FULL PRESCRIBING INFORMATION
NUCYNTA® (tapentadol) is indicated for the relief of moderate to severe acute pain in patients 18 years of age or older.
2 DOSAGE AND ADMINISTRATION
As with many centrally-acting analgesic medications, the dosing regimen should be individualized according to the severity of pain being treated, the previous experience with similar drugs and the ability to monitor the patient.
The dose is 50 mg, 75 mg, or 100 mg every 4 to 6 hours depending upon pain intensity.
On the first day of dosing, the second dose may be administered as soon as one hour after the first dose, if adequate pain relief is not attained with the first dose. Subsequent dosing is 50 mg, 75 mg, or 100 mg every 4 to 6 hours and should be adjusted to maintain adequate analgesia with acceptable tolerability.
Daily doses greater than 700 mg on the first day of therapy and 600 mg on subsequent days have not been studied and are not recommended.
NUCYNTA® may be given with or without food [see Clinical Pharmacology (12.3)].
2.1 Renal Impairment
No dosage adjustment is recommended in patients with mild or moderate renal impairment [see Clinical Pharmacology (12.3)].
NUCYNTA® has not been studied in patients with severe renal impairment. The use in this population is not recommended.
2.2 Hepatic Impairment
No dosage adjustment is recommended in patients with mild hepatic impairment [see Clinical Pharmacology (12.3)].
NUCYNTA® should be used with caution in patients with moderate hepatic impairment. Treatment in these patients should be initiated at 50 mg with the interval between doses no less than every 8 hours (maximum of three doses in 24 hours). Further treatment should reflect maintenance of analgesia with acceptable tolerability, to be achieved by either shortening or lengthening the dosing interval [see Clinical Pharmacology (12.3)].
NUCYNTA® has not been studied in patients with severe hepatic impairment and use in this population is not recommended [see Warnings and Precautions (5.10)].
2.3 Elderly Patients
In general, recommended dosing for elderly patients with normal renal and hepatic function is the same as for younger adult patients with normal renal and hepatic function. Because elderly patients are more likely to have decreased renal and hepatic function, consideration should be given to starting elderly patients with the lower range of recommended doses.
3 DOSAGE FORMS AND STRENGTHS
NUCYNTA® Tablets are round, biconvex and film-coated and are available in the following strengths, colors, and debossings: 50 mg of tapentadol (yellow with "O-M" on one side and "50" on the other side), 75 mg of tapentadol (yellow-orange with "O-M" on one side and "75" on the other side), and 100 mg of tapentadol (orange with "O-M" on one side and "100" on the other side).
4 CONTRAINDICATIONS
4.1 Impaired Pulmonary Function
Like other drugs with mu-opioid agonist activity, NUCYNTA® is contraindicated in patients with significant respiratory depression in unmonitored settings or the absence of resuscitative equipment. NUCYNTA® is also contraindicated in patients with acute or severe bronchial asthma or hypercapnia in unmonitored settings or the absence of resuscitative equipment [see Warnings and Precautions (5.1)].
4.2 Paralytic Ileus
Like drugs with mu-opioid agonist activity, NUCYNTA® is contraindicated in any patient who has or is suspected of having paralytic ileus.
4.3 Monoamine Oxidase Inhibitors
NUCYNTA® is contraindicated in patients who are receiving monoamine oxidase (MAO) inhibitors or who have taken them within the last 14 days due to potential additive effects on norepinephrine levels which may result in adverse cardiovascular events [see Drug Interactions (7.4)].
5 WARNINGS AND PRECAUTIONS
5.1 Respiratory Depression
Respiratory depression is the primary risk of mu-opioid agonists. Respiratory depression occurs more frequently in elderly or debilitated patients and in those suffering from conditions accompanied by hypoxia, hypercapnia, or upper airway obstruction, in whom even moderate therapeutic doses may significantly decrease pulmonary ventilation.
NUCYNTA® should be administered with caution to patients with conditions accompanied by hypoxia, hypercapnia or decreased respiratory reserve such as: asthma, chronic obstructive pulmonary disease or cor pulmonale, severe obesity, sleep apnea syndrome, myxedema, kyphoscoliosis, central nervous system (CNS) depression, or coma. In such patients, even usual therapeutic doses of NUCYNTA® may increase airway resistance and decrease respiratory drive to the point of apnea. Alternative non-mu-opioid agonist analgesics should be considered and NUCYNTA® should be employed only under careful medical supervision at the lowest effective dose in such patients. If respiratory depression occurs, it should be treated as any mu-opioid agonist-induced respiratory depression [see Overdosage (10.2)].
5.2 CNS Depression
Patients receiving other mu-opioid agonist analgesics, general anesthetics, phenothiazines, other tranquilizers, sedatives, hypnotics, or other CNS depressants (including alcohol) concomitantly with NUCYNTA® may exhibit additive CNS depression. Interactive effects resulting in respiratory depression, hypotension, profound sedation, coma or death may result if these drugs are taken in combination with NUCYNTA®. When such combined therapy is contemplated, a dose reduction of one or both agents should be considered.
5.3 Head Injury and Increased Intracranial Pressure
Opioid analgesics can raise cerebrospinal fluid pressure as a result of respiratory depression with carbon dioxide retention. Therefore, NUCYNTA® should not be used in patients who may be susceptible to the effects of raised cerebrospinal fluid pressure such as those with evidence of head injury and increased intracranial pressure. Opioid analgesics may obscure the clinical course of patients with head injury due to effects on pupillary response and consciousness. NUCYNTA® should be used with caution in patients with head injury, intracranial lesions, or other sources of preexisting increased intracranial pressure.
5.4 Misuse and Abuse
Tapentadol is a mu-opioid agonist and is a Schedule II controlled substance. Such drugs are sought by drug abusers and people with addiction disorders. Diversion of Schedule II products is an act subject to criminal penalty.
NUCYNTA® can be abused in a manner similar to other opioid agonists, legal or illicit. This should be considered when prescribing or dispensing NUCYNTA® in situations where the physician or pharmacist is concerned about an increased risk of misuse and abuse. Concerns about abuse and addiction should not prevent the proper management of pain. However, all patients treated with mu-opioid agonists require careful monitoring for signs of abuse and addiction, since use of mu-opioid agonist analgesic products carry the risk of addiction even under appropriate medical use [see Drug Abuse and Dependence (9.2)].
NUCYNTA® may be abused by crushing, chewing, snorting or injecting the product. These practices pose a significant risk to the abuser that could result in overdose and death [see Drug Abuse and Dependence (9)].
5.5 Driving and Operating Machinery
Patients should be cautioned that NUCYNTA® may impair the mental and/or physical abilities required for the performance of potentially hazardous tasks such as driving a car or operating machinery. This is to be expected especially at the beginning of treatment, at any change of dosage as well as in combination with alcohol or tranquilizers [see Drug Interactions (7.3)].
5.6 Interactions with Alcohol and Drugs of Abuse
Due to its mu-opioid agonist activity, NUCYNTA® may be expected to have additive effects when used in conjunction with alcohol, opioids, or illicit drugs that cause central nervous system depression, respiratory depression, hypotension, and profound sedation, coma or death [see Drug Interactions (7.3)].
5.7 Seizures
NUCYNTA® has not been systematically evaluated in patients with a seizure disorder, and such patients were excluded from clinical studies. NUCYNTA® should be prescribed with care in patients with a history of a seizure disorder or any condition that would put the patient at risk of seizures.
5.8 Serotonin Syndrome Risk
The development of a potentially life-threatening serotonin syndrome may occur with use of Serotonin and Norepinephrine Reuptake Inhibitor (SNRI) products, including NUCYNTA®, particularly with concomitant use of serotonergic drugs such as Selective Serotonin Reuptake Inhibitors (SSRIs), SNRIs, tricyclic antidepressants (TCAs), MAOIs and triptans, and with drugs that impair metabolism of serotonin (including MAOIs). This may occur within the recommended dose. Serotonin syndrome may include mental-status changes (e.g., agitation, hallucinations, coma), autonomic instability (e.g., tachycardia, labile blood pressure, hyperthermia), neuromuscular aberrations (e.g., hyperreflexia, incoordination) and/or gastrointestinal symptoms (e.g., nausea, vomiting, diarrhea).
5.9 Withdrawal
Withdrawal symptoms may occur if NUCYNTA® is discontinued abruptly. These symptoms may include: anxiety, sweating, insomnia, rigors, pain, nausea, tremors, diarrhea, upper respiratory symptoms, piloerection, and rarely, hallucinations. Withdrawal symptoms may be reduced by tapering NUCYNTA® [see Drug Abuse and Dependence (9.3)].
5.10 Hepatic Impairment
A study of NUCYNTA® in subjects with hepatic impairment showed higher serum concentrations than in those with normal hepatic function. NUCYNTA® should be used with caution in patients with moderate hepatic impairment [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)].
NUCYNTA® has not been studied in patients with severe hepatic impairment and, therefore, use in this population is not recommended.
6 ADVERSE REACTIONS
The following treatment-emergent adverse events are discussed in more detail in other sections of the labeling:
- Respiratory Depression [see Contraindications (4.1) and Warnings and Precautions (5.1)]
- CNS Depression [see Warnings and Precautions (5.2)]
Because clinical studies are conducted under widely varying conditions, adverse event rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in clinical practice. A treatment-emergent adverse event refers to any untoward medical event associated with the use of the drug in humans, whether or not considered drug-related.
Based on data from nine Phase 2/3 studies that administered multiple doses (seven placebo- and/or active-controlled, one noncontrolled and one Phase 3 active-controlled safety study) the most common adverse events (reported by ≥10% in any NUCYNTA® dose group) were: nausea, dizziness, vomiting and somnolence.
The most common reasons for discontinuation due to adverse events in the studies described above (reported by ≥1% in any NUCYNTA® dose group) were dizziness (2.6% vs. 0.5%), nausea (2.3% vs. 0.6%), vomiting (1.4% vs. 0.2%), somnolence (1.3% vs. 0.2%) and headache (0.9% vs. 0.2%) for NUCYNTA®- and placebo-treated patients, respectively.
Seventy-six percent of NUCYNTA®-treated patients from the nine studies experienced adverse events.
NUCYNTA® was studied in multiple-dose, active- or placebo-controlled studies, or noncontrolled studies (n = 2178), in single-dose studies (n = 870), in open-label study extension (n = 483) and in Phase 1 studies (n = 597). Of these, 2034 patients were treated with doses of 50 mg to 100 mg of NUCYNTA® dosed every 4 to 6 hours.
The data described below reflect exposure to NUCYNTA® in 3161 patients, including 449 exposed for 45 days. NUCYNTA® was studied primarily in placebo- and active-controlled studies (n = 2266, and n = 2944, respectively). The population was 18 to 85 years old (mean age 46 years), 68% were female, 75% white and 67% were postoperative. Most patients received NUCYNTA® doses of 50 mg, 75 mg, or 100 mg every 4 to 6 hours.
6.1 Commonly-Observed Treatment-Emergent Adverse Events in Double-Blind Controlled Clinical Trials
Table 1 lists the adverse events reported in ≥1% or more of NUCYNTA®-treated patients with acute moderate to severe pain in the pooled safety data from nine Phase 2/3 studies that administered multiple doses (seven placebo- and/or active-controlled, one noncontrolled, and one Phase 3 active-controlled safety study).
| System/Organ Class | NUCYNTA® | Placebo | |
|---|---|---|---|
| MedDRA Preferred Term | 21 mg – 120 mg | (n=619) | |
| (n=2178) | % | ||
| % | |||
|
|||
| Gastrointestinal disorders | |||
| Nausea | 30 | 13 | |
| Vomiting | 18 | 4 | |
| Constipation | 8 | 3 | |
| Dry mouth | 4 | <1 | |
| Dyspepsia | 2 | <1 | |
| General disorders and administration site conditions | |||
| Fatigue | 3 | <1 | |
| Feeling hot | 1 | <1 | |
| Infections and infestations | |||
| Nasopharyngitis | 1 | <1 | |
| Upper respiratory tract infection | 1 | <1 | |
| Urinary tract infection | 1 | <1 | |
| Metabolism and nutrition disorders | |||
| Decreased appetite | 2 | 0 | |
| Musculoskeletal and connective tissue disorders | |||
| Arthralgia | 1 | <1 | |
| Nervous system disorders | |||
| Dizziness | 24 | 8 | |
| Somnolence | 15 | 3 | |
| Tremor | 1 | <1 | |
| Lethargy | 1 | <1 | |
| Psychiatric disorders | |||
| Insomnia | 2 | <1 | |
| Confusional state | 1 | 0 | |
| Abnormal dreams | 1 | <1 | |
| Anxiety | 1 | <1 | |
| Skin and subcutaneous tissue disorders | |||
| Pruritus | 5 | 1 | |
| Hyperhidrosis | 3 | <1 | |
| Pruritus generalized | 3 | <1 | |
| Rash | 1 | <1 | |
| Vascular disorders | |||
| Hot flush | 1 | <1 | |
6.2 Other Adverse Reactions Observed During the Premarketing Evaluation of NUCYNTA®
The following adverse drug reactions occurred in <1% of NUCYNTA®-treated patients in the pooled safety data from nine Phase 2/3 studies that administered multiple doses (seven were placebo- and/or active-controlled, one noncontrolled, and one Phase 3 active-controlled safety study):
Cardiac disorders: heart rate increased, heart rate decreased
Eye disorders: visual disturbance
Gastrointestinal disorders: abdominal discomfort, impaired gastric emptying
General disorders and administration site conditions: irritability, edema, drug withdrawal syndrome, feeling drunk
Immune system disorders: hypersensitivity
Investigations: gamma-glutamyltransferase increased, alanine aminotransferase increased, aspartate aminotransferase increased
Musculoskeletal and connective tissue disorders: involuntary muscle contractions, sensation of heaviness
Nervous system disorders: hypoesthesia, paresthesia, disturbance in attention, sedation, dysarthria, depressed level of consciousness, memory impairment, ataxia, presyncope, syncope, coordination abnormal, seizure
Psychiatric disorders: euphoric mood, disorientation, restlessness, agitation, nervousness, thinking abnormal
Renal and urinary disorders: urinary hesitation, pollakiuria
Respiratory, thoracic and mediastinal disorders: oxygen saturation decreased, cough, dyspnea, respiratory depression
Skin and subcutaneous tissue disorders: urticaria
Vascular disorders: blood pressure decreased
In the pooled safety data, the overall incidence of adverse reactions increased with increased dose of NUCYNTA®, as did the percentage of patients with adverse reactions of nausea, dizziness, vomiting, somnolence, and pruritus.
6.3 Post-marketing Experience
The following additional adverse reactions have been identified during post-approval use of NUCYNTA®. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to estimate their frequency reliably.
Nervous system disorders: headache
Psychiatric disorders: hallucination
7 DRUG INTERACTIONS
NUCYNTA® is mainly metabolized by glucuronidation. The following substances have been included in a set of interaction studies without any clinically significant finding: acetaminophen, acetylsalicylic acid, naproxen and probenecid [see Clinical Pharmacology (12.3)].
The pharmacokinetics of tapentadol were not affected when gastric pH or gastrointestinal motility were increased by omeprazole and metoclopramide, respectively [see Clinical Pharmacology (12.3)].
7.1 Drugs Metabolized by Cytochrome P450 Enzymes
In vitro investigations indicate that NUCYNTA® does not inhibit or induce P450 enzymes. Thus, clinically relevant interactions mediated by the cytochrome P450 system are unlikely to occur [see Clinical Pharmacology (12.3)].
7.2 Drugs That Inhibit or Induce Cytochrome P450 Enzymes
The major pathway of tapentadol metabolism is conjugation with glucuronic acid to produce glucuronides. To a lesser extent, tapentadol is additionally metabolized to N-desmethyl tapentadol (13%) by CYP2C9 and CYP2C19 to hydroxy tapentadol (2%) by CYP2D6, which are further metabolized by conjugation. Since only a minor amount of NUCYNTA® is metabolized via the oxidative pathway clinically relevant interactions mediated by the cytochrome P450 system are unlikely to occur [see Clinical Pharmacology (12.3)].
7.3 Centrally-Acting Drugs and Alcohol
Patients receiving other opioid agonist analgesics, general anesthetics, phenothiazines, antiemetics, other tranquilizers, sedatives, hypnotics, or other CNS depressants (including alcohol) concomitantly with NUCYNTA® may exhibit an additive CNS depression. Interactive effects resulting in respiratory depression, hypotension, profound sedation, or coma may result if these drugs are taken in combination with NUCYNTA®. When such combined therapy is contemplated, a dose reduction of one or both agents should be considered [see Warnings and Precautions (5.2) and (5.6)].
7.4 Monoamine Oxidase Inhibitors
NUCYNTA® is contraindicated in patients who are receiving monoamine oxidase (MAO) inhibitors or who have taken them within the last 14 days due to potential additive effects on norepinephrine levels which may result in adverse cardiovascular events [see Contraindications (4.3)].
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category C.
Tapentadol HCl was evaluated for teratogenic effects in pregnant rats and rabbits following intravenous and subcutaneous exposure during the period of embryofetal organogenesis. When tapentadol was administered twice daily by the subcutaneous route in rats at dose levels of 10, 20, or 40 mg/kg/day [producing up to 1 times the plasma exposure at the maximum recommended human dose (MRHD) of 700 mg/day based on an area under the time-curve (AUC) comparison], no teratogenic effects were observed. Evidence of embryofetal toxicity included transient delays in skeletal maturation (i.e. reduced ossification) at the 40 mg/kg/day dose which was associated with significant maternal toxicity. Administration of tapentadol HCl in rabbits at doses of 4, 10, or 24 mg/kg/day by subcutaneous injection [producing 0.2, 0.6, and 1.85 times the plasma exposure at the MRHD based on an AUC comparison] revealed embryofetal toxicity at doses ≥ 10 mg/kg/day. Findings included reduced fetal viability, skeletal delays and other variations. In addition, there were multiple malformations including gastroschisis/thoracogastroschisis, amelia/phocomelia, and cleft palate at doses ≥ 10 mg/kg/day and above, and ablepharia, encephalopathy, and spina bifida at the high dose of 24 mg/kg/day. Embryofetal toxicity, including malformations, may be secondary to the significant maternal toxicity observed in the study.
In a study of pre- and postnatal development in rats, oral administration of tapentadol at doses of 20, 50, 150, or 300 mg/kg/day to pregnant and lactating rats during the late gestation and early postnatal period [resulting in up to 1.7 times the plasma exposure at the MRHD on an AUC basis] did not influence physical or reflex development, the outcome of neurobehavioral tests or reproductive parameters. Treatment-related developmental delay was observed, including incomplete ossification, and significant reductions in pup body weights and body weight gains at doses associated with maternal toxicity (150 mg/kg/day and above). At maternal tapentadol doses ≥ 150 mg/kg/day, a dose-related increase in pup mortality was observed through postnatal Day 4.
There are no adequate and well controlled studies of NUCYNTA® in pregnant women. NUCYNTA® should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
8.2 Labor and Delivery
The effect of tapentadol on labor and delivery in humans is unknown. NUCYNTA® is not recommended for use in women during and immediately prior to labor and delivery. Due to the mu-opioid receptor agonist activity of NUCYNTA®, neonates whose mothers have been taking NUCYNTA® should be monitored for respiratory depression. A specific opioid antagonist, such as naloxone, should be available for reversal of opioid induced respiratory depression in the neonate.
8.3 Nursing Mothers
There is insufficient/limited information on the excretion of tapentadol in human or animal breast milk. Physicochemical and available pharmacodynamic/toxicological data on tapentadol point to excretion in breast milk and risk to the suckling child cannot be excluded. NUCYNTA® should not be used during breast-feeding.
8.4 Pediatric Use
The safety and effectiveness of NUCYNTA® in pediatric patients less than 18 years of age have not been established. NUCYNTA® is not recommended in this population.
8.5 Geriatric Use
Of the total number of patients in Phase 2/3 double-blind, multiple-dose clinical studies of NUCYNTA®, 19% were 65 and over, while 5% were 75 and over. No overall differences in effectiveness were observed between these patients and younger patients. The rate of constipation was higher in subjects greater than or equal to 65 years than those less than 65 years (12% vs. 7%).
In general, recommended dosing for elderly patients with normal renal and hepatic function is the same as for younger adult patients with normal renal and hepatic function. Because elderly patients are more likely to have decreased renal and hepatic function, consideration should be given to starting elderly patients with the lower range of recommended doses [see Clinical Pharmacology (12.3)].
8.6 Renal Impairment
In patients with severe renal impairment, the safety and effectiveness of NUCYNTA® has not been established. NUCYNTA® is not recommended in this population [see Dosage and Administration (2.1)].
8.7 Hepatic Impairment
Administration of NUCYNTA® resulted in higher exposures and serum levels to tapentadol in subjects with impaired hepatic function compared to subjects with normal hepatic function [see Clinical Pharmacology (12.3)]. NUCYNTA® should be used with caution in patients with moderate hepatic impairment [see Dosage and Administration (2.2)].
NUCYNTA® has not been studied in patients with severe hepatic impairment, therefore, use of NUCYNTA® is not recommended in this population [see Warnings and Precautions (5.10)].
9 DRUG ABUSE AND DEPENDENCE
9.1 Controlled Substance
NUCYNTA® contains tapentadol, a mu-opioid agonist and is a Schedule II controlled substance. NUCYNTA® has an abuse potential similar to hydromorphone, can be abused and is subject to criminal diversion.
9.2 Abuse
Addiction is a primary, chronic, neurobiologic disease, with genetic, psychosocial, and environmental factors influencing its development and manifestations. It is characterized by behaviors that include one or more of the following: impaired control over drug use, compulsive use, continued use despite harm, and craving. Drug addiction is a treatable disease, utilizing a multidisciplinary approach, but relapse is common.
Concerns about abuse and addiction should not prevent the proper management of pain. However, all patients treated with opioids require careful monitoring for signs of abuse and addiction, because use of opioid analgesic products carries the risk of addiction even under appropriate medical use.
"Drug seeking" behavior is very common in addicts, and drug abusers. Drug-seeking tactics include emergency calls or visits near the end of office hours, refusal to undergo appropriate examination, testing or referral, repeated claims of loss of prescriptions, tampering with prescriptions and reluctance to provide prior medical records or contact information for other treating physician(s). "Doctor shopping" (visiting multiple prescribers) to obtain additional prescriptions is common among drug abusers and people suffering from untreated addiction. Preoccupation with achieving adequate pain relief can be appropriate behavior in a patient with poor pain control.
Abuse and addiction are separate and distinct from physical dependence and tolerance. Physicians should be aware that addiction may not be accompanied by concurrent tolerance and symptoms of physical dependence in all addicts. In addition, abuse of mu-opioid agonists can occur in the absence of true addiction and is characterized by misuse for non-medical purposes, often in combination with other psychoactive substances. Careful recordkeeping of prescribing information, including quantity, frequency, and renewal requests is strongly advised.
Abuse of NUCYNTA® poses a risk of overdose and death. This risk is increased with concurrent abuse of NUCYNTA® with alcohol and other substances. In addition, parenteral drug abuse is commonly associated with transmission of infectious diseases such as hepatitis and HIV.
Proper assessment of the patient, proper prescribing practices, periodic re-evaluation of therapy, and proper dispensing and storage are appropriate measures that help to limit abuse of drugs with mu-opioid agonist properties.
Infants born to mothers physically dependent on opioids will also be physically dependent and may exhibit respiratory difficulties and withdrawal symptoms [see Warnings and Precautions (5.1)]. Use of NUCYNTA® in this population has not been characterized. As NUCYNTA® has mu-opioid agonist activity, infants whose mothers have taken NUCYNTA®, should be carefully monitored.
9.3 Dependence
Tolerance is the need for increasing doses of opioids to maintain a defined effect such as analgesia (in the absence of disease progression or other external factors). Physical dependence is manifested by withdrawal symptoms after abrupt discontinuation of a drug or upon administration of an antagonist.
The opioid abstinence or withdrawal syndrome is characterized by some or all of the following: restlessness, lacrimation, rhinorrhea, yawning, perspiration, chills, myalgia, and mydriasis. Other symptoms also may develop, including irritability, anxiety, backache, joint pain, weakness, abdominal cramps, insomnia, nausea, anorexia, vomiting, diarrhea, increased blood pressure, respiratory rate, or heart rate.
Generally, tolerance and/or withdrawal are more likely to occur the longer a patient is on continuous opioid therapy. In a safety study where drug was administered up to 90 days, 82.7% of patients taking NUCYNTA® who stopped abruptly without initiating alternative therapy and were assessed 2 to 4 days after discontinuation, did not have objective signs of opioid withdrawal using the Clinical Opiate Withdrawal Scale. Moderate withdrawal symptoms were seen in 0.3% of patients with the rest (17%) experiencing mild symptoms. Withdrawal symptoms may be reduced by tapering NUCYNTA®.
10 OVERDOSAGE
10.1 Human Experience
Experience with NUCYNTA® overdose is very limited. Preclinical data suggest that symptoms similar to those of other centrally acting analgesics with mu-opioid agonist activity are to be expected upon intoxication with tapentadol. In principle, these symptoms may particularly appear in the clinical setting: miosis, vomiting, cardiovascular collapse, consciousness disorders up to coma, convulsions and respiratory depression up to respiratory arrest.
10.2 Management of Overdose
Management of overdose should be focused on treating symptoms of mu-opioid agonism. Primary attention should be given to re-establishment of a patent airway and institution of assisted or controlled ventilation when overdose of NUCYNTA® is suspected. Supportive measures (including oxygen and vasopressors) should be employed in the management of circulatory shock and pulmonary edema accompanying overdose as indicated. Cardiac arrest or arrhythmias may require cardiac massage or defibrillation.
Pure opioid antagonists, such as naloxone, are specific antidotes to respiratory depression resulting from opioid overdose. Respiratory depression following an overdose may outlast the duration of action of the opioid antagonist. Administration of an opioid antagonist is not a substitute for continuous monitoring of airway, breathing, and circulation following an opioid overdose. If the response to opioid antagonists is suboptimal or only brief in nature, an additional antagonist should be administered as directed by the manufacturer of the product.
Gastrointestinal decontamination may be considered in order to eliminate unabsorbed drug. Gastrointestinal decontamination with activated charcoal or by gastric lavage is only recommended within 2 hours after intake. Gastrointestinal decontamination at a later time point may be useful in case of intoxication with exceptionally large quantities. Before attempting gastrointestinal decontamination, care should be taken to secure the airway.
11 DESCRIPTION
NUCYNTA® (tapentadol) Tablets are immediate-release film-coated tablets for oral administration. The chemical name is 3-[(1R,2R)-3-(dimethylamino)-1-ethyl-2-methylpropyl]phenol monohydrochloride. The structural formula is:
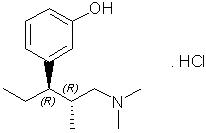
The molecular weight of tapentadol HCl is 257.80, and the molecular formula is C14H23NO•HCl. The n-octanol:water partition coefficient log P value is 2.87. The pKa values are 9.34 and 10.45. In addition to the active ingredient tapentadol HCl, tablets also contain the following inactive ingredients: microcrystalline cellulose, lactose monohydrate, croscarmellose sodium, povidone, magnesium stearate, and Opadry® II, a proprietary film-coating mixture containing polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, and aluminum lake coloring.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Tapentadol is a centrally-acting synthetic analgesic. Although its exact mechanism is unknown, analgesic efficacy is thought to be due to mu-opioid agonist activity and the inhibition of norepinephrine reuptake.
12.2 Pharmacodynamics
Tapentadol is a centrally-acting synthetic analgesic. It is 18 times less potent than morphine in binding to the human mu-opioid receptor and is 2–3 times less potent in producing analgesia in animal models. Tapentadol has been shown to inhibit norepinephrine reuptake in the brains of rats resulting in increased norepinephrine concentrations. In preclinical models, the analgesic activity due to the mu-opioid receptor agonist activity of tapentadol can be antagonized by selective mu-opioid antagonists (e.g., naloxone), whereas the norepinephrine reuptake inhibition is sensitive to norepinephrine modulators. Tapentadol exerts its analgesic effects without a pharmacologically active metabolite.
Effects on the cardiovascular system: There was no effect of therapeutic and supratherapeutic doses of tapentadol on the QT interval. In a randomized, double-blind, placebo- and positive-controlled crossover study, healthy subjects were administered five consecutive doses of NUCYNTA® 100 mg every 6 hours, NUCYNTA® 150 mg every 6 hours, placebo and a single oral dose of moxifloxacin. Similarly, NUCYNTA® had no relevant effect on other ECG parameters (heart rate, PR interval, QRS duration, T-wave or U-wave morphology).
12.3 Pharmacokinetics
Absorption
Mean absolute bioavailability after single-dose administration (fasting) is approximately 32% due to extensive first-pass metabolism. Maximum serum concentrations of tapentadol are typically observed at around 1.25 hours after dosing.
Dose-proportional increases in the Cmax and AUC values of tapentadol have been observed over the 50 to 150 mg dose range.
A multiple (every 6 hour) dose study with doses ranging from 75 to 175 mg tapentadol showed a mean accumulation factor of 1.6 for the parent drug and 1.8 for the major metabolite tapentadol-O-glucuronide, which are primarily determined by the dosing interval and apparent half-life of tapentadol and its metabolite.
Food Effect
The AUC and Cmax increased by 25% and 16%, respectively, when NUCYNTA® was administered after a high-fat, high-calorie breakfast. NUCYNTA® may be given with or without food.
Distribution
Tapentadol is widely distributed throughout the body. Following intravenous administration, the volume of distribution (Vz) for tapentadol is 540 +/- 98 L. The plasma protein binding is low and amounts to approximately 20%.
Metabolism and Elimination
In humans, the metabolism of tapentadol is extensive. About 97% of the parent compound is metabolized. Tapentadol is mainly metabolized via Phase 2 pathways, and only a small amount is metabolized by Phase 1 oxidative pathways. The major pathway of tapentadol metabolism is conjugation with glucuronic acid to produce glucuronides. After oral administration approximately 70% (55% O-glucuronide and 15% sulfate of tapentadol) of the dose is excreted in urine in the conjugated form. A total of 3% of drug was excreted in urine as unchanged drug. Tapentadol is additionally metabolized to N-desmethyl tapentadol (13%) by CYP2C9 and CYP2C19 and to hydroxy tapentadol (2%) by CYP2D6, which are further metabolized by conjugation. Therefore, drug metabolism mediated by cytochrome P450 system is of less importance than phase 2 conjugation.
None of the metabolites contributes to the analgesic activity.
Tapentadol and its metabolites are excreted almost exclusively (99%) via the kidneys. The terminal half-life is on average 4 hours after oral administration. The total clearance is 1530 +/- 177 ml/min.
Special Populations
Elderly
The mean exposure (AUC) to tapentadol was similar in elderly subjects compared to young adults, with a 16% lower mean Cmax observed in the elderly subject group compared to young adult subjects.
Renal Impairment
AUC and Cmax of tapentadol were comparable in subjects with varying degrees of renal function (from normal to severely impaired). In contrast, increasing exposure (AUC) to tapentadol-O-glucuronide was observed with increasing degree of renal impairment. In subjects with mild, moderate, and severe renal impairment, the AUC of tapentadol-O-glucuronide are 1.5-, 2.5-, and 5.5-fold higher compared with normal renal function, respectively.
Hepatic Impairment
Administration of NUCYNTA® resulted in higher exposures and serum levels to tapentadol in subjects with impaired hepatic function compared to subjects with normal hepatic function. The ratio of tapentadol pharmacokinetic parameters for the mild and moderate hepatic impairment groups in comparison to the normal hepatic function group were 1.7 and 4.2, respectively, for AUC; 1.4 and 2.5, respectively, for Cmax; and 1.2 and 1.4, respectively, for t1/2. The rate of formation of tapentadol-O-glucuronide was lower in subjects with increased liver impairment.
Pharmacokinetic Drug Interactions
Tapentadol is mainly metabolized by Phase 2 glucuronidation, a high capacity/low affinity system, therefore, clinically relevant interactions caused by Phase 2 metabolism are unlikely to occur. Naproxen and probenecid increased the AUC of tapentadol by 17% and 57%, respectively. These changes are not considered clinically relevant and no change in dose is required.
No changes in the pharmacokinetic parameters of tapentadol were observed when acetaminophen and acetylsalicylic acid were given concomitantly.
In vitro studies did not reveal any potential of tapentadol to either inhibit or induce cytochrome P450 enzymes. Thus, clinically relevant interactions mediated by the cytochrome P450 system are unlikely to occur.
The pharmacokinetics of tapentadol were not affected when gastric pH or gastrointestinal motility were increased by omeprazole and metoclopramide, respectively.
Plasma protein binding of tapentadol is low (approximately 20%). Therefore, the likelihood of pharmacokinetic drug-drug interactions by displacement from the protein binding site is low.
13 NON-CLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Tapentadol was administered to rats (diet) and mice (oral gavage) for two years.
In mice, tapentadol HCl was administered by oral gavage at dosages of 50, 100 and 200 mg/kg/day for 2 years (up to 0.2 times the plasma exposure at the maximum recommended human dose [MRHD] on an area under the time-curve [AUC] basis). No increase in tumor incidence was observed at any dose level.
In rats, tapentadol HCl was administered in diet at dosages of 10, 50, 125 and 250 mg/kg/day for two years (up to 0.2 times in the male rats and 0.6 times in the female rats the MRHD on an AUC basis). No increase in tumor incidence was observed at any dose level.
Mutagenesis
Tapentadol did not induce gene mutations in bacteria, but was clastogenic with metabolic activation in a chromosomal aberration test in V79 cells. The test was repeated and was negative in the presence and absence of metabolic activation. The one positive result for tapentadol was not confirmed in vivo in rats, using the two endpoints of chromosomal aberration and unscheduled DNA synthesis, when tested up to the maximum tolerated dose.
Impairment of Fertility
Tapentadol HCl was administered intravenously to male or female rats at dosages of 3, 6, or 12 mg/kg/day (representing exposures of up to approximately 0.4 times the exposure at the MRHD on an AUC basis, based on extrapolation from toxicokinetic analyses in a separate 4-week intravenous study in rats). Tapentadol did not alter fertility at any dose level. Maternal toxicity and adverse effects on embryonic development, including decreased number of implantations, decreased numbers of live conceptuses, and increased pre- and post-implantation losses occurred at dosages ≥6 mg/kg/day.
13.2 Animal Toxicology and/or Pharmacology
In toxicological studies with tapentadol, the most common systemic effects of tapentadol were related to the mu-opioid receptor agonist and norepinephrine reuptake inhibition pharmacodynamic properties of the compound. Transient, dose-dependent and predominantly CNS-related findings were observed, including impaired respiratory function and convulsions, the latter occurring in the dog at plasma levels (Cmax) which are in the range associated with the maximum recommended human dose (MRHD).
14 CLINICAL STUDIES
The efficacy and safety of NUCYNTA® in the treatment of moderate to severe acute pain has been established in two randomized, double-blind, placebo- and active-controlled studies of moderate to severe pain from first metatarsal bunionectomy and end-stage degenerative joint disease.
14.1 Orthopedic Surgery – Bunionectomy
A randomized, double-blind, parallel-group, active- and placebo-controlled, multiple-dose study demonstrated the efficacy of 50 mg, 75 mg, and 100 mg NUCYNTA® given every 4 to 6 hours for 72 hours in patients aged 18 to 80 years experiencing moderate to severe pain following unilateral, first metatarsal bunionectomy surgery. Patients who qualified for the study with a baseline pain score of ≥4 on an 11-point rating scale ranging from 0 to 10 were randomized to 1 of 5 treatments. Patients were allowed to take a second dose of study medication as soon as 1 hour after the first dose on study Day 1, with subsequent dosing every 4 to 6 hours. If rescue analgesics were required, the patients were discontinued for lack of efficacy. Efficacy was evaluated by comparing the sum of pain intensity difference over the first 48 hours (SPID48) versus placebo. NUCYNTA® at each dose provided a greater reduction in pain compared to placebo based on SPID48 values.
For various degrees of improvement from baseline to the 48-hour endpoint, Figure 1 shows the fraction of patients achieving that level of improvement. The figures are cumulative, such that every patient that achieves a 50% reduction in pain from baseline is included in every level of improvement below 50%. Patients who did not complete the 48-hour observation period in the study were assigned 0% improvement.
| Figure 1: Percentage of Patients Achieving Various Levels of Pain Relief as Measured by Pain Severity at 48 Hours Compared to Baseline- Post Operative Bunionectomy |
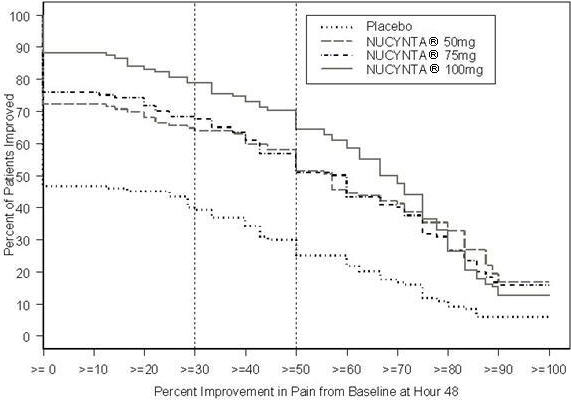 |
The proportions of patients who showed reduction in pain intensity at 48 hours of 30% or greater, or 50% or greater were significantly higher in patients treated with NUCYNTA® at each dose versus placebo.
14.2 End-Stage Degenerative Joint Disease
A randomized, double-blind, parallel-group, active- and placebo-controlled, multiple-dose study evaluated the efficacy and safety of 50 mg and 75 mg NUCYNTA® given every 4 to 6 hours during waking hours for 10 days in patients aged 18 to 80 years, experiencing moderate to severe pain from end stage degenerative joint disease of the hip or knee, defined as a 3-day mean pain score of ≥5 on an 11-point pain intensity scale, ranging from 0 to 10. Pain scores were assessed twice daily and assessed the pain the patient had experienced over the previous 12 hours. Patients were allowed to continue non-opioid analgesic therapy for which they had been on a stable regimen before screening throughout the study. Eighty-three percent (83%) of patients in the tapentadol treatment groups and the placebo group took such analgesia during the study. The 75 mg treatment group was dosed at 50 mg for the first day of the study, followed by 75 mg for the remaining nine days. Patients requiring rescue analgesics other than study medication were discontinued for lack of efficacy. Efficacy was evaluated by comparing the sum of pain intensity difference (SPID) versus placebo over the first five days of treatment. NUCYNTA® 50 mg and 75 mg provided improvement in pain compared with placebo based on the 5-Day SPID.
For various degrees of improvement from baseline to the Day 5 endpoint, Figure 2 shows the fraction of patients achieving that level of improvement. The figures are cumulative, such that every patient that achieves a 50% reduction in pain from baseline is included in every level of improvement below 50%. Patients who did not complete the 5-day observation period in the study were assigned 0% improvement.
| Figure 2: Percentage of Patients Achieving Various Levels of Pain Relief as Measured by Average Pain Severity for the Previous 12 hours, Measured on Study Day 5 Compared to Baseline -- End Stage Degenerative Joint Disease |
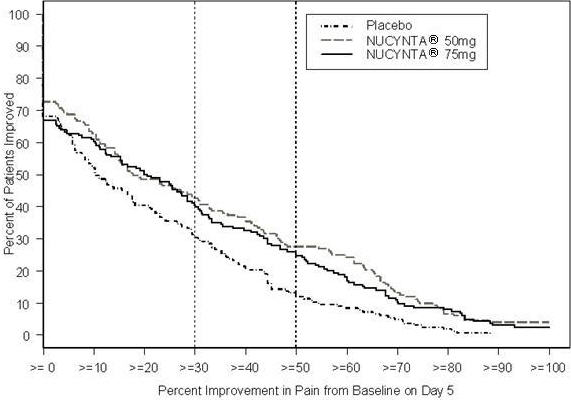 |
The proportions of patients who showed reduction in pain intensity at 5 days of 30% or greater, or 50% or greater were significantly higher in patients treated with NUCYNTA® at each dose versus placebo.
16 HOW SUPPLIED/STORAGE AND HANDLING
NUCYNTA® Tablets are available in the following strengths and packages. All tablets are round and biconvex-shaped.
50 mg tablets are yellow and debossed with "O-M" on one side and "50" on the other side, and are available in bottles of 100 (NDC 50458-820-04) and hospital unit dose blister packs of 10 (NDC 50458-820-02).
75 mg tablets are yellow-orange and debossed with "O-M" on one side and "75" on the other side, and are available in bottles of 100 (NDC 50458-830-04) and hospital unit dose blister packs of 10 (NDC 50458-830-02).
100 mg tablets are orange and debossed with "O-M" on one side and "100" on the other side, and are available in bottles of 100 (NDC 50458-840-04) and hospital unit dose blister packs of 10 (NDC 50458-840-02).
Store up to 25ºC (77ºF); excursions permitted to 15º – 30ºC (59º – 86ºF) [see USP Controlled Room Temperature]. Protect from moisture.
Keep out of reach of children.
17 PATIENT COUNSELING INFORMATION
Physicians are advised to discuss the following issues with patients for whom they prescribe NUCYNTA®:
17.1 Instructions for Use
Patients should be advised NUCYNTA® should be taken only as directed and to report episodes of breakthrough pain and adverse experiences occurring during therapy to their physician. Individualization of dosage is essential to make optimal use of this medication. Patients should be advised not to adjust the dose of NUCYNTA® without consulting their physician [see Dosage and Administration (2)]. Patients should be advised that it may be appropriate to taper dosing when discontinuing treatment with NUCYNTA® as withdrawal symptoms may occur [see Drug Abuse and Dependence (9.3)]. The physician can provide a dose schedule to accomplish a gradual discontinuation of the medication.
17.2 Misuse and Abuse
Patients should be advised that NUCYNTA® is a potential drug of abuse. Patients should protect NUCYNTA® from theft, and NUCYNTA® should never be given to anyone other than the individual for whom NUCYNTA® was prescribed [see Warnings and Precautions (5.4)].
17.3 Interference with Cognitive and Motor Performance
As NUCYNTA® has the potential to impair judgment, thinking, or motor skills, patients should be cautioned about operating hazardous machinery, including automobiles [see Warnings and Precautions (5.5)].
17.4 Pregnancy
Patients should be advised to notify their physician if they become pregnant or intend to become pregnant during treatment with NUCYNTA® [see Use in Specific Populations (8.1)].
17.5 Nursing
Patients should be advised not to breast-feed an infant during treatment with NUCYNTA® [see Use in Specific Populations (8.3)].
17.6 Monoamine Oxidase Inhibitors
Patients should be informed not to take NUCYNTA® while using any drugs that inhibit monoamine oxidase. Patients should not start any new medications while taking NUCYNTA® until they are assured by their healthcare provider that the new medication is not a monoamine oxidase inhibitor.
17.7 Seizures
Patients should be informed that NUCYNTA® could cause seizures if they are at risk for seizures or have epilepsy. Such patients should be advised to use NUCYNTA® with care [see Warnings and Precautions (5.7)]. Patients should be advised to stop taking NUCYNTA® if they have a seizure while taking NUCYNTA® and call their healthcare provider right away.
17.8 Serotonin Syndrome
Patients should be informed that NUCYNTA® could cause rare but potentially life-threatening conditions resulting from concomitant administration of serotonergic drugs (including Serotonin Reuptake Inhibitors, Serotonin and Norepinephrine Reuptake Inhibitors and tricyclic antidepressants) [see Warnings and Precautions (5.8)].
Patients should be advised to inform their physicians if they are taking, or plan to take, any prescription or over-the-counter drugs as there is a potential for interactions [see Drug Interactions (7)].
17.9 Alcohol
Patients should be advised to avoid alcohol while taking NUCYNTA® [see Drug Interactions (7.3)].
Revised: June 2010
Manufactured by:
Janssen Ortho, LLC
Gurabo, PR 00778
Manufactured for:
PriCara®, Division of Ortho-McNeil-Janssen Pharmaceuticals, Inc.
Raritan, NJ 08869
© Ortho-McNeil-Janssen Pharmaceuticals, Inc. 2009
MEDICATION GUIDE
NUCYNTA® (new-SINN-tah)
(tapentadol)
immediate-release oral tablets C-II
|
Read the Medication Guide that comes with NUCYNTA® before you start taking it and each time you get a new prescription. There may be new information. This Medication Guide does not take the place of talking to your doctor about your medical condition or your treatment. Talk to your doctor if you have any questions.
What is the most important information I should know about NUCYNTA®?
NUCYNTA® is a tablet that contains tapentadol, a strong medicine that is a pain medicine.
Use NUCYNTA® exactly how your doctor tells you to. Do not use NUCYNTA® if it has not been prescribed for you.
You should not take NUCYNTA® if your pain is mild and can be controlled with other pain medicines such as non-steroidal anti-inflammatory medicines (NSAIDS) or acetaminophen.
What is NUCYNTA®?
- NUCYNTA® is a prescription medicine that is used in adults 18 years of age or older to treat moderate to severe pain that is expected to last a short time.
NUCYNTA® is for short-term use only because the risks for withdrawal symptoms, abuse and addiction are higher when NUCYNTA® is used longer.
Who should not take NUCYNTA®?
Do not take NUCYNTA® if you:
- have severe lung problems
- have a gastrointestinal problem called paralytic ileus in which the intestines are not working normally.
- take a monoamine oxidase inhibitor (MAOI) medicine or have taken an MAOI within the last 14 days. Ask your doctor or pharmacist if any of your medicines is an MAOI.
What should I tell my doctor before taking NUCYNTA®?
NUCYNTA® may not be right for you. Tell your doctor about all your medical conditions, including if you have:
- trouble breathing or lung problems
- or had a head injury
- liver or kidney problems
- convulsions or seizures
- dependency problems with alcohol
- pancreas or gall bladder problems
- past or present substance abuse or drug addiction. There is a risk of abuse or addiction with narcotic pain medicines. If you have abused drugs in the past, you may have a higher chance of developing abuse or addiction again while using NUCYNTA®.
- are pregnant or plan to become pregnant
- are breast-feeding. You should not breast-feed while taking NUCYNTA®.
Tell your doctor about all the medicines you take, including prescription and nonprescription medicines, vitamins, and herbal supplements. Using NUCYNTA® with other medicines can cause serious side effects. The doses of some other medicines may need to be changed. Your doctor can tell you what medicines can be safely taken with NUCYNTA®. Especially tell your doctor if you take:
- Monoamine Oxidase Inhibitors (MAOIs). See "Who should not take NUCYNTA®."
- any medicine that makes you sleepy. NUCYNTA® can make you sleepy and affect your breathing. Taking these medicines together can be dangerous.
How should I take NUCYNTA®?
- Do not take NUCYNTA® unless it has been prescribed for you by your doctor.
- Take NUCYNTA® exactly as prescribed by your doctor.
- Do not change the dose of NUCYNTA® unless your doctor tells you to. Your doctor may change your dose after seeing how the medicine affects you. Do not use NUCYNTA® more often than prescribed. Call your doctor if your pain is not well controlled while taking NUCYNTA®.
- Follow your doctor's instructions about how to slowly stop taking NUCYNTA® to help lessen withdrawal symptoms.
- NUCYNTA® can be taken with or without food.
What should I avoid while taking NUCYNTA®?
- Do not drive, operate machinery, or participate in any other possibly dangerous activities until you know how you react to this medicine. NUCYNTA® can make you sleepy.
- You should not drink alcohol while using NUCYNTA®. Alcohol increases your chance of having dangerous side effects.
What are the possible side effects of NUCYNTA®?
NUCYNTA® can cause serious side effects including:
-
Life-threatening breathing problems. Call your doctor right away or get emergency medical help if you:
- have trouble breathing, or have slow or shallow breathing
- have a slow heartbeat
- have severe sleepiness
- have cold, clammy skin
- feel faint, dizzy, confused, or can not think, walk or talk normally
- have a seizure
- have hallucinations
- Physical Dependence. NUCYNTA® can cause physical dependence. Talk to your doctor about slowly stopping NUCYNTA® to avoid getting sick with withdrawal symptoms. You could become sick with uncomfortable symptoms because your body has become used to the medicine. Tell your doctor if you have any of these symptoms of withdrawal: feeling anxious, sweating, sleep problems, shivering, pain, nausea, tremors, diarrhea, upper respiratory symptoms, hallucinations, hair "standing on end." Physical dependence is not the same as drug addiction. Your doctor can tell you more about the differences between physical dependence and drug addiction.
- Serotonin syndrome. Serotonin syndrome is a rare, life-threatening problem that could happen if you take NUCYNTA® with Selective Serotonin Reuptake Inhibitors (SSRIs), Serotonin and Norepinephrine Reuptake Inhibitors (SNRIs), Monoamine Oxidase Inhibitors (MAOIs), triptans or certain other medicines. Call your doctor or get medical help right away if you have any one or more of the these symptoms: you feel agitated, have hallucinations, coma, rapid heart beat, feel overheated, loss of coordination, over active reflexes, nausea, vomiting, or diarrhea.
- Seizures. NUCYNTA® can cause seizures in people who are at risk for seizures or who have epilepsy. Tell your doctor right away if you have a seizure and stop taking NUCYNTA®.
- Low blood pressure. This can make you feel dizzy if you get up too fast from sitting or lying down.
The common side effects with NUCYNTA® are nausea, dizziness, vomiting, sleepiness, and itching.
Constipation is a common side effect of all opioid medicines. Talk to your doctor about the use of laxatives and stool softeners to prevent or treat constipation while taking NUCYNTA®.
Tell your doctor about any side effect that bothers you or that does not go away. These are not all the possible side effects of NUCYNTA®. For a complete list, ask your doctor or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store NUCYNTA®?
- Store NUCYNTA® at 59ºF to 86ºF (15ºC to 30ºC). Keep NUCYNTA® tablets dry.
- Dispose of NUCYNTA® tablets you no longer need.
Keep NUCYNTA® in a safe place out of the reach of children.
General information about NUCYNTA®
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use NUCYNTA® for a condition for which it was not prescribed. Do not give NUCYNTA® to other people, even if they have the same symptoms you have. Sharing NUCYNTA® could be harmful and is against the law.
This Medication Guide summarizes the most important information about NUCYNTA®. If you would like more information, talk with your doctor. You can ask your doctor or pharmacist for information about NUCYNTA® that is written for doctors. For more information about NUCYNTA® call 1-800-526-7736.
What are the ingredients in NUCYNTA®?
Active Ingredient: tapentadol
Inactive ingredients: microcrystalline cellulose, lactose monohydrate, croscarmellose sodium, povidone, magnesium stearate, and Opadry® II, a proprietary film-coating mixture containing polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, and aluminum lake coloring.
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Revised: June 2010
Manufactured by:
Janssen Ortho, LLC
Gurabo, PR 00778
Manufactured for:
PriCara®, Division of Ortho-McNeil-Janssen Pharmaceuticals, Inc.
Raritan, NJ 08869
© Ortho-McNeil-Janssen Pharmaceuticals, Inc. 2009
