LUCEMYRA- lofexidine hydrochloride tablet, film coated
US WorldMeds, LLC
----------
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use LUCEMYRA safely and effectively. See full prescribing information for LUCEMYRA.
LUCEMYRA™ (lofexidine) tablets, for oral use Initial U.S. Approval: 2018 INDICATIONS AND USAGELUCEMYRA is a central alpha-2 adrenergic agonist indicated for mitigation of opioid withdrawal symptoms to facilitate abrupt opioid discontinuation in adults. ( 1) DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHSTablets: 0.18 mg. ( 3) CONTRAINDICATIONSNone. ( 4) WARNINGS AND PRECAUTIONS
ADVERSE REACTIONSMost common adverse reactions (incidence ≥ 10% and notably more frequent than placebo) are orthostatic hypotension, bradycardia, hypotension, dizziness, somnolence, sedation, and dry mouth. ( 6.1) To report SUSPECTED ADVERSE REACTIONS, contact US WorldMeds at 1-833-LUCEMYRA or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch DRUG INTERACTIONS
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling. Revised: 5/2018 |
FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
LUCEMYRA is indicated for mitigation of opioid withdrawal symptoms to facilitate abrupt opioid discontinuation in adults.
2 DOSAGE AND ADMINISTRATION
2.1 Dosing Information
The usual LUCEMYRA starting dosage is three 0.18 mg tablets taken orally 4 times daily during the period of peak withdrawal symptoms (generally the first 5 to 7 days following last use of opioid) with dosing guided by symptoms and side effects. There should be 5 to 6 hours between each dose. The total daily dosage of LUCEMYRA should not exceed 2.88 mg (16 tablets) and no single dose should exceed 0.72 mg (4 tablets).
LUCEMYRA treatment may be continued for up to 14 days with dosing guided by symptoms.
Discontinue LUCEMYRA with a gradual dose reduction over a 2- to 4-day period to mitigate LUCEMYRA withdrawal symptoms (e.g., reducing by 1 tablet per dose every 1 to 2 days) [see Warnings & Precautions (5.5)] . The LUCEMYRA dose should be reduced, held, or discontinued for individuals who demonstrate a greater sensitivity to LUCEMYRA side effects [see Adverse Reactions (6.1), Warnings and Precautions (5.1)] . Lower doses may be appropriate as opioid withdrawal symptoms wane.
LUCEMYRA can be administered in the presence or absence of food.
2.2 Dosage Recommendations for Patients with Hepatic Impairment
Recommended dosage adjustments based on the degree of hepatic impairment are shown in Table 1. [see Use in Specific Populations (8.6), Clinical Pharmacology (12.3)] .
| Mild Impairment | Moderate Impairment | Severe Impairment | |
|---|---|---|---|
| Child-Pugh score | 5-6 | 7-9 | > 9 |
| Recommended dose | 3 tablets
4 times daily (2.16 mg per day) | 2 tablets
4 times daily (1.44 mg per day) | 1 tablet
4 times daily (0.72 mg per day) |
2.3 Dosage Recommendations for Patients with Renal Impairment
Recommended dosage adjustments based on the degree of renal impairment are shown in Table 2. LUCEMYRA may be administered without regard to the timing of dialysis [see Use in Specific Populations (8.7), Clinical Pharmacology (12.3)] .
| Moderate Impairment | Severe Impairment, End-Stage Renal Disease, or on Dialysis | |
|---|---|---|
| Estimated GFR, mL/min/1.73 m 2 | 30-89.9 | < 30 |
| Recommended dose | 2 tablets
4 times daily (1.44 mg per day) | 1 tablet
4 times daily (0.72 mg per day) |
3 DOSAGE FORMS AND STRENGTHS
LUCEMYRA is available as round, peach-colored, film-coated tablets, imprinted with "LFX" on one side and "18" on the other side. Each tablet contains 0.18 mg lofexidine (equivalent to 0.2 mg of lofexidine hydrochloride).
5 WARNINGS AND PRECAUTIONS
5.1 Risk of Hypotension, Bradycardia, and Syncope
LUCEMYRA can cause a decrease in blood pressure, a decrease in pulse, and syncope [see Adverse Reactions (6.1), Clinical Pharmacology (12.2)] . Monitor vital signs before dosing. Monitor symptoms related to bradycardia and orthostasis.
Patients being given LUCEMYRA in an outpatient setting should be capable of and instructed on self-monitoring for hypotension, orthostasis, bradycardia, and associated symptoms. If clinically significant or symptomatic hypotension and/or bradycardia occur, the next dose of LUCEMYRA should be reduced in amount, delayed, or skipped.
Inform patients that LUCEMYRA may cause hypotension and that patients moving from a supine to an upright position may be at increased risk for hypotension and orthostatic effects. Instruct patients to stay hydrated, on how to recognize symptoms of low blood pressure, and how to reduce the risk of serious consequences should hypotension occur (e.g., sit or lie down, carefully rise from a sitting or lying position). Instruct outpatients to withhold LUCEMYRA doses when experiencing symptoms of hypotension or bradycardia and to contact their healthcare provider for guidance on how to adjust dosing.
Avoid using LUCEMYRA in patients with severe coronary insufficiency, recent myocardial infarction, cerebrovascular disease, chronic renal failure, and in patients with marked bradycardia.
Avoid using LUCEMYRA in combination with medications that decrease pulse or blood pressure to avoid the risk of excessive bradycardia and hypotension.
5.2 Risk of QT Prolongation
LUCEMYRA prolongs the QT interval.
Avoid using LUCEMYRA in patients with congenital long QT syndrome.
Monitor ECG in patients with congestive heart failure, bradyarrhythmias, hepatic impairment, renal impairment, or patients taking other medicinal products that lead to QT prolongation (e.g., methadone). In patients with electrolyte abnormalities (e.g., hypokalemia or hypomagnesemia), correct these abnormalities first, and monitor ECG upon initiation of LUCEMYRA [see Dosing and Administration (2.1), Adverse Reactions (6.1), Special Populations (8.6)(8.7), Clinical Pharmacology (12.2)] .
5.3 Increased Risk of Central Nervous System Depression with Concomitant use of CNS Depressant Drugs
LUCEMYRA potentiates the CNS depressive effects of benzodiazepines and can also be expected to potentiate the CNS depressive effects of alcohol, barbiturates, and other sedating drugs. Advise patients to inform their healthcare provider of other medications they are taking, including alcohol.
Advise patients using LUCEMYRA in an outpatient setting that, until they learn how they respond to LUCEMYRA, they should be careful or avoid doing activities such as driving or operating heavy machinery.
5.4 Increased Risk of Opioid Overdose after Opioid Discontinuation
LUCEMYRA is not a treatment for opioid use disorder. Patients who complete opioid discontinuation are likely to have a reduced tolerance to opioids and are at increased risk of fatal overdose should they resume opioid use. Use LUCEMYRA in patients with opioid use disorder only in conjunction with a comprehensive management program for the treatment of opioid use disorder and inform patients and caregivers of this increased risk of overdose.
5.5 Risk of Discontinuation Symptoms
Stopping LUCEMYRA abruptly can cause a marked rise in blood pressure. Symptoms including diarrhea, insomnia, anxiety, chills, hyperhidrosis, and extremity pain have also been observed with LUCEMYRA discontinuation. Instruct patients not to discontinue therapy without consulting their healthcare provider. When discontinuing therapy with LUCEMYRA tablets, gradually reduce the dose [see Dosing and Administration (2.1)] .
Symptoms related to discontinuation can be managed by administration of the previous LUCEMYRA dose and subsequent taper.
6 ADVERSE REACTIONS
The following serious adverse reactions are described elsewhere in labeling:
- Hypotension, Bradycardia, and Syncope [see Warnings and Precautions (5.1)]
- QT Prolongation [see Warnings and Precautions (5.2)]
- Central Nervous System Depression [see Warnings and Precautions (5.3)]
- Opioid Overdose [see Warnings and Precautions (5.4)]
- Discontinuation Symptoms [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to adverse reaction rates observed for another drug and may not reflect the rates observed in practice.
The safety of LUCEMYRA was supported by three randomized, double-blind, placebo-controlled clinical trials, an open-label study, and clinical pharmacology studies with concomitant administration of either methadone, buprenorphine, or naltrexone.
The three randomized, double-blind, placebo-controlled clinical trials enrolled 935 subjects dependent on short-acting opioids undergoing abrupt opioid withdrawal. Patients were monitored before each dose in an inpatient setting.
Table 3 presents the incidence, rounded to the nearest percent, of adverse events that occurred in at least 10% of subjects treated with LUCEMYRA and for which the incidence in patients treated with LUCEMYRA was greater than the incidence in subjects treated with placebo in a study that tested two doses of LUCEMYRA, 2.16 mg per day and 2.88 mg per day, and placebo. The overall safety profile in the combined dataset was similar.
Orthostatic hypotension, bradycardia, hypotension, dizziness, somnolence, sedation, and dry mouth were notably more common in subjects treated with LUCEMYRA than subjects treated with placebo.
| Adverse Reaction | LUCEMYRA 2.16 mg
* (%)
N=229 | LUCEMYRA 2.88 mg
* (%)
N=222 | Placebo (%)
N=151 |
|---|---|---|---|
|
|||
| Insomnia | 51 | 55 | 48 |
| Orthostatic Hypotension | 29 | 42 | 5 |
| Bradycardia | 24 | 32 | 5 |
| Hypotension | 30 | 30 | 1 |
| Dizziness | 19 | 23 | 3 |
| Somnolence | 11 | 13 | 5 |
| Sedation | 13 | 12 | 5 |
| Dry Mouth | 10 | 11 | 0 |
Other notable adverse reactions associated with the use of LUCEMYRA but reported in <10% of patients in the LUCEMYRA group included:
- Syncope: 0.9%, 1.4% and 0% for LUCEMYRA 2.16 mg/day and 2.88 mg/day and placebo, respectively
- Tinnitus: 0.9%, 3.2% and 0% for LUCEMYRA 2.16 mg/day and 2.88 mg/day and placebo, respectively
Blood pressure changes and adverse reactions after LUCEMYRA cessation
Elevations in blood pressure above normal values (≥ 140 mmHg systolic) and above a subject's pre-treatment baseline are associated with discontinuing LUCEMYRA, and peaked on the second day after discontinuation, as shown in Table 4. Blood pressure values were evaluated for 3 days following the last dose of a 5-day course of LUCEMYRA 2.88 mg/day.
| Abrupt LUCEMYRA Discontinuation
2.88 mg (N = 134) | Placebo
(N = 129) |
|||
|---|---|---|---|---|
| N at risk | n (%) | N at risk | n (%) | |
| Systolic Blood Pressure on Day 2 after Discontinuation | ||||
| ≥ 140 mmHg and ≥ 20 mmHg increase from baseline | 58 | 23 (39.7) | 37 | 6 (16.2) |
| ≥ 170 mmHg and ≥ 20 mmHg increase from baseline | 58 | 5 (8.6) | 37 | 0 |
Blood pressure elevations of a similar magnitude and incidence were observed in a small number of patients (N=10) that had a one-day, 50% dose reduction prior to discontinuation.
After stopping treatment, subjects that were taking LUCEMYRA also had a higher incidence of diarrhea, insomnia, anxiety, chills, hyperhidrosis, and extremity pain compared to subjects who were taking placebo.
Sex-specific adverse event findings
Four out of 101 females (4%) had serious cardiovascular adverse events compared to 3 out of 289 (1%) of males assigned to receive LUCEMYRA 2.88 mg per day.
Discontinuations and dose holds due to bradycardia and orthostatic hypotension, which are the most common adverse reactions associated with LUCEMYRA, occurred with a greater incidence in females assigned to receive the highest studied dose of LUCEMYRA, 2.88 mg per day as shown in Table 5.
| LUCEMYRA 2.16 mg | LUCEMYRA 2.88 mg | |
|---|---|---|
| Male | 22/162 (14%) | 29/158 (18%) |
| Female | 9/67 (13%) | 20/64 (31%) |
6.2 Postmarketing Experience
Lofexidine is marketed in other countries for relief of opioid withdrawal symptoms. The following events have been identified during postmarketing use of lofexidine. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Since lofexidine's initial market introduction in 1992, the most frequently reported postmarketing adverse event with lofexidine has been hypotension [see Warnings and Precautions (5.1)] . There has been one report of QT prolongation, bradycardia, torsades de pointes, and cardiac arrest with successful resuscitation in a patient that received lofexidine and three reports of clinically significant QT prolongation in subjects concurrently receiving methadone with lofexidine.
7 DRUG INTERACTIONS
7.1 Methadone
LUCEMYRA and methadone both prolong the QT interval. ECG monitoring is recommended in patients receiving methadone and LUCEMYRA [see Warnings and Precautions (5.2), Clinical Pharmacology (12.3)] .
7.2 Oral Naltrexone
Coadministration of LUCEMYRA and oral naltrexone resulted in statistically significant differences in the steady-state pharmacokinetics of naltrexone. It is possible that oral naltrexone efficacy may be reduced if used concomitantly within 2 hours of LUCEMYRA. This interaction is not expected if naltrexone is administered by non-oral routes [see Clinical Pharmacology (12.3)] .
7.3 CNS Depressant Drugs
LUCEMYRA potentiates the CNS depressant effects of benzodiazepines and may potentiate the CNS depressant effects of alcohol, barbiturates, and other sedating drugs. Advise patients to inform their healthcare provider of other medications they are taking, including alcohol [see Warnings and Precautions (5.3)] .
7.4 CYP2D6 Inhibitor - Paroxetine
Coadministration of LUCEMYRA and paroxetine resulted in 28% increase in the extent of absorption of LUCEMYRA. Monitor for orthostatic hypotension and bradycardia when an inhibitor of CYP2D6 is used concomitantly with LUCEMYRA [see Clinical Pharmacology (12.3)] .
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
The safety of LUCEMYRA in pregnant women has not been established. In animal reproduction studies, oral administration of lofexidine during organogenesis to pregnant rats and rabbits caused a reduction in fetal weights, increases in fetal resorptions, and litter loss at exposures below that in humans. When oral lofexidine was administered from the beginning of organogenesis through lactation, increased stillbirths and litter loss were noted along with decreased viability and lactation indices. The offspring exhibited delays in sexual maturation, auditory startle, and surface righting. These effects occurred at exposures below that in humans [see Animal Data] .
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies carry some risk of birth defect, loss, or other adverse outcomes. The background risk of major birth defects in the U.S. general population is 2% to 4% and of miscarriage is 15% to 20% of clinically recognized pregnancies.
Data
Animal Data
Increased incidence of resorptions, decreased number of implantations, and a concomitant reduction in the number of fetuses were observed when pregnant rabbits were orally administered lofexidine hydrochloride during organogenesis (from gestation day [GD] 7 to 19) at a daily dose of 5.0 mg/kg/day (approximately 0.08 times the maximum recommended human dose [MRHD] of 2.88 mg lofexidine base on an AUC basis). Maternal toxicity evidenced by increased mortality was noted at the highest tested dose of 15 mg/kg/day (approximately 0.4 times the MRHD on an AUC basis).
Decreased implantations per dam and decreased mean fetal weights were noted in a study in which pregnant rats were treated with oral lofexidine hydrochloride during organogenesis (from GD 7 to 16) at a daily dose of 3.0 mg/kg/day (approximately 0.9 times the MRHD on an AUC basis). This dose was associated with maternal toxicity (decreased body weight gain and mortality). No malformations or evidence of developmental toxicity were evident at 1.0 mg/kg/day (approximately 0.2 times the MRHD on an AUC basis).
A dose-dependent increase in pup mortality was noted in all doses of lofexidine hydrochloride administered orally to pregnant rats from GD 6 through lactation at an exposure less than the human exposure based on AUC comparisons. Doses higher than 1.0 mg/kg/day (approximately 0.2 times the MRHD on an AUC basis) resulted in incidences of total litter loss and maternal toxicity (piloerection and decreased body weight gain). The highest dose tested of 2.0 mg/kg/day (approximately 0.6 times the MRHD on an AUC basis), increased stillbirths as well as decreased viability and lactation indices were reported. Surviving offspring exhibited lower body weights, developmental delays, and increased delays in auditory startle at doses of 1.0 mg/kg/day or higher. Sexual maturation was delayed in male offspring (preputial separation) at 2.0 mg/kg/day and in female offspring (vaginal opening) at 1.0 mg/kg/day or higher.
8.2 Lactation
Risk Summary
There is no information regarding the presence of LUCEMYRA or its metabolites in human milk, the effects on the breastfed infant, or the effects on milk production. Caution should be exercised when LUCEMYRA is administered to a nursing woman.
The developmental and health benefits should be considered along with the mother's clinical need for LUCEMYRA and any other potential adverse effects on breastfed children from LUCEMYRA or from the underlying maternal condition.
8.3 Females and Males of Reproductive Potential
In animal studies that included some fertility endpoints, lofexidine decreased breeding rate and increased resorptions at exposures below human exposures. The impact of lofexidine on male fertility has not been adequately characterized in animal studies [see Impairment of Fertility (13.1)] .
8.4 Pediatric Use
The safety and effectiveness of LUCEMYRA have not been established in pediatric patients.
8.5 Geriatric Use
No studies have been performed to characterize the pharmacokinetics of LUCEMYRA or establish its safety and effectiveness in geriatric patients. Caution should be exercised when it is administered to patients over 65 years of age. Dosing adjustments similar to those recommended in patients with renal impairment should be considered [see Dosage and Administration (2.3), Use in Specific Populations (8.7)] .
8.6 Hepatic Impairment
Hepatic impairment slows the elimination of LUCEMYRA but exhibits less effect on the peak plasma concentration than on AUC values following a single dose. Dosage adjustments are recommended based on the degree of hepatic impairment. [see Dosage and Administration (2.2), Clinical Pharmacology (12.2)] .
Clinically relevant QT prolongation may occur in subjects with hepatic impairment [see Warnings and Precautions (5.2), Clinical Pharmacology (12.2)] .
8.7 Renal Impairment
Renal impairment slows the elimination of LUCEMYRA but exhibits less effect on the peak plasma concentration than on AUC values following a single dose. Dosage adjustments are recommended based on the degree of renal impairment [see Dosage and Administration (2.3), Clinical Pharmacology (12.3)] .
Only a negligible fraction of the LUCEMYRA dose is removed during a typical dialysis session, so no additional dose needs to be administered after a dialysis session; LUCEMYRA may be administered without regard to the timing of dialysis [see Dosage and Administration (2.3), Clinical Pharmacology (12.3)] .
Clinically relevant QT prolongation may occur in subjects with renal impairment [see Warnings and Precautions (5.2), Clinical Pharmacology (12.2)] .
8.8 CYP2D6 Poor Metabolizers
Although the pharmacokinetics of LUCEMYRA have not been systematically evaluated in patients who do not express the drug metabolizing enzyme CYP2D6, it is likely that the exposure to LUCEMYRA would be increased similarly to taking strong CYP2D6 inhibitors (approximately 28%). Monitor adverse events such as orthostatic hypotension and bradycardia in known CYP2D6 poor metabolizers. Approximately 8% of Caucasians and 3–8% of Black/African Americans cannot metabolize CYP2D6 substrates and are classified as poor metabolizers (PM) [see Clinical Pharmacology (12.3)] .
10 OVERDOSAGE
Overdose with LUCEMYRA may manifest as hypotension, bradycardia, and sedation. In the event of acute overdose, perform gastric lavage where appropriate. Dialysis will not remove a substantial portion of the drug. Initiate general symptomatic and supportive measures in cases of overdosage.
11 DESCRIPTION
LUCEMYRA tablets contain lofexidine, a central alpha-2 adrenergic agonist, as the hydrochloride salt. Lofexidine hydrochloride is chemically designated as 2-[1-(2,6-dichlorophenoxy)ethyl]-4,5 dihydro-1 H- imidazole monohydrochloride with a molecular formula of C 11H 12Cl 2N 2O∙HCl. Its molecular weight is 295.6 g/mole and its structural formula is:
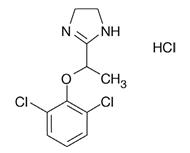
Lofexidine hydrochloride is a white to off-white crystalline powder freely soluble in water, methanol, and ethanol. It is slightly soluble in chloroform and practically insoluble in n-hexane and benzene.
LUCEMYRA is available as round, convex-shaped, peach-colored, film-coated tablets for oral administration. Each tablet contains 0.18 lofexidine, equivalent to 0.2 mg of lofexidine hydrochloride, and the following inactive ingredients: 92.6 mg lactose, 12.3 mg citric acid, 1.1 mg povidone, 5.7 mg microcrystalline cellulose, 1.4 mg calcium stearate, 0.7 mg sodium lauryl sulphate, and Opadry OY S 9480 (contains indigo carmine and sunset yellow).
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Lofexidine is a central alpha-2 adrenergic agonist that binds to receptors on adrenergic neurons. This reduces the release of norepinephrine and decreases sympathetic tone.
12.2 Pharmacodynamics
Cardiac Electrophysiology
Single LUCEMYRA doses of 1.44 to 1.8 mg produced maximum mean change from baseline in QTcF (ΔQTcF) of 14.4 msec (upper two-sided 90% CI: 22.3 msec) and 13.6 msec (17.4 msec) for 1.44 and 1.8 mg respectively in healthy normal volunteers.
In a Phase 3 placebo-controlled, dose response study in opioid dependent subjects, LUCEMYRA was associated with a maximum mean prolongation of the QTcF interval 7.3 (8.8) and 9.3 (10.9) msec at doses of 2.16 and 2.88 mg/day, respectively.
Patients with hepatic impairment
Administration of LUCEMYRA to subjects with hepatic impairment was associated with prolongation of the QTc interval, which was more pronounced in subjects with severe hepatic impairment [see Use in Specific Populations (8.6)] .
Patients with renal impairment
Administration of LUCEMYRA to subjects with renal impairment was associated with prolongation of the QTc interval, which was more pronounced in subjects with severe renal impairment [see Use in Specific Populations (8.7)]
12.3 Pharmacokinetics
Absorption
LUCEMYRA is well absorbed and achieves peak plasma concentration 3 to 5 hours after administration of a single dose.
LUCEMYRA shows approximately dose-proportional pharmacokinetics. Administration of LUCEMYRA with food does not alter its pharmacokinetics.
The absolute bioavailability of a single oral LUCEMYRA dose ( 0.36 mg in solution) compared with an intravenous infusion (0.2 mg infused for 200 minutes) was 72%. Mean LUCEMYRA C max after the oral dose and intravenous infusion was 0.82 ng/mL (at median T max of 3 hours) and 0.64 ng/mL (at median T max of 4 hours), respectively. Mean estimates of overall systemic exposure (AUC inf) were 14.9 ng∙h/mL and 12.0 ng∙h/mL, respectively.
Distribution
Mean LUCEMYRA apparent volume of distribution and volume of distribution values following the administration of an oral dose and an intravenous dose were 480.0 L and 297.9 L, respectively, which are appreciably greater than total body volume, suggesting extensive LUCEMYRA distribution into body tissue.
LUCEMYRA protein binding is approximately 55%.
LUCEMYRA is not preferentially taken up by blood cells. In a study comparing LUCEMYRA concentrations in plasma and whole blood at the time of peak LUCEMYRA concentrations in human volunteers, it was determined that red blood cells contain approximately 27% the LUCEMYRA concentration of the plasma.
Elimination
Metabolism
From absolute bioavailability results, approximately 30% of the administered LUCEMYRA dose is converted to inactive metabolites during the first pass effect associated with drug absorption from the gut.
LUCEMYRA and its major metabolites did not induce or inhibit any CYP450 isoforms, with the exception of a slight inhibition of CYP2D6 by LUCEMYRA, with an IC 50 of 4551 nM (approximately 225 times the steady-state C max for LUCEMYRA with 0.72 mg 4 times daily dosing). Any LUCEMYRA interaction with CYP2D6 substrates is not expected to be clinically significant.
LUCEMYRA is metabolized when incubated in vitro with human liver microsomes, the major contributor to the hepatic metabolism of LUCEMYRA is CYP2D6, with CYP1A2 and CYP2C19 also capable of metabolizing LUCEMYRA.
Excretion
The elimination half-life is approximately 12 hours and mean clearance is 17.6 L/h following an IV infusion.
LUCEMYRA has a terminal half-life of approximately 11 to 13 hours following the first dose. At steady-state, the terminal half-life is approximately 17 to 22 hours. Accumulation occurs up to 4 days with repeat dosing, following the recommended dosing regimen.
A mass balance study of LUCEMYRA showed nearly complete recovery of radiolabel in urine (93.5%) over 144 hours postdose, with an additional 0.92% recovered in the feces over 216 hours postdose. Thus, it appears that all, or nearly all, of the dose was absorbed, and that the primary route of elimination of the parent drug and its metabolites is via the kidney. Renal elimination of unchanged drug accounts for approximately 15% to 20% of the administered dose.
Specific Populations
Hepatic Impairment
Hepatic impairment slows the elimination of LUCEMYRA, but exhibits less effect on the peak plasma concentration following a single dose. In a study comparing the pharmacokinetics of LUCEMYRA (0.36 mg) in mild, moderate, and severe hepatically impaired subjects to subjects with normal hepatic function (6 subjects in each hepatic function group), mean C max values were similar for subjects with normal, mild, and moderate hepatic impairment as shown in Table 6.
| Normal | Mild Impairment | Moderate Impairment | Severe Impairment | |
|---|---|---|---|---|
| Child-Pugh Class & Score | Normal Function | Class A
5-6 | Class B
7-9 | Class C
10-15 |
| C max % of normal | 100 | 114 | 117 | 166 |
| AUC last % of normal | 100 | 127 | 190 | 304 |
| AUC ∞ % of normal | 100 | 117 | 185 | 260 |
| t 1/2 % of normal | 100 | 139 | 281 | 401 |
Renal Impairment
Renal impairment slows the elimination of LUCEMYRA but exhibits less effect on the peak plasma concentration following a single dose. In a study comparing the pharmacokinetics of LUCEMYRA (0.36 mg) in 8 end-stage renal disease subjects on 3 times weekly hemodialysis to 8 subjects with normal renal function matched for sex, age, and body mass index, mean C max values were similar for end-stage renal disease and normal renal function subjects, indicating no change in maximum LUCEMYRA exposure with renal impairment as shown in Table 7.
The impact of dialysis on the overall pharmacokinetics of LUCEMYRA during a typical 4-hour dialysis was minimal; the drop in LUCEMYRA plasma concentrations produced during the dialysis session was transient, with a rebound to nearly predialysis concentrations after re-equilibration within a few hours following completion of the dialysis cycle [see Dosage and Administration (2.3), Use in Specific Populations (8.7)] .
In a study comparing the pharmacokinetics of LUCEMYRA (0.36 mg) in 6 subjects each with normal renal function, mild renal impairment, and moderate renal impairment as well as 5 subjects with severe renal impairment but not requiring dialysis, there were similar increases in mean C max values in subjects with mild and moderate renal impairment in comparison to subjects with normal renal function with additional increase in mean C max values in subjects with severe renal impairment. Mean AUC last, AUC ∞, and t 1/2 increased with severity of renal impairment as shown in Table 7.
| Normal | Mild Impairment | Moderate Impairment | Severe Impairment | ESRD or on dialysis | |
|---|---|---|---|---|---|
| eGFR (mL/min/1.73 m 2) | ≥ 90 | 60-89 | 30-59 | 15-29 | < 15 |
| C max % of normal | 100 | 124 | 117 | 154 | 104 |
| AUC last % of normal | 100 | 157 | 187 | 272 | 181 |
| AUC ∞ % of normal | 100 | 144 | 173 | 243 | 171 |
| t 1/2 % of normal | 100 | 111 | 145 | 157 | 137 |
Drug Interaction Studies
LUCEMYRA coadministered with methadone
In a double-blind placebo-controlled study of 23 patients maintained on a methadone dose of 80-120 mg/day and concomitantly administered LUCEMYRA up to 2.88 mg/day, LUCEMYRA did not alter the pharmacokinetics of methadone. LUCEMYRA concentrations may be slightly increased when coadministered with methadone; however, the increase at concentrations expected with recommended dosing is not clinically meaningful [see Drug Interactions (7.1)] .
LUCEMYRA coadministered with buprenorphine
In a double-blind placebo-controlled study of 30 subjects maintained on buprenorphine (16-24 mg/day) concomitantly administered LUCEMYRA up to 2.88 mg/day, no pharmacokinetic or pharmacodynamic interactions between LUCEMYRA and buprenorphine were seen.
LUCEMYRA coadministered with oral naltrexone
In an open-label, single-arm study of 24 healthy subjects, oral naltrexone (50 mg/day) did not significantly alter the single-dose pharmacokinetics of LUCEMYRA (0.36 mg). The alteration in steady-state pharmacokinetics of oral naltrexone was statistically significant in the presence of LUCEMYRA. The t max was delayed for both naltrexone and 6ß-naltrexol (2-3 hours), and overall exposure was slightly reduced when naltrexone was administered with LUCEMYRA [see Drug Interactions (7.2)] .
LUCEMYRA coadministered with paroxetine
In an open-label, single-sequence study of 24 healthy subjects, the strong CYP2D6 inhibitor paroxetine (40 mg/day) increased LUCEMYRA (0.36 mg) C max and AUC ∞ by approximately 11% and 28%, respectively [see Drug Interactions (7.4)] .
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
No adequate long-term animal studies have been completed to evaluate the carcinogenic potential of lofexidine.
Mutagenesis
Lofexidine tested positive in the in vitro mouse lymphoma assay. Lofexidine tested negative in the in vitro bacterial reverse mutation assay (Ames assay) and in the in vivo rat micronucleus assay.
Impairment of Fertility
In a female fertility study in rabbits, fertility was not adversely impacted by administration of lofexidine hydrochloride up to 6.4 mg/kg/day (approximately 0.1 times the MRHD of 2.88 mg on an AUC basis) when administered orally to female rabbits starting 2 weeks prior to mating and through gestation and lactation. However, decreased breeding rate and higher post-implantation loss was observed at this dose, which correlated with higher resorptions and reduced litter size. Maternal toxicity, which included increased mortality rate, reduced body weight gain, and moderate sedation was observed at 6.4 mg/kg/day. The NOAEL for female fertility was 6.4 mg/kg/day and the NOAEL for female-mediated developmental parameters was 0.4 mg/kg/day (approximately 0.005 times the MRHD on an AUC basis).
In a fertility study in rats, fertility was unaffected by administration of lofexidine up to 0.88 mg/kg/day (approximately 0.2 times the MRHD on an AUC basis) via diet to male and female rats prior to mating and to the dams through gestation and lactation. No evidence of maternal toxicity was observed. However, no assessment of sperm or reproductive organs were performed in this study.
Reduced testes, epididymis, and seminiferous tubule weights, as well as delayed sexual maturation of males and females and decreases in the number of corpora lutea and implantations after mating, were noted in offspring of pregnant rats administered lofexidine hydrochloride orally from GD 6 through lactation at exposures less than the human exposure based on AUC comparisons.
14 CLINICAL STUDIES
Two randomized, double-blind, placebo-controlled trials supported the efficacy of LUCEMYRA.
Study 1, NCT01863186
Study 1 was a 2-part efficacy, safety, and dose-response study conducted in the United States in patients meeting DSM-IV criteria for opioid dependence who were physically dependent on short-acting opioids (e.g., heroin, hydrocodone, oxycodone). The first part of the study was an inpatient, randomized, double-blind, placebo-controlled design consisting of 7 days of inpatient treatment (Days 1 – 7) with LUCEMYRA 2.16 mg total daily dose (0.54 mg 4 times daily) (n=229), LUCEMYRA 2.88 mg total daily dose (0.72 mg 4 times daily) (n=222), or matching placebo (n=151). Patients also had access to a variety of support medications for withdrawal symptoms (guaifenesin, antacids, dioctyl sodium sulfosuccinate, psyllium hydrocolloid suspension, bismuth sulfate, acetaminophen, and zolpidem). The second part of the study (Days 8 – 14) was an open-label design where all patients who successfully completed Days 1 – 7 were eligible to receive open-label treatment with variable dose LUCEMYRA treatment (as determined by the investigator, but not to exceed 2.88 mg total daily dose) for up to an additional 7 days (Days 8 – 14) in either an inpatient or outpatient setting as determined by the investigator and the patient. No patient received LUCEMYRA for more than 14 days.
The two endpoints to support efficacy were the mean Short Opiate Withdrawal Scale of Gossop (SOWS-Gossop) total score on Days 1 – 7 of treatment and the proportion of patients that completed 7 days of treatment. The SOWS-Gossop, a patient-reported outcome (PRO) instrument, evaluates the following opioid withdrawal symptoms: feeling sick, stomach cramps, muscle spasms/twitching, feeling of coldness, heart pounding, muscular tension, aches and pains, yawning, runny eyes and insomnia/problems sleeping. For each opioid withdrawal symptom, patients are asked to rate their symptom severity using four response options (none, mild, moderate, and severe). The SOWS-Gossop total score ranges from 0 to 30 where a higher score indicates a greater withdrawal symptom severity. The SOWS-Gossop was administered at baseline and once daily 3.5 hours after the first morning dose on Days 1 – 7.
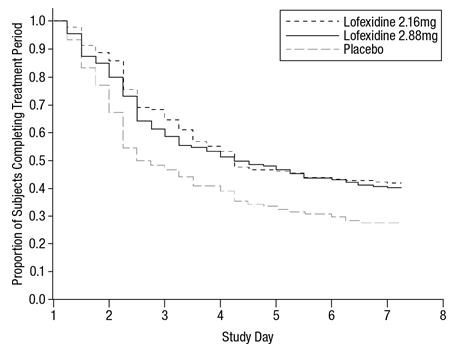
Of the randomized and treated patients, 28% of placebo patients, 41% of LUCEMYRA 2.16 mg and 40% of LUCEMYRA 2.88 mg patients completed 7 days of treatment. The difference in proportion in both LUCEMYRA groups was significant compared to placebo. See Figure 1. Patients in the placebo group were more likely to drop out of the study prematurely due to lack of efficacy than patients treated with LUCEMYRA.
Figure 1: Completion of treatment period for Study 1
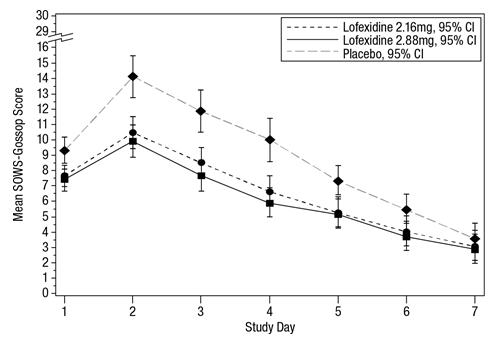
The mean SOWS-Gossop scores for Days 1 – 7 were 8.8, 6.5, and 6.1 for placebo, LUCEMYRA 2.16 mg and LUCEMYRA 2.88 mg, respectively. Results are shown in Figure 2. The mean difference between LUCEMYRA 2.16 mg and placebo was -2.3 with a 95% CI of (-3.4, -1.2). The mean difference between LUCEMYRA 2.88 mg and placebo was -2.7 with a 95% CI of (-3.9, -1.6). They were both significant. Symptoms assessed on the SOWS-Gossop were recorded as absent or mild for almost all patients remaining to the end of the assessment period.
Figure 2: Mean SOWS-Gossop Scores for Days 1 – 7 in Study 1
Study 2, NCT00235729
Study 2 was an inpatient, randomized, multicenter, double-blind, placebo-controlled study carried out in the United States in patients meeting DSM-IV criteria for opioid dependence who were physically dependent on short-acting opioids (e.g., heroin, hydrocodone, oxycodone). Patients were treated with LUCEMYRA tablets (2.88 mg/day [0.72 mg four times daily]) or matching placebo for 5 days (Days 1 – 5). Patients also had access to a variety of support medications for withdrawal symptoms (guaifenesin, antacids, dioctyl sodium sulfosuccinate, psyllium hydrocolloid suspension, bismuth sulfate, acetaminophen, and zolpidem). All patients then received placebo on Days 6 and 7 and were discharged on Day 8.
The two endpoints to support efficacy were the mean SOWS-Gossop total score on Days 1 – 5 of treatment and the proportion of patients that completed 5 days of treatment. The SOWS-Gossop was administered at baseline and once daily 3.5 hours after the first morning dose on Days 1 – 5.
A total of 264 patients were randomized into the study. Of these, 134 patients were randomized to LUCEMYRA 2.88 mg/day and 130 patients to placebo.
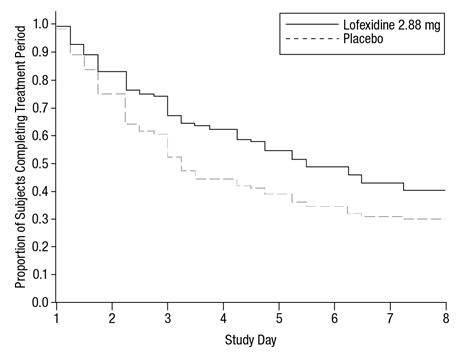
Of the randomized and treated patients, 33% of placebo patients and 49% of LUCEMYRA patients completed 5 days of treatment. The difference in proportion between the two groups was significant. See Figure 3. Patients in the placebo group were more likely to drop out of the study prematurely due to lack of efficacy than patients treated with LUCEMYRA.
Figure 3: Completion of treatment period in Study 2
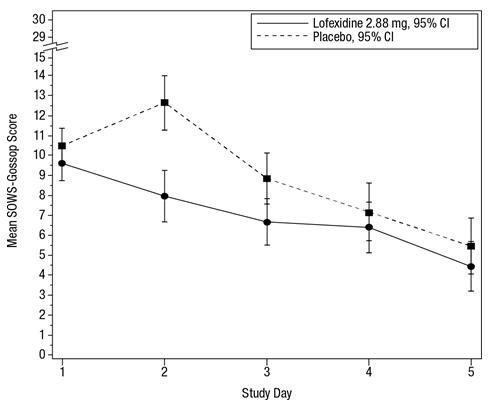
The mean SOWS-Gossop scores for Days 1 – 5 were 8.9 and 7.0 for placebo and LUCEMYRA 2.88 mg, respectively. Results are shown in Figure 4. The mean difference was -1.9 with a 95% CI of (-3.2, -0.6) and was statistically significant.
Figure 4: Mean SOWS-Gossop Scores for Days 1 – 5 in Study 2
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
Available as 0.18 mg round, convex-shaped, peach colored, film-coated tablets, imprinted with "LFX" on one side and "18" on the other side; approximately 7 mm in diameter.
| Bottles of 36 tablets | NDC 27505-050-36 |
| Bottles of 96 tablets | NDC 27505-050-96 |
Storage
Store in original container at controlled room temperature, 25°C (77°F); with excursions permitted between 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature]. Keep LUCEMYRA away from excess heat and moisture both in the pharmacy and after dispensing. Do not remove desiccant packs from bottles until all tablets are used. Keep LUCEMYRA and all medicines out of the reach of children.
17 PATIENT COUNSELING INFORMATION
Advise patients to read the FDA-approved patient labeling (Patient Information).
LUCEMYRA may mitigate, but not completely prevent, the symptoms associated with opioid withdrawal syndrome, which may include feeling sick, stomach cramps, muscle spasms or twitching, feeling of cold, heart pounding, muscular tension, aches and pains, yawning, runny eyes and sleep problems (insomnia). Patients should be advised that withdrawal will not be easy. Additional supportive measures should be clearly advised, as needed.
Hypotension and Bradycardia
Inform patients to be alert for any symptoms of low blood pressure or pulse (e.g., dizziness, lightheadedness, or feelings of faintness at rest or on abruptly standing). Advise patients on how to reduce the risk of serious consequences should hypotension occur (sit or lie down, carefully rise from a sitting or lying position).
Patients being given LUCEMYRA in an outpatient setting should be capable of and instructed on self-monitoring for hypotension, orthostasis and bradycardia and advised to withhold LUCEMYRA doses and contact their healthcare provider for instructions if they experience these signs or related symptoms [see Warnings and Precautions (5.1)] .
Advise patients to avoid becoming dehydrated or overheated, which may potentially increase the risks of hypotension and syncope [see Warnings and Precautions (5.1)] .
Concomitant Medications
Review with patients all concomitant medications being taken and request that they immediately inform their healthcare provider of any changes in concomitant medications, including any other medications that may be used to treat individual symptoms of withdrawal.
Increased Risk of CNS Depression with Concomitant use of CNS Depressant Drugs
Inform patients of the increased risk of CNS depression with concomitant use of benzodiazepines, alcohol, barbiturates, or other sedating drugs [see Warnings and Precautions (5.3)] .
Advise patients using LUCEMYRA in an outpatient setting that, until they learn how they respond to LUCEMYRA, they should be careful or avoid doing activities such as driving or operating heavy machinery.
Sudden Discontinuation of LUCEMYRA
Inform patients not to discontinue LUCEMYRA without consulting their healthcare provider [see Warnings and Precautions (5.5)] .
Risk of Opioid Overdose After Discontinuation of Opioids
Advise patients that after a period of not using opioid drugs, they may be more sensitive to the effects of opioids and at greater risk of overdosing [see Warnings and Precautions (5.4)] .
Distributed by:
US WorldMeds, LLC
4441 Springdale Road
Louisville, KY 40241
Under License from Britannia Pharmaceuticals Limited.
US WorldMeds, LLC is the exclusive licensee and distributor of LUCEMYRA™ in the United States and Its territories. ©2018. LUCEMYRA™ is a trademark of US WorldMeds, LLC.
360-10020
| This Patient Information has been approved by the U.S. Food and Drug Administration. | Issued: 05/2018 | |
| 360-10020 | ||
| PATIENT INFORMATION
LUCEMYRA™ (LEW-sem-EER-uh) (lofexidine) tablets |
||
| What is the most important information I should know about LUCEMYRA and discontinuing opioid drugs?
LUCEMYRA can cause serious side effects, including low blood pressure (hypotension), slow heart rate (bradycardia), and fainting. If you get any of the following signs or symptoms, tell your healthcare provider right away: |
||
|
|
|
| If you take LUCEMYRA at home and have any of these signs and symptoms, do not take your next dose of LUCEMYRA until you have talked to your healthcare provider. You should avoid becoming dehydrated or overheated during treatment with LUCEMYRA, which may increase your risk of low blood pressure and fainting. You should also be careful not to stand up too suddenly from lying down or sitting.
When your treatment is complete you will need to stop taking LUCEMYRA gradually or your blood pressure could increase. For more information about side effects, see " What are the possible side effects of LUCEMYRA?" Increased risk of opioid overdose. After a period of time of not using opioid drugs, you can become more sensitive to the effects of opioids if you start using opioids again. This may increase your risk of overdose and death. |
||
| What is LUCEMYRA?
LUCEMYRA is a non-opioid prescription medicine used in adults to help with the symptoms of opioid withdrawal that may happen when you stop taking an opioid suddenly. LUCEMYRA will not completely prevent the symptoms of opioid withdrawal, which may include feeling sick, stomach cramps, muscle spasms or twitching, feeling of cold, heart pounding, muscular tension, aches and pains, yawning, runny eyes and sleep problems (insomnia). LUCEMYRA is not a treatment for opioid use disorder. If you have been diagnosed with opioid use disorder (opioid addiction), your healthcare provider may prescribe LUCEMYRA as part of a complete treatment program for your opioid use disorder (opioid addiction). It is not known if LUCEMYRA is safe and effective in children. |
||
Before taking LUCEMYRA, tell your healthcare provider about all of your medical conditions, including if you:
Especially tell your healthcare provider if you take benzodiazepines, barbiturates, tranquilizers, or sleeping pills. Taking LUCEMYRA with these medicines can cause serious side effects. Ask your healthcare provider or pharmacist if you are not sure if you are taking any of these medicines. |
||
How should I take LUCEMYRA?
|
||
| What should I avoid while taking LUCEMYRA?
Do not drive, operate heavy machinery, or perform any other dangerous activities until you know how LUCEMYRA affects you. |
||
| What are the possible side effects of LUCEMYRA?
The most common side effects of LUCEMYRA include: |
||
|
|
|
| These are not all the possible side effects of LUCEMYRA.
Call your healthcare provider for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. You may also report side effects to US WorldMeds at 1-833-LUCEMYRA. |
||
How should I store LUCEMYRA?
|
||
| General information about the safe and effective use of LUCEMYRA.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use LUCEMYRA for a condition for which it was not prescribed. Do not give LUCEMYRA to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about LUCEMYRA that is written for health professionals. |
||
| What are the ingredients of LUCEMYRA?
Active ingredient: lofexidine. Inactive ingredients: lactose, citric acid, povidone, microcrystalline cellulose, calcium stearate, sodium lauryl sulphate, and Opadry OY S 9480 (contains indigo carmine and sunset yellow). |
||
| Distributed by: US WorldMeds, LLC, 4441 Springdale Road, Louisville, KY 40241
Under License from Britannia Pharmaceuticals Limited. US WorldMeds, LLC is the exclusive licensee and distributor of LUCEMYRA™ in the United States and Its territories. ©2018. LUCEMYRA™ is a trademark of US WorldMeds, LLC. For more information, go to www.LUCEMYRA.com or call 1-833-LUCEMYRA |
||
PRINCIPAL DISPLAY PANEL - 0.18 mg Tablet Bottle Carton
Rx only
NDC 27505-050-36
Lucemyra™
(lofexidine) tablets
0.18 mg
Store and dispense
in original container.
Protect from heat and moisture.
Do not remove desiccants.
Keep the bottle tightly closed.
Keep out of reach of children.
36 tablets
US WorldMeds™
| LUCEMYRA
lofexidine hydrochloride tablet, film coated |
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
| Labeler - US WorldMeds, LLC (087875626) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| SGS Life Science Services | 062491980 | analysis(27505-050) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Acceleration Laboratory Services, Inc. | 187562629 | analysis(27505-050) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Apace Packaging LLC | 361961142 | pack(27505-050) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Helsinn Advanced Synthesis SA | 481296960 | api manufacture(27505-050) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| UFAG Laboratoriien AG | 486383151 | analysis(27505-050) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Catalent Pharma Solutions, LLC | 829672745 | manufacture(27505-050) , analysis(27505-050) , pack(27505-050) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| A+ Secrure Packaging, LLC | 963589036 | label(27505-050) , pack(27505-050) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Helsinn Birex Pharmaceuticals LTD | 985084409 | pack(27505-050) | |