FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
1.1 Acute Ischemic Stroke
Activase is indicated for the treatment of acute ischemic stroke.
Exclude intracranial hemorrhage as the primary cause of stroke signs and symptoms prior to initiation of treatment [see Contraindications (4.1)]. Initiate treatment as soon as possible but within 3 hours after symptom onset.
1.2 Acute Myocardial Infarction
Activase is indicated for use in acute myocardial infarction (AMI) for the reduction of mortality and reduction of the incidence of heart failure.
1.3 Pulmonary Embolism
Activase is indicated for the lysis of acute massive pulmonary embolism, defined as:
- Acute pulmonary emboli obstructing blood flow to a lobe or multiple lung segments.
- Acute pulmonary emboli accompanied by unstable hemodynamics, e.g., failure to maintain blood pressure without supportive measures.
2 DOSAGE AND ADMINISTRATION
2.1 Acute Ischemic Stroke
Administer Activase as soon as possible but within 3 hours after onset of symptoms.
The recommended dose is 0.9 mg/kg (not to exceed 90 mg total dose), with 10% of the total dose administered as an initial intravenous bolus over 1 minute and the remainder infused over 60 minutes.
During and following Activase administration for the treatment of acute ischemic stroke, frequently monitor and control blood pressure.
In patients without recent use of oral anticoagulants or heparin, Activase treatment can be initiated prior to the availability of coagulation study results. Discontinue Activase if the pretreatment International Normalized Ratio (INR) is greater than 1.7 or the activated partial thromboplastin time (aPTT) is elevated [see Contraindications (4.1)].
2.2 Acute Myocardial Infarction
Administer Activase as soon as possible after the onset of symptoms.
The recommended total doses for acute myocardial infarction (AMI) is based on patient weight, not to exceed 100 mg, regardless of the selected administration regimen (accelerated or 3 hour, described below).
There are two Activase dose regimens (accelerated and 3-hour) for use in the management of AMI; there are no controlled studies to compare clinical outcomes with these regimens [see Clinical Studies (14.2)].
Accelerated Infusion
The recommended accelerated infusion dose consists of an IV bolus [see Dosage and Administration (2.4, 2.5)] followed by an IV infusion as set forth in Table 1.
| Patient weight | Intravenous Bolus | First 30 min | Next 60 min |
|---|---|---|---|
| > 67 kg | 15 mg | 50 mg | 35 mg |
| ≤ 67 kg | 15 mg | 0.75 mg/kg | 0.50 mg/kg |
The safety and efficacy of accelerated infusion of Activase have only been investigated with concomitant administration of heparin and aspirin [see Clinical Studies (14.2)].
3-Hour Infusion
For patients weighing ≥ 65 kg, the recommended dose is 100 mg administered as 60 mg in the first hour (6-10 mg administered as a bolus), 20 mg over the second hour, and 20 mg over the third hour. For smaller patients (< 65 kg), a dose of 1.25 mg/kg administered over 3 hours may be used. Weight-based doses are shown in Table 2.
| Patient weight | Bolus | Rest of 1st hour | 2nd hour | 3rd hour |
|---|---|---|---|---|
| ≥ 65 kg | 6-10 mg | 50-54 mg | 20 mg | 20 mg |
| < 65 kg | 0.075 mg/kg | 0.675 mg/kg | 0.25 mg/kg | 0.25 mg/kg |
2.3 Pulmonary Embolism (PE)
The recommended dose is 100 mg administered by IV infusion over 2 hours.
Institute parenteral anticoagulation near the end of or immediately following the Activase infusion when the partial thromboplastin time or thrombin time returns to twice normal or less.
2.4 Activase 50 mg Reconstitution and Administration Instructions
- Activase is for intravenous administration only.
- Do not add any other medication to infusion solutions containing Activase.
- Extravasation of Activase infusion can cause ecchymosis or inflammation. If extravasation occurs, terminate the infusion at that intravenous site and apply local therapy.
- Use within 8 hours following reconstitution (when stored at 2–30°C). Activase contains no antibacterial preservatives.
Activase 50 mg Reconstitution Notes
- Use only the accompanying Sterile Water for Injection (SWFI), USP without preservatives. Do not use Bacteriostatic Water for Injection, USP.
- Reconstitute using aseptic technique.
- Slight foaming is not unusual; let stand undisturbed for several minutes to allow large bubbles to dissipate. Inspect parenteral drug products for particulate matter and discoloration prior to administration whenever solution and container permit.
- Activase may be administered as reconstituted at 1 mg/mL or further diluted immediately before administration in an equal volume of 0.9% Sodium Chloride Injection, USP, or 5% Dextrose Injection, USP, to yield a concentration of 0.5 mg/mL, using either polyvinyl chloride bags or glass vials.
- Avoid excessive agitation during dilution; mix by gently swirling and/or slow inversion.
Activase 50 mg Reconstitution Instructions
- DO NOT USE IF VACUUM IS NOT PRESENT.
- Using a large bore needle (e.g., 18 gauge) and a syringe, reconstitute by adding the contents of the accompanying 50 mL vial of SWFI to the 50 mg vial of Activase, directing the SWFI stream into the lyophilized cake.
Activase 50 mg Preparation of Bolus Dose
- Prepare the bolus dose in one of the following ways:
- Remove the appropriate volume from the vial of reconstituted (1 mg/mL) Activase using a syringe and needle. The syringe should not be primed with air and the needle should be inserted into the Activase vial stopper.
- Remove the appropriate volume from a port (second injection site) on the infusion line after the infusion set is primed.
- Program an infusion pump to deliver the appropriate volume as a bolus at the initiation of the infusion.
Activase 50 mg Administration
- Following bolus dose, if indicated [see Dosage and Administration (2.1, 2.2)]:
- Administer using either a polyvinyl chloride bag or glass vial and infusion set.
2.5 Activase 100 mg Instructions for Use
See the enclosed Instructions for Use for Activase 100 mg enclosed in the carton for reconstitution and administration instructions.
Alternative Dilution Instructions
Activase may be administered as reconstituted at 1 mg/mL or further diluted immediately before administration in an equal volume of 0.9% Sodium Chloride Injection, USP, or 5% Dextrose Injection, USP, to yield a concentration of 0.5 mg/mL, using either polyvinyl chloride bags or glass vials.
3 DOSAGE FORMS AND STRENGTHS
- 50 mg lyophilized powder in single-dose vial with 50 mL SWFI USP for reconstitution
- 100 mg lyophilized powder in single-dose vial with 100 mL SWFI USP for reconstitution
4 CONTRAINDICATIONS
4.1 Acute Ischemic Stroke
Do not administer Activase to treat acute ischemic stroke in the following situations in which the risk of bleeding is greater than the potential benefit [see Warnings and Precautions (5.1)]:
- Current intracranial hemorrhage
- Subarachnoid hemorrhage
- Active internal bleeding
- Recent (within 3 months) intracranial or intraspinal surgery or serious head trauma
- Presence of intracranial conditions that may increase the risk of bleeding (e.g., some neoplasms, arteriovenous malformations, or aneurysms)
- Bleeding diathesis
- Current severe uncontrolled hypertension.
4.2 Acute Myocardial Infarction or Pulmonary Embolism
Do not administer Activase for treatment of AMI or PE in the following situations in which the risk of bleeding is greater than the potential benefit [see Warnings and Precautions (5.1)]:
- Active internal bleeding
- History of recent stroke
- Recent (within 3 months) intracranial or intraspinal surgery or serious head trauma
- Presence of intracranial conditions that may increase the risk of bleeding (e.g. some neoplasms, arteriovenous malformations, or aneurysms)
- Bleeding diathesis
- Current severe uncontrolled hypertension.
5 WARNINGS AND PRECAUTIONS
5.1 Bleeding
Activase can cause significant, sometimes fatal, internal or external bleeding, especially at arterial and venous puncture sites. Avoid intramuscular injections and trauma to the patient while on Activase. Perform venipunctures carefully and only as required. To minimize bleeding from noncompressible sites, avoid internal jugular and subclavian venous punctures. If an arterial puncture is necessary during Activase infusion, use an upper extremity vessel that is accessible to manual compression, apply pressure for at least 30 minutes, and monitor the puncture site closely.
Because of the higher risk of intracranial hemorrhage in patients treated for acute ischemic stroke, limit treatment to facilities that can provide timely access to appropriate evaluation and management of intracranial hemorrhage.
Fatal cases of hemorrhage associated with traumatic intubation in patients administered Activase have been reported.
Aspirin and heparin have been administered concomitantly with and following infusions of Activase in the management of acute myocardial infarction and pulmonary embolism, but the concomitant administration of heparin and aspirin with and following infusions of Activase for the treatment of acute ischemic stroke during the first 24 hours after symptom onset has not been investigated. Because heparin, aspirin, or Activase may cause bleeding complications, carefully monitor for bleeding, especially at arterial puncture sites. Hemorrhage can occur 1 or more days after administration of Activase, while patients are still receiving anticoagulant therapy.
If serious bleeding occurs, terminate the Activase infusion and treat appropriately. In the following conditions, the risks of bleeding with Activase therapy for all approved indications are increased and should be weighed against the anticipated benefits:
- Recent major surgery or procedure, (e.g., coronary artery bypass graft, obstetrical delivery, organ biopsy, previous puncture of noncompressible vessels)
- Cerebrovascular disease
- Recent intracranial hemorrhage
- Recent gastrointestinal or genitourinary bleeding
- Recent trauma
- Hypertension: systolic BP above 175 mm Hg or diastolic BP above 110 mm Hg
- Acute pericarditis
- Subacute bacterial endocarditis
- Hemostatic defects including those secondary to severe hepatic or renal disease
- Significant hepatic dysfunction
- Pregnancy
- Diabetic hemorrhagic retinopathy, or other hemorrhagic ophthalmic conditions
- Septic thrombophlebitis or occluded AV cannula at seriously infected site
- Advanced age [see Use in Specific Populations (8.5)]
- Patients currently receiving anticoagulants (e.g., warfarin sodium)
Any other condition in which bleeding constitutes a significant hazard or would be particularly difficult to manage because of its location.
5.2 Hypersensitivity
Hypersensitivity, including urticarial / anaphylactic reactions, have been reported after administration of Activase (e.g., laryngeal edema, rash and shock). Rare fatal outcome for hypersensitivity was reported. Angioedema has been observed during and up to 2 hours after Activase infusion in patients treated for acute ischemic stroke and acute myocardial infarction. In many cases, patients received concomitant angiotensin-converting enzyme inhibitors [see Drug Interactions (7)].
Monitor patients treated with Activase during and for several hours after infusion for hypersensitivity. If signs of hypersensitivity occur, e.g. anaphylactoid reaction or angioedema develops, discontinue the Activase infusion and promptly institute appropriate therapy (e.g., antihistamines, intravenous corticosteroids, epinephrine).
5.3 Thromboembolism
The use of thrombolytics can increase the risk of thrombo-embolic events in patients with high likelihood of left heart thrombus, such as patients with mitral stenosis or atrial fibrillation. Activase has not been shown to treat adequately underlying deep vein thrombosis in patients with PE. Consider the possible risk of re-embolization due to the lysis of underlying deep venous thrombi in this setting.
5.4 Cholesterol Embolization
Cholesterol embolism has been reported rarely in patients treated with thrombolytic agents; the true incidence is unknown. Cholesterol embolism may present with livedo reticularis, "purple toe" syndrome, acute renal failure, gangrenous digits, hypertension, pancreatitis, myocardial infarction, cerebral infarction, spinal cord infarction, retinal artery occlusion, bowel infarction, or rhabdomyolysis and can be fatal. It is associated with invasive vascular procedures (e.g., cardiac catheterization, angiography, vascular surgery) and/or anticoagulant therapy.
5.5 Coagulation Tests May Be Unreliable during Activase Therapy
Coagulation tests and measures of fibrinolytic activity may be unreliable during Activase therapy, unless specific precautions are taken to prevent in vitro artifacts. When present in blood at pharmacologic concentrations, Activase remains active under in vitro conditions, which can result in degradation of fibrinogen in blood samples removed for analysis.
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in the other sections of the label:
- Bleeding [see Contraindications (4), Warnings and Precautions (5.1)]
- Hypersensitivity [see Warnings and Precautions (5.2)]
- Thromboembolism [see Warnings and Precautions (5.3)]
- Cholesterol Embolization [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The most frequent adverse reaction associated with Activase in all approved indications is bleeding.
Bleeding
Acute Ischemic Stroke (AIS)
In clinical studies in patients with AIS (Studies 1 and 2) the incidence of intracranial hemorrhage, especially symptomatic intracranial hemorrhage, was higher in Activase-treated patients than in placebo patients. A dose-finding study of Activase suggested that doses greater than 0.9 mg/kg may be associated with an increased incidence of intracranial hemorrhage.
The incidence of all-cause 90-day mortality, intracranial hemorrhage, and new ischemic stroke following Activase treatment compared to placebo are presented in Table 3 as a combined safety analysis (n=624) for Studies 1 and 2. These data indicate a significant increase in intracranial hemorrhage following Activase treatment, particularly symptomatic intracranial hemorrhage within 36 hours. There was no increase in the incidences of 90-day mortality or severe disability in Activase-treated patients compared to placebo.
| Placebo (n= 312) | Activase (n=312) |
p-Value* |
|
|---|---|---|---|
|
|||
| All-Cause 90-day Mortality | 64 (20.5%) | 54 (17.3%) | 0.36 |
| Total ICH† | 20 (6.4%) | 48 (15.4%) | <0.01 |
| Symptomatic | 4 (1.3%) | 25 (8.0%) | <0.01 |
| Asymptomatic | 16 (5.1%) | 23 (7.4%) | 0.32 |
| Symptomatic Intracranial Hemorrhage within 36 hours | 2 (0.6%) | 20 (6.4%) | <0.01 |
| New Ischemic Stroke (3-months) | 17 (5.4%) | 18 (5.8%) | 1.00 |
Bleeding events other than intracranial hemorrhage were noted in the studies of AIS and were consistent with the general safety profile of Activase. In Studies 1 and 2, the frequency of bleeding requiring red blood cell transfusions was 6.4% for Activase-treated patients compared to 3.8% for placebo (p = 0.19).
Although exploratory analyses of Studies 1 and 2 suggest that severe neurological deficit (National Institutes of Health Stroke Scale [NIHSS > 22]) at presentation was associated with an increased risk of intracranial hemorrhage, efficacy results suggest a reduced but still favorable clinical outcome for these patients.
Acute Myocardial Infarction (AMI)
For the 3-hour infusion regimen in the treatment of AMI, the incidence of significant internal bleeding (estimated as > 250 mL blood loss) has been reported in studies in over 800 patients (Table 4). These data do not include patients treated with the Activase accelerated infusion.
| Total Dose ≤100 mg | |
|---|---|
| Gastrointestinal | 5% |
| Genitourinary | 4% |
| Ecchymosis | 1% |
| Retroperitoneal | <1% |
| Epistaxis | <1% |
| Gingival | <1% |
The incidence of intracranial hemorrhage in AMI patients treated with Activase is presented in Table 5.
| Dose | Number of Patients | Intracranial Hemorrhage (%) |
|---|---|---|
| 100 mg, 3-hour | 3272 | 0.4 |
| ≤ 100 mg, accelerated | 10,396 | 0.7 |
| 150 mg | 1779 | 1.3 |
| 1-1.4 mg/kg | 237 | 0.4 |
A dose of 150 mg or greater should not be used in the treatment of AMI because it has been associated with an increase in intracranial bleeding.
6.2 Post-Marketing Experience
The following adverse reactions have been identified during post-approval use of Activase. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure. These reactions are frequent sequelae of the underlying disease, and the effect of Activase on the incidence of these events is unknown.
Acute Ischemic Stroke: Cerebral edema, cerebral herniation, seizure, new ischemic stroke, embolism. These events may be life threatening and may lead to death.
Acute Myocardial Infarction: Arrhythmias, AV block, cardiogenic shock, heart failure, cardiac arrest, recurrent ischemia, myocardial reinfarction, myocardial rupture, electromechanical dissociation, pericardial effusion, pericarditis, mitral regurgitation, cardiac tamponade, thromboembolism, pulmonary edema. These events may be life threatening and may lead to death. Nausea and/or vomiting, hypotension and fever have also been reported.
7 DRUG INTERACTIONS
The interaction of Activase with other cardioactive or cerebroactive drugs has not been studied. Anticoagulants and antiplatelet drugs increase the risk of bleeding if administered prior to, during, or after Activase therapy.
In the post-marketing setting, there have been reports of angioedema in patients (primarily patients with AIS) receiving concomitant angiotensin-converting enzyme inhibitors. [see Warnings and Precautions (5.2)].
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Published studies and case reports on alteplase use in pregnant women are insufficient to inform a drug associated risk of adverse developmental outcomes. Alteplase is embryocidal in rabbits when intravenously administered during organogenesis at the clinical exposure for AMI, but no maternal or fetal toxicity was evident at lower exposure in pregnant rats or rabbits (see Data).
All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. The estimated background risk of major birth defects and miscarriage for the indicated populations is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Clinical Considerations
Maternal Adverse Reactions
The most common complication of thrombolytic therapy is bleeding. Pregnancy may increase this risk [see Warnings and Precautions (5.1)].
Data
Animal Data
Alteplase is embryocidal in rabbits when administered intravenously during organogenesis in doses (3 mg/kg) approximately equal to the human exposure (based on AUC) at the dose for AMI. No maternal or fetal toxicity was evident at doses (1 mg/kg) approximately 0.3 times the human exposure. In pregnant rats, no maternal or fetal toxicity was evident at doses (1 mg/kg) approximately 0.6 times the human dose for AMI (based on body weight) dosed during the period of organogenesis.
8.4 Pediatric Use
Safety and effectiveness of Activase in pediatric patients have not been established.
8.5 Geriatric Use
Acute Ischemic Stroke
In exploratory, multivariate analyses of Studies 1 and 2, age greater than 77 years was one of several interrelated baseline characteristics associated with an increased risk of intracranial hemorrhage. Efficacy results suggest a reduced but still favorable clinical outcome for Activase-treated elderly [see Clinical Studies (14.1)].
Acute Myocardial Infarction
In a large trial of accelerated-infusion Activase that enrolled 41,021 patients with AMI to one of four thrombolytic regimens [see Clinical Studies (14.2)], patients over 75 years of age, a predefined subgroup, comprised 12% of enrollment. In these patients, the incidence of stroke was 4.0% for the Activase accelerated infusion group, 2.8% for streptokinase IV [SK (IV)], and 3.2% for streptokinase SQ [SK (SQ)]. The incidence of combined 30-day mortality or nonfatal stroke was 20.6% for accelerated infusion of Activase, 21.5% for SK (IV), and 22.0% for SK (SQ).
11 DESCRIPTION
Alteplase is a tissue plasminogen activator produced by recombinant DNA technology. It is a sterile, purified glycoprotein of 527 amino acids. It is synthesized using the complementary DNA (cDNA) for natural human tissue-type plasminogen activator obtained from a human melanoma cell line. Biological potency is determined by an in vitro clot lysis assay and is expressed in International Units (IU).
Activase (alteplase) is a sterile, white to off-white, lyophilized powder for intravenous administration after reconstitution with Sterile Water for Injection, USP.
| 100 mg Vial | 50 mg Vial | |
|---|---|---|
| Alteplase | 100 mg (58 million IU) | 50 mg (29 million IU) |
| L-Arginine | 3.5 g | 1.7 g |
| Phosphoric Acid | 1 g | 0.5 g |
| Polysorbate 80 | 10 mg | 5 mg |
| Vacuum | No | Yes |
The reconstituted preparation results in a colorless to pale yellow transparent solution containing Activase 1 mg/mL at approximately pH 7.3. The osmolality of this solution is approximately 215 mOsm/kg.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Alteplase is a serine protease responsible for fibrin-enhanced conversion of plasminogen to plasmin. It produces limited conversion of plasminogen in the absence of fibrin.
When introduced into the systemic circulation at pharmacologic concentration, alteplase binds to fibrin in a thrombus and converts the entrapped plasminogen to plasmin. This initiates local fibrinolysis with limited systemic proteolysis.
12.2 Pharmacodynamics
Following administration of 100 mg Activase, there is a decrease (16%-36%) in circulating fibrinogen. In a controlled trial, 8 of 73 patients (11%) receiving Activase (1.25 mg/kg body weight over 3 hours) experienced a decrease in fibrinogen to below 100 mg/dL.
12.3 Pharmacokinetics
Alteplase in acute myocardial infarction (AMI) patients is rapidly cleared from the plasma with an initial half-life of less than 5 minutes. There is no difference in the dominant initial plasma half-life between the 3-hour and accelerated regimens for AMI. The plasma clearance of alteplase is 380-570 mL/min, primarily mediated by the liver. The initial volume of distribution approximates plasma volume.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term studies in animals have not been performed to evaluate the carcinogenic potential or the effect on fertility. Short-term studies, which evaluated tumorigenicity of Activase and effect on tumor metastases in rodents, were negative.
Studies to determine mutagenicity (Ames test) and chromosomal aberration assays in human lymphocytes were negative at all concentrations tested. Cytotoxicity, as reflected by a decrease in mitotic index, was evidenced only after prolonged exposure and only at the highest concentrations tested.
14 CLINICAL STUDIES
14.1 Acute Ischemic Stroke (AIS)
Two placebo-controlled, double-blind trials (Studies 1 and 2) were conducted in patients with AIS. Both studies enrolled patients with measurable neurological deficit who could complete screening and begin study treatment within 3 hours from symptom onset. A cranial computerized tomography (CT) scan was performed prior to treatment to rule out the presence of intracranial hemorrhage. Blood pressure was actively controlled (185/110 mm Hg or lower) for 24 hours.
Patients were randomized (1:1) to receive either 0.9 mg/kg Activase (maximum of 90 mg) or placebo. Activase was administered as a 10% initial IV bolus over 1 minute followed by continuous IV infusion of the remainder over 60 minutes. Study treatment was initiated prior to the availability of coagulation study results in patients without recent use of oral anticoagulants and/or heparin and was discontinued if the pretreatment prothrombin time (PT) was greater than 15 seconds or the activated partial thromboplastin time (aPTT) was elevated. Patients with prior aspirin use were included. Administration of anticoagulants and antiplatelet agents was prohibited for the first 24 hours following symptom onset.
Study 1 (n=291) evaluated neurological improvement at 24 hours after stroke onset. The primary endpoint, the proportion of patients with a 4 point or greater improvement in the National Institutes of Health Stroke Scale (NIHSS) score or complete recovery (NIHSS score of 0), was not significantly different between treatment groups. A prespecified secondary analysis suggested improved 3-month outcome associated with Activase treatment using the following stroke assessment scales: Barthel Index, Modified Rankin Scale, Glasgow Outcome Scale, and the NIHSS.
Study 2 (n=333) assessed clinical outcome at 3 months. A favorable outcome was defined as minimal or no disability using four stroke assessment scales: Barthel Index (score of 95 or greater), Modified Rankin Scale (score of 1 or less), Glasgow Outcome Scale (score of 1), and NIHSS (score of 1 or less). The results comparing Activase- and placebo-treated patients for the four outcome scales together (Generalized Estimating Equations) and individually are presented in Table 7. In this study, depending upon the scale, the favorable outcome of minimal or no disability occurred in at least 11 per 100 more patients treated with Activase than those receiving placebo. Study results demonstrated consistent functional and neurological improvement within all four stroke scales as indicated by median scores. These results were consistent with the 3-month outcome treatment effects observed in Study 1.
| Analysis | Frequency of Favorable Outcome* | ||||
|---|---|---|---|---|---|
| Placebo (n=165) | Activase (n=168) | Absolute Difference (95% CI) | Odds Ratio†
(95% Cl) | p-Value‡ | |
| Generalized Estimating Equations (Multivariate) | - | - | - | 1.71 (1.15, 2.56) | 0.02 |
| Barthel Index | 37.6% | 50.0% | 12.4% (3.0, 21.9) | 1.66 (1.07, 2.57) | 0.02 |
| Modified Rankin Scale | 26.1% | 38.7% | 12.6% (3.7, 21.6) | 1.79 (1.12, 2.85) | 0.02 |
| Glasgow Outcome Scale | 31.5% | 44.0% | 12.5% (3.3, 21.8) | 1.71 (1.09, 2.68) | 0.02 |
| NIHSS | 20.0% | 31.0% | 11.0% (2.6, 19.3) | 1.79 (1.06, 2.96) | 0.02 |
In a prespecified subgroup analysis of patients receiving aspirin prior to onset of stroke symptoms, the favorable outcome for Activase-treated patients was preserved.
14.2 Acute Myocardial Infarction (AMI)
Two Activase dose regimens have been studied in patients experiencing acute myocardial infarction [see Dosage and Administration (2.2)]. The comparative efficacy of these two regimens has not been evaluated.
Accelerated Infusion in AMI Patients
Accelerated infusion of Activase was studied in an international, multi-center trial that randomized 41,021 patients with AMI to four thrombolytic regimens (Study 3). Entry criteria included onset of chest pain within 6 hours of treatment and ST-segment elevation of ECG. The four treatment regimens included accelerated infusion of Activase (≤100 mg over 90 minutes) plus intravenous (IV) heparin (n = 10,396); Streptokinase (1.5 million units over 60 minutes) plus IV heparin (SK [IV], n =10,410); Streptokinase plus subcutaneous (SQ) heparin (SK [SQ] n= 9841). A fourth regimen combined Activase and Streptokinase (n =10,374). All patients received 160 mg chewable aspirin administered as soon as possible, followed by 160-325 mg daily. Bolus IV heparin 5000 U was initiated as soon as possible, followed by a 1000 U/hour continuous IV infusion for at least 48 hours; subsequent heparin therapy was at the physician's discretion. Heparin SQ 12,500 U was administered 4 hours after initiation of SK therapy, followed by 12,500 U twice daily for 7 days or until discharge, whichever came first. Many of the patients randomized to receive SQ heparin received some IV heparin, usually in response to recurrent chest pain and/or the need for a medical procedure. Some received IV heparin on arrival to the emergency room prior to enrollment and randomization.
Key results from Study 3 are shown in Table 8. The incidence of 30-day mortality for Activase accelerated infusion was 1.0% lower than for either Streptokinase plus heparin regimen. The incidence of combined 30-day mortality or nonfatal stroke for the Activase accelerated infusion was 1.0% lower than for SK (IV) and 0.8% lower than for SK (SQ).
| Event | Accelerated Activase | SK (IV) | p-Value* | SK (SQ) | p-Value* |
|---|---|---|---|---|---|
|
|||||
| 30-Day Mortality | 6.3% | 7.3% | 0.003 | 7.3% | 0.007 |
| 30-Day Mortality or Nonfatal Stroke | 7.2% | 8.2% | 0.006 | 8.0% | 0.036 |
| 24-Hour Mortality | 2.4% | 2.9% | 0.009 | 2.8% | 0.029 |
| Any Stroke | 1.6% | 1.4% | 0.32 | 1.2% | 0.03 |
| Intracerebral Hemorrhage | 0.7% | 0.6% | 0.22 | 0.5% | 0.02 |
Subgroup analysis of patients by age, infarct location, time from symptom onset to thrombolytic treatment, and treatment in the U.S. or elsewhere showed consistently lower 30-day mortality on Activase.
For patients who were over 75 years of age, a predefined subgroup consisting of 12% of patients enrolled, the incidence of stroke was 4.0% for the Activase accelerated infusion group, 2.8% for SK (IV), and 3.2% for SK (SQ); the incidence of combined 30-day mortality or nonfatal stroke was 20.6% for accelerated infusion of Activase, 21.5% for SK (IV), and 22.0% for SK (SQ).
3-Hour Infusion in AMI Patients
In a double-blind, randomized trial (n = 138) comparing 3-hour infusion of Activase to placebo (Study 4), patients infused with Activase within 4 hours of onset of symptoms experienced improved left ventricular function at Day 10 compared to the placebo group, when ejection fraction was measured by gated blood pool scan (53.2% vs. 46.4%, p =0.018). Relative to baseline (Day 1) values, the net changes in ejection fraction were + 3.6% and -4.7% for the treated and placebo groups, respectively (p=0.0001). The treated group had a reduced incidence of clinical heart failure (14%) compared to the placebo group (33%) (p = 0.009).
In a double-blind, randomized trial (n =5013) comparing 3-hour infusion of Activase to placebo (Study 5), patients infused with Activase within 5 hours of AMI symptom onset experienced improved 30-day survival compared to the placebo arm. At 1 month, the overall mortality rates were 7.2% for the Activase group and 9.8% for the placebo group (p = 0.001). At 6 months, the overall mortality rate for Activase-treated patients was 10.4% compared to the placebo arm (13.1%, p= 0.008).
14.3 Acute Massive Pulmonary Embolism (PE)
Study 6 was a comparative randomized trial (n =45) in which 59% of patients (n =22) treated with Activase (100 mg over 2 hours) experienced moderate or marked lysis of pulmonary emboli when assessed by pulmonary angiography 2 hours after treatment initiation. Activase-treated patients also experienced a significant reduction in pulmonary embolism-induced pulmonary hypertension within 2 hours of treatment (p=0.003). Pulmonary perfusion at 24 hours, as assessed by radionuclide scan, was significantly improved (p= 0.002).
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
Activase is supplied as a sterile, lyophilized powder in 50 mg single-dose vials containing vacuum and in 100 mg single-dose vials without vacuum.
Each 50 mg Activase vial (29 million IU) is packaged with diluent for reconstitution (50 mL Sterile Water for Injection, USP): NDC 50242-044-13.
Each 100 mg Activase vial (58 million IU) is packaged with diluent for reconstitution (100 mL Sterile Water for Injection, USP), and one transfer device: NDC 50242-085-27.
16.2 Stability and Storage
Store lyophilized Activase at controlled room temperature not to exceed 30°C (86°F), or under refrigeration at 2° to 8°C (36° to 46°F). Protect the lyophilized material during extended storage from excessive exposure to light. If stored between 2-30°C (36-86°F), Activase may be used within 8 hours following reconstitution. Discard any unused solution after administration is complete.
Do not use beyond the expiration date stamped on the vial.
17 PATIENT COUNSELING INFORMATION
Following Activase administration, patients are at increased risk of bleeding internally or externally. Advise patients to contact a health-care professional if they experience symptoms or signs consistent with bleeding (e.g., unusual bruising, pink or brown urine, red or black or tarry stools, coughing up blood, vomiting blood or blood that looks like coffee grounds), headache, or stroke symptoms.
Activase® (alteplase)
Manufactured by:
Genentech, Inc.
A Member of the Roche Group
1 DNA Way
South San Francisco, CA
94080-4990
U.S. License No. 1048
Activase® is a registered trademark of Genentech, Inc.
©2022 Genentech, Inc.
INSTRUCTIONS FOR USE
Activase®
(alteplase) for injection
for intravenous use
100 mg (58 million IU)

Read before preparing Activase®
See also enclosed, full prescribing information
Instructions for Use should accompany reconstituted Activase to patient bedside.
| Kit Contents | |
| Transfer device |
|
| Activase vial (no vacuum) |
|
| Activase vial stopper parts: | |
|
| |
| Sterile Water for Injection (water) vial Note:Do not use Bacteriostatic Water for Injection, USP. |
|

Prescribing Information
Instructions for Use
Also Required
(not included in kit)
1 Luer syringe for removing bolus dose, as needed
1 Luer syringe for removing excess volume, as needed
2 large bore needles
2 Alcohol swabs
IV infusion set
| Reconstitution (use aseptic technique) | ||
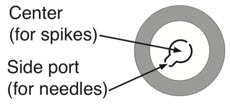
| Step 1: Cleaning | Step 2: Spiking Water vial | Step 3: Spiking Activase vial |
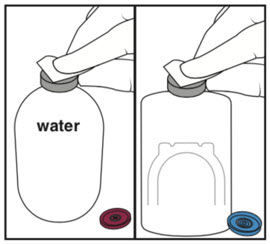 | 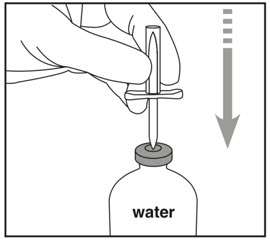 | 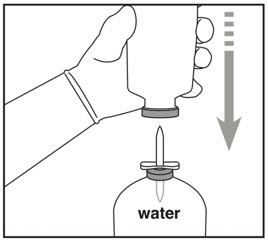 |
|
|
|
| Reconstitution (use aseptic technique) | Administration Warning | |
| Step 4: Inverting and transferring | Step 5: Inspecting | |
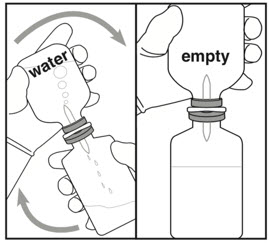 |  |  |
|
|
Review important information below before continuing to Step 6.
|
| Administration (use aseptic technique) | ||
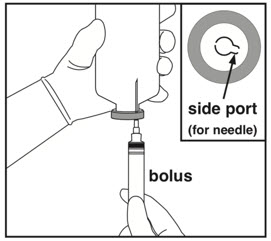
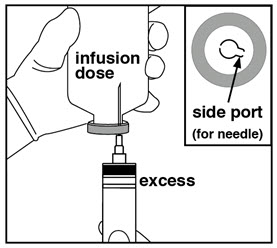
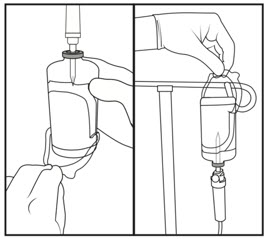
| Step 6: Preparing bolus | Step 7: Removing excess volume | Step 8: Spiking and hanging |
 |  |  |
|
|
|
|
|
|
|
|
|
|








Administration Notes
- Activase is for intravenous administration only.
- Do not add any other medication to infusion solutions containing Activase.
- Extravasation of Activase infusion can cause ecchymosis or inflammation. If extravasation occurs, terminate the infusion at that IV site and apply local therapy.
- See full prescribing information for alternative dilution instructions.
Storage & Stability
- Store lyophilized Activase at controlled room temperature not to exceed 30°C (86°F), or under refrigeration at 2° to 8° C (36° to 46° F). Protect the lyophilized material during extended storage from excessive exposure to light.
- Activase contains no antibacterial preservatives and must be used within 8 hours following reconstitution (when stored at 2-30°C).
Manufactured by:
Genentech, Inc.
A Member of the Roche Group
1 DNA Way
South San Francisco, CA 94080-4990
U.S. License No. 1048
Activase® (alteplase) is a registered trademark of Genentech, Inc.
©2022 Genentech, Inc. All rights reserved.
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Revised: September/2022
Representative sample of labeling (see the HOW SUPPLIED section for complete listing):

