DESCRIPTION
Rocaltrol (calcitriol) is a synthetic vitamin D analog which is active in the regulation of the absorption of calcium from the gastrointestinal tract and its utilization in the body. Rocaltrol is available as capsules containing 0.25 mcg or 0.5 mcg calcitriol and as an oral solution containing 1 mcg/mL of calcitriol. All dosage forms contain butylated hydroxyanisole (BHA) and butylated hydroxytoluene (BHT) as antioxidants. The capsules contain a fractionated triglyceride of coconut oil, and the oral solution contains a fractionated triglyceride of palm seed oil. Gelatin capsule shells contain glycerin, and sorbitol, with the following dye systems: 0.25 mcg - FD&C Yellow No. 6 and titanium dioxide; 0.5 mcg - FD&C Red No. 3, FD&C Yellow No. 6 and titanium dioxide. The oral solution contains no additional adjuvants or coloring principles.
Calcitriol is a white, crystalline compound which occurs naturally in humans. It has a calculated molecular weight of 416.65 and is soluble in organic solvents but relatively insoluble in water. Chemically, calcitriol is 9,10-seco(5Z,7E)-5,7,10(19)-cholestatriene-1α, 3β, 25-triol and has the following structural formula:
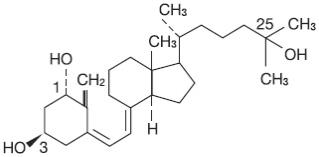
The other names frequently used for calcitriol are 1α,25-dihydroxycholecalciferol, 1,25-dihydroxyvitamin D3, 1,25-DHCC, 1,25(OH)2D3 and 1,25-diOHC.
CLINICAL PHARMACOLOGY
Man's natural supply of vitamin D depends mainly on exposure to the ultraviolet rays of the sun for conversion of 7-dehydrocholesterol in the skin to vitamin D3 (cholecalciferol). Vitamin D3 must be metabolically activated in the liver and the kidney before it is fully active as a regulator of calcium and phosphorus metabolism at target tissues. The initial transformation of vitamin D3 is catalyzed by a vitamin D3-25-hydroxylase enzyme (25-OHase) present in the liver, and the product of this reaction is 25-hydroxyvitamin D3 [25-(OH) D3]. Hydroxylation of 25-(OH) D3 occurs in the mitochondria of kidney tissue, activated by the renal 25-hydroxyvitamin D3-1 alpha-hydroxylase (alpha-OHase), to produce 1,25-(OH)2D3 (calcitriol), the active form of vitamin D3. Endogenous synthesis and catabolism of calcitriol, as well as physiological control mechanisms affecting these processes, play a critical role regulating the serum level of calcitriol. Physiological daily production is normally 0.5 to 1.0 mcg and is somewhat higher during periods of increased bone synthesis (e.g., growth or pregnancy).
Pharmacodynamics
The two known sites of action of calcitriol are intestine and bone. A calcitriol receptor-binding protein appears to exist in the mucosa of human intestine. Additional evidence suggests that calcitriol may also act on the kidney and the parathyroid glands. Calcitriol is the most active known form of vitamin D3 in stimulating intestinal calcium transport. In acutely uremic rats calcitriol has been shown to stimulate intestinal calcium absorption.
The kidneys of uremic patients cannot adequately synthesize calcitriol, the active hormone formed from precursor vitamin D. Resultant hypocalcemia and secondary hyperparathyroidism are a major cause of the metabolic bone disease of renal failure. However, other bone-toxic substances which accumulate in uremia (e.g., aluminum) may also contribute.
The beneficial effect of Rocaltrol in renal osteodystrophy appears to result from correction of hypocalcemia and secondary hyperparathyroidism. It is uncertain whether Rocaltrol produces other independent beneficial effects. Rocaltrol treatment is not associated with an accelerated rate of renal function deterioration. No radiographic evidence of extraskeletal calcification has been found in predialysis patients following treatment. The duration of pharmacologic activity of a single dose of calcitriol is about 3 to 5 days.
Pharmacokinetics
Absorption
Calcitriol is rapidly absorbed from the intestine. Peak serum concentrations (above basal values) were reached within 3 to 6 hours following oral administration of single doses of 0.25 to 1.0 mcg of Rocaltrol. Following a single oral dose of 0.5 mcg, mean serum concentrations of calcitriol rose from a baseline value of 40.0±4.4 (SD) pg/mL to 60.0±4.4 pg/mL at 2 hours, and declined to 53.0±6.9 at 4 hours, 50±7.0 at 8 hours, 44±4.6 at 12 hours, and 41.5±5.1 at 24 hours.
Following multiple-dose administration, serum calcitriol levels reached steady-state within 7 days.
Distribution
Calcitriol is approximately 99.9% bound in blood. Calcitriol and other vitamin D metabolites are transported in blood, by an alpha-globulin vitamin D binding protein. There is evidence that maternal calcitriol may enter the fetal circulation. Calcitriol is transferred into human breast milk at low levels (i.e., 2.2±0.1 pg/mL).
Metabolism
In vivo and in vitro studies indicate the presence of two pathways of metabolism for calcitriol. The first pathway involves the 24-hydroxylase as the first step in catabolism of calcitriol. There is definite evidence of 24-hydroxylase activity in the kidney; this enzyme is also present in many target tissues which possess the vitamin D receptor such as the intestine. The end product of this pathway is a side chain shortened metabolite, calcitroic acid. The second pathway involves the conversion of calcitriol via the stepwise hydroxylation of carbon-26 and carbon-23, and cyclization to yield ultimately 1α, 25R(OH)2-26, 23S-lactone D3. The lactone appears to be the major metabolite circulating in humans, with mean serum concentrations of 131±17 pg/mL. In addition, several other metabolites of calcitriol have been identified: 1α, 25(OH)2-24-oxo-D3; 1α, 23,25(OH)3-24-oxo-D3; 1α, 24R,25(OH)3D3; 1α, 25S,26(OH)3D3; 1α, 25(OH)2-23-oxo-D3; 1α, 25R,26(OH)3-23-oxo-D3; 1α, (OH)24,25,26,27-tetranor-COOH-D3.
Excretion
Enterohepatic recycling and biliary excretion of calcitriol occur. The metabolites of calcitriol are excreted primarily in feces. Following intravenous administration of radiolabeled calcitriol in normal subjects, approximately 27% and 7% of the radioactivity appeared in the feces and urine, respectively, within 24 hours. When a 1-mcg oral dose of radiolabeled calcitriol was administered to normal subjects, approximately 10% of the total radioactivity appeared in urine within 24 hours. Cumulative excretion of radioactivity on the sixth day following intravenous administration of radiolabeled calcitriol averaged 16% in urine and 49% in feces. The elimination half-life of calcitriol in serum after single oral doses is about 5 to 8 hours in normal subjects.
Special Populations
Pediatric Pharmacokinetics
The steady-state pharmacokinetics of oral Rocaltrol were determined in a small group of pediatric patients (age range: 1.8 to 16 years) undergoing peritoneal dialysis. Rocaltrol was administered for 2 months at an average dose of 10.2 ng/kg (SD 5.5 ng/kg). In this pediatric population, mean Cmax was 116 pmol/L, mean serum half-life was 27.4 hours, and mean clearance was 15.3 mL/hr/kg.1
Geriatric
No studies have examined the pharmacokinetics of calcitriol in geriatric patients.
Gender
Controlled studies examining the influence of gender on calcitriol have not been conducted.
Hepatic Insufficiency
Controlled studies examining the influence of hepatic disease on calcitriol have not been conducted.
Renal Insufficiency
Lower predose and peak calcitriol levels in serum were observed in patients with nephrotic syndrome and in patients undergoing hemodialysis compared with healthy subjects. The elimination half-life of calcitriol increased by at least twofold in chronic renal failure and hemodialysis patients compared with healthy subjects. Peak serum levels in patients with nephrotic syndrome were reached in 4 hours. For patients requiring hemodialysis peak serum levels were reached in 8 to 12 hours; half-lives were estimated to be 16.2 and 21.9 hours, respectively.
INDICATIONS AND USAGE
Predialysis Patients
Rocaltrol is indicated in the management of secondary hyperparathyroidism and resultant metabolic bone disease in patients with moderate to severe chronic renal failure (Ccr 15 to 55 mL/min) not yet on dialysis. In children, the creatinine clearance value must be corrected for a surface area of 1.73 square meters. A serum iPTH level of ≥ 100 pg/mL is strongly suggestive of secondary hyperparathyroidism.
Dialysis Patients
Rocaltrol is indicated in the management of hypocalcemia and the resultant metabolic bone disease in patients undergoing chronic renal dialysis. In these patients, Rocaltrol administration enhances calcium absorption, reduces serum alkaline phosphatase levels, and may reduce elevated parathyroid hormone levels and the histological manifestations of osteitis fibrosa cystica and defective mineralization.
Hypoparathyroidism Patients
Rocaltrol is also indicated in the management of hypocalcemia and its clinical manifestations in patients with postsurgical hypoparathyroidism, idiopathic hypoparathyroidism, and pseudohypoparathyroidism.
CONTRAINDICATIONS
Rocaltrol should not be given to patients with hypercalcemia or evidence of vitamin D toxicity. Use of Rocaltrol in patients with known hypersensitivity to Rocaltrol (or drugs of the same class) or any of the inactive ingredients is contraindicated.
WARNINGS
Overdosage of any form of vitamin D is dangerous (see OVERDOSAGE). Progressive hypercalcemia due to overdosage of vitamin D and its metabolites may be so severe as to require emergency attention. Chronic hypercalcemia can lead to generalized vascular calcification, nephrocalcinosis and other soft-tissue calcification. The serum calcium times phosphate (Ca x P) product should not be allowed to exceed 70 mg2/dL2. Radiographic evaluation of suspect anatomical regions may be useful in the early detection of this condition.
Rocaltrol is the most potent metabolite of vitamin D available. The administration of Rocaltrol to patients in excess of their daily requirements can cause hypercalcemia, hypercalciuria, and hyperphosphatemia. Therefore, pharmacologic doses of vitamin D and its derivatives should be withheld during Rocaltrol treatment to avoid possible additive effects and hypercalcemia. If treatment is switched from ergocalciferol (vitamin D2) to calcitriol, it may take several months for the ergocalciferol level in the blood to return to the baseline value (see OVERDOSAGE).
Calcitriol increases inorganic phosphate levels in serum. While this is desirable in patients with hypophosphatemia, caution is called for in patients with renal failure because of the danger of ectopic calcification. A nonaluminum phosphate-binding compound and a low-phosphate diet should be used to control serum phosphorus levels in patients undergoing dialysis.
Magnesium-containing preparations (e.g., antacids) and Rocaltrol should not be used concomitantly in patients on chronic renal dialysis because such use may lead to the development of hypermagnesemia.
Studies in dogs and rats given calcitriol for up to 26 weeks have shown that small increases of calcitriol above endogenous levels can lead to abnormalities of calcium metabolism with the potential for calcification of many tissues in the body.
PRECAUTIONS
General
Excessive dosage of Rocaltrol induces hypercalcemia and in some instances hypercalciuria; therefore, early in treatment during dosage adjustment, serum calcium should be determined twice weekly. In dialysis patients, a fall in serum alkaline phosphatase levels usually antedates the appearance of hypercalcemia and may be an indication of impending hypercalcemia. An abrupt increase in calcium intake as a result of changes in diet (e.g., increased consumption of dairy products) or uncontrolled intake of calcium preparations may trigger hypercalcemia.
Should hypercalcemia develop, treatment with Rocaltrol should be stopped immediately. During periods of hypercalcemia, serum calcium and phosphate levels must be determined daily. When normal levels have been attained, treatment with Rocaltrol can be continued, at a daily dose 0.25 mcg lower than that previously used. An estimate of daily dietary calcium intake should be made and the intake adjusted when indicated. Rocaltrol should be given cautiously to patients on digitalis, because hypercalcemia in such patients may precipitate cardiac arrhythmias.
Immobilized patients, e.g., those who have undergone surgery, are particularly exposed to the risk of hypercalcemia.
In patients with normal renal function, chronic hypercalcemia may be associated with an increase in serum creatinine. While this is usually reversible, it is important in such patients to pay careful attention to those factors which may lead to hypercalcemia. Rocaltrol therapy should always be started at the lowest possible dose and should not be increased without careful monitoring of the serum calcium. An estimate of daily dietary calcium intake should be made and the intake adjusted when indicated.
Patients with normal renal function taking Rocaltrol should avoid dehydration. Adequate fluid intake should be maintained.
Information for Patients
The patient and his or her caregivers should be informed about compliance with dosage instructions, adherence to instructions about diet and calcium supplementation, and avoidance of the use of unapproved nonprescription drugs. Patients and their caregivers should also be carefully informed about the symptoms of hypercalcemia (see ADVERSE REACTIONS).
The effectiveness of Rocaltrol therapy is predicated on the assumption that each patient is receiving an adequate daily intake of calcium. Patients are advised to have a dietary intake of calcium at a minimum of 600 mg daily. The U.S. RDA for calcium in adults is 800 mg to 1200 mg.
Laboratory Tests
For dialysis patients, serum calcium, phosphorus, magnesium, and alkaline phosphatase should be determined periodically. For hypoparathyroid patients, serum calcium, phosphorus, and 24-hour urinary calcium should be determined periodically. For predialysis patients, serum calcium, phosphorus, alkaline phosphatase, creatinine, and intact PTH (iPTH) should be determined initially. Thereafter, serum calcium, phosphorus, alkaline phosphatase, and creatinine should be determined monthly for a 6-month period and then determined periodically. Intact PTH (iPTH) should be determined periodically every 3 to 4 months at the time of visits. During the titration period of treatment with Rocaltrol, serum calcium levels should be checked at least twice weekly (see DOSAGE AND ADMINISTRATION).
Drug Interactions
Cholestyramine
Cholestyramine has been reported to reduce intestinal absorption of fat-soluble vitamins; as such it may impair intestinal absorption of Rocaltrol (see WARNINGS and PRECAUTIONS: General).
Phenytoin/Phenobarbital
The coadministration of phenytoin or phenobarbital will not affect plasma concentrations of calcitriol, but may reduce endogenous plasma levels of 25(OH)D3 by accelerating metabolism. Since blood level of calcitriol will be reduced, higher doses of Rocaltrol may be necessary if these drugs are administered simultaneously.
Thiazides
Thiazides are known to induce hypercalcemia by the reduction of calcium excretion in urine. Some reports have shown that the concomitant administration of thiazides with Rocaltrol causes hypercalcemia. Therefore, precaution should be taken when coadministration is necessary.
Digitalis
Calcitriol dosage must be determined with care in patients undergoing treatment with digitalis, as hypercalcemia in such patients may precipitate cardiac arrhythmias (see PRECAUTIONS: General).
Ketoconazole
Ketoconazole may inhibit both synthetic and catabolic enzymes of calcitriol. Reductions in serum endogenous calcitriol concentrations have been observed following the administration of 300 mg/day to 1200 mg/day ketoconazole for a week to healthy men. However, in vivo drug interaction studies of ketoconazole with Rocaltrol have not been investigated.
Corticosteroids
A relationship of functional antagonism exists between vitamin D analogues, which promote calcium absorption, and corticosteroids, which inhibit calcium absorption.
Phosphate-Binding Agents
Since Rocaltrol also has an effect on phosphate transport in the intestine, kidneys and bones, the dosage of phosphate-binding agents must be adjusted in accordance with the serum phosphate concentration.
Vitamin D
Since calcitriol is the most potent active metabolite of vitamin D3, pharmacological doses of vitamin D and its derivatives should be withheld during treatment with Rocaltrol to avoid possible additive effects and hypercalcemia (see WARNINGS).
Calcium Supplements
Uncontrolled intake of additional calcium-containing preparations should be avoided (see PRECAUTIONS: General).
Magnesium
Magnesium-containing preparations (e.g., antacids) may cause hypermagnesemia and should therefore not be taken during therapy with Rocaltrol by patients on chronic renal dialysis.
Carcinogenesis, Mutagenesis and Impairment of Fertility
Long-term studies in animals have not been conducted to evaluate the carcinogenic potential of Rocaltrol. Rocaltrol is not mutagenic in vitro in the Ames Test, nor is it genotoxic in vivo in the Mouse Micronucleus Test. No significant effects of Rocaltrol on fertility and/or general reproductive performances were observed in a Segment I study in rats at doses of up to 0.3 mcg/kg (approximately 3 times the maximum recommended dose based on body surface area).
Pregnancy
Teratogenic Effects
Rocaltrol has been found to be teratogenic in rabbits when given at doses of 0.08 and 0.3 mcg/kg (approximately 2 and 6 times the maximum recommended dose based on mg/m2). All 15 fetuses in 3 litters at these doses showed external and skeletal abnormalities. However, none of the other 23 litters (156 fetuses) showed external and skeletal abnormalities compared with controls.
Teratogenicity studies in rats at doses up to 0.45 mcg/kg (approximately 5 times maximum recommended dose based on mg/m2) showed no evidence of teratogenic potential. There are no adequate and well-controlled studies in pregnant women. Rocaltrol should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Nonteratogenic Effects
In the rabbit, dosages of 0.3 mcg/kg/day (approximately 6 times maximum recommended dose based on surface area) administered on days 7 to 18 of gestation resulted in 19% maternal mortality, a decrease in mean fetal body weight, and a reduced number of newborn surviving to 24 hours. A study of perinatal and postnatal development in rats resulted in hypercalcemia in the offspring of dams given Rocaltrol at doses of 0.08 or 0.3 mcg/kg/day (approximately 1 and 3 times the maximum recommended dose based on mg/m2), hypercalcemia and hypophosphatemia in dams given Rocaltrol at a dose of 0.08 or 0.3 mcg/kg/day, and increased serum urea nitrogen in dams given Rocaltrol at a dose of 0.3 mcg/kg/day. In another study in rats, maternal weight gain was slightly reduced at a dose of 0.3 mcg/kg/day (approximately 3 times the maximum recommended dose based on mg/m2) administered on days 7 to 15 of gestation. The offspring of a woman administered 17 mcg/day to 36 mcg/day of Rocaltrol (approximately 17 to 36 times the maximum recommended dose) during pregnancy manifested mild hypercalcemia in the first 2 days of life which returned to normal at day 3.
Nursing Mothers
Calcitriol from ingested Rocaltrol may be excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions from Rocaltrol in nursing infants, a mother should not nurse while taking Rocaltrol.
Pediatric Use
Safety and effectiveness of Rocaltrol in pediatric patients undergoing dialysis have not been established. The safety and effectiveness of Rocaltrol in pediatric predialysis patients is based on evidence from adequate and well-controlled studies of Rocaltrol in adults with predialysis chronic renal failure and additional supportive data from nonplacebo-controlled studies in pediatric patients. Dosing guidelines have not been established for pediatric patients under 1 year of age with hypoparathyroidism or for pediatric patients less than 6 years of age with pseudohypoparathyroidism (see DOSAGE AND ADMINISTRATION: Hypoparathyroidism).
Oral doses of Rocaltrol ranging from 10 to 55 ng/kg/day have been shown to improve calcium homeostasis and bone disease in pediatric patients with chronic renal failure for whom hemodialysis is not yet required (predialysis). Long-term calcitriol therapy is well tolerated by pediatric patients. The most common safety issues are mild, transient episodes of hypercalcemia, hyperphosphatemia, and increases in the serum calcium times phosphate (Ca x P) product which are managed effectively by dosage adjustment or temporary discontinuation of the vitamin D derivative.
Geriatric Use
Clinical studies of Rocaltrol did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
ADVERSE REACTIONS
Since Rocaltrol is believed to be the active hormone which exerts vitamin D activity in the body, adverse effects are, in general, similar to those encountered with excessive vitamin D intake, i.e., hypercalcemia syndrome or calcium intoxication, depending on the severity and duration of hypercalcemia (see WARNINGS). Because of the short biological half-life of calcitriol, pharmacokinetic investigations have shown normalization of elevated serum calcium within a few days of treatment withdrawal, i.e., much faster than in treatment with vitamin D3 preparations.
The early and late signs and symptoms of vitamin D intoxication associated with hypercalcemia include:
Early: weakness, headache, somnolence, nausea, vomiting, dry mouth, constipation, muscle pain, bone pain, metallic taste, and anorexia, abdominal pain or stomach ache.
Late: polyuria, polydipsia, anorexia, weight loss, nocturia, conjunctivitis (calcific), pancreatitis, photophobia, rhinorrhea, pruritus, hyperthermia, decreased libido, elevated BUN, albuminuria, hypercholesterolemia, elevated SGOT (AST) and SGPT (ALT), ectopic calcification, nephrocalcinosis, hypertension, cardiac arrhythmias, dystrophy, sensory disturbances, dehydration, apathy, arrested growth, urinary tract infections, and, rarely, overt psychosis.
In clinical studies on hypoparathyroidism and pseudohypoparathyroidism, hypercalcemia was noted on at least one occasion in about 1 in 3 patients and hypercalciuria in about 1 in 7 patients. Elevated serum creatinine levels were observed in about 1 in 6 patients (approximately one half of whom had normal levels at baseline).
In concurrent hypercalcemia and hyperphosphatemia, soft-tissue calcification may occur; this can be seen radiographically (see WARNINGS).
In patients with normal renal function, chronic hypercalcemia may be associated with an increase in serum creatinine (see PRECAUTIONS: General).
Hypersensitivity reactions (pruritus, rash, urticaria, and very rarely severe erythematous skin disorders) may occur in susceptible individuals. One case of erythema multiforme and one case of allergic reaction (swelling of lips and hives all over the body) were confirmed by rechallenge.
To report SUSPECTED ADVERSE REACTIONS, contact Validus Pharmaceuticals LLC at 1-866-982-5438 (1-866-9VALIDUS) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
OVERDOSAGE
Administration of Rocaltrol to patients in excess of their daily requirements can cause hypercalcemia, hypercalciuria, and hyperphosphatemia. Since calcitriol is a derivative of vitamin D, the signs and symptoms of overdose are the same as for an overdose of vitamin D (see ADVERSE REACTIONS). High intake of calcium and phosphate concomitant with Rocaltrol may lead to similar abnormalities. The serum calcium times phosphate (Ca x P) product should not be allowed to exceed 70 mg2/dL2. High levels of calcium in the dialysate bath may contribute to the hypercalcemia (see WARNINGS).
Treatment of Hypercalcemia and Overdosage in Dialysis Patients and Hypoparathyroidism Patients
General treatment of hypercalcemia (greater than 1 mg/dL above the upper limit of the normal range) consists of immediate discontinuation of Rocaltrol therapy, institution of a low-calcium diet and withdrawal of calcium supplements. Serum calcium levels should be determined daily until normocalcemia ensues. Hypercalcemia frequently resolves in 2 to 7 days. When serum calcium levels have returned to within normal limits, Rocaltrol therapy may be reinstituted at a dose of 0.25 mcg/day less than prior therapy. Serum calcium levels should be obtained at least twice weekly after all dosage changes and subsequent dosage titration. In dialysis patients, persistent or markedly elevated serum calcium levels may be corrected by dialysis against a calcium-free dialysate.
Treatment of Hypercalcemia and Overdosage in Predialysis Patients
If hypercalcemia ensues (greater than 1 mg/dL above the upper limit of the normal range), adjust dosage to achieve normocalcemia by reducing Rocaltrol therapy from 0.5 mcg to 0.25 mcg daily. If the patient is receiving a therapy of 0.25 mcg daily, discontinue Rocaltrol until patient becomes normocalcemic. Calcium supplements should also be reduced or discontinued. Serum calcium levels should be determined 1 week after withdrawal of calcium supplements. If serum calcium levels have returned to normal, Rocaltrol therapy may be reinstituted at a dosage of 0.25 mcg/day if previous therapy was at a dosage of 0.5 mcg/day. If Rocaltrol therapy was previously administered at a dosage of 0.25 mcg/day, Rocaltrol therapy may be reinstituted at a dosage of 0.25 mcg every other day. If hypercalcemia is persistent at the reduced dosage, serum PTH should be measured. If serum PTH is normal, discontinue Rocaltrol therapy and monitor patient in 3 months' time.
Treatment of Hyperphosphatemia in Predialysis Patients
If serum phosphorus levels exceed 5.0 mg/dL to 5.5 mg/dL, a calcium-containing phosphate-binding agent (i.e., calcium carbonate or calcium acetate) should be taken with meals. Serum phosphorus levels should be determined as described earlier (see PRECAUTIONS: Laboratory Tests). Aluminum-containing gels should be used with caution as phosphate-binding agents because of the risk of slow aluminum accumulation.
Treatment of Accidental Overdosage of Rocaltrol
The treatment of acute accidental overdosage of Rocaltrol should consist of general supportive measures. If drug ingestion is discovered within a relatively short time, induction of emesis or gastric lavage may be of benefit in preventing further absorption. If the drug has passed through the stomach, the administration of mineral oil may promote its fecal elimination. Serial serum electrolyte determinations (especially calcium), rate of urinary calcium excretion, and assessment of electrocardiographic abnormalities due to hypercalcemia should be obtained. Such monitoring is critical in patients receiving digitalis. Discontinuation of supplemental calcium and a low-calcium diet are also indicated in accidental overdosage. Due to the relatively short duration of the pharmacological action of calcitriol, further measures are probably unnecessary. Should, however, persistent and markedly elevated serum calcium levels occur, there are a variety of therapeutic alternatives which may be considered, depending on the patient's underlying condition. These include the use of drugs such as phosphates and corticosteroids as well as measures to induce an appropriate forced diuresis. The use of peritoneal dialysis against a calcium-free dialysate has also been reported.
DOSAGE AND ADMINISTRATION
The optimal daily dose of Rocaltrol must be carefully determined for each patient. Rocaltrol can be administered orally either as a capsule (0.25 mcg or 0.50 mcg) or as an oral solution (1 mcg/mL). Rocaltrol therapy should always be started at the lowest possible dose and should not be increased without careful monitoring of serum calcium.
The effectiveness of Rocaltrol therapy is predicated on the assumption that each patient is receiving an adequate but not excessive daily intake of calcium. Patients are advised to have a dietary intake of calcium at a minimum of 600 mg daily. The U.S. RDA for calcium in adults is 800 mg to 1200 mg. To ensure that each patient receives an adequate daily intake of calcium, the physician should either prescribe a calcium supplement or instruct the patient in proper dietary measures.
Because of improved calcium absorption from the gastrointestinal tract, some patients on Rocaltrol may be maintained on a lower calcium intake. Patients who tend to develop hypercalcemia may require only low doses of calcium or no supplementation at all.
During the titration period of treatment with Rocaltrol, serum calcium levels should be checked at least twice weekly. When the optimal dosage of Rocaltrol has been determined, serum calcium levels should be checked every month (or as given below for individual indications). Samples for serum calcium estimation should be taken without a tourniquet.
Dialysis Patients
The recommended initial dose of Rocaltrol is 0.25 mcg/day. If a satisfactory response in the biochemical parameters and clinical manifestations of the disease state is not observed, dosage may be increased by 0.25 mcg/day at 4- to 8-week intervals. During this titration period, serum calcium levels should be obtained at least twice weekly, and if hypercalcemia is noted, the drug should be immediately discontinued until normocalcemia ensues (see PRECAUTIONS: General). Phosphorus, magnesium, and alkaline phosphatase should be determined periodically.
Patients with normal or only slightly reduced serum calcium levels may respond to Rocaltrol doses of 0.25 mcg every other day. Most patients undergoing hemodialysis respond to doses between 0.5 and 1 mcg/day.
Oral Rocaltrol may normalize plasma-ionized calcium in some uremic patients, yet fail to suppress parathyroid hyperfunction. In these individuals with autonomous parathyroid hyper-function, oral Rocaltrol may be useful to maintain normocalcemia, but has not been shown to be adequate treatment for hyperparathyroidism.
Hypoparathyroidism
The recommended initial dosage of Rocaltrol is 0.25 mcg/day given in the morning. If a satisfactory response in the biochemical parameters and clinical manifestations of the disease is not observed, the dose may be increased at 2- to 4-week intervals. During the dosage titration period, serum calcium levels should be obtained at least twice weekly and, if hypercalcemia is noted, Rocaltrol should be immediately discontinued until normocalcemia ensues (see PRECAUTIONS: General). Careful consideration should also be given to lowering the dietary calcium intake. Serum calcium, phosphorus, and 24-hour urinary calcium should be determined periodically.
Most adult patients and pediatric patients age 6 years and older have responded to dosages in the range of 0.5 mcg to 2 mcg daily. Pediatric patients in the 1- to 5-year age group with hypoparathyroidism have usually been given 0.25 mcg to 0.75 mcg daily. The number of treated patients with pseudohypoparathyroidism less than 6 years of age is too small to make dosage recommendations.
Malabsorption is occasionally noted in patients with hypoparathyroidism; hence, larger doses of Rocaltrol may be needed.
Predialysis Patients
The recommended initial dosage of Rocaltrol is 0.25 mcg/day in adults and pediatric patients 3 years of age and older. This dosage may be increased if necessary to 0.5 mcg/day.
For pediatric patients less than 3 years of age, the recommended initial dosage of Rocaltrol is 10 to 15 ng/kg/day.
HOW SUPPLIED
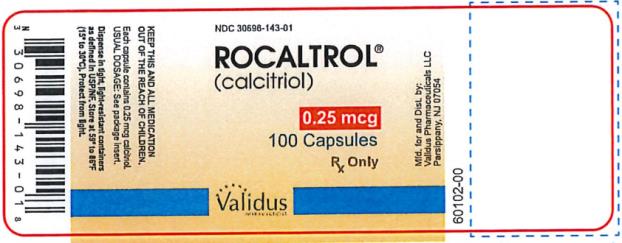
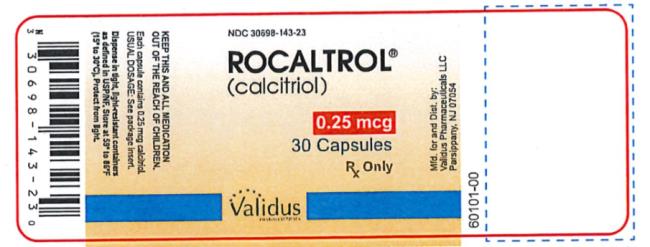
Capsules: 0.25 mcg calcitriol in soft gelatin, light orange, oval capsules, imprinted with R25; bottles of 30 (NDC 30698-143-23), and bottles of 100 (NDC 30698-143-01).
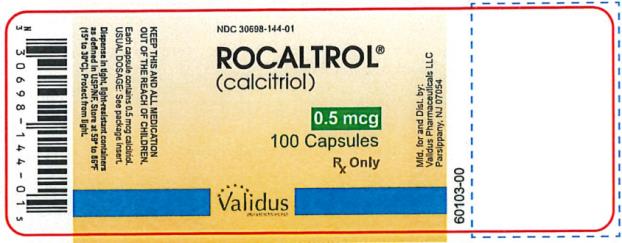
Capsules: 0.5 mcg calcitriol in soft gelatin, dark orange, oblong capsules, imprinted with R50; bottles of 100 (NDC 30698-144-01).
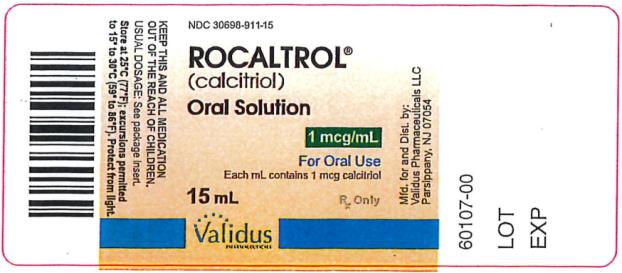
Oral Solution: a clear, colorless to pale yellow oral solution containing 1 mcg/mL of calcitriol; each amber glass bottle of 15 mL of oral solution supplied with 20 single-use, graduated oral dispensers (NDC 30698-911-15).
Rocaltrol Capsules and Oral Solution should be protected from light.
Store at 59° to 86°F (15° to 30°C) [See USP Controlled Room Temperature].
Dispense in a tight, light-resistant container as defined in USP.
REFERENCE
- Jones CL, et al. Comparisons between oral and intraperitoneal 1,25-dihydroxyvitamin D3 therapy in children treated with peritoneal dialysis. Clin Nephrol. 1994; 42:44-49.
Manufactured for and Distributed by:
Validus Pharmaceuticals LLC
119 Cherry Hill Road, Suite 310
Parsippany, NJ 07054
info@validuspharma.com
www.validuspharma.com
1-866-982-5438 (1-866-9VALIDUS)
© 2018 Validus Pharmaceuticals LLC
60110-02 May 2018