including anaphylaxis. Tell your healthcare provider or get emergency medical help right away if you have any of the following symptoms of an allergic reaction:
|
|
Do not use NATPARA if you are allergic to parathyroid hormone or any of the ingredients in NATPARA. See the end of this Medication Guide for a complete list of ingredients in NATPARA.
The most common side effects of NATPARA include tingling, tickling, or a burning feeling of your skin (paresthesia), headache and nausea.
These are not all the possible side effects of NATPARA. For more information, ask your doctor. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store NATPARA?
- Unmixed NATPARA medicine cartridges: Refrigerate NATPARA between 36°F to 46°F (2°C to 8°C). Do not freeze.
-
Mixed NATPARA medicine cartridges:
- Refrigerate between 36°F to 46°F (2°C to 8°C). Do not freeze.
- You can use the Q-Cliq pen for up to 14 days after mixing the medicine cartridge.
- Throw away the mixed NATPARA medicine cartridges 14 days after mixing the medicine cartridge.
- Store NATPARA away from heat and light.
- Do not freeze or shake NATPARA. Do not use NATPARA if it was frozen or shaken.
Keep NATPARA and all medicines out of the reach of children.
General information about the safe and effective use of NATPARA.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use NATPARA for a condition for which it was not prescribed. Do not give NATPARA to other people, even if they have the same symptoms you have. It may harm them.
This Medication Guide summarizes the most important information about NATPARA. If you would like more information, talk with your healthcare provider. You can ask your pharmacist or healthcare provider for information about NATPARA that is written for health professionals.
What are the ingredients in NATPARA?
Active ingredient: recombinant human parathyroid hormone
Inactive ingredients: sodium chloride, mannitol, citric acid monohydrate, m-cresol in sterile water
Manufactured by: Takeda Pharmaceuticals U.S.A., Inc., Lexington, MA 02421, USA
U.S. License Number 1898
NATPARA® is a registered trademark of Takeda Pharmaceuticals U.S.A., Inc.
Q-Cliq® is a registered trademark of Takeda Pharmaceuticals U.S.A., Inc.
©2023 Takeda Pharmaceuticals U.S.A., Inc. All rights reserved.
For more information, go to www.NATPARA.com or call 1-800-828-2088.
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Revised: 2/2023
Instructions for Use
NATPARA® (nat-PAH-rah)
(parathyroid hormone)
for injection, for subcutaneous use
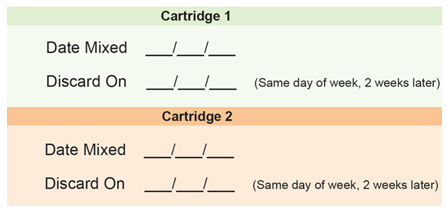
Medicine Cartridge Tracker
Instructions:
1. Enter today's date in the space next to "Date Mixed."
2. Enter the date 14 days from today in the "Discard On" section.
3. Dispose of your medicine cartridge on the "Discard On" date even if you have medicine left in the cartridge.
Read this Instructions for Use before you start using NATPARA and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or your treatment. Before you use NATPARA for the first time, make sure your healthcare provider shows you the right way to use it.
Table of Contents
Getting to Know the Parts of Your Q-Cliq Pen and Your NATPARA Medicine
Preparing and Mixing Your NATPARA
Preparing Your Q-Cliq Pen
Giving Your Daily NATPARA
Frequently Asked Questions
Getting to Know the Parts of Your Q-Cliq Pen and Your NATPARA Medicine
Your Q-Cliq pen can be re-used for up to 2 years. See Figure A.
The rod protector protects the rod during shipment from the factory. Throw away the rod protector when you are ready to use your Q-Cliq pen.
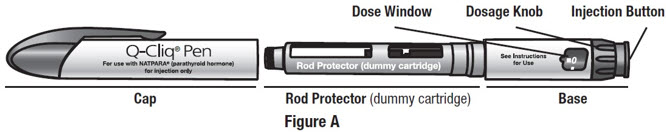
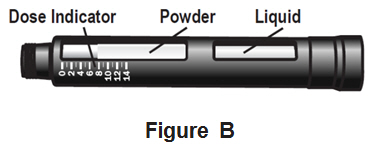
Your NATPARA Medicine Cartridge:
- Your NATPARA medicine cartridge contains medicine powder and a liquid to mix the powder in. See Figure B. You must mix the powder and the liquid in the medicine cartridge before using your Q-Cliq pen.
- Each medicine cartridge contains 14 doses.
- The dose indicator shows you the number of doses left in the medicine cartridge.
| Other supplies you will need to give your NATPARA. See Figure C. | 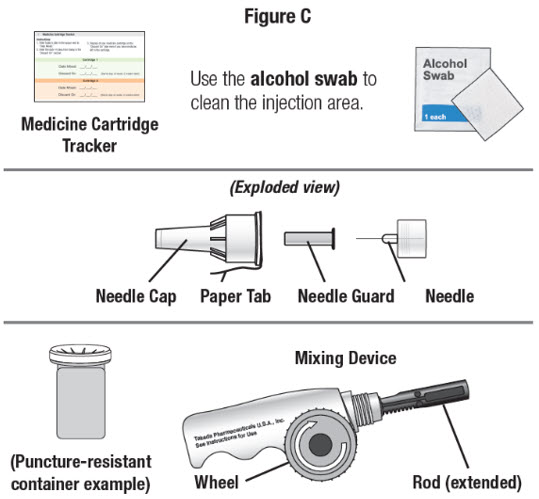 |
|
Preparing and Mixing Your NATPARA
- Mix your NATPARA 1 time every 14 days.
- If this is your first time using NATPARA by yourself, a trainer will guide you through how to mix your first NATPARA medicine cartridge and also your second medicine cartridge.
Step 1. Wash and dry your hands.
Step 2. Gather your supplies, including:
- your Q-Cliq pen
- your mixing device
- new NATPARA medicine cartridge from the refrigerator
- new disposable Pen Needle
- puncture-resistant sharps container
- pencil or pen
- Medicine Cartridge Tracker
Step 3. Look at the Medicine Cartridge Tracker in the front of your NATPARA booklet.
| Step 4. Write the date in the space next to "Date Mixed." See Figure D. |  |
| Step 5. Write the date 14 days from the day you mixed NATPARA in the space next to "Discard on." This will be the same day of the week 2 weeks later. See Figure E. |  |
| |
| Step 6. Remove the paper tab from the needle cap. See Figure F. |  |
| Step 7. While keeping the needle cap straight, screw it firmly onto the cartridge in a clockwise direction. Do not remove the needle cap or guard until you are ready to give your medicine. See Figure G. |  |
| Step 8. Turn the wheel of the mixing device in a counterclockwise direction to lower the rod if it is not already lowered. See Figure H. | 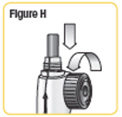 |
| 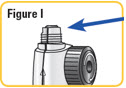 |
| Step 9. Screw the cartridge onto your NATPARA mixing device in a clockwise direction. The Pen Needle must be attached. See Figure J. | 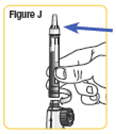 |
| Step 10. With the needle cap pointing up, turn the wheel slowly in a clockwise direction until the stoppers no longer move. Make sure the wheel turns easily. See Figure K. | 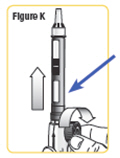 |
| Step 11. Make sure the stoppers look like this and stay together. See Figure L. |  |
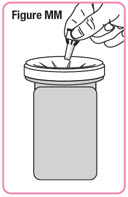
| Step 12. Mix the powder by slowly moving the cartridge back and forth about 10 times. Do not shake the cartridge. See Figure M. |  |
Step 13. Set the mixing device down with the medicine cartridge attached. Wait until the powder is dissolved and the liquid is colorless, or 5 minutes have passed. It is normal to see small particles in the liquid.
Step 14. If the liquid is colorless, go to Step 15. If the liquid is not colorless, call 1-800-828-2088 for help.
Preparing Your Q-Cliq Pen
You will prepare your Q-Cliq pen 1 time every 14 days
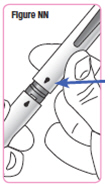
| Step 15. Pick up your Q-Cliq pen and remove the cap. Save the cap for later use. See Figure N. | 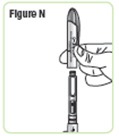 |
| Step 16. Unscrew the rod protector or the empty medicine cartridge in a counterclockwise direction and throw it away in a puncture-resistant sharps container. See Figure O. | 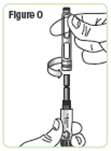 |
| Step 17. Press the injection button. You should see the "0" line up with the notch in the dose window. If you do not see the "0" line up, press the injection button until it is lined up. See Figure P. | 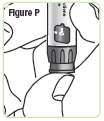 |
| Step 18. Lower the rod. If the rod is extended, turn the dark red ring counterclockwise to lower it. Do not tighten the ring too much. See Figure Q. | 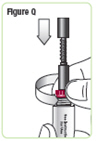 |
| Step 19. Check the rod. It will have a small space when done the right way. See Figure R. | 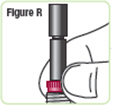 |
| Step 20. Pick up the mixing device with the needle cap pointing up. Unscrew the cartridge from the mixing device in a counterclockwise direction and set the mixing device down. See Figure S. | 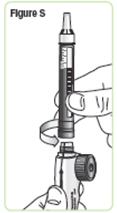 |
| Step 21. Attach the medicine cartridge to the pen. Pick up the Q-Cliq pen base and hold it with the rod pointed upright. See Figure T. | 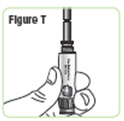 |
| Step 22. With the needle cap pointing up, screw the medicine cartridge onto the Q-Cliq pen in a clockwise direction until there is no space between the medicine cartridge and the Q-Cliq pen. See Figure U. | 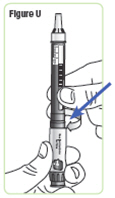 |
Priming your Q-Cliq pen.
|
Step 23. Turn the dosage knob until "GO" lines up with the notch in the dose window. See Figure V. |  |
| Step 24. Hold the Q-Cliq pen with the needle cap pointing up. See Figure W. | 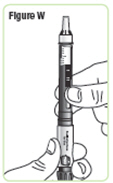 |
| Step 25. Press the injection button on a flat surface, such as a table top, until the "0" lines up with the notch in the dose window. See Figure X. | 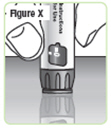 |
- It is normal for 1 or 2 drops of liquid to leak out during this step.
- Do not remove the medicine cartridge from the Q-Cliq pen until the "Discard on" date or the medicine cartridge is empty.
- Prime your Q-Cliq pen only 1 time for each new medicine cartridge.
Giving Your Daily NATPARA
- If you have just finished mixing your medicine and preparing your Q-Cliq pen and the Pen Needle is on, go to Step 33.
If you need help at any time, call 1-800-828-2088
Step 26. Wash and dry your hands.
Step 27. Gather your supplies, including:
- your Q-Cliq pen from the refrigerator
- new disposable Pen Needle
- puncture resistant sharps container
| Step 28. Remove the cap from the Q-Cliq pen. The mixed medicine cartridge should be inside. |  |
Step 29. Check the medicine inside the cartridge. See Figure Y.
Step 30. If the liquid is colorless, go to Step 31. It is normal to see small particles in the liquid. If the liquid is not colorless, call 1-800-828-2088 for help.
Attaching a new Pen Needle
| Step 31. Remove the paper tab from the needle cap. See Figure Z. | 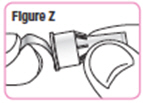 |
| Step 32. While keeping the needle cap straight, screw it firmly onto the medicine cartridge in a clockwise direction. Do not remove the needle cap or needle guard until you are ready to give your NATPARA. See Figure AA. | 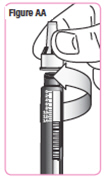 |
| Step 33. Clean the area of your thigh where you will give NATPARA with an alcohol swab. Use a different thigh every time you give your NATPARA. See Figure BB. | 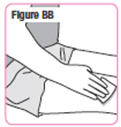 |
Make sure the needle cap is pointing downward at all times during Step 34 to Step 39.
| Step 34. Hold the Q-Cliq pen with the needle cap pointing down until after your injection. See Figure CC. |  |
| Step 35. Hold the Q-Cliq pen so you can see the dose window. See Figure DD. | 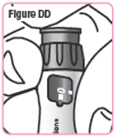 |
| Step 36. Turn the dosage knob until "GO" lines up with the notch in the window. Do not turn the dosage knob past "GO." See Figure EE. |  |
-
If the dosage knob is hard to turn, you may not have enough liquid left.
Check the dose indicator on the medicine cartridge to see if there are any doses left or check the "Discard on" date on the Medicine Cartridge Tracker to see if it has been more than 14 days.
| Step 37. Gently tap the medicine cartridge 3 to 5 times. See Figure FF. | 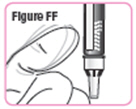 |
Prepare the Pen Needle for giving the injection:
| Step 38. Pull the needle cap off and set it aside. Make sure you do not unscrew the needle cap. Pull the needle guard off and throw it away. See Figure GG. | 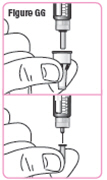 |
| Step 39. Hold the Q-Cliq pen so you can see "GO" in the dose window with the Pen Needle pointing down. See Figure HH. | 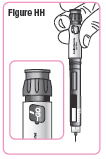 |
Giving your NATPARA Injection
| Step 40. Insert the Pen Needle fully into your thigh. Make sure you can see "GO" in the window. See Figure II. | 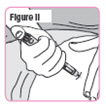 |
| Step 41. Press the injection button until the "0" lines up with the notch in the dose window. You should see and feel the dosage knob turn back to "0." See Figure JJ. | 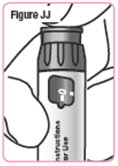 |
|  |
Step 42. Remove the Pen Needle from your skin by pulling it straight out.
- It is normal for 1 or 2 drops of liquid to leak out during this step.
- If you do not think you received your full dose, do not take another dose. Call your healthcare provider. You may need to take calcium and active vitamin D.
| Step 43. Carefully put the large needle cap back on the Pen Needle by scooping the cap back on the needle using only 1 hand. See Figure KK. | 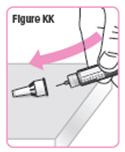 |
| Step 44. Unscrew the needle cap (with Pen Needle inside) in a counterclockwise direction while holding the medicine cartridge. See Figure LL. |  |
- Do not share your Q-Cliq pen or Pen Needles with anyone else. You may give an infection to them or get an infection from them.
After your injection:
Step 45. Discard your used needles and medicine cartridges
|  |
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: http://www.fda.gov/safesharpsdisposal.
- Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Do not recycle your used sharps disposal container.
Step 46. Put the cap back on your Q-Cliq pen
|  |
Step 47. Put your Q-Cliq pen in the refrigerator.
How should I store NATPARA?
- Unmixed NATPARA medicine cartridges: Refrigerate NATPARA between 36°F to 46°F (2°C to 8°C). Do not freeze.
-
Mixed NATPARA medicine cartridges:
- Refrigerate between 36°F to 46°F (2°C to 8°C). Do not freeze.
- You can use the Q-Cliq pen for up to 14 days after mixing the medicine cartridge.
- Throw away the mixed NATPARA medicine cartridges 14 days after mixing the medicine cartridge.
- Store NATPARA away from heat and light.
- Do not freeze or shake NATPARA. Do not use NATPARA if it was frozen or shaken.
Keep NATPARA and all medicines out of the reach of children.
Frequently Asked Questions
There are 4 ways to get your questions answered:
- Call your healthcare provider.
- Visit our Website at www.NATPARA.com.
- Call 1-800-828-2088.
- Find it in the following list of questions.
What does the dose window in the Q-Cliq pen tell me?
The dose window tells you if the Q-Cliq pen is ready for the injection:
- "0" means not ready
- "GO" means ready
- The dose window does not count the number of doses left or given
Why does the dose indicator scale on the medicine cartridge show "14" after I have given an injection using a newly mixed NATPARA medicine cartridge?
You might have forgotten to prime the Q-Cliq pen. See Step 23 in the "Priming your Q-Cliq pen." section of the Instructions for Use. Call 1-800-828-2088 for help.
What if the dosage knob is hard to turn to "GO"?
- Do not use force if the knob will not easily turn to "GO." You may have used your last dose.
- Check the dose indicator on the medicine cartridge to see if there are any doses left, or check the "Discard on" date on the Medicine Cartridge Tracker to see if it has been more than 14 days.
- If the cartridge contains at least 1 dose, call 1-800-828-2088 for help.
What should I do if the NATPARA medicine cartridge is frozen, whether or not it is attached to the Q-Cliq pen?
Throw away the frozen medicine cartridge and mix a new medicine cartridge.
Why do I not throw away the medicine cartridge on day 14 after giving the last injection?
A medicine cartridge or the "dummy" rod protector is needed to put the cap on the Q-Cliq pen. After injecting yourself on day 14, leave the current medicine cartridge on the Q-Cliq pen, put the pen cap back on, and store the pen in the refrigerator. The next day, which will be day 15, throw away the old medicine cartridge and mix a new one.
What if the newly mixed NATPARA medicine cartridge is hard to screw onto the Q-Cliq pen?
- The rod in the Q-Cliq pen may be extended.
- Remove the cartridge and make sure that the rod is fully lowered. If it is not fully lowered, turn the dark red ring to lower it until the ring stops. Do not tighten it too much. Reattach the cartridge and see if it is easier to attach.
- If it is still too hard to screw onto the Q-Cliq pen, check to see if the stoppers in the cartridge window are together.
- If the stoppers are together, call 1-800-828-2088 for help.
What if the stoppers do not stay together after mixing?
- The Pen Needle may not be on correctly.
- Make sure that the Pen Needle is on the right way and that the threads are lined up. Be sure that the needle is firmly attached. You may need to use a new Pen Needle.
What if I have more than a few drops on the tip of the Pen Needle after injection?
This may mean that you did not hold the needle in your thigh for the full 10 seconds. When you give your next scheduled injection, be sure that you hold the needle in your thigh for at least 10 seconds.
What should I do if there are many small bubbles after mixing the NATPARA medicine cartridge?
It is normal to see small air bubbles in the liquid after you finish mixing your NATPARA medicine.
What should I do if the liquid is colored?
Call 1-800-828-2088 for help.
What should I do if the liquid has small particles in it?
It is normal to sometimes see small particles.
How do I clean my Q-Cliq pen and mixing device?
If needed, clean the Q-Cliq pen and mixing device by wiping them with a damp cloth.
Do not place the Q-Cliq pen and mixing device in water, or wash them with any liquid, such as alcohol.
Can I reuse the Pen Needle?
Do not reuse your Pen Needle. You must use a new Pen Needle for each injection.
Manufactured by:
Takeda Pharmaceuticals U.S.A., Inc.
Lexington, MA 02421
USA
1-800-828-2088
U.S. License Number 1898
NATPARA® is a registered trademark of Takeda Pharmaceuticals U.S.A., Inc.
Q-Cliq® is a registered trademark of Takeda Pharmaceuticals U.S.A., Inc.
©2023 Takeda Pharmaceuticals U.S.A., Inc. All rights reserved.
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Revised: 2/2023
PRINCIPAL DISPLAY PANEL - 25 mcg Cartridge Carton
NDC 68875-0202-2
For subcutaneous use only
Natpara®
(parathyroid hormone)
for Injection
25
mcg/dose
Contains two 14-dose medication
cartridges of Natpara®
(parathyroid hormone) for Injection
Use only after training by a
healthcare provider
- For use with the Mixing Device,
Q-Cliq® pen, and 31G x 8 mm
BD Ultra-Fine™ Pen Needles - See enclosed Full Prescribing
Information, Medication Guide,
and Instructions for Use - Protect from light
- Do not shake or freeze
- Must be refrigerated.
Store at 36 to 46°F (2 to 8°C) - Discard mixed solution after 14 days
Rx only
Takeda
PRINCIPAL DISPLAY PANEL - 50 mcg Cartridge Carton
NDC 68875-0203-2
For subcutaneous use only
Natpara®
(parathyroid hormone)
for Injection
50
mcg/dose
Contains two 14-dose medication
cartridges of Natpara®
(parathyroid hormone) for Injection
Use only after training by a
healthcare provider
- For use with the Mixing Device,
Q-Cliq® pen, and 31G x 8 mm
BD Ultra-Fine™ Pen Needles - See enclosed Full Prescribing
Information, Medication Guide,
and Instructions for Use - Protect from light
- Do not shake or freeze
- Must be refrigerated.
Store at 36 to 46°F (2 to 8°C) - Discard mixed solution after 14 days
Rx only
Takeda

PRINCIPAL DISPLAY PANEL - 75 mcg Cartridge Carton
NDC 68875-0204-2
For subcutaneous use only
Natpara®
(parathyroid hormone)
for Injection
75
mcg/dose
Contains two 14-dose medication
cartridges of Natpara®
(parathyroid hormone) for Injection
Use only after training by a
healthcare provider
- For use with the Mixing Device,
Q-Cliq® pen, and 31G x 8 mm
BD Ultra-Fine™ Pen Needles - See enclosed Full Prescribing
Information, Medication Guide,
and Instructions for Use - Protect from light
- Do not shake or freeze
- Must be refrigerated.
Store at 36 to 46°F (2 to 8°C) - Discard mixed solution after 14 days
Rx only
Takeda

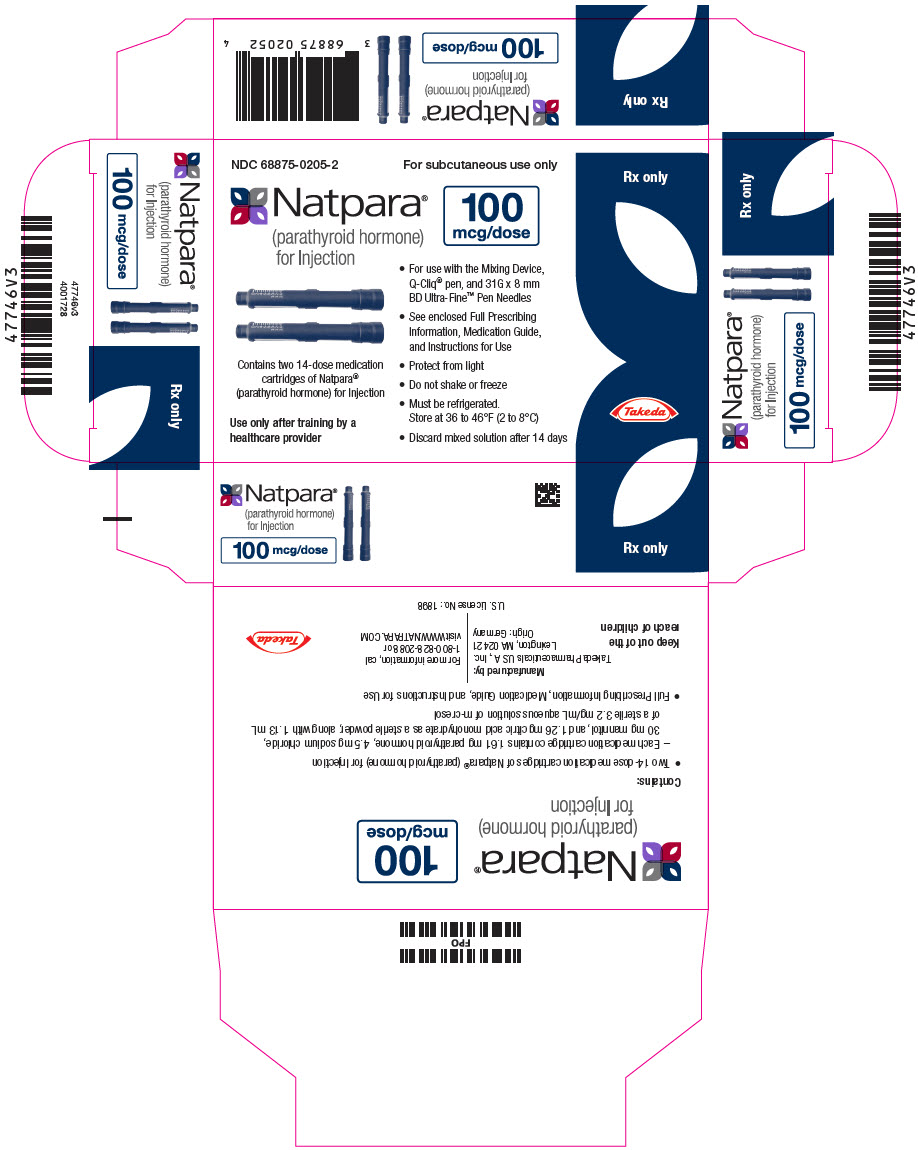
PRINCIPAL DISPLAY PANEL - 100 mcg Cartridge Carton
NDC 68875-0205-2
For subcutaneous use only
Natpara®
(parathyroid hormone)
for Injection
100
mcg/dose
Contains two 14-dose medication
cartridges of Natpara®
(parathyroid hormone) for Injection
Use only after training by a
healthcare provider
- For use with the Mixing Device,
Q-Cliq® pen, and 31G x 8 mm
BD Ultra-Fine™ Pen Needles - See enclosed Full Prescribing
Information, Medication Guide,
and Instructions for Use - Protect from light
- Do not shake or freeze
- Must be refrigerated.
Store at 36 to 46°F (2 to 8°C) - Discard mixed solution after 14 days
Rx only
Takeda