Suicidality and Antidepressant Drugs
Antidepressants increased the risk compared to placebo of suicidal thinking and behavior (suicidality) in children, adolescents, and young adults in short-term studies of major depressive disorder (MDD) and other psychiatric disorders. Anyone considering the use of mirtazapine tablets or any other antidepressant in a child, adolescent, or young adult must balance this risk with the clinical need. Short-term studies did not show an increase in the risk of suicidality with antidepressants compared to placebo in adults beyond age 24; there was a reduction in risk with antidepressants compared to placebo in adults aged 65 and older. Depression and certain other psychiatric disorders are themselves associated with increases in the risk of suicide. Patients of all ages who are started on antidepressant therapy should be monitored appropriately and observed closely for clinical worsening, suicidality, or unusual changes in behavior. Families and caregivers should be advised of the need for close observation and communication with the prescriber. Mirtazapine tablets are not approved for use in pediatric patients. (See WARNINGS: Clinical Worsening and Suicide Risk, PRECAUTIONS: Information for Patients, and PRECAUTIONS: Pediatric Use)
DESCRIPTION
Mirtazapine tablets, USP are an orally administered drug. Mirtazapine has a tetracyclic chemical structure and belongs to the piperazino-azepine group of compounds. It is designated 1,2,3,4,10,14b-hexahydro-2-methylpyrazino [2,1-α] pyrido [2,3-c] [2] benzazepine and has the molecular formula of C17H19N3. Its molecular weight is 265.36. The structural formula is the following and it is the racemic mixture:
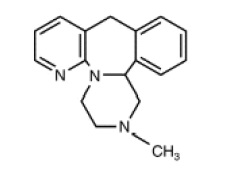
Mirtazapine is a white to creamy white crystalline powder, which is sparingly soluble in water.
Mirtazapine tablets, USP are supplied for oral administration as scored film-coated tablets containing 15 or 30 mg of mirtazapine, and unscored film-coated tablets containing 45 mg of mirtazapine. Each tablet also contains the following inactive ingredients: croscarmellose sodium, hydroxypropyl cellulose, hypromellose, lactose monohydrate, magnesium stearate, microcrystalline cellulose, polyethylene glycol and titanium dioxide. In addition, mirtazapine tablets, USP 15 mg and 30 mg contains iron oxide yellow and mirtazapine tablets, USP 30 mg contains iron oxide red.
CLINICAL PHARMACOLOGY
Pharmacodynamics
The mechanism of action of mirtazapine tablets, as with other drugs effective in the treatment of major depressive disorder, is unknown.
Evidence gathered in preclinical studies suggests that mirtazapine enhances central noradrenergic and serotonergic activity. These studies have shown that mirtazapine acts as an antagonist at central presynaptic α2-adrenergic inhibitory autoreceptors and heteroreceptors, an action that is postulated to result in an increase in central noradrenergic and serotonergic activity.
Mirtazapine is a potent antagonist of 5-HT2 and 5-HT3 receptors. Mirtazapine has no significant affinity for the 5-HT1A and 5-HT1B receptors.
Mirtazapine is a potent antagonist of histamine (H1) receptors, a property that may explain its prominent sedative effects.
Mirtazapine is a moderate peripheral α1-adrenergic antagonist, a property that may explain the occasional orthostatic hypotension reported in association with its use.
Mirtazapine is a moderate antagonist at muscarinic receptors, a property that may explain the relatively low incidence of anticholinergic side effects associated with its use.
Pharmacokinetics
Mirtazapine tablets are rapidly and completely absorbed following oral administration and have a half-life of about 20 to 40 hours. Peak plasma concentrations are reached within about 2 hours following an oral dose. The presence of food in the stomach has a minimal effect on both the rate and extent of absorption and does not require a dosage adjustment.
Mirtazapine is extensively metabolized after oral administration. Major pathways of biotransformation are demethylation and hydroxylation followed by glucuronide conjugation. In vitro data from human liver microsomes indicate that cytochrome 2D6 and 1A2 are involved in the formation of the 8-hydroxy metabolite of mirtazapine, whereas cytochrome 3A is considered to be responsible for the formation of the N-desmethyl and N-oxide metabolite. Mirtazapine has an absolute bioavailability of about 50%. It is eliminated predominantly via urine (75%) with 15% in feces. Several unconjugated metabolites possess pharmacological activity but are present in the plasma at very low levels. The (–) enantiomer has an elimination half-life that is approximately twice as long as the (+) enantiomer and therefore achieves plasma levels that are about three times as high as that of the (+) enantiomer.
Plasma levels are linearly related to dose over a dose range of 15 to 80 mg. The mean elimination half-life of mirtazapine after oral administration ranges from approximately 20 to 40 hours across age and gender subgroups, with females of all ages exhibiting significantly longer elimination half-lives than males (mean half-life of 37 hours for females vs. 26 hours for males). Steady state plasma levels of mirtazapine are attained within 5 days, with about 50% accumulation (accumulation ratio = 1.5).
Mirtazapine is approximately 85% bound to plasma proteins over a concentration range of 0.01 to 10 mcg/mL.
Special Populations
Geriatric
Following oral administration of mirtazapine tablets 20 mg/day for 7 days to subjects of varying ages (range, 25 to 74), oral clearance of mirtazapine was reduced in the elderly compared to the younger subjects. The differences were most striking in males, with a 40% lower clearance in elderly males compared to younger males, while the clearance in elderly females was only 10% lower compared to younger females. Caution is indicated in administering mirtazapine tablets to elderly patients (see PRECAUTIONS and DOSAGE AND ADMINISTRATION).
Pediatrics
Safety and effectiveness of mirtazapine in the pediatric population have not been established (see PRECAUTIONS).
Gender
The mean elimination half-life of mirtazapine after oral administration ranges from approximately 20 to 40 hours across age and gender subgroups, with females of all ages exhibiting significantly longer elimination half-lives than males (mean half-life of 37 hours for females vs. 26 hours for males) (see Pharmacokinetics).
Race
There have been no clinical studies to evaluate the effect of race on the pharmacokinetics of mirtazapine tablets.
Renal Insufficiency
The disposition of mirtazapine was studied in patients with varying degrees of renal function. Elimination of mirtazapine is correlated with creatinine clearance. Total body clearance of mirtazapine was reduced approximately 30% in patients with moderate (Clcr = 11 to 39 mL/min/1.73 m2) and approximately 50% in patients with severe (Clcr =<10 mL/min/1.73 m2) renal impairment when compared to normal subjects. Caution is indicated in administering mirtazapine tablets to patients with compromised renal function (see PRECAUTIONS and DOSAGE AND ADMINISTRATION).
Hepatic Insufficiency
Following a single 15-mg oral dose of mirtazapine tablets, the oral clearance of mirtazapine was decreased by approximately 30% in hepatically impaired patients compared to subjects with normal hepatic function. Caution is indicated in administering mirtazapine tablets to patients with compromised hepatic function (see PRECAUTIONS and DOSAGE AND ADMINISTRATION).
Clinical Trials Showing Effectiveness
The efficacy of mirtazapine tablets as a treatment for major depressive disorder was established in four placebo-controlled, 6-week trials in adult outpatients meeting DSM-III criteria for major depressive disorder. Patients were titrated with mirtazapine from a dose range of 5 mg up to 35 mg/day. Overall, these studies demonstrated mirtazapine to be superior to placebo on at least three of the following four measures: 21-Item Hamilton Depression Rating Scale (HDRS) total score; HDRS Depressed Mood Item; CGI Severity score; and Montgomery and Asberg Depression Rating Scale (MADRS). Superiority of mirtazapine over placebo was also found for certain factors of the HDRS, including anxiety/somatization factor and sleep disturbance factor. The mean mirtazapine dose for patients who completed these four studies ranged from 21 to 32 mg/day. A fifth study of similar design utilized a higher dose (up to 50 mg) per day and also showed effectiveness.
Examination of age and gender subsets of the population did not reveal any differential responsiveness on the basis of these subgroupings.
In a longer-term study, patients meeting (DSM-IV) criteria for major depressive disorder who had responded during an initial 8 to 12 weeks of acute treatment on mirtazapine tablets were randomized to continuation of mirtazapine tablets or placebo for up to 40 weeks of observation for relapse. Response during the open phase was defined as having achieved a HAM-D 17 total score of ≤8 and a CGI-Improvement score of 1 or 2 at two consecutive visits beginning with week 6 of the 8 to 12 weeks in the open-label phase of the study. Relapse during the double-blind phase was determined by the individual investigators. Patients receiving continued mirtazapine tablets treatment experienced significantly lower relapse rates over the subsequent 40 weeks compared to those receiving placebo. This pattern was demonstrated in both male and female patients.
INDICATIONS AND USAGE
Mirtazapine tablets are indicated for the treatment of major depressive disorder.
The efficacy of mirtazapine tablets in the treatment of major depressive disorder was established in 6-week controlled trials of outpatients whose diagnoses corresponded most closely to the Diagnostic and Statistical Manual of Mental Disorders – 3rd edition (DSM-III) category of major depressive disorder (see CLINICAL PHARMACOLOGY).
A major depressive episode (DSM-IV) implies a prominent and relatively persistent (nearly every day for at least 2 weeks) depressed or dysphoric mood that usually interferes with daily functioning, and includes at least five of the following nine symptoms: depressed mood, loss of interest in usual activities, significant change in weight and/or appetite, insomnia or hypersomnia, psychomotor agitation or retardation, increased fatigue, feelings of guilt or worthlessness, slowed thinking or impaired concentration, a suicide attempt, or suicidal ideation.
The effectiveness of mirtazapine tablets in hospitalized depressed patients has not been adequately studied.
The efficacy of mirtazapine tablets in maintaining a response in patients with major depressive disorder for up to 40 weeks following 8 to 12 weeks of initial open-label treatment was demonstrated in a placebo-controlled trial. Nevertheless, the physician who elects to use mirtazapine tablets for extended periods should periodically re-evaluate the long-term usefulness of the drug for the individual patient (see CLINICAL PHARMACOLOGY).
CONTRAINDICATIONS
Hypersensitivity
Mirtazapine tablets are contraindicated in patients with a known hypersensitivity to mirtazapine or to any of the excipients.
Monoamine Oxidase Inhibitors
The use of monoamine oxidase inhibitors (MAOIs) intended to treat psychiatric disorders with mirtazapine tablets, USP or within 14 days of stopping treatment with mirtazapine tablets is contraindicated because of an increased risk of serotonin syndrome. The use of mirtazapine tablets within 14 days of stopping an MAOI intended to treat psychiatric disorders is also contraindicated (see WARNINGS and DOSAGE AND ADMINISTRATION).
Starting mirtazapine tablets in a patient who is being treated with MAOIs such as linezolid or intravenous methylene blue is also contraindicated because of an increased risk of serotonin syndrome (see WARNINGS and DOSAGE AND ADMINISTRATION).
WARNINGS
Clinical Worsening and Suicide Risk
Patients with major depressive disorder (MDD), both adult and pediatric, may experience worsening of their depression and/or the emergence of suicidal ideation and behavior (suicidality) or unusual changes in behavior, whether or not they are taking antidepressant medications, and this risk may persist until significant remission occurs. Suicide is a known risk of depression and certain other psychiatric disorders, and these disorders themselves are the strongest predictors of suicide. There has been a long-standing concern, however, that antidepressants may have a role in inducing worsening of depression and the emergence of suicidality in certain patients during the early phases of treatment. Pooled analyses of short-term placebo-controlled trials of antidepressant drugs (SSRIs and others) showed that these drugs increase the risk of suicidal thinking and behavior (suicidality) in children, adolescents, and young adults (ages 18 to 24) with major depressive disorder (MDD) and other psychiatric disorders. Short-term studies did not show an increase in the risk of suicidality with antidepressants compared to placebo in adults beyond age 24; there was a reduction in risk with antidepressants compared to placebo in adults aged 65 and older.
The pooled analyses of placebo-controlled trials in children and adolescents with MDD, obsessive compulsive disorder (OCD), or other psychiatric disorders included a total of 24 short-term trials of 9 antidepressant drugs in over 4400 patients. The pooled analyses of placebo-controlled trials in adults with MDD or other psychiatric disorders included a total of 295 short-term trials (median duration of 2 months) of 11 antidepressant drugs in over 77,000 patients. There was considerable variation in risk of suicidality among drugs, but a tendency toward an increase in the younger patients for almost all drugs studied. There were differences in absolute risk of suicidality across different indications, with the highest incidence in MDD. The risk differences (drug vs. placebo), however, were relatively stable within age strata and across indications. These risk differences (drug-placebo difference in the number of cases of suicidality per 1,000 patients treated) are provided in Table 1.
| Table 1 | |
| Age Range | Drug-placebo Difference in Number of Cases of Suicidality per 1,000 Patients Treated |
| Increases Compared to Placebo | |
| <18 | 14 additional cases |
| 18 to 24 | 5 additional cases |
| Decreases Compared to Placebo | |
| 25 to 64 | 1 fewer case |
| ≥65 | 6 fewer cases |
No suicides occurred in any of the pediatric trials. There were suicides in the adult trials, but the number was not sufficient to reach any conclusion about drug effect on suicide. It is unknown whether the suicidality risk extends to longer-term use, i.e., beyond several months. However, there is substantial evidence from placebo-controlled maintenance trials in adults with depression that the use of antidepressants can delay the recurrence of depression.
All patients being treated with antidepressants for any indication should be monitored appropriately and observed closely for clinical worsening, suicidality, and unusual changes in behavior, especially during the initial few months of a course of drug therapy, or at times of dose changes, either increases or decreases.
The following symptoms, anxiety, agitation, panic attacks, insomnia, irritability, hostility, aggressiveness, impulsivity, akathisia (psychomotor restlessness), hypomania, and mania, have been reported in adult and pediatric patients being treated with antidepressants for major depressive disorder as well as for other indications, both psychiatric and nonpsychiatric. Although a causal link between the emergence of such symptoms and either the worsening of depression and/or the emergence of suicidal impulses has not been established, there is concern that such symptoms may represent precursors to emerging suicidality.
Consideration should be given to changing the therapeutic regimen, including possibly discontinuing the medication, in patients whose depression is persistently worse, or who are experiencing emergent suicidality or symptoms that might be precursors to worsening depression or suicidality, especially if these symptoms are severe, abrupt in onset, or were not part of the patient’s presenting symptoms.
Families and caregivers of patients being treated with antidepressants for major depressive disorder or other indications, both psychiatric and nonpsychiatric, should be alerted about the need to monitor patients for the emergence of agitation, irritability, unusual changes in behavior, and the other symptoms described above, as well as the emergence of suicidality, and to report such symptoms immediately to health care providers. Such monitoring should include daily observation by families and caregivers. Prescriptions for mirtazapine tablets should be written for the smallest quantity of tablets consistent with good patient management, in order to reduce the risk of overdose.
Screening Patients for Bipolar Disorder
A major depressive episode may be the initial presentation of bipolar disorder. It is generally believed (though not established in controlled trials) that treating such an episode with an antidepressant alone may increase the likelihood of precipitation of a mixed/manic episode in patients at risk for bipolar disorder. Whether any of the symptoms described above represent such a conversion is unknown. However, prior to initiating treatment with an antidepressant, patients with depressive symptoms should be adequately screened to determine if they are at risk for bipolar disorder; such screening should include a detailed psychiatric history, including a family history of suicide, bipolar disorder, and depression. It should be noted that mirtazapine tablets are not approved for use in treating bipolar depression.
Agranulocytosis
In premarketing clinical trials, two (one with Sjögren’s Syndrome) out of 2796 patients treated with mirtazapine tablets developed agranulocytosis [absolute neutrophil count (ANC) <500/mm3 with associated signs and symptoms, e.g., fever, infection, etc.] and a third patient developed severe neutropenia (ANC <500/mm3 without any associated symptoms). For these three patients, onset of severe neutropenia was detected on days 61, 9, and 14 of treatment, respectively. All three patients recovered after mirtazapine tablets were stopped. These three cases yield a crude incidence of severe neutropenia (with or without associated infection) of approximately 1.1 per thousand patients exposed, with a very wide 95% confidence interval, i.e., 2.2 cases per 10,000 to 3.1 cases per 1,000. If a patient develops a sore throat, fever, stomatitis, or other signs of infection, along with a low WBC count, treatment with mirtazapine tablets should be discontinued and the patient should be closely monitored.
Serotonin Syndrome
The development of a potentially life-threatening serotonin syndrome has been reported with SNRIs and SSRIs, including mirtazapine tablets, alone but particularly with concomitant use of other serotonergic drugs (including triptans, tricyclic antidepressants, fentanyl, lithium, tramadol, tryptophan, buspirone, and St. John's wort), and with drugs that impair metabolism of serotonin (in particular, MAOIs, both those intended to treat psychiatric disorders and also others, such as linezolid and intravenous methylene blue).
Serotonin syndrome symptoms may include mental status changes (e.g., agitation, hallucinations, delirium, and coma), autonomic instability (e.g., tachycardia, labile blood pressure, dizziness, diaphoresis, flushing, hyperthermia), neuromuscular symptoms (e.g., tremor, rigidity, myoclonus, hyperreflexia, incoordination), seizures, and/or gastrointestinal symptoms (e.g., nausea, vomiting, diarrhea). Patients should be monitored for the emergence of serotonin syndrome.
The concomitant use of mirtazapine tablets with MAOIs intended to treat psychiatric disorders is contraindicated. Mirtazapine tablets should also not be started in a patient who is being treated with MAOIs such as linezolid or intravenous methylene blue. All reports with methylene blue that provided information on the route of administration involved intravenous administration in the dose range of 1 mg/kg to 8 mg/kg. No reports involved the administration of methylene blue by other routes (such as oral tablets or local tissue injection) or at lower doses. There may be circumstances when it is necessary to initiate treatment with an MAOI such as linezolid or intravenous methylene blue in a patient taking mirtazapine tablets. Mirtazapine tablets should be discontinued before initiating treatment with the MAOI (see CONTRAINDICATIONS and DOSAGE AND ADMINISTRATION).
If concomitant use of mirtazapine with other serotonergic drugs, including triptans, tricyclic antidepressants, fentanyl, lithium, tramadol, buspirone, tryptophan, and St. John's wort, is clinically warranted, be aware of a potential increased risk for serotonin syndrome, particularly during treatment initiation and dose increases.
Treatment with mirtazapine tablets and any concomitant serotonergic agents should be discontinued immediately if the above events occur and supportive symptomatic treatment should be initiated.
Angle-closure Glaucoma
The pupillary dilation that occurs following use of many antidepressant drugs including mirtazapine may trigger an angle-closure attack in a patient with anatomically narrow angles who does not have a patent iridectomy.
QT Prolongation and Torsades de Pointes
The effect of mirtazapine on QTc interval was assessed in a clinical randomized trial with placebo and positive (moxifloxacin) controls involving 54 healthy volunteers using exposure response analysis. This trial showed a positive relationship between mirtazapine concentrations and prolongation of the QTc interval. However, the degree of QT prolongation observed with both 45 mg (therapeutic) and 75 mg (supratherapeutic) doses of mirtazapine was not at a level generally considered to be clinically meaningful. During the postmarketing use of mirtazapine, cases of QT prolongation, Torsades de Pointes, ventricular tachycardia, and sudden death, have been reported (see ADVERSE REACTIONS). The majority of reports occurred in association with overdose or in patients with other risk factors for QT prolongation, including concomitant use of QTc-prolonging medicines (see PRECAUTIONS, Drug Interactions and OVERDOSE sections). Caution should be exercised when mirtazapine is prescribed in patients with known cardiovascular disease or family history of QT prolongation, and in concomitant use with other medicinal products thought to prolong the QTc interval.
PRECAUTIONS
General
Discontinuation Symptoms
There have been reports of adverse reactions upon the discontinuation of mirtazapine tablets, USP (particularly when abrupt), including but not limited to the following: dizziness, abnormal dreams, sensory disturbances (including paresthesia and electric shock sensations), agitation, anxiety, fatigue, confusion, headache, tremor, nausea, vomiting, and sweating, or other symptoms which may be of clinical significance. The majority of the reported cases are mild and self-limiting. Even though these have been reported as adverse reactions, it should be realized that these symptoms may be related to underlying disease.
Patients currently taking mirtazapine tablets should NOT discontinue treatment abruptly, due to risk of discontinuation symptoms. At the time that a medical decision is made to discontinue treatment with mirtazapine tablets, a gradual reduction in the dose, rather than an abrupt cessation, is recommended.
Akathisia/Psychomotor Restlessness
The use of antidepressants has been associated with the development of akathisia, characterized by a subjectively unpleasant or distressing restlessness and need to move, often accompanied by an inability to sit or stand still. This is most likely to occur within the first few weeks of treatment. In patients who develop these symptoms, increasing the dose may be detrimental.
Hyponatremia
Hyponatremia has been reported very rarely with the use of mirtazapine. Caution should be exercised in patients at risk, such as elderly patients or patients concomitantly treated with medications known to cause hyponatremia.
Somnolence
In US controlled studies, somnolence was reported in 54% of patients treated with mirtazapine tablets, compared to 18% for placebo and 60% for amitriptyline. In these studies, somnolence resulted in discontinuation for 10.4% of mirtazapine tablets-treated patients, compared to 2.2% for placebo. It is unclear whether or not tolerance develops to the somnolent effects of mirtazapine tablets. Because of the potentially significant effects of mirtazapine tablets on impairment of performance, patients should be cautioned about engaging in activities requiring alertness until they have been able to assess the drug’s effect on their own psychomotor performance (see PRECAUTIONS: Information for Patients).
Dizziness
In US controlled studies, dizziness was reported in 7% of patients treated with mirtazapine tablets, compared to 3% for placebo and 14% for amitriptyline. It is unclear whether or not tolerance develops to the dizziness observed in association with the use of mirtazapine tablets.
Increased Appetite/Weight Gain
In US controlled studies, appetite increase was reported in 17% of patients treated with mirtazapine tablets, compared to 2% for placebo and 6% for amitriptyline. In these same trials, weight gain of ≥7% of body weight was reported in 7.5% of patients treated with mirtazapine, compared to 0% for placebo and 5.9% for amitriptyline. In a pool of premarketing US studies, including many patients for long-term, open-label treatment, 8% of patients receiving mirtazapine tablets discontinued for weight gain. In an 8-week-long pediatric clinical trial of doses between 15 to 45 mg/day, 49% of mirtazapine treated patients had a weight gain of at least 7%, compared to 5.7% of placebo-treated patients (see PRECAUTIONS: Pediatric Use).
Cholesterol/Triglycerides
In US controlled studies, nonfasting cholesterol increases to ≥20% above the upper limits of normal were observed in 15% of patients treated with mirtazapine tablets, compared to 7% for placebo and 8% for amitriptyline. In these same studies, nonfasting triglyceride increases to ≥500 mg/dL were observed in 6% of patients treated with mirtazapine, compared to 3% for placebo and 3% for amitriptyline.
Transaminase Elevations
Clinically significant ALT (SGPT) elevations (≥3 times the upper limit of the normal range) were observed in 2.0% (8/424) of patients exposed to mirtazapine tablets in a pool of short-term US controlled trials, compared to 0.3% (1/328) of placebo patients and 2.0% (3/181) of amitriptyline patients. Most of these patients with ALT increases did not develop signs or symptoms associated with compromised liver function. While some patients were discontinued for the ALT increases, in other cases, the enzyme levels returned to normal despite continued mirtazapine treatment. Mirtazapine tablets should be used with caution in patients with impaired hepatic function (see CLINICAL PHARMACOLOGY and DOSAGE AND ADMINISTRATION).
Activation of Mania/Hypomania
Mania/hypomania occurred in approximately 0.2% (3/1299 patients) of mirtazapine tablet-treated patients in US studies. Although the incidence of mania/hypomania was very low during treatment with mirtazapine, it should be used carefully in patients with a history of mania/hypomania.
Seizure
In premarketing clinical trials, only one seizure was reported among the 2796 US and non-US patients treated with mirtazapine tablets. However, no controlled studies have been carried out in patients with a history of seizures. Therefore, care should be exercised when mirtazapine is used in these patients.
Use in Patients with Concomitant Illness
Clinical experience with mirtazapine tablets in patients with concomitant systemic illness is limited. Accordingly, care is advisable in prescribing mirtazapine for patients with diseases or conditions that affect metabolism or hemodynamic responses.
Mirtazapine tablets have not been systematically evaluated or used to any appreciable extent in patients with a recent history of myocardial infarction or other significant heart disease. Mirtazapine tablets were associated with significant orthostatic hypotension in early clinical pharmacology trials with normal volunteers. Orthostatic hypotension was infrequently observed in clinical trials with depressed patients. Mirtazapine tablets should be used with caution in patients with known cardiovascular or cerebrovascular disease that could be exacerbated by hypotension (history of myocardial infarction, angina, or ischemic stroke) and conditions that would predispose patients to hypotension (dehydration, hypovolemia, and treatment with antihypertensive medication).
Mirtazapine clearance is decreased in patients with moderate [glomerular filtration rate (GFR) =11 to 39 mL/min/1.73 m2] and severe [GFR <10 mL/min/1.73 m2] renal impairment, and also in patients with hepatic impairment. Caution is indicated in administering mirtazapine tablets to such patients (see CLINICAL PHARMACOLOGY and DOSAGE AND ADMINISTRATION).
Information for Patients
Prescribers or other health professionals should inform patients, their families, and their caregivers about the benefits and risks associated with treatment with mirtazapine tablets and should counsel them in its appropriate use. A patient Medication Guide about “Antidepressant Medicines, Depression and other Serious Mental Illnesses, and Suicidal Thoughts or Actions” is available for mirtazapine tablets. The prescriber or health professional should instruct patients, their families, and their caregivers to read the Medication Guide and should assist them in understanding its contents. Patients should be given the opportunity to discuss the contents of the Medication Guide and to obtain answers to any questions they may have. The complete text of the Medication Guide is reprinted at the end of this document.
Patients should be advised of the following issues and asked to alert their prescriber if these occur while taking mirtazapine tablets.
Clinical Worsening and Suicide Risk
Patients, their families, and their caregivers should be encouraged to be alert to the emergence of anxiety, agitation, panic attacks, insomnia, irritability, hostility, aggressiveness, impulsivity, akathisia (psychomotor restlessness), hypomania, mania, other unusual changes in behavior, worsening of depression, and suicidal ideation, especially early during antidepressant treatment and when the dose is adjusted up or down. Families and caregivers of patients should be advised to look for the emergence of such symptoms on a day-to-day basis, since changes may be abrupt. Such symptoms should be reported to the patient's prescriber or health professional, especially if they are severe, abrupt in onset, or were not part of the patient's presenting symptoms. Symptoms such as these may be associated with an increased risk for suicidal thinking and behavior and indicate a need for very close monitoring and possibly changes in the medication.
Agranulocytosis
Patients who are to receive mirtazapine tablets should be warned about the risk of developing agranulocytosis. Patients should be advised to contact their physician if they experience any indication of infection such as fever, chills, sore throat, mucous membrane ulceration, or other possible signs of infection. Particular attention should be paid to any flu-like complaints or other symptoms that might suggest infection.
Interference with Cognitive and Motor Performance
Mirtazapine tablets may impair judgment, thinking, and particularly, motor skills, because of its prominent sedative effect. The drowsiness associated with mirtazapine use may impair a patient’s ability to drive, use machines, or perform tasks that require alertness. Thus, patients should be cautioned about engaging in hazardous activities until they are reasonably certain that mirtazapine tablet therapy does not adversely affect their ability to engage in such activities.
Completing Course of Therapy
While patients may notice improvement with mirtazapine tablet therapy in 1 to 4 weeks, they should be advised to continue therapy as directed.
Concomitant Medication
Patients should be advised to inform their physician if they are taking, or intend to take, any prescription or over-the-counter drugs, since there is a potential for mirtazapine tablets to interact with other drugs.
Patients should be made aware of a potential increased risk for serotonin syndrome if concomitant use of mirtazapine tablets with other serotonergic drugs, including triptans, tricyclic antidepressants, fentanyl, lithium, tramadol, buspirone, tryptophan, and St. John's wort, is clinically warranted, particularly during treatment initiation and dose increases.
Alcohol
The impairment of cognitive and motor skills produced by mirtazapine tablets has been shown to be additive with those produced by alcohol. Accordingly, patients should be advised to avoid alcohol while taking mirtazapine.
Drug Interactions
As with other drugs, the potential for interaction by a variety of mechanisms (e.g., pharmacodynamic, pharmacokinetic inhibition or enhancement, etc.) is a possibility (see CLINICAL PHARMACOLOGY).
Drugs Affecting Hepatic Metabolism
The metabolism and pharmacokinetics of mirtazapine tablets may be affected by the induction or inhibition of drug-metabolizing enzymes.
Drugs that are Metabolized by and/or Inhibit Cytochrome P450 Enzymes
CYP Enzyme Inducers (these studies used both drugs at steady state)
Phenytoin: In healthy male patients (n=18), phenytoin (200 mg daily) increased mirtazapine (30 mg daily) clearance about 2-fold, resulting in a decrease in average plasma mirtazapine concentrations of 45%. Mirtazapine did not significantly affect the pharmacokinetics of phenytoin.
Carbamazepine: In healthy male patients (n=24), carbamazepine (400 mg b.i.d.) increased mirtazapine (15 mg b.i.d.) clearance about 2-fold, resulting in a decrease in average plasma mirtazapine concentrations of 60%.
When phenytoin, carbamazepine, or another inducer of hepatic metabolism (such as rifampicin) is added to mirtazapine therapy, the mirtazapine dose may have to be increased. If treatment with such a medicinal product is discontinued, it may be necessary to reduce the mirtazapine dose.
CYP Enzyme Inhibitors
Cimetidine: In healthy male patients (n=12), when cimetidine, a weak inhibitor of CYP1A2, CYP2D6, and CYP3A4, given at 800 mg b.i.d. at steady state was coadministered with mirtazapine (30 mg daily) at steady state, the Area Under the Curve (AUC) of mirtazapine increased more than 50%. Mirtazapine did not cause relevant changes in the pharmacokinetics of cimetidine. The mirtazapine dose may have to be decreased when concomitant treatment with cimetidine is started, or increased when cimetidine treatment is discontinued.
Ketoconazole: In healthy, male, Caucasian patients (n=24), coadministration of the potent CYP3A4 inhibitor ketoconazole (200 mg b.i.d. for 6.5 days) increased the peak plasma levels and the AUC of a single 30-mg dose of mirtazapine by approximately 40% and 50%, respectively.
Caution should be exercised when coadministering mirtazapine with potent CYP3A4 inhibitors, HIV protease inhibitors, azole antifungals, erythromycin, or nefazodone.
Paroxetine: In an in vivo interaction study in healthy, CYP2D6 extensive metabolizer patients (n=24), mirtazapine (30 mg/day), at steady state, did not cause relevant changes in the pharmacokinetics of steady state paroxetine (40 mg/day), a CYP2D6 inhibitor.
Other Drug-drug Interactions
Amitriptyline
In healthy, CYP2D6 extensive metabolizer patients (n=32), amitriptyline (75 mg daily), at steady state, did not cause relevant changes in the pharmacokinetics of steady state mirtazapine (30 mg daily); mirtazapine also did not cause relevant changes to the pharmacokinetics of amitriptyline.
Warfarin
In healthy male subjects (n=16), mirtazapine (30 mg daily), at steady state, caused a small (0.2) but statistically significant increase in the International Normalized Ratio (INR) in subjects treated with warfarin. As at a higher dose of mirtazapine, a more pronounced effect can not be excluded, it is advisable to monitor the INR in case of concomitant treatment of warfarin with mirtazapine.
Lithium
No relevant clinical effects or significant changes in pharmacokinetics have been observed in healthy male subjects on concurrent treatment with subtherapeutic levels of lithium (600 mg/day for 10 days) at steady state and a single 30-mg dose of mirtazapine. The effects of higher doses of lithium on the pharmacokinetics of mirtazapine are unknown.
Risperidone
In an in vivo, nonrandomized, interaction study, subjects (n=6) in need of treatment with an antipsychotic and antidepressant drug, showed that mirtazapine (30 mg daily) at steady state did not influence the pharmacokinetics of risperidone (up to 3 mg b.i.d.).
Alcohol
Concomitant administration of alcohol (equivalent to 60 g) had a minimal effect on plasma levels of mirtazapine (15 mg) in 6 healthy male subjects. However, the impairment of cognitive and motor skills produced by mirtazapine tablets were shown to be additive with those produced by alcohol. Accordingly, patients should be advised to avoid alcohol while taking mirtazapine tablets.
Diazepam
Concomitant administration of diazepam (15 mg) had a minimal effect on plasma levels of mirtazapine (15 mg) in 12 healthy subjects. However, the impairment of motor skills produced by mirtazapine tablets has been shown to be additive with those caused by diazepam. Accordingly, patients should be advised to avoid diazepam and other similar drugs while taking mirtazapine tablets.
QTc-Prolonging Drugs
The risk of QT prolongation and/or ventricular arrhythmias (e.g., Torsades de Pointes) may be increased with concomitant use of medicines which prolong the QTc interval (e.g., some antipsychotics and antibiotics) and in case of mirtazapine overdose (see ADVERSE REACTIONS and OVERDOSE sections).
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Carcinogenicity studies were conducted with mirtazapine given in the diet at doses of 2, 20, and 200 mg/kg/day to mice and 2, 20, and 60 mg/kg/day to rats. The highest doses used are approximately 20 and 12 times the maximum recommended human dose (MRHD) of 45 mg/day on an mg/m2 basis in mice and rats, respectively. There was an increased incidence of hepatocellular adenoma and carcinoma in male mice at the high dose. In rats, there was an increase in hepatocellular adenoma in females at the mid and high doses and in hepatocellular tumors and thyroid follicular adenoma/cystadenoma and carcinoma in males at the high dose. The data suggest that the above effects could possibly be mediated by non-genotoxic mechanisms, the relevance of which to humans is not known.
The doses used in the mouse study may not have been high enough to fully characterize the carcinogenic potential of mirtazapine tablets.
Mutagenesis
Mirtazapine was not mutagenic or clastogenic and did not induce general DNA damage as determined in several genotoxicity tests: Ames test, in vitro gene mutation assay in Chinese hamster V 79 cells, in vitro sister chromatid exchange assay in cultured rabbit lymphocytes, in vivo bone marrow micronucleus test in rats, and unscheduled DNA synthesis assay in HeLa cells.
Impairment of Fertility
In a fertility study in rats, mirtazapine was given at doses up to 100 mg/kg [20 times the maximum recommended human dose (MRHD) on an mg/m2 basis]. Mating and conception were not affected by the drug, but estrous cycling was disrupted at doses that were 3 or more times the MRHD, and pre-implantation losses occurred at 20 times the MRHD.
Pregnancy
Teratogenic Effects - Pregnancy Category C
Reproduction studies in pregnant rats and rabbits at doses up to 100 mg/kg and 40 mg/kg, respectively [20 and 17 times the maximum recommended human dose (MRHD) on an mg/m2 basis, respectively], have revealed no evidence of teratogenic effects. However, in rats, there was an increase in postimplantation losses in dams treated with mirtazapine. There was an increase in pup deaths during the first 3 days of lactation and a decrease in pup birth weights. The cause of these deaths is not known. The effects occurred at doses that were 20 times the MRHD, but not at 3 times the MRHD, on an mg/m2 basis. There are no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed.
Nursing Mothers
Because some mirtazapine may be excreted into breast milk, caution should be exercised when mirtazapine tablets are administered to nursing women.
Pediatric Use
Safety and effectiveness in the pediatric population have not been established (see BOXED WARNING and WARNINGS: Clinical Worsening and Suicide Risk). Two placebo-controlled trials in 258 pediatric patients with MDD have been conducted with mirtazapine tablets, and the data were not sufficient to support a claim for use in pediatric patients. Anyone considering the use of mirtazapine tablets in a child or adolescent must balance the potential risks with the clinical need.
In an 8-week-long pediatric clinical trial of doses between 15 to 45 mg/day, 49% of mirtazapine-treated patients had a weight gain of at least 7%, compared to 5.7% of placebo-treated patients. The mean increase in weight was 4 kg (2 kg SD) for mirtazapine-treated patients versus 1 kg (2 kg SD) for placebo-treated patients (see PRECAUTIONS, Increased Appetite/Weight Gain).
Geriatric Use
Approximately 190 elderly individuals (≥65 years of age) participated in clinical studies with mirtazapine tablets. This drug is known to be substantially excreted by the kidney (75%), and the risk of decreased clearance of this drug is greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection. Sedating drugs may cause confusion and over-sedation in the elderly. No unusual adverse age-related phenomena were identified in this group. Pharmacokinetic studies revealed a decreased clearance in the elderly. Caution is indicated in administering mirtazapine tablets to elderly patients (see CLINICAL PHARMACOLOGY and DOSAGE AND ADMINISTRATION).
ADVERSE REACTIONS
Associated with Discontinuation of Treatment
Approximately 16% of the 453 patients who received mirtazapine tablets in US 6-week controlled clinical trials discontinued treatment due to an adverse experience, compared to 7% of the 361 placebo-treated patients in those studies. The most common events (≥1%) associated with discontinuation and considered to be drug related (i.e., those events associated with dropout at a rate at least twice that of placebo) are included in Table 2.
| Table 2 : Common Adverse Events Associated with Discontinuation of Treatment in 6-week US Mirtazapine Tablets Trials | ||
| Adverse Event | Percentage of Patients Discontinuing with Adverse Event | |
| Mirtazapine Tablets (n=453) | Placebo (n=361) |
|
| Somnolence | 10.4% | 2.2% |
| Nausea | 1.5% | 0% |
Commonly Observed Adverse Events in US Controlled Clinical Trials
The most commonly observed adverse events associated with the use of mirtazapine tablets (incidence of 5% or greater) and not observed at an equivalent incidence among placebo-treated patients (mirtazapine tablets incidence at least twice that for placebo) are listed in Table 3.
| Table 3: Common Treatment-emergent Adverse Events Associated with the Use of Mirtazapine Tablets in 6-week US Trials | ||
| Adverse Event | Percentage of Patients Reporting Adverse Event | |
| Mirtazapine Tablets (n=453) | Placebo (n=361) |
|
| Somnolence | 54% | 18% |
| Increased Appetite | 17% | 2% |
| Weight Gain | 12% | 2% |
| Dizziness | 7% | 3% |
Adverse Events Occurring at an Incidence of 1% or More Among Mirtazapine Tablet-treated Patients
Table 4 enumerates adverse events that occurred at an incidence of 1% or more, and were more frequent than in the placebo group, among mirtazapine tablet-treated patients who participated in short-term US placebo-controlled trials in which patients were dosed in a range of 5 to 60 mg/day. This table shows the percentage of patients in each group who had at least one episode of an event at some time during their treatment. Reported adverse events were classified using a standard COSTART-based dictionary terminology.
The prescriber should be aware that these figures cannot be used to predict the incidence of side effects in the course of usual medical practice where patient characteristics and other factors differ from those which prevailed in the clinical trials. Similarly, the cited frequencies cannot be compared with figures obtained from other investigations involving different treatments, uses, and investigators. The cited figures, however, do provide the prescribing physician with some basis for estimating the relative contribution of drug and nondrug factors to the side-effect incidence rate in the population studied.
| Table 4: Incidence of Adverse Clinical Experiences* (≥ 1%) in Short-term US Controlled Studies | |||
| Body System Adverse Clinical Experience | Mirtazapine Tablets (n=453) | Placebo (n=361) |
|
| Body as a Whole | |||
| Asthenia | 8% | 5% | |
| Flu Syndrome | 5% | 3% | |
| Back Pain | 2% | 1% | |
| Digestive System | |||
| Dry Mouth | 25% | 15% | |
| Increased Appetite | 17% | 2% | |
| Constipation | 13% | 7% | |
| Metabolic and Nutritional Disorders | |||
| Weight Gain | 12% | 2% | |
| Peripheral Edema | 2% | 1% | |
| Edema | 1% | 0% | |
| Musculoskeletal System | |||
| Myalgia | 2% | 1% | |
| Nervous System | |||
| Somnolence | 54% | 18% | |
| Dizziness | 7% | 3% | |
| Abnormal Dreams | 4% | 1% | |
| Thinking Abnormal | 3% | 1% | |
| Tremor | 2% | 1% | |
| Confusion | 2% | 0% | |
| Respiratory System | |||
| Dyspnea | 1% | 0% | |
| Urogenital System | |||
| Urinary Frequency | 2% | 1% | |
*Events reported by at least 1% of patients treated with mirtazapine tablets are included, except the following events, which had an incidence on placebo greater than or equal to mirtazapine tablets: headache, infection, pain, chest pain, palpitation, tachycardia, postural hypotension, nausea, dyspepsia, diarrhea, flatulence, insomnia, nervousness, libido decreased, hypertonia, pharyngitis, rhinitis, sweating, amblyopia, tinnitus, taste perversion.
ECG Changes
The electrocardiograms for 338 patients who received mirtazapine tablets and 261 patients who received placebo in 6-week, placebo-controlled trials were analyzed. Prolongation in QTc ≥500 msec was not observed among mirtazapine-treated patients; mean change in QTc was +1.6 msec for mirtazapine and -3.1 msec for placebo. Mirtazapine was associated with a mean increase in heart rate of 3.4 bpm, compared to 0.8 bpm for placebo. The clinical significance of these changes is unknown.
The effect of mirtazapine on QTc interval was assessed in a clinical randomized trial with placebo and positive (moxifloxacin) controls involving 54 healthy volunteers using exposure response analysis. This trial showed a positive relationship between mirtazapine concentrations and prolongation of the QTc interval. However, the degree of QT prolongation observed with both 45 mg (therapeutic) and 75 mg (supratherapeutic) doses of mirtazapine was not at a level generally considered to be clinically meaningful.
Other Adverse Events Observed During the Premarketing Evaluation of Mirtazapine Tablets
During its premarketing assessment, multiple doses of mirtazapine tablets were administered to 2796 patients in clinical studies. The conditions and duration of exposure to mirtazapine varied greatly, and included (in overlapping categories) open and double-blind studies, uncontrolled and controlled studies, inpatient and outpatient studies, fixed-dose and titration studies. Untoward events associated with this exposure were recorded by clinical investigators using terminology of their own choosing. Consequently, it is not possible to provide a meaningful estimate of the proportion of individuals experiencing adverse events without first grouping similar types of untoward events into a smaller number of standardized event categories.
In the tabulations that follow, reported adverse events were classified using a standard COSTART-based dictionary terminology. The frequencies presented, therefore, represent the proportion of the 2796 patients exposed to multiple doses of mirtazapine tablets who experienced an event of the type cited on at least one occasion while receiving mirtazapine tablets. All reported events are included except those already listed in Table 4, those adverse experiences subsumed under COSTART terms that are either overly general or excessively specific so as to be uninformative, and those events for which a drug cause was very remote.
It is important to emphasize that, although the events reported occurred during treatment with mirtazapine tablets, they were not necessarily caused by it.
Events are further categorized by body system and listed in order of decreasing frequency according to the following definitions: frequent adverse events are those occurring on one or more occasions in at least 1/100 patients; infrequent adverse events are those occurring in 1/100 to 1/1,000 patients; rare events are those occurring in fewer than 1/1,000 patients. Only those events not already listed in Table 4 appear in this listing. Events of major clinical importance are also described in the WARNINGS and PRECAUTIONS sections.
Body as a Whole
Frequent: malaise, abdominal pain, abdominal syndrome acute; infrequent: chills, fever, face edema, ulcer, photosensitivity reaction, neck rigidity, neck pain, abdomen enlarged; rare: cellulitis, chest pain substernal.
Cardiovascular System
Frequent: hypertension, vasodilatation; infrequent: angina pectoris, myocardial infarction, bradycardia, ventricular extrasystoles, syncope, migraine, hypotension; rare: atrial arrhythmia, bigeminy, vascular headache, pulmonary embolus, cerebral ischemia, cardiomegaly, phlebitis, left heart failure.
Digestive System
Frequent: vomiting, anorexia; infrequent: eructation, glossitis, cholecystitis, nausea and vomiting, gum hemorrhage, stomatitis, colitis, liver function tests abnormal; rare: tongue discoloration, ulcerative stomatitis, salivary gland enlargement, increased salivation, intestinal obstruction, pancreatitis, aphthous stomatitis, cirrhosis of liver, gastritis, gastroenteritis, oral moniliasis, tongue edema.
Hemic and Lymphatic System
Rare: lymphadenopathy, leukopenia, petechia, anemia, thrombocytopenia, lymphocytosis, pancytopenia.
Metabolic and Nutritional Disorders
Frequent: thirst; infrequent: dehydration, weight loss; rare: gout, SGOT increased, healing abnormal, acid phosphatase increased, SGPT increased, diabetes mellitus, hyponatremia.
Musculoskeletal System
Frequent: myasthenia, arthralgia; infrequent: arthritis, tenosynovitis; rare: pathologic fracture, osteoporosis fracture, bone pain, myositis, tendon rupture, arthrosis, bursitis.
Nervous System
Frequent: hypesthesia, apathy, depression, hypokinesia, vertigo, twitching, agitation, anxiety, amnesia, hyperkinesia, paresthesia; infrequent: ataxia, delirium, delusions, depersonalization, dyskinesia, extrapyramidal syndrome, libido increased, coordination abnormal, dysarthria, hallucinations, manic reaction, neurosis, dystonia, hostility, reflexes increased, emotional lability, euphoria, paranoid reaction; rare: aphasia, nystagmus, akathisia (psychomotor restlessness), stupor, dementia, diplopia, drug dependence, paralysis, grand mal convulsion, hypotonia, myoclonus, psychotic depression, withdrawal syndrome, serotonin syndrome.
Respiratory System
Frequent: cough increased, sinusitis; infrequent: epistaxis, bronchitis, asthma, pneumonia; rare: asphyxia, laryngitis, pneumothorax, hiccup.
Skin and Appendages
Frequent: pruritus, rash; infrequent: acne, exfoliative dermatitis, dry skin, herpes simplex, alopecia; rare: urticaria, herpes zoster, skin hypertrophy, seborrhea, skin ulcer.
Special Senses
Infrequent: eye pain, abnormality of accommodation, conjunctivitis, deafness, keratoconjunctivitis, lacrimation disorder, angle-closure glaucoma, hyperacusis, ear pain; rare: blepharitis, partial transitory deafness, otitis media, taste loss, parosmia.
Urogenital System
Frequent: urinary tract infection; infrequent: kidney calculus, cystitis, dysuria, urinary incontinence, urinary retention, vaginitis, hematuria, breast pain, amenorrhea, dysmenorrhea, leukorrhea, impotence; rare: polyuria, urethritis, metrorrhagia, menorrhagia, abnormal ejaculation, breast engorgement, breast enlargement, urinary urgency.
Other Adverse Events Observed During Postmarketing Evaluation of Mirtazapine Tablets
Adverse events reported since market introduction, which were temporally (but not necessarily causally) related to mirtazapine therapy, include cases of the ventricular arrhythmia Torsades de Pointes. In the majority of these cases, however, concomitant drugs were implicated.
Cases of severe skin reactions, including Stevens-Johnson syndrome, bullous dermatitis, erythema multiforme and toxic epidermal necrolysis have also been reported.
Increased creatine kinase blood levels and rhabdomyolysis have also been reported.
DRUG ABUSE AND DEPENDENCE
Physical and Psychologic Dependence
Mirtazapine tablets have not been systematically studied in animals or humans for its potential for abuse, tolerance, or physical dependence. While the clinical trials did not reveal any tendency for any drug-seeking behavior, these observations were not systematic and it is not possible to predict on the basis of this limited experience the extent to which a CNS-active drug will be misused, diverted and/or abused once marketed. Consequently, patients should be evaluated carefully for history of drug abuse, and such patients should be observed closely for signs of mirtazapine tablets misuse or abuse (e.g., development of tolerance, incrementations of dose, drug-seeking behavior).
OVERDOSAGE
Human Experience
There is very limited experience with mirtazapine tablets overdose. In premarketing clinical studies, there were eight reports of mirtazapine tablets overdose alone or in combination with other pharmacological agents. The only drug overdose death reported while taking mirtazapine tablets was in combination with amitriptyline and chlorprothixene in a non-US clinical study. Based on plasma levels, the mirtazapine tablets dose taken was 30 to 45 mg, while plasma levels of amitriptyline and chlorprothixene were found to be at toxic levels. All other premarketing overdose cases resulted in full recovery. Signs and symptoms reported in association with overdose included disorientation, drowsiness, impaired memory, and tachycardia. There were no reports of ECG abnormalities, coma, or convulsions following overdose with mirtazapine tablets alone.
However, based on postmarketing reports, there is a possibility of more serious outcomes (including fatalities) at dosages much higher than the therapeutic dose, especially with mixed overdoses. In these cases, QT prolongation and Torsades de Pointes have also been reported (see PRECAUTIONS, Drug Interactions and ADVERSE REACTIONS sections).
Overdose Management
Treatment should consist of those general measures employed in the management of overdose with any drug effective in the treatment of major depressive disorder. Ensure an adequate airway, oxygenation, and ventilation. Monitor ECG parameters (including cardiac rhythm) and vital signs. General supportive and symptomatic measures are also recommended. Induction of emesis is not recommended. Gastric lavage with a large-bore orogastric tube with appropriate airway protection, if needed, may be indicated if performed soon after ingestion, or in symptomatic patients. Activated charcoal should be administered. There is no experience with the use of forced diuresis, dialysis, hemoperfusion, or exchange transfusion in the treatment of mirtazapine overdosage. No specific antidotes for mirtazapine are known.
In managing overdosage, consider the possibility of multiple-drug involvement. The physician should consider contacting a poison control center for additional information on the treatment of any overdose. Telephone numbers for certified poison control centers are listed in the Physicians’ Desk Reference (PDR).
DOSAGE AND ADMINISTRATION
Initial Treatment
The recommended starting dose for mirtazapine tablets is 15 mg/day, administered in a single dose, preferably in the evening prior to sleep. In the controlled clinical trials establishing the efficacy of mirtazapine in the treatment of major depressive disorder, the effective dose range was generally 15 to 45 mg/day. While the relationship between dose and satisfactory response in the treatment of major depressive disorder for mirtazapine tablets has not been adequately explored, patients not responding to the initial 15-mg dose may benefit from dose increases up to a maximum of 45 mg/day. Mirtazapine tablets has an elimination half-life of approximately 20 to 40 hours; therefore, dose changes should not be made at intervals of less than one to two weeks in order to allow sufficient time for evaluation of the therapeutic response to a given dose.
Elderly and Patients with Renal or Hepatic Impairment
The clearance of mirtazapine is reduced in elderly patients and in patients with moderate to severe renal or hepatic impairment. Consequently, the prescriber should be aware that plasma mirtazapine levels may be increased in these patient groups, compared to levels observed in younger adults without renal or hepatic impairment (see PRECAUTIONS and CLINICAL PHARMACOLOGY).
Maintenance/Extended Treatment
It is generally agreed that acute episodes of depression require several months or longer of sustained pharmacological therapy beyond response to the acute episode. Systematic evaluation of mirtazapine tablets has demonstrated that its efficacy in major depressive disorder is maintained for periods of up to 40 weeks following 8 to 12 weeks of initial treatment at a dose of 15 to 45 mg/day (see CLINICAL PHARMACOLOGY). Based on these limited data, it is unknown whether or not the dose of mirtazapine tablets needed for maintenance treatment is identical to the dose needed to achieve an initial response. Patients should be periodically reassessed to determine the need for maintenance treatment and the appropriate dose for such treatment.
Switching a Patient To or From a Monoamine Oxidase Inhibitor (MAOI) Intended to Treat Psychiatric Disorders
At least 14 days should elapse between discontinuation of an MAOI intended to treat psychiatric disorders and initiation of therapy with mirtazapine tablets. Conversely, at least 14 days should be allowed after stopping mirtazapine tablets before starting an MAOI intended to treat psychiatric disorders (see CONTRAINDICATIONS).
Use of Mirtazapine Tablets With Other MAOIs, Such as Linezolid or Methylene Blue
Do not start mirtazapine tablets in a patient who is being treated with linezolid or intravenous methylene blue because there is an increased risk of serotonin syndrome. In a patient who requires more urgent treatment of a psychiatric condition, other interventions, including hospitalization, should be considered (see CONTRAINDICATIONS).
In some cases, a patient already receiving therapy with mirtazapine may require urgent treatment with linezolid or intravenous methylene blue. If acceptable alternatives to linezolid or intravenous methylene blue treatment are not available and the potential benefits of linezolid or intravenous methylene blue treatment are judged to outweigh the risks of serotonin syndrome in a particular patient, Mirtazapine tablets should be stopped promptly, and linezolid or intravenous methylene blue can be administered. The patient should be monitored for symptoms of serotonin syndrome for 2 weeks or until 24 hours after the last dose of linezolid or intravenous methylene blue, whichever comes first. Therapy with mirtazapine tablets may be resumed 24 hours after the last dose of linezolid or intravenous methylene blue (see WARNINGS).
The risk of administering methylene blue by non-intravenous routes (such as oral tablets or by local injection) or in intravenous doses much lower than 1 mg/kg with mirtazapine tablets is unclear. The clinician should, nevertheless, be aware of the possibility of emergent symptoms of serotonin syndrome with such use (see WARNINGS).
Discontinuation of Mirtazapine Tablets, USP Treatment
Symptoms associated with the discontinuation or dose reduction of mirtazapine tablets have been reported. Patients should be monitored for these and other symptoms when discontinuing treatment or during dosage reduction. A gradual reduction in the dose over several weeks, rather than abrupt cessation, is recommended whenever possible. If intolerable symptoms occur following a decrease in the dose or upon discontinuation of treatment, dose titration should be managed on the basis of the patient’s clinical response (see PRECAUTIONS and ADVERSE REACTIONS).
Information for Patients
Patients should be advised that taking mirtazapine tablets can cause mild pupillary dilation, which in susceptible individuals, can lead to an episode of angle-closure glaucoma. Pre-existing glaucoma is almost always open-angle glaucoma because angle-closure glaucoma, when diagnosed, can be treated definitively with iridectomy. Open-angle glaucoma is not a risk factor for angle-closure glaucoma. Patients may wish to be examined to determine whether they are susceptible to angle-closure, and have a prophylactic procedure (e.g., iridectomy), if they are susceptible.
HOW SUPPLIED
Product: 50090-4222
NDC: 50090-4222-0 1 TABLET, FILM COATED in a BLISTER PACK / 33 in a BOX, UNIT-DOSE
MEDICATION GUIDE
Mirtazapine Tablets, USP
(mir taz’ a peen)
What is the most important information I should know about mirtazapine tablets, USP?
Mirtazapine tablets and other antidepressant medicines may cause serious side effects, including:
1. Suicidal thoughts or actions:
- Mirtazapine tablets and other antidepressant medicines may increase suicidal thoughts or actions in some children, teenagers, or young adults within the first few months of treatment or when the dose is changed.
- Depression or other serious mental illnesses are the most important causes of suicidal thoughts or actions.
- Watch for these changes and call your healthcare provider right away if you notice:
- New or sudden changes in mood, behavior, actions, thoughts, or feelings, especially if severe.
- Pay particular attention to such changes when mirtazapine tablets are started or when the dose is changed.
Keep all follow-up visits with your healthcare provider and call between visits if you are worried about symptoms.
Call your healthcare provider right away if you have any of the following symptoms, or call 911 if an emergency, especially if they are new, worse, or worry you:
- attempts to commit suicide
- acting on dangerous impulses
- acting aggressive or violent
- thoughts about suicide or dying
- new or worse depression
- new or worse anxiety or panic attacks
- feeling agitated, restless, angry or irritable
- trouble sleeping
- an increase in activity or talking more than what is normal for you
- other unusual changes in behavior or mood
Call your healthcare provider right away if you have any of the following symptoms, or call 911 if an emergency. Mirtazapine tablets may be associated with these serious side effects:
2. Manic episodes:
- greatly increased energy
- severe trouble sleeping
- racing thoughts
- reckless behavior
- unusually grand ideas
- excessive happiness or irritability
- talking more or faster than usual
3. Decreased White Blood Cells called neutrophils, which are needed to fight infections. Tell your doctor if you have any indication of infection such as fever, chills, sore throat, or mouth or nose sores, especially symptoms which are flu-like.
4. Serotonin Syndrome. This condition can be life-threatening and may include:
- agitation, hallucinations, coma or other changes in mental status
- coordination problems or muscle twitching (overactive reflexes)
- racing heartbeat, high or low blood pressure
- sweating or fever
- nausea, vomiting, or diarrhea
- muscle rigidity
5. Visual problems
- eye pain
- changes in vision
- swelling or redness in or around the eye
Only some people are at risk for these problems. You may want to undergo an eye examination to see if you are at risk and receive preventative treatment if you are.
6. Seizures
7. Low salt (sodium) levels in the blood. Elderly people may be at greater risk for this. Symptoms may include:
- headache
- weakness or feeling unsteady
- confusion, problems concentrating or thinking or memory problems
8. Sleepiness. It is best to take mirtazapine tablets close to bedtime.
9. Severe skin reactions: Call your doctor right away if you have any or all of the following symptoms:
- severe rash with skin swelling (including on the palms of the hands and soles of the feet)
- painful reddening of the skin, blisters, or ulcers on the body or in the mouth
10. Severe allergic reactions: trouble breathing, swelling of the face, tongue, eyes or mouth
- rash, itchy welts (hives) or blisters, alone or with fever or joint pain
11. Increases in appetite or weight. Children and adolescents should have height and weight monitored during treatment.
12. Increased cholesterol and triglyceride levels in your blood
Do not stop mirtazapine tablets without first talking to your healthcare provider. Stopping mirtazapine tablets too quickly may cause potentially serious symptoms including:
- dizziness
- abnormal dreams
- agitation
- anxiety
- fatigue
- confusion
- headache
- shaking
- tingling sensation
- nausea, vomiting
- sweating
What are Mirtazapine Tablets?
Mirtazapine tablets are a prescription medicine used to treat depression. It is important to talk with your healthcare provider about the risks of treating depression and also the risks of not treating it. You should discuss all treatment choices with your healthcare provider.
Talk to your healthcare provider if you do not think that your condition is getting better with mirtazapine tablets treatment.
Who should not take mirtazapine tablets?
Do not take mirtazapine tablets:
- if you are allergic to mirtazapine or any of the ingredients in mirtazapine tablets. See the end of this Medication Guide for a complete list of ingredients in mirtazapine tablets.
- if you take a monoamine oxidase inhibitor (MAOI). Ask your healthcare provider or pharmacist if you are not sure if you take an MAOI, including the antibiotic linezolid.
- Do not take an MAOI within 2 weeks of stopping mirtazapine tablets unless directed to do so by your healthcare provider.
- Do not start mirtazapine tablets if you stopped taking an MAOI in the last 2 weeks unless directed to do so by your healthcare provider.
People who take mirtazapine tablets close in time to an MAOI may have serious or even life-threatening side effects. Get medical help right away if you have any of these symptoms:
- high fever
- uncontrolled muscle spasms
- stiff muscles
- rapid changes in heart rate or blood pressure
- confusion
- loss of consciousness (pass out)
What should I tell my healthcare provider before taking mirtazapine tablets?
Before you take mirtazapine tablets, tell your healthcare provider about all of your medical conditions, including if you:
- are taking certain drugs such as:
- Triptans used to treat migraine headache
- Medicines used to treat mood, anxiety, psychotic or thought disorders, including tricyclics, lithium, SSRIs, SNRIs, or antipsychotics
- Tramadol used to treat pain
- Over-the-counter supplements such as tryptophan or St. John’s wort
- Phenytoin, carbamazepine, or rifampicin (these drugs can decrease your blood level of mirtazapine tablets)
- Cimetidine or ketoconazole (these drugs can increase your blood level of mirtazapine tablets)
- Medicines that may affect your hearts rhythm (such as certain antibiotics and some antipsychotics)
- have or had:
- liver problems
- kidney problems
- heart problems or certain conditions that may change your heart rhythm
- seizures or convulsions
- bipolar disorder or mania
- a tendency to get dizzy or faint
- are pregnant or plan to become pregnant. It is not known if mirtazapine tablets will harm your unborn baby. Talk to your healthcare provider about the benefits and risks of treating depression during pregnancy
- are breastfeeding or plan to breastfeed. Some mirtazapine may pass into your breast milk. Talk to your healthcare provider about the best way to feed your baby while taking mirtazapine tablets
Tell your healthcare provider about all the medicines that you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Mirtazapine tablets and some medicines may interact with each other, may not work as well, or may cause serious side effects.
Your healthcare provider or pharmacist can tell you if it is safe to take mirtazapine tablets with your other medicines. Do not start or stop any medicine while taking mirtazapine tablets without talking to your healthcare provider first. If you take mirtazapine tablets, you should not take any other medicines that contain mirtazapine including mirtazapine tablets SolTab.
How should I take mirtazapine tablets?
- Take mirtazapine tablets exactly as prescribed. Your healthcare provider may need to change the dose of mirtazapine tablets until it is the right dose for you.
- Take mirtazapine tablets at the same time each day, preferably in the evening at bedtime.
- Swallow mirtazapine tablets as directed.
- It is common for antidepressant medicines such as mirtazapine tablets to take up to a few weeks before you start to feel better. Do not stop taking mirtazapine tablets if you do not feel results right away.
- Do not stop taking or change the dose of mirtazapine tablets without first talking to your doctor, even if you feel better.
- Mirtazapine tablets may be taken with or without food.
- If you miss a dose of mirtazapine tablets, take the missed dose as soon as you remember. If it is almost time for the next dose, skip the missed dose and take your next dose at the regular time. Do not take two doses of mirtazapine tablets at the same time.
- If you take too much mirtazapine tablets, call your healthcare provider or poison control center right away, or get emergency treatment. The signs of an overdose of mirtazapine tablets (without other medicines or alcohol) include:
- confusion,
- drowsiness
- memory problems
- increased heart rate.
The symptoms of a possible overdose may include changes to your heart rhythm (fast, irregular heartbeat) or fainting, which could be symptoms of a life-threatening condition known as Torsades de Pointes.
What should I avoid while taking mirtazapine tablets?
- Mirtazapine tablets can cause sleepiness or may affect your ability to make decisions, think clearly, or react quickly. You should not drive, operate heavy machinery, or do other dangerous activities until you know how mirtazapine tablets affect you.
- Avoid drinking alcohol or taking diazepam (a medicine used for anxiety, insomnia and seizures, for example) or similar medicines while taking mirtazapine tablets. If you are uncertain about whether certain medication can be taken with mirtazapine tablets, please discuss with your doctor.
What are the possible side effects of mirtazapine tablets?
Mirtazapine tablets may cause serious side effects:
- See "What is the most important information I should know about mirtazapine tablets?"
The most common side effects of mirtazapine tablets include:
- sleepiness
- increased appetite
- weight gain
- abnormal dreams
- constipation
- dizziness
- dry mouth
These are not all the possible side effects of mirtazepine tablets.
Call your doctor for medical advice about side effects. You may report side effects to the FDA at 1-800-FDA-1088.
How should I store mirtazapine tablets, USP?
- Store mirtazapine tablets at room temperature between 68°F to 77°F (20°C to 25°C).
- Keep mirtazapine tablets away from light.
- Keep mirtazapine tablets bottle closed tightly.
Keep mirtazapine tablets, USP and all medicines out of the reach of children.
General information about the safe and effective use of mirtazapine tablets, USP.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use mirtazapine tablets for a condition for which it was not prescribed. Do not give mirtazapine tablets to other people, even if they have the same symptoms that you have. It may harm them.
You can ask your pharmacist or healthcare provider for information about mirtazapine tablets that is written for healthcare professionals.
What are the ingredients in mirtazapine tablets, USP?
Active ingredient: mirtazapine.
Inactive ingredients:
- 15mg tablets: Croscarmellose sodium, hydroxypropyl cellulose, hypromellose, lactose monohydrate, magnesium stearate, microcrystalline cellulose, polyethylene glycol, titanium dioxide and iron oxide yellow.
- 30mg tablets: Croscarmellose sodium, hydroxypropyl cellulose, hypromellose, lactose monohydrate, magnesium stearate, microcrystalline cellulose, polyethylene glycol, titanium dioxide, iron oxide yellow and iron oxide red.
- 45mg tablets: Croscarmellose sodium, hydroxypropyl cellulose, hypromellose, lactose monohydrate, magnesium stearate, microcrystalline cellulose, polyethylene glycol and titanium dioxide.
For more information about mirtazapine tablets call 1-800-706-5575.
This Medication Guide has been approved by the U.S. Food and Drug Administration.
APOTEX INC.
MIRTAZAPINE TABLETS, USP
15 mg, 30 mg and 45 mg
Manufactured by Manufactured for
Apotex Inc. Apotex Corp.
Toronto, Ontario Weston, Florida
Canada M9L 1T9 33326
Revised: August 2016
