FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
Ramelteon tablets are indicated for the treatment of insomnia characterized by difficulty with sleep onset.
The clinical trials performed in support of efficacy were up to six months in duration. The final formal assessments of sleep latency were performed after two days of treatment during the crossover study (elderly only), at five weeks in the six week studies (adults and elderly), and at the end of the six month study (adults and elderly) [see Clinical Studies (14)]
2 DOSAGE AND ADMINISTRATION
2.1 Dosage in Adults
The recommended dose of ramelteon tablets is 8 mg taken within 30 minutes of going to bed. It is recommended that ramelteon tablets not be taken with or immediately after a high-fat meal.
The total ramelteon tablets dose should not exceed 8 mg per day.
3 DOSAGE FORMS AND STRENGTHS
Ramelteon tablets, 8 mg are available as pale orange-yellow, round, film-coated tablets, debossed with “R” on one side and “8” on the other.
4 CONTRAINDICATIONS
Patients who develop angioedema after treatment with ramelteon tablets should not be rechallenged with the drug.
Patients should not take ramelteon tablets in conjunction with fluvoxamine [see Drug Interaction (7)].
5 WARNINGS AND PRECAUTIONS
5.1 Severe Anaphylactic and Anaphylactoid Reactions
Rare cases of angioedema involving the tongue, glottis or larynx have been reported in patients after taking the first or subsequent doses of ramelteon. Some patients have had additional symptoms such as dyspnea, throat closing, or nausea and vomiting that suggest anaphylaxis. Some patients have required medical therapy in the emergency department. If angioedema involves the tongue, glottis or larynx, airway obstruction may occur and be fatal. Patients who develop angioedema after treatment with ramelteon should not be rechallenged with the drug.
5.2 Need to Evaluate for Comorbid Diagnoses
Since sleep disturbances may be the presenting manifestation of a physical and/or psychiatric disorder, symptomatic treatment of insomnia should be initiated only after a careful evaluation of the patient. The failure of insomnia to remit after 7 to 10 days of treatment may indicate the presence of a primary psychiatric and/or medical illness that should be evaluated. Worsening of insomnia, or the emergence of new cognitive or behavioral abnormalities, may be the result of an unrecognized underlying psychiatric or physical disorder and requires further evaluation of the patient. Exacerbation of insomnia and emergence of cognitive and behavioral abnormalities were seen with ramelteon during the clinical development program.
5.3 Abnormal Thinking and Behavioral Changes
A variety of cognitive and behavior changes have been reported to occur in association with the use of hypnotics. In primarily depressed patients, worsening of depression (including suicidal ideation and completed suicides) has been reported in association with the use of hypnotics.
Hallucinations, as well as behavioral changes such as bizarre behavior, agitation and mania have been reported with ramelteon tablets use. Amnesia, anxiety and other neuro-psychiatric symptoms may also occur unpredictably.
Complex behaviors such as "sleep-driving" (i.e., driving while not fully awake after ingestion of a hypnotic) and other complex behaviors (e.g., preparing and eating food, making phone calls, or having sex), with amnesia for the event, have been reported in association with hypnotic use. The use of alcohol and other CNS depressants may increase the risk of such behaviors. These events can occur in hypnotic-naive as well as in hypnotic-experienced persons. Complex behaviors have been reported with the use of ramelteon. Discontinuation of ramelteon should be strongly considered for patients who report any complex sleep behavior.
5.4 CNS Effects
Patients should avoid engaging in hazardous activities that require concentration (such as operating a motor vehicle or heavy machinery) after taking ramelteon.
After taking ramelteon tablets, patients should confine their activities to those necessary to prepare for bed.
Patients should be advised not to consume alcohol in combination with ramelteon as alcohol and ramelteon may have additive effects when used in conjunction.
5.5 Reproductive Effects
Ramelteon tablets has been associated with an effect on reproductive hormones in adults, e.g., decreased testosterone levels and increased prolactin levels. It is not known what effect chronic or even chronic intermittent use of ramelteon may have on the reproductive axis in developing humans [see Clinical Trials (14.3)].
5.6 Use in Patients with Concomitant Illness
Ramelteon tablets has not been studied in subjects with severe sleep apnea and is not recommended for use in this population [see Use in Specific Populations (8.7)].
Ramelteon tablets should not be used by patients with severe hepatic impairment [see Clinical Pharmacology (12.4)].
5.7 Laboratory Tests
Monitoring
No standard monitoring is required.
For patients presenting with unexplained amenorrhea, galactorrhea, decreased libido, or problems with fertility, assessment of prolactin levels and testosterone levels should be considered as appropriate.
Interference with Laboratory Tests
Ramelteon tablets is not known to interfere with commonly used clinical laboratory tests. In addition, in vitro data indicate that ramelteon does not cause false-positive results for benzodiazepines, opiates, barbiturates, cocaine, cannabinoids, or amphetamines in two standard urine drug screening methods in vitro.
6 ADVERSE REACTIONS
The following serious adverse reactions are discussed in greater detail in other sections:
• Severe anaphylactic and anaphylactoid reactions [see Warnings and Precautions (5.1)]
• Abnormal thinking, behavior changes, and complex behaviors [see Warnings and Precautions (5.3)]
• CNS effects [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Adverse Reactions Resulting in Discontinuation of Treatment
The data described in this section reflect exposure to ramelteon in 5373 subjects, including 722 exposed for six months or longer, and 448 subjects for one year.
Six percent of the 5373 individual subjects exposed to ramelteon in clinical studies discontinued treatment owing to an adverse event, compared with 2% of the 2279 subjects receiving placebo. The most frequent adverse events leading to discontinuation in subjects receiving ramelteon were somnolence, dizziness, nausea, fatigue, headache, and insomnia; all of which occurred in 1% of the patients or less.
Ramelteon Most Commonly Observed Adverse Events
Table 1 displays the incidence of adverse events reported by the 2861 patients with chronic insomnia who participated in placebo-controlled trials of ramelteon. Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in clinical trials of other drugs, and may not reflect the rates observed in practice. The adverse reaction information from clinical trials does, however, provide a basis for identifying the adverse events that appear to be related to drug use and for approximating rates.
Table 1. Incidence (% of subjects) of Treatment-Emergent Adverse Events
| MedDRA Preferred Term | Placebo (n = 1456) | Ramelteon 8 mg (n = 1405) |
| Somnolence | 2% | 3% |
| Fatigue | 2% | 3% |
| Dizziness | 3% | 4% |
| Nausea | 2% | 3% |
| Insomnia exacerbated | 2% | 3% |
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on Ramelteon tablets
Fluvoxamine (strong CYP1A2 inhibitor)
AUC0-inf for ramelteon increased approximately 190-fold, and the Cmax increased approximately 70-fold upon coadministration of fluvoxamine and ramelteon, compared to ramelteon administered alone. Ramelteon should not be used in combination with fluvoxamine [see Contraindications (4), Clinical Pharmacology (12.5)]. Other less strong CYP1A2 inhibitors have not been adequately studied. Ramelteon tablets should be administered with caution to patients taking less strong CYP1A2 inhibitors.
Rifampin (strong CYP enzyme inducer)
Administration of multiple doses of rifampin resulted in a mean decrease of approximately 80% in total exposure to ramelteon tablets and metabolite M-II. Efficacy may be reduced when ramelteon tablets is used in combination with strong CYP enzyme inducers such as rifampin [see Clinical Pharmacology (12.5)].
Ketoconazole (strong CYP3A4 inhibitor)
The AUC0-inf and Cmax of ramelteon tablets increased by approximately 84% and 36% upon coadministration of ketoconazole with ramelteon tablets. Ramelteon tablets should be administered with caution in subjects taking strong CYP3A4 inhibitors such as ketoconazole [see Clinical Pharmacology (12.5)].
Fluconazole (strong CYP2C9 inhibitor)
The AUC0-inf and Cmax of ramelteon tablets was increased by approximately 150% when ramelteon tablets was coadministered with fluconazole. Ramelteon tablets should be administered with caution in subjects taking strong CYP2C9 inhibitors such as fluconazole [see Clinical Pharmacology (12.5)].
Donepezil
The AUC0-inf and Cmax of ramelteon increased by approximately 100% and 87%, respectively upon coadministration of donepezil with ramelteon tablets. Patients should be closely monitored when ramelteon tablets is coadministered with donepezil [see Clinical Pharmacology (12.5)].
Doxepin
The AUC0-inf and Cmax of ramelteon increased by approximately 66% and 69%, respectively, upon coadministration of doxepin with ramelteon tablets. Patients should be closely monitored when ramelteon tablets is coadministered with doxepin [see Clinical Pharmacology (12.5)].
7.2 Effect of Alcohol on Ramelteon tablets
Alcohol by itself impairs performance and can cause sleepiness. Since the intended effect of ramelteon is to promote sleep, patients should be cautioned not to consume alcohol when using ramelteon [see Clinical Pharmacology (12.5)]. Use of the products in combination may have an additive effect.
7.3 Drug/Laboratory Test Interactions
Ramelteon is not known to interfere with commonly used clinical laboratory tests. In addition, in vitro data indicate that ramelteon does not cause false-positive results for benzodiazepines, opiates, barbiturates, cocaine, cannabinoids, or amphetamines in two standard urine drug screening methods in vitro.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Available data from postmarketing reports with ramelteon use in pregnant women have not identified a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. In animal studies, ramelteon produced evidence of developmental toxicity, including teratogenic effects, in rats at doses greater than 36 times the recommended human dose (RHD) of 8 mg/day based on body surface area (mg/m2) (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Animal Data
Oral administration of ramelteon tablets (10, 40, 150 or 600 mg/kg/day) to pregnant rats during the period of organogenesis was associated with increased incidences of fetal structural abnormalities (malformations and variations) at doses greater than 40 mg/kg/day. The no-effect dose is approximately 50 times the RHD based on mg/m2. Treatment of pregnant rabbits during the period of organogenesis produced no evidence of embryo-fetal toxicity at oral doses of up to 300 mg/kg/day (or up to 720 times the RHD based on mg/m2.
When rats were orally administered ramelteon tablets (30, 100, or 300 mg/kg/day) throughout gestation and lactation, growth retardation, developmental delay, and behavioral changes were observed in the offspring at doses greater than 30 mg/kg/day. The no-effect dose is 36 times the RHD based on mg/m2. Increased incidences of malformation and death among offspring were seen at the highest dose.
8.2 Lactation
Risk Summary
There are no data regarding the presence of ramelteon tablets or its metabolites in human milk, the effects on the breastfed infant, or the effects on milk production. Ramelteon and/or its metabolites are present in rat milk. When a drug is present in animal milk, it is likely that the drug will be present in human milk. Because of the mechanism of action of ramelteon, there is a potential risk for somnolence in a breastfed infant (see Clinical Considerations). The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for ramelteon tablets and any potential adverse effects on the breastfed infant from ramelteon tablets or from the underlying maternal condition.
Clinical Considerations
Infants exposed to ramelteon through breastmilk should be monitored for somnolence and feeding problems. A lactating woman may consider interrupting breastfeeding and pumping and discarding breast milk during treatment and for 25 hours (approximately 5 elimination half-lives) after ramelteon tablets administration in order to minimize drug exposure to a breastfed infant.
8.4 Pediatric Use
Safety and effectiveness of ramelteon tablets in pediatric patients have not been established. Further study is needed prior to determining that this product may be used safely in prepubescent and pubescent patients.
8.5 Geriatric Use
A total of 654 subjects in double-blind, placebo-controlled, efficacy trials who received ramelteon tablets were at least 65 years of age; of these, 199 were 75 years of age or older. No overall differences in safety or efficacy were observed between elderly and younger adult subjects.
A double-blind, randomized, placebo-controlled study in elderly subjects with insomnia (n=33) evaluated the effect of a single dose of ramelteon tablets on balance, mobility, and memory functions after middle of the night awakening. There is no information on the effect of multiple dosing. Night time dosing of ramelteon tablets 8 mg did not impair middle of the night balance, mobility, or memory functions relative to placebo. The effects on night balance in the elderly cannot be definitively known from this study.
8.6 Chronic Obstructive Pulmonary Disease
The respiratory depressant effect of ramelteon tablets was evaluated in a crossover design study of subjects (n=26) with mild to moderate COPD after administering a single 16 mg dose or placebo, and in a separate study (n=25), the effects of ramelteon tablets on respiratory parameters were evaluated after administering an 8 mg dose or placebo in a crossover design to patients with moderate to severe COPD, defined as patients who had forced expiratory volume at one second (FEV1)/forced vital capacity ratio of <70%, and a FEV1 <80% of predicted with <12% reversibility to albuterol. Treatment with a single dose of ramelteon has no demonstrable respiratory depressant effects in subjects with mild to severe COPD, as measured by arterial O2 saturation (SaO2). There is no available information on the respiratory effects of multiple doses of ramelteon in patients with COPD. The respiratory depressant effects in patients with COPD cannot be definitively known from this study.
8.7 Sleep Apnea
The effects of ramelteon tablets were evaluated after administering a 16 mg dose or placebo in a crossover design to subjects (n=26) with mild to moderate obstructive sleep apnea. Treatment with ramelteon tablets 16 mg for one night showed no difference compared with placebo on the Apnea/Hypopnea Index (the primary outcome variable), apnea index, hypopnea index, central apnea index, mixed apnea index, and obstructive apnea index. Treatment with a single dose of ramelteon tablets does not exacerbate mild to moderate obstructive sleep apnea. There is no available information on the respiratory effects of multiple doses of ramelteon tablets in patients with sleep apnea. The effects on exacerbation in patients with mild to moderate sleep apnea cannot be definitively known from this study.
Ramelteon tablets has not been studied in subjects with severe obstructive sleep apnea; use of ramelteon is not recommended in such patients.
8.8 Hepatic Impairment
Exposure to ramelteon tablets was increased by four-fold in subjects with mild hepatic impairment and by more than ten-fold in subjects with moderate hepatic impairment. Ramelteon tablets should be used with caution in patients with moderate hepatic impairment [see Clinical Pharmacology (12.4)]. Ramelteon tablets is not recommended in patients with severe hepatic impairment.
8.9 Renal Impairment
No effects on Cmax and AUC0-t of parent drug or M-II were seen. No adjustment of ramelteon tablets dosage is required in patients with renal impairment [see Clinical Pharmacology (12.4)].
9 DRUG ABUSE AND DEPENDENCE
Ramelteon tablets is not a controlled substance.
Discontinuation of ramelteon in animals or in humans after chronic administration did not produce withdrawal signs. Ramelteon does not appear to produce physical dependence.
Human Data
A laboratory abuse potential study was performed with ramelteon tablets [see Clinical Studies (14.2)].
Animal Data
Ramelteon did not produce any signals from animal behavioral studies indicating that the drug produces rewarding effects. Monkeys did not self-administer ramelteon and the drug did not induce a conditioned place preference in rats. There was no generalization between ramelteon and midazolam. Ramelteon did not affect rotorod performance, an indicator of disruption of motor function, and it did not potentiate the ability of diazepam to interfere with rotorod performance.
10 OVERDOSAGE
General symptomatic and supportive measures should be used, along with immediate gastric lavage where appropriate. Intravenous fluids should be administered as needed. As in all cases of drug overdose, respiration, pulse, blood pressure, and other appropriate vital signs should be monitored, and general supportive measures employed.
Hemodialysis does not effectively reduce exposure to ramelteon tablets. Therefore, the use of dialysis in the treatment of overdosage is not appropriate.
Poison Control Center
As with the management of all overdosage, the possibility of multiple drug ingestion should be considered. Contact a poison control center for current information on the management of overdosage.
11 DESCRIPTION
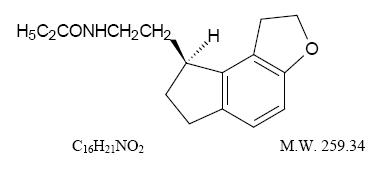
Ramelteon tablets is an orally active hypnotic chemically designated as (S)-N-[2-(1,6,7,8-tetrahydro-2H-indeno-[5,4-b]furan-8-yl)ethyl]propionamide and containing one chiral center. The compound is produced as the (S)-enantiomer, and has the following structural formula:
Ramelteon is freely soluble in organic solvents, such as methanol, ethanol, and dimethyl sulfoxide; soluble in 1-octanol and acetonitrile; and very slightly soluble in water and in aqueous buffers from pH 3 to pH 11.
Each ramelteon tablet for oral administration contains 8 mg ramelteon, and has the following inactive ingredients: hydroxypropyl cellulose, hypromellose, iron oxide red, iron oxide yellow, lactose monohydrate, magnesium stearate, pregelatinized corn starch, titanium dioxide, and triacetin.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Ramelteon is a melatonin receptor agonist with both high affinity for melatonin MT1 and MT2 receptors and relative selectivity over the MT3 receptor.
The activity of ramelteon at the MT1 and MT2 receptors is believed to contribute to its sleep-promoting properties, as these receptors, acted upon by endogenous melatonin, are thought to be involved in the maintenance of the circadian rhythm underlying the normal sleep-wake cycle.
Ramelteon has no appreciable affinity for the GABA receptor complex or for receptors that bind neuropeptides, cytokines, serotonin, dopamine, noradrenaline, acetylcholine, and opiates. Ramelteon also does not interfere with the activity of a number of selected enzymes in a standard panel.
The major metabolite of ramelteon, M-II, is pharmacologically active and has approximately one tenth and one fifth the binding affinity of the parent molecule for the human MT1 and MT2 receptors, respectively. However, M-II circulates at higher concentrations than the parent producing 20- to 100-fold greater mean systemic exposure when compared to ramelteon. Similar to ramelteon, M-II does not interfere with the activity of a number of endogenous enzymes.
All other known metabolites of ramelteon are inactive.
12.3 Pharmacokinetics
The pharmacokinetic profile of ramelteon tablets has been evaluated in healthy subjects as well as in subjects with hepatic or renal impairment. When administered orally to humans in doses ranging from 4 to 64 mg, ramelteon tablets undergoes rapid, high first-pass metabolism, and exhibits linear pharmacokinetics. Maximal serum concentration (Cmax) and area under the concentration-time curve (AUC) data show substantial intersubject variability, consistent with the high first-pass effect; the coefficient of variation for these values is approximately 100%. Several metabolites have been identified in human serum and urine.
Absorption
Ramelteon tablets is absorbed rapidly, with median peak concentrations occurring at approximately 0.75 hour (range, 0.5 to 1.5 hours) after fasted oral administration. Although the total absorption of ramelteon tablet is at least 84%, the absolute oral bioavailability is only 1.8% due to extensive first-pass metabolism.
Distribution
In vitro protein binding of ramelteon tablets is approximately 82% in human serum, independent of concentration. Binding to albumin accounts for most of that binding, since 70% of the drug is bound in human serum albumin. Ramelteon is not distributed selectively to red blood cells.
Ramelteon has a mean volume of distribution after intravenous administration of 73.6 L, suggesting substantial tissue distribution.
Metabolism
Metabolism of ramelteon consists primarily of oxidation to hydroxyl and carbonyl derivatives, with secondary metabolism producing glucuronide conjugates. CYP1A2 is the major isozyme involved in the hepatic metabolism of ramelteon; the CYP2C subfamily and CYP3A4 isozymes are also involved to a minor degree.
The rank order of the principal metabolites by prevalence in human serum is M-II, M-IV, M-I, and M-III. These metabolites are formed rapidly and exhibit a monophasic decline and rapid elimination. The overall mean systemic exposure of M-II is approximately 20- to 100-fold higher than parent drug.
Elimination
Following oral administration of radiolabeled ramelteon, 84% of total radioactivity was excreted in urine and approximately 4% in feces, resulting in a mean recovery of 88%. Less than 0.1% of the dose was excreted in urine and feces as the parent compound. Elimination was essentially complete by 96 hours postdose.
Repeated once daily dosing with ramelteon tablets does not result in significant accumulation owing to the short elimination half-life of ramelteon tablets (on average, approximately one to 2.6 hours).
The half-life of M-II is two to five hours and independent of dose. Serum concentrations of the parent drug and its metabolites in humans are at or below the lower limits of quantitation within 24 hours.
Effect of Food
When administered with a high-fat meal, the AUC0-inf for a single 16 mg dose of ramelteon tablets was 31% higher and the Cmax was 22% lower than when given in a fasted state. Median Tmax was delayed by approximately 45 minutes when ramelteon was administered with food. Effects of food on the AUC values for M-II were similar. It is therefore recommended that ramelteon tablets not be taken with or immediately after a high-fat meal [see Dosage and Administration (2.1)].
12.4 Pharmacokinetics in Special Populations
In a group of 24 elderly subjects aged 63 to 79 years administered a single ramelteon tablets 16 mg dose, the mean Cmax and AUC0-inf values were 11.6 ng/mL (SD, 13.8) and 18.7 ng•hr/mL (SD, 19.4), respectively. The elimination half-life was 2.6 hours (SD, 1.1). Compared with younger adults, the total exposure (AUC0-inf) and Cmax of ramelteon were 97 and 86% higher, respectively, in elderly subjects. The AUC0-inf and Cmax of M-II were increased by 30 and 13%, respectively, in elderly subjects.
Gender
There are no clinically meaningful gender-related differences in the pharmacokinetics of ramelteon tablets or its metabolites.
Hepatic Impairment
Exposure to ramelteon tablets was increased almost four-fold in subjects with mild hepatic impairment after seven days of dosing with 16 mg/day; exposure was further increased (more than ten-fold) in subjects with moderate hepatic impairment. Exposure to M-II was only marginally increased in mildly and moderately impaired subjects relative to healthy matched controls. The pharmacokinetics of ramelteon have not been evaluated in subjects with severe hepatic impairment (Child-Pugh Class C). Ramelteon tablets should be used with caution in patients with moderate hepatic impairment [see Warnings and Precautions (5.6)].
Renal Impairment
The pharmacokinetic characteristics of ramelteon tablets were studied after administering a 16 mg dose to subjects with mild, moderate, or severe renal impairment based on predose creatinine clearance (53 to 95, 35 to 49, or 15 to 30 mL/min/1.73 m2, respectively), and in subjects who required chronic hemodialysis. Wide intersubject variability was seen in ramelteon tablets exposure parameters. However, no effects on Cmax or AUC0-t of parent drug or M-II were seen in any of the treatment groups; the incidence of adverse events was similar across groups. These results are consistent with the negligible renal clearance of ramelteon, which is principally eliminated via hepatic metabolism. No adjustment of ramelteon tablets dosage is required in patients with renal impairment, including patients with severe renal impairment (creatinine clearance of ≤30 mL/min/1.73 m2) and patients who require chronic hemodialysis.
12.5 Drug-Drug Interactions
Ramelteon tablets has a highly variable intersubject pharmacokinetic profile (approximately 100% coefficient of variation in Cmax and AUC). As noted above, CYP1A2 is the major isozyme involved in the metabolism of ramelteon tablets; the CYP2C subfamily and CYP3A4 isozymes are also involved to a minor degree.
Effects of Other Drugs on Ramelteon tablets Metabolism
Fluvoxamine (strong CYP1A2 inhibitor)
When fluvoxamine 100 mg twice daily was administered for three days prior to single-dose coadministration of ramelteon tablets 16 mg and fluvoxamine, the AUC0-inf for ramelteon tablets increased approximately 190-fold, and the Cmax increased approximately 70-fold, compared to ramelteon tablets administered alone. Ramelteon tablets should not be used in combination with fluvoxamine. Other less strong CYP1A2 inhibitors have not been adequately studied. Ramelteon tablets should be administered with caution to patients taking less strong CYP1A2 inhibitors [see Contraindications (4), Drug Interactions (7)].
Rifampin (strong CYP enzyme inducer)
Administration of rifampin 600 mg once daily for 11 days resulted in a mean decrease of approximately 80% (40 to 90%) in total exposure to ramelteon tablets and metabolite M-II, (both AUC0-inf and Cmax) after a single 32 mg dose of ramelteon tablets. Efficacy may be reduced when ramelteon tablets is used in combination with strong CYP enzyme inducers such as rifampin [see Drug Interactions (7)].
Ketoconazole (strong CYP3A4 inhibitor)
The AUC0-inf and Cmax of ramelteon tablets increased by approximately 84% and 36%, respectively, when a single 16 mg dose of ramelteon tablets was administered on the fourth day of ketoconazole 200 mg twice daily administration, compared to administration of ramelteon alone. Similar increases were seen in M-II pharmacokinetic variables. Ramelteon tablets should be administered with caution in subjects taking strong CYP3A4 inhibitors such as ketoconazole [see Drug Interactions (7)].
Fluconazole (strong CYP2C9 inhibitor)
The total and peak systemic exposure (AUC0-inf and Cmax) of ramelteon after a single 16 mg dose of ramelteon tablets was increased by approximately 150% when administered with fluconazole. Similar increases were also seen in M-II exposure. Ramelteon should be administered with caution in subjects taking strong CYP2C9 inhibitors such as fluconazole [see Drug Interactions (7)].
Donepezil
Administration of donepezil 10 mg once daily for 26 days resulted in a mean increase of approximately 100% in overall exposure to ramelteon tablets, (AUC0-inf) and a mean increase of approximately 87% in maximum exposure to ramelteon tablets (Cmax) after a single 8 mg dose of ramelteon. No change was seen in M-II exposure. Patients should be closely monitored when ramelteon is coadministered with donepezil [see Drug Interactions (7)].
Doxepin
Administration of doxepin 10 mg once daily for 23 days resulted in a mean increase of approximately 66% in overall exposure to ramelteon tablets, (AUC0-inf) and a mean increase of approximately 69% in maximum exposure to ramelteon (Cmax) after a single 8 mg dose of ramelteon. No change was seen in M-II exposure. Patients should be closely monitored when ramelteon is coadministered with doxepin [see Drug Interactions (7)].
Interaction studies of concomitant administration of ramelteon tablets with fluoxetine (CYP2D6 inhibitor), omeprazole (CYP1A2 inducer/CYP2C19 inhibitor), theophylline (CYP1A2 substrate), dextromethorphan (CYP2D6 substrate), sertraline, venlafaxine, escitalopram, gabapentin, and zolpidem did not produce clinically meaningful changes in either peak or total exposures to ramelteon or the M-II metabolite.
Effects of Ramelteon tablets on Metabolism of Other Drugs
Zolpidem
Administration of ramelteon 8 mg once daily for 11 days resulted in an increase in median Tmax of zolpidem by approximately 20 minutes and exposure to zolpidem (both AUC0-inf and Cmax) was unchanged after a single 10 mg dose of zolpidem. Ordinarily zolpidem should not be given in a patient taking ramelteon tablets.
Concomitant administration of ramelteon tablets with omeprazole (CYP2C19 substrate), dextromethorphan (CYP2D6 substrate), midazolam (CYP3A4 substrate), theophylline (CYP1A2 substrate), digoxin (p-glycoprotein substrate), warfarin (CYP2C9 [S]/CYP1A2 [R] substrate), venlafaxine, fluvoxamine, donepezil, doxepin, sertraline, escitalopram, and gabapentin did not produce clinically meaningful changes in peak and total exposures to these drugs.
Effect of Alcohol on Ramelteon tablets
With single-dose, daytime coadministration of ramelteon tablets 32 mg and alcohol (0.6 g/kg), there were no clinically meaningful or statistically significant effects on peak or total exposure to ramelteon tablets. However, an additive effect was seen on some measures of psychomotor performance (i.e., the Digit Symbol Substitution Test, the Psychomotor Vigilance Task Test, and a Visual Analog Scale of Sedation) at some postdose time points. No additive effect was seen on the Delayed Word Recognition Test. Because alcohol by itself impairs performance, and the intended effect of ramelteon tablets is to promote sleep, patients should be cautioned not to consume alcohol when using ramelteon.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Ramelteon was administered to mice and rats at oral doses of 0, 30, 100, 300, or 1,000 mg/kg/day (mice) and 0, 15, 60, 250, or 1,000 mg/kg/day (rats). Mice and rats were dosed for two years, except at the high dose (94 weeks for male and female mice and female rats). In mice, dose-related increases in the incidence of hepatic tumors (adenomas, carcinomas, hepatoblastomas) were observed in males and females. The no-effect dose for hepatic tumors in mice (30 mg/kg/day) is approximately 20 times the recommended human dose (RHD) of 8 mg/day based on body surface area (mg/m2).
In rats, the incidence of hepatic adenoma and benign Leydig cell tumors of the testis was increased in males at doses ≥250 mg/kg/day. In females, the incidence of hepatic adenoma was increased at doses ≥60 mg/kg/day. The incidence of hepatic carcinoma was increased in males and female rats at 1,000 mg/kg/day. The no-effect dose for tumors in rats (15 mg/kg/day) is approximately 20 times the RHD based on mg/m2 basis.
Mutagenesis
Ramelteon was not genotoxic in the in vitro bacterial reverse mutation (Ames) assay, the in vitro mouse lymphoma TK+/-assay, and in in vivo oral micronucleus assays in mouse and rat. Ramelteon was clastogenic in the in vitro chromosomal aberration assay in Chinese hamster lung cells.
The M-II metabolite was not tested for genotoxicity. However, it was present in the test medium of the parent drug at concentrations higher than those of the parent.
Separate studies indicated that the concentration of the M-II metabolite formed in the presence of metabolic activation exceeded the concentration of ramelteon; therefore, the genotoxic potential of the M-II metabolite was also assessed in the in vitro studies.
Impairment of Fertility
When ramelteon (doses of 6 to 600 mg/kg/day) was administered orally to male and female rats prior to and during mating and early gestation, alterations in estrus cyclicity and decreased numbers of corpora lutea, implantations, and live embryos were observed at doses greater than 20 mg/kg/day. The no-effect dose is approximately 24 times the RHD of 8 mg/day based on mg/m2. Oral administration of ramelteon (up to 600 mg/kg/day) to male rats had no effects on sperm quality or reproductive performance.
14 CLINICAL STUDIES
14.1 Controlled Clinical Trials
Chronic Insomnia
Three randomized, double-blind trials in subjects with chronic insomnia employing polysomnography (PSG) were provided as objective support of ramelteon tablets effectiveness in sleep initiation.
One study enrolled younger adults (aged 18 to 64 years, inclusive) with chronic insomnia and employed a parallel design in which the subjects received a single, nightly dose of ramelteon tablets (8 or 16 mg) or matching placebo for 35 days. PSG was performed on the first two nights in each of Weeks 1, 3, and 5 of treatment. Ramelteon tablets reduced the average latency to persistent sleep at each of the time points when compared to placebo. The 16 mg dose conferred no additional benefit for sleep initiation.
The second study employing PSG was a three-period crossover trial performed in subjects aged 65 years and older with a history of chronic insomnia. Subjects received ramelteon tablets (4 or 8 mg) or placebo and underwent PSG assessment in a sleep laboratory for two consecutive nights in each of the three study periods. Both doses of ramelteon tablets reduced latency to persistent sleep when compared to placebo.
The third study evaluated long-term efficacy and safety in adults with chronic insomnia. Subjects received a single, nightly dose of ramelteon tablets 8 mg or matching placebo for six months. PSG was performed on the first two nights of Week 1 and Months 1, 3, 5, and 6. Ramelteon tablets reduced sleep latency at each time point when compared to placebo. In this study, when the PSG results from nights 1 and 2 of Month 7 were compared to the results from nights 22 and 23 of Month 6, there was a statistically significant increase in LPS of 33% (9.5 minutes) in the ramelteon group. There was no increase in LPS in the placebo group when the same time periods were compared.
A randomized, double-blind, parallel group study was conducted in outpatients aged 65 years and older with chronic insomnia and employed subjective measures of efficacy (sleep diaries). Subjects received ramelteon tablets (4 or 8 mg) or placebo for 35 nights. Ramelteon reduced patient-reported sleep latency compared to placebo. A similarly designed study performed in younger adults (aged 18 to 64 years) using 8 and 16 mg of ramelteon tablets did not replicate this finding of reduced patient-reported sleep latency compared to placebo.
While the 16 mg dose was evaluated as a potential treatment for adults, it was shown to confer no additional benefit for sleep initiation and was associated with higher incidences of fatigue, headache and next-day somnolence.
Transient Insomnia
In a randomized, double-blind, parallel-group trial using a first-night-effect model, healthy adults received placebo or ramelteon tablets before spending one night in a sleep laboratory and being evaluated with PSG. Ramelteon tablets demonstrated a decrease in mean latency to persistent sleep as compared to placebo.
14.2 Studies Pertinent to Safety Concerns for Sleep- promoting Drugs
Results from Human Laboratory Abuse Liability Studies
A human laboratory abuse potential study was performed in 14 subjects with a history of sedative/hypnotic or anxiolytic drug abuse. Subjects received single oral doses of ramelteon (16, 80, or 160 mg), triazolam (0.25, 0.50, or 0.75 mg) or placebo. All subjects received each of the seven treatments separated by a wash-out period and underwent multiple standard tests of abuse potential. No differences in subjective responses indicative of abuse potential were found between ramelteon and placebo at doses up to 20 times the recommended therapeutic dose. The positive control drug, triazolam, consistently showed a dose-response effect on these subjective measures, as demonstrated by the differences from placebo in peak effect and overall 24 hour effect.
Residual Pharmacological Effect in Insomnia Trials
In order to evaluate potential next-day residual effects, the following scales were used: a Memory Recall Test, a Word List Memory Test, a Visual Analog Mood and Feeling Scale, the Digit-Symbol Substitution Test, and a post sleep questionnaire to assess alertness and ability to concentrate. There was no evidence of next-day residual effect seen after two nights of ramelteon use during the crossover studies.
In a 35 night, double-blind, placebo-controlled, parallel-group study in adults with chronic insomnia, measures of residual effects were performed at three time points. Overall, the magnitudes of any observed differences were small. At Week 1, patients who received 8 mg of ramelteon had a mean VAS score (46 mm on a 100 mm scale) indicating more fatigue in comparison to patients who received placebo (42 mm). At Week 3, patients who received 8 mg of ramelteon had a lower mean score for immediate recall (7.5 out of 16 words) compared to patients who received placebo (8.2 words); and the patients treated with ramelteon had a mean VAS score indicating more sluggishness (27 mm on a 100 mm VAS) in comparison to the placebo-treated patients (22 mm). Patients who received ramelteon did not have next-morning residual effects that were different from placebo at Week 5.
Rebound Insomnia/Withdrawal
Potential rebound insomnia and withdrawal effects were assessed in four studies in which subjects received ramelteon or placebo for up to six months; three were 35 day studies, one was a six month study. These studies included a total of 2,533 subjects, of whom 854 were elderly.
Tyrer Benzodiazepine Withdrawal Symptom Questionnaire (BWSQ)
The BWSQ is a self-report questionnaire that solicits specific information on 20 symptoms commonly experienced during withdrawal from benzodiazepine receptor agonists; ramelteon is not a benzodiazepine receptor agonist.
In two of the three 35 day insomnia studies, the questionnaire was administered one week after completion of treatment; in the third study, the questionnaire was administered on Days 1 and 2 after completion. In all three of the 35 day studies, subjects receiving ramelteon 4, 8, or 16 mg daily reported BWSQ scores similar to those of subjects receiving placebo.
In the six month study, there was no evidence of withdrawal from the 8 mg dose as measured by the BWSQ.
Rebound Insomnia
Rebound insomnia was assessed in the 35 day studies by measuring sleep latency after abrupt treatment discontinuation. One of these studies employed PSG in younger adult subjects receiving ramelteon 8 or 16 mg; the other two studies employed subjective measures of sleep-onset insomnia in elderly subjects receiving ramelteon 4 or 8 mg, and in younger adult subjects receiving ramelteon 8 or 16 mg. There was no evidence that ramelteon caused rebound insomnia during the posttreatment period.
14.3 Studies to Evaluate Effect on Endocrine Function
Two controlled studies evaluated the effects of ramelteon tablets on endocrine function.
In the first trial, ramelteon tablets 16 mg once daily or placebo was administered to 99 healthy volunteer subjects for four weeks. This study evaluated the thyroid axis, adrenal axis and reproductive axis. No clinically significant endocrinopathies were demonstrated in this study. However, the study was limited in its ability to detect such abnormalities due to its limited duration.
In the second trial, ramelteon tablets 16 mg once daily or placebo was administered to 122 subjects with chronic insomnia for six months. This study evaluated the thyroid axis, adrenal axis and reproductive axis. There were no significant abnormalities seen in either the thyroid or the adrenal axes. Abnormalities were, however, noted within the reproductive axis. Overall, the mean serum prolactin level change from baseline was 4.9 mcg/L (34% increase) for women in the ramelteon group compared with -0.6 mcg/L (4% decrease) for women in the placebo group (p=0.003). No differences between active- and placebo-treated groups occurred among men. Thirty two percent of all patients who were treated with ramelteon in this study (women and men) had prolactin levels that increased from normal baseline levels compared to 19% of patients who were treated with placebo. Subject-reported menstrual patterns were similar between the two treatment groups.
In a 12 month, open-label study in adult and elderly patients, there were two patients who were noted to have abnormal morning cortisol levels, and subsequent abnormal ACTH stimulation tests. A 29 year old female patient was diagnosed with a prolactinoma. The relationship of these events to ramelteon therapy is not clear.
16 HOW SUPPLIED/STORAGE AND HANDLING
Ramelteon tablets, 8 mg are available as pale orange-yellow, round, film-coated tablets, debossed with “R” on one side and “8” on the other and are supplied in bottles of 30, 100, and 1,000.
Bottles of 30 NDC 43598-741-30
Bottles of 100 NDC 43598-741-01
Bottles of 1,000 NDC 43598-741-10
Store at 20° to 25°C (68° to 77°F) [See USP Controlled Room Temperature]. Keep container tightly closed and protected from moisture and humidity.
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Severe Anaphylactic and Anaphylactoid Reactions
Inform patients that severe anaphylactic and anaphylactoid reactions have occurred with ramelteon. Describe the relevant signs/symptoms and advise seeking immediate medical attention if any such things occur.
Sleep-Driving and other Complex Behaviors
There have been reports of people getting out of bed after taking a sleep medication and driving their cars while not fully awake, often with no memory of the event. If a patient experiences such an episode, it should be reported to his or her doctor immediately, since "sleep-driving" can be dangerous. This behavior is more likely to occur when sleep medications are taken with alcohol or other central nervous system depressants. Other complex behaviors (e.g., preparing and eating food, making phone calls, or having sex) have been reported in patients who are not fully awake after taking a sleep medication. As with sleep-driving, patients usually do not remember these events.
Endocrine Effects
Patients should consult their healthcare providers if they experience one of the following: cessation of menses or galactorrhea in females, decreased libido, or problems with fertility. Describe the relevant signs/symptoms and advise seeking medical attention if any such things occur.
Administration Instructions
• Patients should be advised to take ramelteon tablets within 30 minutes prior to going to bed and should confine their activities to those necessary to prepare for bed.
• Patients should be advised that they should not take ramelteon tablets with or immediately after a high-fat meal.
• Do not break the tablet; it should be swallowed whole.
Lactation
Advise mothers using ramelteon tablets to monitor neonates for signs of somnolence and feeding problems. A lactating woman may consider pumping and discarding breast milk during treatment and for 25 hours after ramelteon administration to minimize drug exposure to a breastfed infant [see Use in Specific Populations (8.2)].
RX only
Manufactured By:
Dr. Reddy’s Laboratories LA LLC
Shreveport, LA 71106 USA
Distributed By:
Dr. Reddy’s Laboratories Inc.,
Princeton, NJ 08540 USA
Issued: 12/2021
Medication Guide
Ramelteon Tablets
rah-MELL-tee-on
Read the Medication Guide that comes with ramelteon tablets before you start taking them and each time you get a refill. There may be new information. This Medication Guide does not take the place of talking to your doctor about your medical condition or treatment.
What is the most important information I should know about ramelteon tablets?
Ramelteon tablets may cause severe allergic reactions. Symptoms include swelling of the tongue or throat, trouble breathing, and nausea and vomiting. Get emergency medical help if you get these symptoms after taking ramelteon tablets.
After taking ramelteon tablets, you may get up out of bed while not being fully awake and do an activity that you do not know you are doing. The next morning, you may not remember that you did anything during the night. You have a higher chance for doing these activities if you drink alcohol or take other medicines that make you sleepy with ramelteon tablets. Activities may include:
- driving a car ("sleep-driving")
- making and eating food
- talking on the phone
- having sex
- sleep-walking
Call your doctor right away if you find out that you have done any of the above activities after taking ramelteon tablets.
Important:
1. Take ramelteon tablets exactly as prescribed
- Do not take more ramelteon tablets than prescribed.
- Take ramelteon tablets within 30 minutes of going to bed, not sooner.
2. Do not take ramelteon tablets if you:
- drink alcohol
- take other medicines that can make you sleepy. Talk to your doctor about all of your medicines. Your doctor will tell you if you can take ramelteon tablets with your other medicines
- cannot get a full night’s sleep
What are ramelteon tablets?
Ramelteon tablets are a hypnotic (sleep) medicine. Ramelteon tablets are used in adults for the treatment of the symptom of trouble falling asleep from insomnia.
Ramelteon tablets are not for children.
Who should not take ramelteon tablets?
Do not take ramelteon tablets if you are allergic to anything in it. See the end of this Medication Guide for a complete list of ingredients in ramelteon tablets.
Do not take ramelteon tablets if you are currently taking Luvox (fluvoxamine).
Ramelteon tablets may not be right for you. Before starting ramelteon tablets, tell your doctor about all of your health conditions, including if you:
- have a history of depression, mental illness, or suicidal thoughts
- have liver disease
- have a lung disease or breathing problems
- are pregnant, or planning to become pregnant, or breastfeeding
- are breastfeeding or plan to breastfeed. Ramelteon may cause somnolence in a breastfed infant. You may consider interrupting breastfeeding and pumping and discarding breastmilk during treatment and for 25 hours after administration of ramelteon tablets.
Tell your doctor about all of the medicines you take including prescription and nonprescription medicines, vitamins and herbal supplements. Medicines can interact with each other, sometimes causing serious side effects.
Do not take ramelteon tablets with:
- other medicines that can make you sleepy
- Luvox (fluvoxamine)
Know the medicines you take. Keep a list of your medicines with you to show your doctor and pharmacist each time you get a new medicine.
How should I take ramelteon tablets?
- Take ramelteon tablets exactly as prescribed. Do not take more ramelteon tablets than prescribed for you.
- Do not break the tablets. They should be swallowed whole.
- Take ramelteon tablets within 30 minutes of going to bed. After taking ramelteon tablets only do activities to get ready for bed.
- Do not take ramelteon tablets with or right after a meal.
- Do not take ramelteon tablets unless you are able to get a full night’s sleep before you must be active again.
- Call your doctor if your insomnia worsens or is not better within 7 to 10 days.
This may mean that there is another condition causing your sleep problems.
- If you take too much ramelteon tablets or overdose, call your doctor or poison control center right away, or get emergency treatment.
What are the possible side effects of ramelteon tablets?
Possible serious side effects of ramelteon tablets include:
- severe allergic reactions. Symptoms include swelling of the tongue or throat, trouble breathing, and nausea and vomiting. Get emergency medical help if you get these symptoms after taking ramelteon tablets.
- getting out of bed while not being fully awake and do an activity that you do not know you are doing. (See “What is the most important information I should know about ramelteon tablets?)
- abnormal thoughts and behavior. Symptoms include worsening of depression, suicidal thoughts or actions, nightmares, and hallucinations.
- hormone effects. Ramelteon tablets can decrease testosterone levels and increase prolactin levels in the blood. Symptoms of low testosterone or high prolactin levels are:
o decreased interest in sex
o problems getting pregnant
o irregular menstrual periods or no menstrual periods
o leakage of milk from the nipples of a person who is not breastfeeding
Call your doctor right away if you have any of the above side effects or any other side effects that worry you while using ramelteon tablets. Call your doctor for medical advice about side effects. You may report side effects to the FDA at 1-800-FDA-1088.
The most common side effects of ramelteon tablets are:
- drowsiness
- tiredness
- dizziness
- You may still feel drowsy the next day after taking ramelteon tablets. Do not drive or do other dangerous activities after taking ramelteon tablets until you feel fully awake.
These are not all the side effects of ramelteon tablets. Ask your doctor or pharmacist for more information.
How should I Store ramelteon tablets?
- Store at 20° to 25°C (68° to 77°F) [See USP Controlled Room Temperature]. Keep container tightly closed and protected from moisture and humidity.
- Keep ramelteon tablets and all medicines out of reach of children.
General Information about ramelteon tablets
- Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide.
- Do not use ramelteon tablets for a condition for which they were not prescribed.
- Do not share ramelteon tablets with other people, even if you think they have the same symptoms that you have. They may harm them.
This Medication Guide summarizes the most important information about ramelteon tablets. If you would like more information, talk with your doctor. You can ask your doctor or pharmacist for information about ramelteon tablets that is written for healthcare professionals. For more information, call Dr. Reddy’s Laboratories, Inc. at 1-888-375-3784.
What are the ingredients in ramelteon tablets?
Active Ingredient: ramelteon
Inactive Ingredients: hydroxypropyl cellulose, hypromellose, iron oxide red, iron oxide yellow, lactose monohydrate, magnesium stearate, pregelatinized corn starch, titanium dioxide, and triacetin.
Rx only
This Medication Guide has been approved by the U.S. Food and Drug Administration.
To reorder additional Medication Guides, contact Dr. Reddy’s Customer Service at 1-866-733-3952.
All other trademark names are the property of their respective owners.
RX only
Manufactured By:
Dr. Reddy’s Laboratories LA LLC
Shreveport, LA 71106 USA
Distributed By:
Dr. Reddy’s Laboratories Inc.,
Princeton, NJ 08540 USA
Issued: 12/2021