FULL PRESCRIBING INFORMATION
WARNING: SERIOUS SKIN REACTIONS
- • PADCEV can cause severe and fatal cutaneous adverse reactions including Stevens-Johnson syndrome (SJS) and Toxic Epidermal Necrolysis (TEN), which occurred predominantly during the first cycle of treatment, but may occur later.
- • Closely monitor patients for skin reactions.
- • Immediately withhold PADCEV and consider referral for specialized care for suspected SJS or TEN or severe skin reactions.
- • Permanently discontinue PADCEV in patients with confirmed SJS or TEN; or Grade 4 or recurrent Grade 3 skin reactions [see Dosage and Administration (2.2), Warnings and Precautions (5.1) and Adverse Reactions (6.1)].
1 INDICATIONS AND USAGE
PADCEV®, in combination with pembrolizumab, is indicated for the treatment of adult patients with locally advanced or metastatic urothelial cancer (mUC).
PADCEV, as a single agent, is indicated for the treatment of adult patients with locally advanced or mUC who:
- •
- have previously received a programmed death receptor-1 (PD-1) or programmed death-ligand 1 (PD-L1) inhibitor and platinum-containing chemotherapy, or
- •
- are ineligible for cisplatin-containing chemotherapy and have previously received one or more prior lines of therapy.
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
When given in combination with pembrolizumab, the recommended dose of PADCEV is 1.25 mg/kg (up to a maximum of 125 mg for patients ≥100 kg) administered as an intravenous infusion over 30 minutes on Days 1 and 8 of a 21-day cycle until disease progression or unacceptable toxicity. Refer to the pembrolizumab Prescribing Information for the recommended dosing information of pembrolizumab.
The recommended dose of PADCEV as a single agent is 1.25 mg/kg (up to a maximum of 125 mg for patients ≥100 kg) administered as an intravenous infusion over 30 minutes on Days 1, 8 and 15 of a 28-day cycle until disease progression or unacceptable toxicity.
2.2 Dose Modifications
| Adverse Reaction | Severity* | Dose Modification* |
|---|---|---|
|
||
|
Skin Reactions [see Boxed Warning, Warnings and Precautions (5.1)] |
For persistent or recurrent Grade 2 skin reactions |
Consider withholding until Grade ≤1, then resume treatment at the same dose level or dose reduce by one dose level. |
|
Grade 3 skin reactions |
Withhold until Grade ≤1, then resume treatment at the same dose level or dose reduce by one dose level. |
|
|
Suspected SJS or TEN |
Immediately withhold, consult a specialist to confirm the diagnosis. If not SJS/TEN, see Grade 2-4 skin reactions. |
|
|
Confirmed SJS or TEN; Grade 4 or recurrent Grade 3 skin reactions |
Permanently discontinue. |
|
|
Hyperglycemia [see Warnings and Precautions (5.2)] |
Blood glucose >250 mg/dL |
Withhold until elevated blood glucose has improved to ≤250 mg/dL, then resume treatment at the same dose level. |
|
Pneumonitis/Interstitial Lung Disease (ILD) [see Warnings and Precautions (5.3)] |
Grade 2 |
Withhold until Grade ≤1, then resume treatment at the same dose level or consider dose reduction by one dose level. |
|
Grade ≥3 |
Permanently discontinue. |
|
|
Peripheral Neuropathy [see Warnings and Precautions (5.4)] |
Grade 2 |
Withhold until Grade ≤1, then resume treatment at the same dose level (if first occurrence). For a recurrence, withhold until Grade ≤1, then resume treatment reduced by one dose level. |
|
Grade ≥3 |
Permanently discontinue. |
|
|
Other nonhematologic toxicity [see Adverse Reactions (6)] |
Grade 3 |
Withhold until Grade ≤1, then resume treatment at the same dose level or consider dose reduction by one dose level. |
|
Grade 4 |
Permanently discontinue. |
|
|
Hematologic toxicity [see Adverse Reactions (6)] |
Grade 3, or Grade 2 thrombocytopenia |
Withhold until Grade ≤1, then resume treatment at the same dose level or consider dose reduction by one dose level. |
|
Grade 4 |
Withhold until Grade ≤1, then reduce dose by one dose level or discontinue treatment. |
|
|
Dose Level |
|
|
Starting dose |
1.25 mg/kg up to 125 mg |
|
First dose reduction |
1.0 mg/kg up to 100 mg |
|
Second dose reduction |
0.75 mg/kg up to 75 mg |
|
Third dose reduction |
0.5 mg/kg up to 50 mg |
2.3 Instructions for Preparation and Administration
- •
- Administer PADCEV as an intravenous infusion only.
- •
- PADCEV is a hazardous drug. Follow applicable special handling and disposal procedures.1
Prior to administration, the PADCEV vial is reconstituted with Sterile Water for Injection (SWFI). The reconstituted solution is subsequently diluted in an intravenous infusion bag containing either 5% Dextrose Injection, USP, 0.9% Sodium Chloride Injection, USP, or Lactated Ringer’s Injection, USP.
Reconstitution in Single-dose Vial
- 1.
- Follow procedures for proper handling and disposal of anticancer drugs.
- 2.
- Use appropriate aseptic technique for reconstitution and preparation of dosing solutions.
- 3.
- Calculate the recommended dose based on the patient’s weight to determine the number and strength (20 mg or 30 mg) of vials needed.
- 4.
- Reconstitute each vial as follows and, if possible, direct the stream of SWFI along the walls of the vial and not directly onto the lyophilized powder:
- a.
- 20 mg vial: Add 2.3 mL of SWFI, resulting in 10 mg/mL PADCEV.
- b.
- 30 mg vial: Add 3.3 mL of SWFI, resulting in 10 mg/mL PADCEV.
- 5.
- Slowly swirl each vial until the contents are completely dissolved. Allow the reconstituted vial(s) to settle for at least 1 minute until the bubbles are gone. DO NOT SHAKE THE VIAL. Do not expose to direct sunlight.
- 6.
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. The reconstituted solution should be clear to slightly opalescent, colorless to light yellow and free of visible particles. Discard any vial with visible particles or discoloration.
- 7.
- Based upon the calculated dose amount, the reconstituted solution from the vial(s) should be added to the infusion bag immediately. This product does not contain a preservative. If not used immediately, reconstituted vials may be stored for up to 24 hours in refrigeration at 2°C to 8°C (36°F to 46°F). DO NOT FREEZE. Discard unused vials with reconstituted solution beyond the recommended storage time.
Dilution in Infusion Bag
- 8.
- Withdraw the calculated dose amount of reconstituted solution from the vial(s) and transfer into an infusion bag.
- 9.
- Dilute PADCEV with either 5% Dextrose Injection, 0.9% Sodium Chloride Injection, or Lactated Ringer's Injection. The infusion bag size should allow enough diluent to achieve a final concentration of 0.3 mg/mL to 4 mg/mL PADCEV.
- 10.
- Mix diluted solution by gentle inversion. DO NOT SHAKE THE BAG. Do not expose to direct sunlight.
- 11.
- Visually inspect the infusion bag for any particulate matter or discoloration prior to use. The reconstituted solution should be clear to slightly opalescent, colorless to light yellow and free of visible particles. DO NOT USE the infusion bag if particulate matter or discoloration is observed.
- 12.
- Discard any unused portion left in the single-dose vials.
Administration
- 13.
- Immediately administer the infusion over 30 minutes through an intravenous line.
- 14.
- If the infusion is not administered immediately, the prepared infusion bag should not be stored longer than 8 hours at 2°C to 8°C (36°F to 46°F). DO NOT FREEZE.
DO NOT administer PADCEV as an intravenous push or bolus.
DO NOT mix PADCEV with, or administer as an infusion with, other medicinal products.
3 DOSAGE FORMS AND STRENGTHS
For Injection: 20 mg and 30 mg of enfortumab vedotin-ejfv as a white to off-white lyophilized powder in a single-dose vial for reconstitution.
5 WARNINGS AND PRECAUTIONS
5.1 Skin Reactions
Severe cutaneous adverse reactions, including fatal cases of SJS or TEN occurred in patients treated with PADCEV. SJS and TEN occurred predominantly during the first cycle of treatment but may occur later.
Skin reactions occurred in 70% (all grades) of the 564 patients treated with PADCEV in combination with pembrolizumab in clinical trials. When PADCEV was given in combination with pembrolizumab, the incidence of skin reactions, including severe events, occurred at a higher rate compared to PADCEV as a single agent. The majority of the skin reactions that occurred with combination therapy included maculo-papular rash, macular rash and papular rash. Grade 3-4 skin reactions occurred in 17% of patients (Grade 3: 16%, Grade 4: 1%), including maculo-papular rash, bullous dermatitis, dermatitis, exfoliative dermatitis, pemphigoid, rash, erythematous rash, macular rash, and papular rash. A fatal reaction of bullous dermatitis occurred in one patient (0.2%). The median time to onset of severe skin reactions was 1.7 months (range: 0.1 to 17.2 months). Skin reactions led to discontinuation of PADCEV in 6% of patients [see Adverse Reactions (6.1)]. Of the patients who experienced a skin reaction and had data regarding resolution (N = 391), 59% had complete resolution and 41% had residual skin reactions at their last evaluation. Of the patients with residual skin reactions at last evaluation, 27% (43/159) had Grade ≥2 skin reactions.
Skin reactions occurred in 58% (all grades) of the 720 patients treated with PADCEV as a single agent in clinical trials. Twenty-three percent (23%) of patients had maculo-papular rash and 34% had pruritus. Grade 3-4 skin reactions occurred in 14% of patients, including maculo-papular rash, erythematous rash, rash or drug eruption, symmetrical drug-related intertriginous and flexural exanthema (SDRIFE), bullous dermatitis, exfoliative dermatitis, and palmar-plantar erythrodysesthesia. The median time to onset of severe skin reactions was 0.6 months (range: 0.1 to 8 months). Among patients experiencing a skin reaction leading to dose interruption who then restarted PADCEV (n=75), 24% of patients restarting at the same dose and 24% of patients restarting at a reduced dose experienced recurrent severe skin reactions. Skin reactions led to discontinuation of PADCEV in 3.1% of patients [see Adverse Reactions (6.1)]. Of the patients who experienced a skin reaction and had data regarding resolution (N =328), 58% had complete resolution and 42% had residual skin reactions at their last evaluation. Of the patients with residual skin reactions at last evaluation, 39% (53/137) had Grade ≥2 skin reactions.
Monitor patients closely throughout treatment for skin reactions. Consider topical corticosteroids and antihistamines, as clinically indicated.
For persistent or recurrent Grade 2 skin reactions, consider withholding PADCEV until Grade ≤1. Withhold PADCEV and refer for specialized care for suspected SJS, TEN or for Grade 3 skin reactions.
Permanently discontinue PADCEV in patients with confirmed SJS or TEN; or Grade 4 or recurrent Grade 3 skin reactions [see Dosage and Administration (2.2)].
5.2 Hyperglycemia
Hyperglycemia and diabetic ketoacidosis (DKA), including fatal events, occurred in patients with and without pre-existing diabetes mellitus, treated with PADCEV.
Patients with baseline hemoglobin A1C ≥8% were excluded from clinical trials.
In clinical trials of PADCEV as a single agent, 17% of the 720 patients treated with PADCEV developed hyperglycemia of any grade; 7% of patients developed Grade 3-4 hyperglycemia (Grade 3: 6.5%, Grade 4: 0.6%). Fatal events of hyperglycemia and diabetic ketoacidosis occurred in one patient each (0.1%). The incidence of Grade 3-4 hyperglycemia increased consistently in patients with higher body mass index and in patients with higher baseline A1C. The median time to onset of hyperglycemia was 0.5 months (range: 0 to 20 months). Hyperglycemia led to discontinuation of PADCEV in 0.7% of patients [see Adverse Reactions (6.1)]. Five percent (5%) of patients required initiation of insulin therapy for treatment of hyperglycemia. Of the patients who initiated insulin therapy for treatment of hyperglycemia, 66% (23/35) discontinued insulin by the time of last evaluation.
Closely monitor blood glucose levels in patients with, or at risk for, diabetes mellitus or hyperglycemia.
If blood glucose is elevated (>250 mg/dL), withhold PADCEV [see Dosage and Administration (2.2)].
5.3 Pneumonitis/Interstitial Lung Disease (ILD)
Severe, life-threatening or fatal pneumonitis/ILD occurred in patients treated with PADCEV.
When PADCEV was given in combination with pembrolizumab, 10% of the 564 patients treated with combination therapy had pneumonitis/ILD of any grade and 4% had Grade 3-4. A fatal event of pneumonitis/ILD occurred in two patients (0.4%). The incidence of pneumonitis/ILD, including severe events, occurred at a higher rate when PADCEV was given in combination with pembrolizumab compared to PADCEV as a single agent. The median time to onset of any grade pneumonitis/ILD was 4 months (range: 0.3 to 26 months).
In clinical trials of PADCEV as a single agent, 3% of the 720 patients treated with PADCEV had pneumonitis/ILD of any grade and 0.8% had Grade 3-4. The median time to onset of any grade pneumonitis/ILD was 2.9 months (range: 0.6 to 6 months).
Monitor patients for signs and symptoms indicative of pneumonitis/ILD such as hypoxia, cough, dyspnea or interstitial infiltrates on radiologic exams. Evaluate and exclude infectious, neoplastic and other causes for such signs and symptoms through appropriate investigations.
Withhold PADCEV for patients who develop Grade 2 pneumonitis/ILD and consider dose reduction. Permanently discontinue PADCEV in all patients with Grade 3 or 4 pneumonitis/ILD [see Dosage and Administration (2.2)].
5.4 Peripheral Neuropathy
When PADCEV was given in combination with pembrolizumab, 67% of the 564 patients treated with combination therapy had peripheral neuropathy of any grade, 36% had Grade 2 neuropathy, and 7% had Grade 3 neuropathy. The incidence of peripheral neuropathy occurred at a higher rate when PADCEV was given in combination with pembrolizumab compared to PADCEV as a single agent. The median time to onset of Grade ≥2 peripheral neuropathy was 6 months (range: 0.3 to 25 months) [see Adverse Reactions (6.1)]. Of the patients who experienced neuropathy and had data regarding resolution (N = 373), 13% had complete resolution, and 87% of patients had residual neuropathy at last evaluation. Of the patients with residual neuropathy at last evaluation, 45% (146/326) had Grade ≥2 neuropathy.
Peripheral neuropathy occurred in 53% of the 720 patients treated with PADCEV as a single agent in clinical trials including 38% with sensory neuropathy, 8% with muscular weakness and 7% with motor neuropathy. Thirty percent of patients experienced Grade 2 reactions and 5% experienced Grade 3-4 reactions. Peripheral neuropathy occurred in patients treated with PADCEV with or without preexisting peripheral neuropathy. The median time to onset of Grade ≥2 peripheral neuropathy was 4.9 months (range: 0.1 to 20 months). Neuropathy led to treatment discontinuation in 6% of patients [see Adverse Reactions (6.1)]. Of the patients who experienced neuropathy who had data regarding resolution (N = 296), 11% had complete resolution, and 89% had residual neuropathy at the time of their last evaluation. Of the patients with residual neuropathy at last evaluation, 50% (132/262) had Grade ≥2 neuropathy.
Monitor patients for symptoms of new or worsening peripheral neuropathy and consider dose interruption or dose reduction of PADCEV when peripheral neuropathy occurs.
Permanently discontinue PADCEV in patients who develop Grade >3 peripheral neuropathy [see Dosage and Administration (2.2)].
5.5 Ocular Disorders
Ocular disorders were reported in 40% of the 384 patients treated with PADCEV as a single agent in clinical trials in which ophthalmologic exams were scheduled. The majority of these events involved the cornea and included events associated with dry eye such as keratitis, blurred vision, increased lacrimation, conjunctivitis, limbal stem cell deficiency, and keratopathy.
Dry eye symptoms occurred in 30% of patients, and blurred vision occurred in 10% of patients, during treatment with PADCEV. The median time to onset to symptomatic ocular disorder was 1.7 months (range: 0 to 30.6 months). Monitor patients for ocular disorders. Consider artificial tears for prophylaxis of dry eyes and ophthalmologic evaluation if ocular symptoms occur or do not resolve. Consider treatment with ophthalmic topical steroids, if indicated after an ophthalmic exam. Consider dose interruption or dose reduction of PADCEV for symptomatic ocular disorders.
5.6 Infusion Site Extravasation
Skin and soft tissue reactions secondary to extravasation have been observed after administration of PADCEV. Of the 720 patients treated with PADCEV as a single agent in clinical trials, 1% of patients experienced skin and soft tissue reactions, including 0.3% who experienced Grade 3-4 reactions. Reactions may be delayed. Erythema, swelling, increased temperature, and pain worsened until 2-7 days after extravasation and resolved within 1-4 weeks of peak. Two patients (0.3%) developed extravasation reactions with secondary cellulitis, bullae, or exfoliation. Ensure adequate venous access prior to starting PADCEV and monitor for possible extravasation during administration. If extravasation occurs, stop the infusion and monitor for adverse reactions.
5.7 Embryo-Fetal Toxicity
Based on the mechanism of action and findings in animals, PADCEV can cause fetal harm when administered to a pregnant woman. In animal reproduction studies, administration of enfortumab vedotin-ejfv to pregnant rats during the period of organogenesis caused maternal toxicity, embryo-fetal lethality, structural malformations and skeletal anomalies at maternal exposures similar to the clinical exposures at the recommended human dose of 1.25 mg/kg.
Advise patients of the potential risk to the fetus. Advise female patients of reproductive potential to use effective contraception during treatment with PADCEV and for 2 months after the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with PADCEV and for 4 months after the last dose [see Use in Specific Populations (8.1, 8.3) and Clinical Pharmacology (12.1)].
6 ADVERSE REACTIONS
The following serious adverse reactions are described elsewhere in the labeling:
- •
- Skin Reactions [see Boxed Warning, Warnings and Precautions (5.1)]
- •
- Hyperglycemia [see Warnings and Precautions (5.2)]
- •
- Pneumonitis/Interstitial Lung Disease (ILD) [see Warnings and Precautions (5.3)]
- •
- Peripheral Neuropathy [see Warnings and Precautions (5.4)]
- •
- Ocular Disorders [see Warnings and Precautions (5.5)]
- •
- Infusion Site Extravasation [see Warnings and Precautions (5.6)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The pooled safety population described in the WARNINGS AND PRECAUTIONS reflect exposure to PADCEV in combination with pembrolizumab at 1.25 mg/kg in 564 patients in EV-302 and EV-103 and PADCEV as a single agent at 1.25 mg/kg in 720 patients in EV-301, EV-201, EV-203 (NCT04995419), EV-101 (NCT02091999), and EV-102 (NCT03070990). Ocular disorders reflect 384 patients in EV-201, EV-101, and EV-102. Among 564 patients receiving PADCEV in combination with pembrolizumab, 59% were exposed for >6 months, and 24% were exposed for ≥12 months. In this pooled population, the most common (>20%) adverse reactions, including laboratory abnormalities, were increased aspartate aminotransferase, increased creatinine, rash, increased glucose, peripheral neuropathy, increased lipase, decreased lymphocytes, increased alanine aminotransferase, decreased hemoglobin, fatigue, decreased sodium, decreased phosphate, decreased albumin, pruritus, diarrhea, alopecia, decreased weight, decreased appetite, increased urate, decreased neutrophils, decreased potassium, dry eye, nausea, constipation, increased potassium, dysgeusia, urinary tract infection and decreased platelets. Among 720 patients receiving PADCEV as a single agent, 37% were exposed for >6 months, and 14% were exposed for ≥12 months. In this pooled population, the most common (>20%) adverse reactions, including laboratory abnormalities, were increased glucose, increased aspartate aminotransferase, decreased lymphocytes, increased creatinine, rash, fatigue, peripheral neuropathy, decreased albumin, decreased hemoglobin, alopecia, decreased appetite, decreased neutrophils, decreased sodium, increased alanine aminotransferase, decreased phosphate, diarrhea, nausea, pruritus, increased urate, dry eye, dysgeusia, constipation, increased lipase, decreased weight, decreased platelets, abdominal pain, dry skin.
The data described in the following section reflects exposure to PADCEV in combination with pembrolizumab from EV-302 and the dose escalation cohort, Cohort A and Cohort K of EV-103. Patients received PADCEV 1.25 mg/kg in combination with pembrolizumab until disease progression or unacceptable toxicity.
The data described in the following section also reflects exposure to PADCEV as a single agent from an open-label, randomized, trial (EV‑301) and Cohort 1 and Cohort 2 of an open-label, single arm, two cohort trial (EV-201). Patients received PADCEV 1.25 mg/kg until disease progression or unacceptable toxicity.
Previously Untreated Locally Advanced or Metastatic Urothelial Cancer
EV-302
The safety of PADCEV in combination with pembrolizumab was evaluated in an open-label, randomized, multicenter trial (EV-302) in patients with locally advanced or metastatic urothelial cancer. Patients received either PADCEV 1.25 mg/kg and pembrolizumab (n=440) or gemcitabine and platinum chemotherapy (either cisplatin or carboplatin) (n=433). Among patients who received PADCEV and pembrolizumab, the median duration of exposure for PADCEV was 7 months (range: 0.3 to 31.9 months).
Serious adverse reactions occurred in 50% of patients treated with PADCEV in combination with pembrolizumab. The most common serious adverse reactions (≥2%) were rash (6%), acute kidney injury (5%), pneumonitis/ILD (4.5%), urinary tract infection (3.6%), diarrhea (3.2%) pneumonia (2.3%), pyrexia (2%), and hyperglycemia (2%).
Fatal adverse reactions occurred in 3.9% of patients treated with PADCEV in combination with pembrolizumab including acute respiratory failure (0.7%), pneumonia (0.5%), and pneumonitis/ILD (0.2%).
Adverse reactions leading to discontinuation of PADCEV occurred in 35% of patients. The most common adverse reactions (≥2%) leading to discontinuation of PADCEV were peripheral neuropathy (15%), rash (4.1%) and pneumonitis/ILD (2.3%).
Adverse reactions leading to dose interruption of PADCEV occurred in 73% of patients. The most common adverse reactions (≥2%) leading to dose interruption of PADCEV were peripheral neuropathy (22%), rash (16%), COVID-19 (10%), diarrhea (5%), pneumonitis/ILD (4.8%), fatigue (3.9%), hyperglycemia (3.6%), increased alanine aminotransferase (3%) and pruritus (2.5%).
Adverse reactions leading to dose reduction of PADCEV occurred in 42% of patients. The most common adverse reactions (≥2%) leading to dose reduction of PADCEV were rash (16%), peripheral neuropathy (13%) and fatigue (2.7%).
Table 3 summarizes the most common (≥15%) adverse reactions in EV-302.
| Adverse Reaction | PADCEV in combination with pembrolizumab
n=440 | Chemotherapy
n=433 |
||
|---|---|---|---|---|
| All Grades
% | Grade 3-4
% | All Grades
% | Grade 3-4
% |
|
|
||||
|
Skin and subcutaneous tissue disorders |
||||
|
Rash* |
68 |
15 |
15 |
0 |
|
Pruritus |
41 |
1.1 |
7 |
0 |
|
Alopecia |
35 |
0.5 |
8 |
0.2 |
|
Dry skin |
17 |
0.2 |
1 |
0 |
|
General disorders and administration site conditions |
||||
|
Fatigue* |
51 |
6 |
57 |
7 |
|
Pyrexia |
18 |
0.7 |
16 |
1.2 |
|
Nervous system disorders |
||||
|
Peripheral neuropathy* |
67 |
8 |
14 |
0 |
|
Dysgeusia |
21 |
0 |
9 |
0 |
|
Metabolism and nutrition disorders |
||||
|
Decreased appetite |
33 |
1.8 |
26 |
1.8 |
|
Gastrointestinal disorders |
||||
|
Diarrhea |
38 |
4.5 |
16 |
1.4 |
|
Nausea |
26 |
1.6 |
41 |
2.8 |
|
Constipation |
26 |
0 |
34 |
0.7 |
|
Investigations |
||||
|
Decreased weight |
33 |
3.6 |
9 |
0.2 |
|
Eye disorders |
||||
|
Dry eye* |
24 |
0 |
2.1 |
0 |
|
Infections and infestations |
||||
|
Urinary tract infection |
21 |
5 |
19 |
8 |
Clinically relevant adverse reactions (<15%) include vomiting (12%), pneumonitis/ILD (10%), hypothyroidism (10%), blurred vision (6%), infusion site extravasation (2%) and myositis (0.5%).
| Laboratory Abnormality | PADCEV in combination with pembrolizumab | Chemotherapy | ||
|---|---|---|---|---|
| All Grades*
% | Grade 3-4*
% | All Grades*
% | Grade 3-4*
% |
|
|
||||
|
Chemistry |
||||
|
Increased aspartate aminotransferase |
75 |
5 |
39 |
3 |
|
Increased creatinine |
71 |
3 |
68 |
3 |
|
Increased glucose |
66 |
14 |
54 |
5 |
|
Increased alanine aminotransferase |
59 |
5 |
49 |
3 |
|
Decreased sodium |
46 |
13 |
47 |
13 |
|
Decreased phosphate |
44 |
9 |
36 |
9 |
|
Decreased albumin |
39 |
2 |
35 |
0.5 |
|
Decreased potassium |
26 |
5 |
16 |
3 |
|
Increased potassium |
24 |
1 |
36 |
4 |
|
Increased calcium |
21 |
1 |
14 |
0.2 |
|
Hematology |
||||
|
Decreased lymphocytes |
58 |
15 |
59 |
17 |
|
Decreased hemoglobin |
53 |
7 |
89 |
33 |
|
Decreased neutrophils |
30 |
9 |
80 |
50 |
Previously Untreated Cisplatin Ineligible Patients with Locally Advanced or Metastatic Urothelial Cancer
EV-103
The safety of PADCEV was evaluated in combination with pembrolizumab in a multi cohort trial (EV-103) in 121 patients with locally advanced or metastatic urothelial cancer who were not eligible for cisplatin-containing chemotherapy and received at least one dose of PADCEV 1.25 mg/kg and pembrolizumab [see Clinical Studies (14)]. The median duration of exposure to PADCEV was 7 months (range: 0.6 to 33 months).
Serious adverse reactions occurred in 50% of patients treated with PADCEV in combination with pembrolizumab. The most common serious adverse reactions (≥2%) were acute kidney injury (7%), urinary tract infection (7%), urosepsis (5%), sepsis (3.3%), pneumonia (3.3%), hematuria (3.3%), pneumonitis/ILD (3.3%), urinary retention (2.5%), diarrhea (2.5%), myasthenia gravis (2.5%), myositis (2.5%), anemia (2.5%), and hypotension (2.5%).
Fatal adverse reactions occurred in 5% of patients treated with PADCEV in combination with pembrolizumab including sepsis (1.6%), bullous dermatitis (0.8%), myasthenia gravis (0.8%), and pneumonitis/ILD (0.8%).
Adverse reactions leading to discontinuation of PADCEV occurred in 36% of patients. The most common adverse reactions (≥2%) leading to discontinuation of PADCEV were peripheral neuropathy (20%) and rash (6%).
Adverse reactions leading to dose interruption of PADCEV occurred in 69% of patients. The most common adverse reactions (≥2%) leading to dose interruption of PADCEV were peripheral neuropathy (18%), rash (12%), increased lipase (6%), pneumonitis/ILD (6%), diarrhea (4.1%), acute kidney injury (3.3%), increased alanine aminotransferase (3.3%), fatigue (3.3%), neutropenia (3.3%), urinary tract infection (3.3%), increased amylase (2.5%), anemia (2.5%), COVID-19 (2.5%), hyperglycemia (2.5%), and hypotension (2.5%).
Adverse reactions leading to dose reduction of PADCEV occurred in 45% of patients. The most common adverse reactions (≥2%) leading to dose reduction of PADCEV were peripheral neuropathy (17%), rash (12%), fatigue (5%), neutropenia (5%), and diarrhea (4.1%).
Table 5 summarizes the most common (≥20%) adverse reactions in EV-103.
| Adverse Reaction | PADCEV in combination with pembrolizumab
n=121 |
|
|---|---|---|
| All Grades
% | Grade 3-4
% |
|
|
||
|
Skin and subcutaneous tissue disorders |
||
|
Rash* |
71 |
21 |
|
Alopecia |
52 |
0 |
|
Pruritus |
40 |
3.3 |
|
Dry skin |
21 |
0.8 |
|
Nervous system disorders |
||
|
Peripheral neuropathy* |
65 |
3.3 |
|
Dysgeusia |
35 |
0 |
|
Dizziness |
23 |
0 |
|
General disorders and administration site conditions |
||
|
Fatigue |
60 |
11 |
|
Peripheral edema |
26 |
0 |
|
Investigations |
||
|
Decreased weight |
48 |
5 |
|
Gastrointestinal disorders |
||
|
Diarrhea |
45 |
7 |
|
Nausea |
36 |
0.8 |
|
Constipation |
27 |
0 |
|
Metabolism and nutrition disorders |
||
|
Decreased appetite |
38 |
0.8 |
|
Infections and infestations |
||
|
Urinary tract infection |
30 |
12 |
|
Eye disorders |
||
|
Dry eye |
25 |
0 |
|
Musculoskeletal and connective tissue disorders |
||
|
Arthralgia |
23 |
1.7 |
Clinically relevant adverse reactions (<20%) include vomiting (19.8%), pyrexia (18%), hypothyroidism (11%), pneumonitis/ILD (10%), myasthenia gravis (2.5%), myositis (3.3%), and infusion site extravasation (0.8%).
| Laboratory Abnormality | PADCEV in combination with pembrolizumab | |
|---|---|---|
| All Grades*
% | Grade 3-4*
% |
|
|
||
|
Chemistry |
||
|
Increased glucose |
74 |
13 |
|
Increased aspartate aminotransferase |
73 |
9 |
|
Increased creatinine |
69 |
3.3 |
|
Decreased sodium |
60 |
19 |
|
Increased alanine aminotransferase |
60 |
7 |
|
Increased lipase |
59 |
32 |
|
Decreased albumin |
59 |
4.2 |
|
Decreased phosphate |
51 |
15 |
|
Decreased potassium |
35 |
8 |
|
Increased potassium |
27 |
1.7 |
|
Increased calcium |
27 |
4.2 |
|
Hematology |
||
|
Decreased hemoglobin |
69 |
15 |
|
Decreased lymphocytes |
64 |
17 |
|
Decreased neutrophils |
32 |
12 |
Previously Treated Locally Advanced or Metastatic Urothelial Cancer
EV-301
The safety of PADCEV was evaluated as a single agent in EV-301 in patients with locally advanced or metastatic urothelial cancer (n=296) who received at least one dose of PADCEV 1.25 mg/kg and who were previously treated with a PD-1 or PD-L1 inhibitor and a platinum-based chemotherapy [see Clinical Studies (14)]. Routine ophthalmologic exams were not conducted in EV-301. The median duration of exposure to PADCEV was 5 months (range: 0.5 to 19 months).
Serious adverse reactions occurred in 47% of patients treated with PADCEV. The most common serious adverse reactions (≥2%) were urinary tract infection, acute kidney injury (7% each) and pneumonia (5%). Fatal adverse reactions occurred in 3% of patients, including multiorgan dysfunction (1%), hepatic dysfunction, septic shock, hyperglycemia, pneumonitis/ILD and pelvic abscess (0.3% each).
Adverse reactions leading to discontinuation occurred in 17% of patients; the most common adverse reactions (≥2%) leading to discontinuation were peripheral neuropathy (5%) and rash (4%).
Adverse reactions leading to dose interruption occurred in 61% of patients; the most common adverse reactions (≥4%) leading to dose interruption were peripheral neuropathy (23%), rash (11%) and fatigue (9%).
Adverse reactions leading to dose reduction occurred in 34% of patients; the most common adverse reactions (≥2%) leading to dose reduction were peripheral neuropathy (10%), rash (8%), decreased appetite (3%) and fatigue (3%).
Table 7 summarizes the most common (≥15%) adverse reactions in EV-301.
| Adverse Reaction | PADCEV
n=296 | Chemotherapy
n=291 |
||
|---|---|---|---|---|
| All Grades
% | Grade 3-4
% | All Grades
% | Grade 3-4
% |
|
|
||||
|
Skin and subcutaneous tissue disorders | ||||
|
Rash* |
54 |
14 |
20 |
0.3 |
|
Alopecia |
47 |
0 |
38 |
0 |
|
Pruritus |
34 |
2 |
7 |
0 |
|
Dry skin |
17 |
0 |
4 |
0 |
|
General disorders and administration site conditions |
||||
|
Fatigue* |
50 |
9 |
40 |
7 |
|
Pyrexia* |
22 |
2 |
14 |
0 |
|
Nervous system disorders | ||||
|
Peripheral neuropathy* |
50 |
5 |
34 |
3 |
|
Dysgeusia* |
26 |
0 |
8 |
0 |
|
Metabolism and nutrition disorders | ||||
|
Decreased appetite |
41 |
5 |
27 |
2 |
|
Gastrointestinal disorders | ||||
|
Diarrhea* |
35 |
4 |
23 |
2 |
|
Nausea |
30 |
1 |
25 |
2 |
|
Constipation |
28 |
1 |
25 |
2 |
|
Abdominal Pain* |
20 |
1 |
14 |
3 |
|
Musculoskeletal and connective tissue disorders |
||||
|
Musculoskeletal Pain* |
25 |
2 |
35 |
5 |
|
Eye Disorders |
||||
|
Dry eye* |
24 |
0.7 |
6 |
0.3 |
|
Infections and infestations |
||||
|
Urinary Tract Infection* |
17 |
6 |
13 |
3 |
|
Vascular disorders |
||||
|
Hemorrhage* |
17 |
3 |
13 |
2 |
|
Investigations |
||||
|
Decreased weight |
16 |
0.3 |
7 |
0 |
Clinically relevant adverse reactions (<15%) include vomiting (14%), increased aspartate aminotransferase (12%), hyperglycemia (10%), increased alanine aminotransferase (9%), pneumonitis/ILD (3%) and infusion site extravasation (0.7%).
| Laboratory Abnormality | PADCEV* | Chemotherapy* | ||
|---|---|---|---|---|
| Grades 2-4
% | Grade 3-4
% | Grades 2-4
% | Grade 3-4
% |
|
|
||||
|
Hematology |
||||
|
Decreased lymphocytes |
41 |
14 |
34 |
18 |
|
Decreased hemoglobin |
28 |
4 |
42 |
14 |
|
Decreased neutrophils |
27 |
12 |
25 |
17 |
|
Chemistry |
||||
|
Decreased phosphate |
39 |
8 |
24 |
6 |
|
Increased glucose (non-fasting) |
33 |
9 |
27 |
6 |
|
Increased creatinine |
18 |
2 |
13 |
0 |
|
Decreased potassium |
16 |
2 |
7 |
3 |
|
Increased lipase |
13 |
8 |
7 |
4 |
|
Decreased sodium |
8 |
8 |
5 |
5 |
EV-201, Cohort 1
The safety of PADCEV was evaluated as a single agent in EV-201, Cohort 1 in patients (n=125) with locally advanced or metastatic urothelial cancer who had received prior treatment with a PD-1 or PD-L1 inhibitor and platinum-based chemotherapy [see Clinical Studies (14)]. Patients received PADCEV 1.25 mg/kg on Days 1, 8 and 15 of a 28-day cycle until disease progression or unacceptable toxicity. The median duration of exposure to PADCEV was 4.6 months (range: 0.5-15.6).
Serious adverse reactions occurred in 46% of patients treated with PADCEV. The most common serious adverse reactions (≥3%) were urinary tract infection (6%), cellulitis (5%), febrile neutropenia (4%), diarrhea (4%), sepsis (3%), acute kidney injury (3%), dyspnea (3%), and rash (3%). Fatal adverse reactions occurred in 3.2% of patients, including acute respiratory failure, aspiration pneumonia, cardiac disorder, sepsis and pneumonitis/ILD (each 0.8%).
Adverse reactions leading to discontinuation occurred in 16% of patients; the most common adverse reaction leading to discontinuation was peripheral neuropathy (6%).
Adverse reactions leading to dose interruption occurred in 64% of patients; the most common adverse reactions leading to dose interruption were peripheral neuropathy (18%), rash (9%) and fatigue (6%).
Adverse reactions leading to dose reduction occurred in 34% of patients; the most common adverse reactions leading to dose reduction were peripheral neuropathy (12%), rash (6%) and fatigue (4%).
Table 9 summarizes the All Grades and Grades 3-4 adverse reactions reported in patients in EV-201, Cohort 1.
| Adverse Reaction | PADCEV
n=125 |
|
|---|---|---|
| All Grades
% | Grade 3-4
% |
|
|
||
|
General disorders and administration site conditions |
||
|
Fatigue* |
56 |
6 |
|
Nervous system disorders |
||
|
Peripheral neuropathy* |
56 |
4 |
|
Dysgeusia |
42 |
0 |
|
Metabolism and nutrition disorders |
||
|
Decreased appetite |
52 |
2 |
|
Skin and subcutaneous tissue disorders |
||
|
Rash* |
52 |
13 |
|
Alopecia |
50 |
0 |
|
Dry skin |
26 |
0 |
|
Pruritus* |
26 |
2 |
|
Gastrointestinal disorders |
||
|
Nausea |
45 |
3 |
|
Diarrhea* |
42 |
6 |
|
Vomiting |
18 |
2 |
|
Eye disorders |
||
|
Dry eye* |
40 |
0 |
Clinically relevant adverse reactions (<15%) include herpes zoster (3%), pneumonitis/ILD (2%) and infusion site extravasation (2%).
| Laboratory Abnormality | PADCEV | |
|---|---|---|
| Grades 2-4*
% | Grade 3-4*
% |
|
|
Hematology |
||
|
Decreased hemoglobin |
34 |
10 |
|
Decreased lymphocytes |
32 |
10 |
|
Decreased neutrophils |
14 |
5 |
|
Chemistry |
||
|
Decreased phosphate |
34 |
10 |
|
Increased glucose (non-fasting) |
27 |
8 |
|
Increased creatinine |
20 |
2 |
|
Decreased potassium |
19† |
1 |
|
Increased lipase |
14 |
9 |
|
Decreased sodium |
8 |
8 |
|
Increased urate |
7 |
7 |
EV-201, Cohort 2
The safety of PADCEV was evaluated as a single agent in EV-201, Cohort 2 in patients with locally advanced or metastatic urothelial cancer (n=89) who received at least one dose of PADCEV 1.25 mg/kg and had prior treatment with a PD-1 or PD-L1 inhibitor and were not eligible for cisplatin-based chemotherapy. The median duration of exposure was 5.98 months (range: 0.3 to 24.6 months).
Serious adverse reactions occurred in 39% of patients treated with PADCEV. The most common serious adverse reactions (≥3%) were pneumonia, sepsis and diarrhea (5% each). Fatal adverse reactions occurred in 8% of patients, including acute kidney injury (2.2%), metabolic acidosis, sepsis, multiorgan dysfunction, pneumonia and pneumonitis/ILD (1.1% each).
Adverse reactions leading to discontinuation occurred in 20% of patients; the most common adverse reaction (≥2%) leading to discontinuation was peripheral neuropathy (7%).
Adverse reactions leading to dose interruption occurred in 60% of patients; the most common adverse reactions (≥3%) leading to dose interruption were peripheral neuropathy (19%), rash (9%), fatigue (8%), diarrhea (5%), increased aspartate aminotransferase (3%) and hyperglycemia (3%).
Adverse reactions leading to dose reduction occurred in 49% of patients; the most common adverse reactions (≥3%) leading to dose reduction were peripheral neuropathy (19%), rash (11%) and fatigue (7%).
Table 11 summarizes the All Grades and Grades 3-4 adverse reactions reported in patients in EV-201, Cohort 2.
| Adverse Reaction | PADCEV
n=89 |
|
|---|---|---|
| All Grades
(%) | Grades 3-4
(%) |
|
|
||
|
Skin and subcutaneous tissue disorders |
||
|
Rash* |
66 |
17 |
|
Alopecia |
53 |
0 |
|
Pruritus |
35 |
3 |
|
Dry skin |
19 |
1 |
|
Nervous system disorders |
||
|
Peripheral neuropathy* |
58 |
8 |
|
Dysgeusia* |
29 |
0 |
|
General disorders and administration site conditions |
||
|
Fatigue* |
48 |
11 |
|
Metabolism and nutrition disorders |
||
|
Decreased appetite |
40 |
6 |
|
Hyperglycemia |
16 |
9 |
|
Gastrointestinal disorders | ||
|
Diarrhea* |
36 |
8 |
|
Nausea |
30 |
1 |
|
Investigations | ||
|
Decreased weight |
35 |
1 |
|
Eye disorders | ||
|
Dry eye* |
30 |
0 |
Clinically relevant adverse reactions (<15%) include vomiting (13%), increased aspartate aminotransferase (12%), increased lipase (11%), increased alanine aminotransferase (10%), pneumonitis/ILD (4%) and infusion site extravasation (1%).
|
||
|
Laboratory Abnormality |
PADCEV n=88* |
|
|
Grades 2-4* % |
Grade 3-4* % |
|
|
Hematology |
||
|
Decreased lymphocytes |
43 |
15 |
|
Decreased hemoglobin |
34 |
5 |
|
Decreased neutrophils |
20 |
9 |
|
Chemistry |
||
|
Increased glucose (non-fasting) |
36 |
13 |
|
Decreased phosphate |
25 |
7 |
|
Increased creatinine |
23 |
3 |
|
Increased lipase |
18 |
11 |
|
Increased urate |
9 |
9 |
|
Increased potassium |
8 |
6 |
|
Decreased sodium |
7 |
7 |
6.2 Post Marketing Experience
The following adverse reactions have been identified during post-approval use of PADCEV. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Skin and subcutaneous tissue disorders: Epidermal necrosis, Stevens-Johnson syndrome, toxic epidermal necrolysis [see Warnings and Precautions (5.1)].
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on PADCEV
Dual P-gp and Strong CYP3A4 Inhibitors
Concomitant use with dual P-gp and strong CYP3A4 inhibitors may increase unconjugated MMAE exposure [see Clinical Pharmacology (12.3)], which may increase the incidence or severity of PADCEV toxicities. Closely monitor patients for signs of toxicity when PADCEV is given concomitantly with dual P-gp and strong CYP3A4 inhibitors.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on the mechanism of action and findings in animals, PADCEV can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. There are no available human data on PADCEV use in pregnant women to inform a drug-associated risk. In an animal reproduction study, administration of enfortumab vedotin-ejfv to pregnant rats during organogenesis caused maternal toxicity, embryo-fetal lethality, structural malformations and skeletal anomalies at maternal exposures similar to the exposures at the recommended human dose of 1.25 mg/kg (see Data). Advise patients of the potential risk to the fetus.
The background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2%-4% and 15%-20%, respectively.
Data
Animal Data
In a rat pilot embryo-fetal development study, administration of enfortumab vedotin-ejfv on gestation day 6 and 13 during the period of organogenesis resulted in a complete litter loss in all pregnant rats at the maternally toxic dose of 5 mg/kg (approximately 3 times the exposure at the recommended human dose). A dose of 2 mg/kg (similar to the exposure at the recommended human dose) resulted in maternal toxicity, embryo-fetal lethality and structural malformations that included gastroschisis, malrotated hindlimb, absent forepaw, malpositioned internal organs and fused cervical arch. Additionally, skeletal anomalies (asymmetric, fused, incompletely ossified, and misshapen sternebrae, misshapen cervical arch, and unilateral ossification of the thoracic centra) and decreased fetal weight were observed.
8.2 Lactation
Risk Summary
There are no data on the presence of enfortumab vedotin-ejfv in human milk, the effects on the breastfed child, or the effects on milk production. Because of the potential for serious adverse reactions in a breastfed child, advise lactating women not to breastfeed during treatment with PADCEV and for 3 weeks after the last dose.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Verify pregnancy status in females of reproductive potential prior to initiating PADCEV treatment [see Use in Specific Populations (8.1)].
Contraception
Females
PADCEV can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception during treatment with PADCEV and for 2 months after the last dose.
Males
Advise male patients with female partners of reproductive potential to use effective contraception during treatment with PADCEV and for 4 months after the last dose.
Infertility
Females
Based on findings in animal studies with MMAE-containing antibody-drug conjugates (ADCs), PADCEV may impair female fertility. The effect on fertility is reversible [see Nonclinical Toxicology (13.1)].
Males
Based on findings from animal studies, PADCEV may impair male fertility [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
Safety and effectiveness of PADCEV in pediatric patients have not been established.
8.5 Geriatric Use
Of the 564 patients treated with PADCEV in combination with pembrolizumab, 44% (n=247) were 65-74 years and 26% (n=144) were 75 years or older. Of the 720 patients treated with PADCEV as a single agent in clinical trials, 39% (n=282) were 65-74 years and 24% (n=170) were 75 years or older. No overall differences in effectiveness were observed between patients 65 years of age or older and younger patients.
Patients 75 years of age or older treated with PADCEV in combination with pembrolizumab experienced a higher incidence of fatal adverse reactions than younger patients. The incidence of fatal adverse reactions was 4% in patients younger than 75 and 7% in patients 75 years or older.
Patients 75 years of age or older treated with PADCEV as a single agent experienced a higher incidence of fatal adverse reactions than younger patients. The incidence of fatal adverse reactions was 6% in patients younger than 75 years, and 11% in patients 75 years or older.
No significant difference was observed in the pharmacokinetics of PADCEV between patients 65 years and older and younger patients [see Clinical Pharmacology (12.3)].
8.6 Hepatic Impairment
Avoid the use of PADCEV in patients with moderate or severe hepatic impairment (total bilirubin >1.5 x ULN and AST any). PADCEV has only been studied in a limited number of patients with moderate hepatic impairment (n=3) and has not been evaluated in patients with severe hepatic impairment [see Clinical Pharmacology (12.3)]. In another ADC that contains MMAE, the frequency of ≥ Grade 3 adverse reactions and deaths was greater in patients with moderate (Child-Pugh B) or severe (Child-Pugh C) hepatic impairment compared to patients with normal hepatic function.
11 DESCRIPTION
Enfortumab vedotin-ejfv is a Nectin-4 directed antibody-drug conjugate (ADC) comprised of a fully human anti-Nectin-4 IgG1 kappa monoclonal antibody (AGS-22C3) conjugated to the small molecule microtubule disrupting agent, monomethyl auristatin E (MMAE) via a protease-cleavable maleimidocaproyl valine-citrulline (vc) linker (SGD-1006). Conjugation takes place on cysteine residues that comprise the interchain disulfide bonds of the antibody to yield a product with a drug-to-antibody ratio of approximately 3.8:1. The molecular weight is approximately 152 kDa.
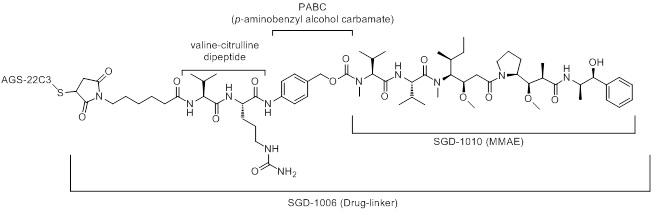
Approximately 4 molecules of MMAE are attached to each antibody molecule. Enfortumab vedotin-ejfv is produced by chemical conjugation of the antibody and small molecule components. The antibody is produced by mammalian (Chinese hamster ovary) cells and the small molecule components are produced by chemical synthesis.
PADCEV (enfortumab vedotin-ejfv) for injection is provided as a sterile, preservative-free, white to off-white lyophilized powder in single-dose vials for intravenous use. PADCEV is supplied as a 20 mg per vial and a 30 mg per vial and requires reconstitution with Sterile Water for Injection, USP, (2.3 mL and 3.3 mL, respectively) resulting in a clear to slightly opalescent, colorless to slightly yellow solution with a final concentration of 10 mg/mL [see Dosage and Administration (2.3)]. After reconstitution, each vial allows the withdrawal of 2 mL (20 mg) and 3 mL (30 mg). Each mL of reconstituted solution contains 10 mg of enfortumab vedotin-ejfv, histidine (1.4 mg), histidine hydrochloride monohydrate (2.31 mg), polysorbate 20 (0.2 mg) and trehalose dihydrate (55 mg) with a pH of 6.0.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Enfortumab vedotin-ejfv is an ADC. The antibody is a human IgG1 kappa directed against Nectin-4, an adhesion protein located on the surface of cells. The small molecule, MMAE, is a microtubule-disrupting agent, attached to the antibody via a protease-cleavable linker. Nonclinical data suggest that the anticancer activity of enfortumab vedotin-ejfv is due to the binding of the ADC to Nectin-4-expressing cells, followed by internalization of the ADC-Nectin-4 complex, and the release of MMAE via proteolytic cleavage. Release of MMAE disrupts the microtubule network within the cell, subsequently inducing cell cycle arrest and apoptosis. The combination of enfortumab vedotin-ejfv with a PD-1 blocking antibody resulted in up-regulation of immune function and increased anti-tumor activity in syngeneic mouse tumor models expressing Nectin-4.
12.2 Pharmacodynamics
In an exposure-response analysis for safety, higher enfortumab vedotin-ejfv exposure was associated with higher incidence of some adverse reactions (e.g., Grade ≥2 peripheral neuropathy, Grade ≥3 hyperglycemia). The exposure-response relationship for efficacy has not been fully characterized.
Cardiac Electrophysiology
At the recommended dose, PADCEV had no large QTc prolongation (>20 msec).
12.3 Pharmacokinetics
Enfortumab vedotin-ejfv (ADC) pharmacokinetics were characterized after single and multiple doses in patients with locally advanced or metastatic urothelial carcinoma and other solid tumors.
The pharmacokinetics of the ADC and unconjugated MMAE were consistent when assessed following PADCEV administration as a single agent and in combination with pembrolizumab after 1 treatment cycle.
The exposure parameters of the ADC and unconjugated MMAE (the cytotoxic component of enfortumab vedotin-ejfv) are summarized in Table 13 below. Peak ADC concentrations were observed near the end of intravenous infusion while peak unconjugated MMAE concentrations were observed approximately 2 days after PADCEV dosing. Minimal accumulation of the ADC and unconjugated MMAE was observed following repeat administration of PADCEV in patients. Steady-state concentrations of the ADC were reached after 1 treatment cycle for the ADC as a single agent and in combination with pembrolizumab.
| Cmax = maximum concentration, AUC0-28d = area under the concentration-time curve from time zero to 28 days, Ctrough,0-28d = pre-dose concentration on day 28 | ||
|
ADC Mean (± SD) |
Unconjugated MMAE Mean (± SD) |
|
|
Cmax |
28 (6.1) µg/mL |
5.5 (3.0) ng/mL |
|
AUC0-28d |
110 (26) µg∙d/mL |
85 (50) ng∙d/mL |
|
Ctrough,0-28d |
0.31 (0.18) µg/mL |
0.81 (0.88) ng/mL |
Distribution
The estimated mean steady-state volume of distribution of the ADC was 12.8 L following administration of PADCEV. In vitro, plasma protein binding of unconjugated MMAE ranged from 68% to 82%.
Elimination
The ADC and unconjugated MMAE exhibited multi-exponential declines with an elimination half-life of 3.6 days and 2.6 days, respectively. The mean clearance (CL) of the ADC and unconjugated MMAE was 0.11 L/h and 2.11 L/h, respectively. Elimination of unconjugated MMAE appeared to be limited by its rate of release from the ADC.
Metabolism
Catabolism of the ADC has not been studied in humans; however, it is expected to undergo catabolism to small peptides, amino acids, unconjugated MMAE, and unconjugated MMAE-related catabolites. The ADC releases MMAE via proteolytic cleavage, and unconjugated MMAE is primarily metabolized by CYP3A4 in vitro.
Excretion
The excretion of the ADC is not fully characterized. Following a single-dose of another ADC that contains unconjugated MMAE, 17% of the total unconjugated MMAE administered was recovered in feces and 6% in urine over a 1-week period, primarily as unchanged form. A similar excretion profile of unconjugated MMAE is expected after PADCEV administration.
Specific Populations
No clinically significant differences in the pharmacokinetics of the ADC or unconjugated MMAE were identified based on age (24 to 90 years), sex, race (Caucasian, Asian, or Black), renal impairment and mild hepatic impairment (total bilirubin of 1 to 1.5 × ULN and AST any, or total bilirubin ≤ULN and AST >ULN). The effect of end stage renal disease with or without dialysis and moderate or severe hepatic impairment (total bilirubin >1.5 x ULN and AST any) on the pharmacokinetics of the ADC or unconjugated MMAE is unknown.
Drug Interaction Trials
No clinical trials evaluating the drug-drug interaction potential of the ADC have been conducted.
Physiologically Based Pharmacokinetic (PBPK) Modeling Predictions:
Dual P-gp and Strong CYP3A4 Inhibitor: Concomitant use of PADCEV with ketoconazole (a dual P-gp and strong CYP3A4 inhibitor) is predicted to increase unconjugated MMAE Cmax by 15% and AUC by 38%.
Dual P-gp and Strong CYP3A4 Inducer: Concomitant use of PADCEV with rifampin (a dual P-gp and strong CYP3A4 inducer) is predicted to decrease unconjugated MMAE Cmax by 28% and AUC by 53%.
Sensitive CYP3A substrates: Concomitant use of PADCEV is predicted not to affect exposure to midazolam (a sensitive CYP3A substrate).
In Vitro Studies
Transporter Systems: MMAE is a substrate of P-glycoprotein (P-gp) and is not an inhibitor of P-gp.
12.6 Immunogenicity
The observed incidence of anti-drug antibody (ADA) is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of ADA in the studies described below with the incidence of ADA in other studies, including those of PADCEV or of other enfortumab vedotin products.
In the 0.3-to-55.7-month treatment periods with ADA sampling in eight clinical studies of PADCEV 1.25 mg/kg as a single agent on Days 1, 8 and 15 of a 28-day cycle and in combination with pembrolizumab on Days 1 and 8 of a 21-day cycle in patients with locally advanced or metastatic urothelial cancer [see Clinical Studies (14)], the incidence of anti-enfortumab vedotin-ejfv antibody formation was 3.6% (22 of 617 patients who were tested for ADA) for PADCEV as a single agent and 3.0% (14 of 466 patients who were tested for ADA) for PADCEV in combination with pembrolizumab.
Because of the low occurrence of ADA, the effect of the ADA on the pharmacokinetics, pharmacodynamics, safety and/or effectiveness of PADCEV is unknown.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies with enfortumab vedotin-ejfv or the small molecule cytotoxic agent (MMAE) have not been conducted.
MMAE was genotoxic in the rat bone marrow micronucleus study through an aneugenic mechanism. This effect is consistent with the pharmacological effect of MMAE as a microtubule-disrupting agent. MMAE was not mutagenic in the bacterial reverse mutation assay (Ames test) or the L5178Y mouse lymphoma forward mutation assay.
Fertility studies with enfortumab vedotin-ejfv or MMAE have not been conducted. However, results of repeat-dose toxicity studies indicate the potential for enfortumab vedotin-ejfv to impair female and male reproductive function and fertility.
In repeat-dose toxicology studies conducted in rats for up to 13 weeks, doses ≥2 mg/kg enfortumab vedotin-ejfv (at exposures similar to the exposures at the recommended human dose) resulted in decreases in testes and epididymis weights, seminiferous tubule degeneration, spermatid/spermatocyte depletion in the testes and cell debris, sperm granuloma and hypospermia/abnormal spermatids in the epididymis. Findings in the testes and epididymis did not reverse by the end of the recovery period.
MMAE-containing ADCs have been associated with adverse ovarian effects when administered to sexually immature animals. Adverse effects included decrease in, or absence of, secondary and tertiary ovarian follicles after weekly administration to cynomolgus monkeys in studies of 4-week duration. These effects showed a trend towards recovery 6 weeks after the end of dosing; no changes were observed in primordial follicles.
14 CLINICAL STUDIES
14.1 Metastatic Urothelial Cancer
Previously Untreated Locally Advanced or Metastatic Urothelial Cancer
EV-302
The efficacy of PADCEV in combination with pembrolizumab was evaluated in EV-302 (NCT04223856), an open label, randomized, multicenter trial that enrolled 886 patients with locally advanced or metastatic urothelial cancer who received no prior systemic therapy for locally advanced or metastatic disease. Patients with active CNS metastases, ongoing sensory or motor neuropathy Grade ≥2, or uncontrolled diabetes defined as hemoglobin A1C (HbA1c) ≥8% or HbA1c ≥7% with associated diabetes symptoms were excluded.
Patients were randomized 1:1 to receive either:
- •
- PADCEV 1.25 mg/kg on Days 1 and 8 of a 21-day cycle followed by pembrolizumab 200 mg on Day 1 of a 21-day cycle approximately 30 minutes after PADCEV. Treatment was continued until disease progression or unacceptable toxicity. In the absence of disease progression or unacceptable toxicity, pembrolizumab was continued for up to 2 years.
- •
- Gemcitabine 1000 mg/m2 on Days 1 and 8 of a 21-day cycle with cisplatin 70 mg/m2 or carboplatin (AUC = 4.5 or 5) on Day 1 of a 21-day cycle. Treatment was continued until disease progression or unacceptable toxicity for up to 6 cycles.
Randomization was stratified by cisplatin eligibility, PD-L1 expression, and presence of liver metastases.
The median age was 69 years (range: 22 to 91); 77% were male; 67% were White, 22% were Asian, 1% were Black or African American, and 10% were unknown or other; 12% were Hispanic or Latino. Patients had a baseline Eastern Cooperative Oncology Group (ECOG) performance status of 0 (49%), 1 (47%) or 2 (3%). Forty-seven percent of patients had a documented baseline HbA1c of <5.7%. At baseline, 95% of patients had metastatic urothelial cancer, including 72% with visceral and 22% with liver metastases, and 5% had locally advanced urothelial cancer. Eighty-five percent of patients had urothelial carcinoma (UC) histology including 6% with UC mixed squamous differentiation and 2% with UC mixed other histologic variants. Forty-six percent of patients were considered cisplatin-ineligible and 54% were considered cisplatin-eligible at time of randomization.
The major efficacy outcome measures were overall survival (OS) and progression-free survival (PFS) as assessed by blinded independent central review (BICR) according to Response Evaluation Criteria in Solid Tumors (RECIST) v1.1. Additional efficacy outcome measures included objective response rate (ORR) as assessed by BICR.
The trial demonstrated statistically significant improvements in OS, PFS, and ORR for patients randomized to PADCEV in combination with pembrolizumab as compared to platinum-based chemotherapy. Efficacy results were consistent across all stratified patient subgroups.
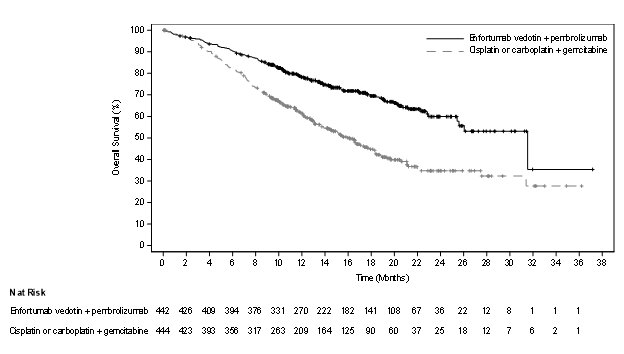
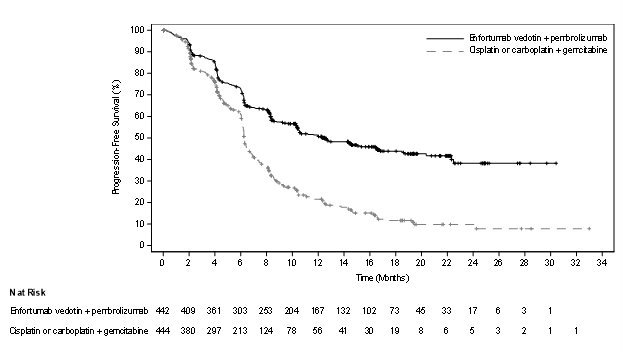
Table 14 and Figures 2-3 summarize the efficacy results for EV-302.
| Endpoint | PADCEV
with pembrolizumab n=442 | Cisplatin or carboplatin with gemcitabine
n=444 |
|---|---|---|
| NE = Not estimable. | ||
|
||
|
Overall Survival |
||
|
Number (%) of patients with events |
133 (30.1) |
226 (50.9) |
|
Median in months (95% CI) |
31.5 (25.4, NE) |
16.1 (13.9, 18.3) |
|
Hazard ratio (95% CI)* |
0.47 (0.38, 0.58) |
|
|
<0.0001 |
||
|
Progression Free Survival |
||
|
Number (%) of patients with events |
223 (50.5) |
307 (69.1) |
|
Median in months (95% CI) |
12.5 (10.4, 16.6) |
6.3 (6.2, 6.5) |
|
Hazard ratio (95% CI)* |
0.45 (0.38, 0.54) |
|
|
<0.0001 |
||
|
Confirmed Objective Response Rate§ |
||
|
ORR (%) (95% CI) |
67.7 (63.1, 72.1) |
44.4 (39.7, 49.2) |
|
<0.0001 |
||
|
Complete response rate (%) |
29.1 |
12.5 |
|
Partial response rate (%) |
38.7 |
32.0 |
Cisplatin Ineligible Patients with Previously Untreated Locally Advanced or Metastatic Urothelial Cancer
EV-103
The efficacy of PADCEV in combination with pembrolizumab was evaluated in EV-103 (NCT03288545), an open-label, multi-cohort (dose escalation cohort, Cohort A, Cohort K) trial in patients with locally advanced or metastatic urothelial cancer who were ineligible for cisplatin-containing chemotherapy and received no prior systemic therapy for locally advanced or metastatic disease. Patients with active CNS metastases, ongoing sensory or motor neuropathy Grade ≥2, or uncontrolled diabetes defined as hemoglobin A1C (HbA1c) ≥8% or HbA1c ≥7% with associated diabetes symptoms were excluded from participating in the trial.
Patients in the dose escalation cohort (n=5), Cohort A (n=40), and Cohort K (n=76) received PADCEV 1.25 mg/kg as an IV infusion over 30 minutes on Days 1 and 8 of a 21-day cycle followed by pembrolizumab 200 mg as an IV infusion on Day 1 of a 21-day cycle approximately 30 minutes after PADCEV. Patients were treated until disease progression or unacceptable toxicity.
A total of 121 patients received PADCEV in combination with pembrolizumab. The median age was 71 years (range: 51 to 91); 74% were male; 85% were White, 5% were Black, 4% were Asian and 6% were other, unknown or not reported. Ten percent of patients were Hispanic or Latino. Forty-five percent of patients had an ECOG performance status of 1 and 15% had an ECOG performance status of 2. Forty-seven percent of patients had a documented baseline HbA1c of <5.7%. Reasons for cisplatin ineligibility included: 60% with baseline creatinine clearance of 30 ̶ 59 mL/min, 10% with ECOG PS of 2, 13% with Grade 2 or greater hearing loss, and 16% with more than one cisplatin-ineligibility criteria.
At baseline, 97.5% of patients had metastatic urothelial cancer and 2.5% of patients had locally advanced urothelial cancer. Thirty-seven percent of patients had upper tract disease. Eighty-four percent of patients had visceral metastasis at baseline including 22% with liver metastases. Thirty-nine percent of patients had transitional cell carcinoma (TCC) histology; 13% had TCC with squamous differentiation and 48% had TCC with other histologic variants.
The major efficacy outcome measures were ORR and DoR as assessed by BICR according to RECIST v1.1.
The median follow-up time for the dose escalation cohort + Cohort A was 44.7 months (range: 0.7 to 52.4) and for Cohort K was 14.8 months (range: 0.6 to 26.2).
Efficacy results are presented in Table 15 below.
| PADCEV in combination with pembrolizumab
n=121 |
|
|---|---|
|
Confirmed ORR (95% CI) |
68% (58.7, 76.0) |
|
12% |
|
55% |
The median duration of response for the dose escalation cohort + Cohort A was 22.1 months (range: 1.0+ to 46.3+) and for Cohort K was not reached (range: 1.2 to 24.1+).
Previously Treated Locally Advanced or Metastatic Urothelial Cancer
EV-301
The efficacy of PADCEV as a single agent was evaluated in EV-301 (NCT03474107), an open-label, randomized, multicenter trial that enrolled 608 patients with locally advanced or metastatic urothelial cancer who received prior treatment with a PD-1 or PD-L1 inhibitor and platinum-based chemotherapy. Patients were randomized 1:1 to receive either PADCEV 1.25 mg/kg on Days 1, 8 and 15 of a 28-day cycle or investigator’s choice of chemotherapy. Randomization was stratified by ECOG PS (0 vs 1), region of world (Western Europe vs US vs Rest of World), and presence of liver metastasis.
Patients were excluded if they had active central nervous system (CNS) metastases, ongoing sensory or motor neuropathy ≥Grade 2, or uncontrolled diabetes defined as hemoglobin A1C (HbA1c) ≥8% or HbA1c ≥7% with associated diabetes symptoms.
The median age was 68 years (range: 30 to 88 years) and 77% were male. Racial demographics were reported as White (52%), Asian (33%), Black (0.7%), Native Hawaiian or Other Pacific Islander (0.2%) or not reported (15%). Nine percent of patients were Hispanic or Latino. All patients had a baseline ECOG performance status of 0 (40%) or 1 (60%). Thirty-four percent of patients had tumors located in the upper tract that included the renal pelvis and ureter. Eighty percent of patients had visceral metastases including 31% with liver metastases. Seventy-six percent of patients had pure TCC histology; 14% had TCC with other histologic variants; and 10% had other tumor histologies including adenocarcinoma and squamous cell carcinoma. The median number of prior therapies was 2 (range 1 to ≥3). Sixty-three percent of patients received prior cisplatin-based regimens, 26% received prior carboplatin-based regimens, and an additional 11% received both cisplatin and carboplatin-based regimens. Patients on the control arm received docetaxel (38%), paclitaxel (36%) or vinflunine (25%).
The major efficacy outcome measures were OS, PFS, and ORR assessed by investigator using RECIST v1.1. Efficacy results were consistent across all stratified patient subgroups.
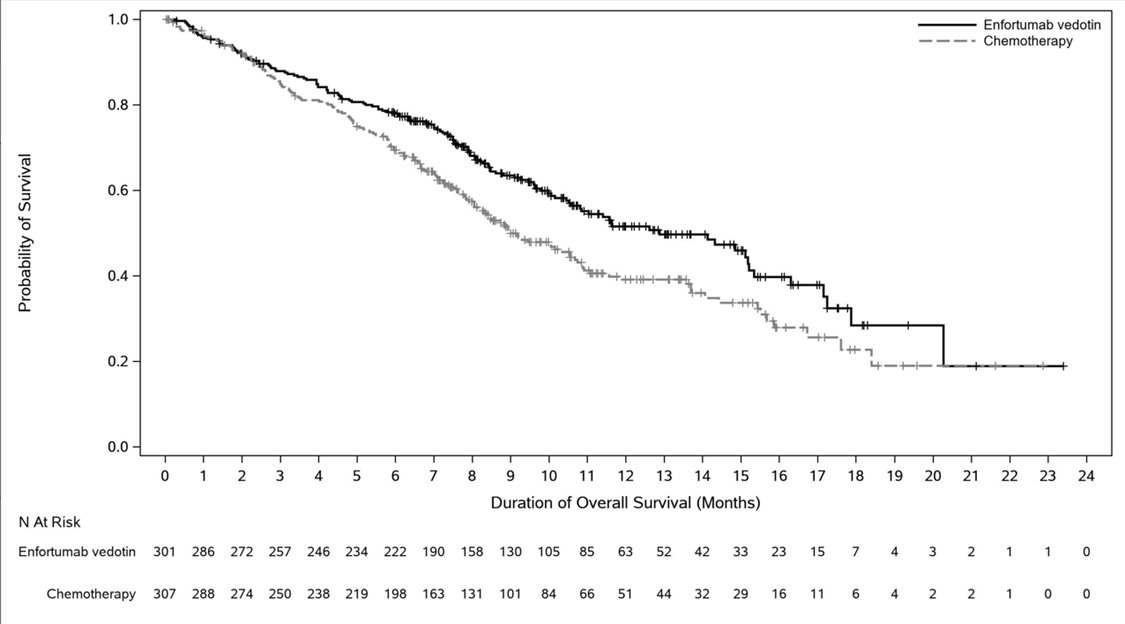
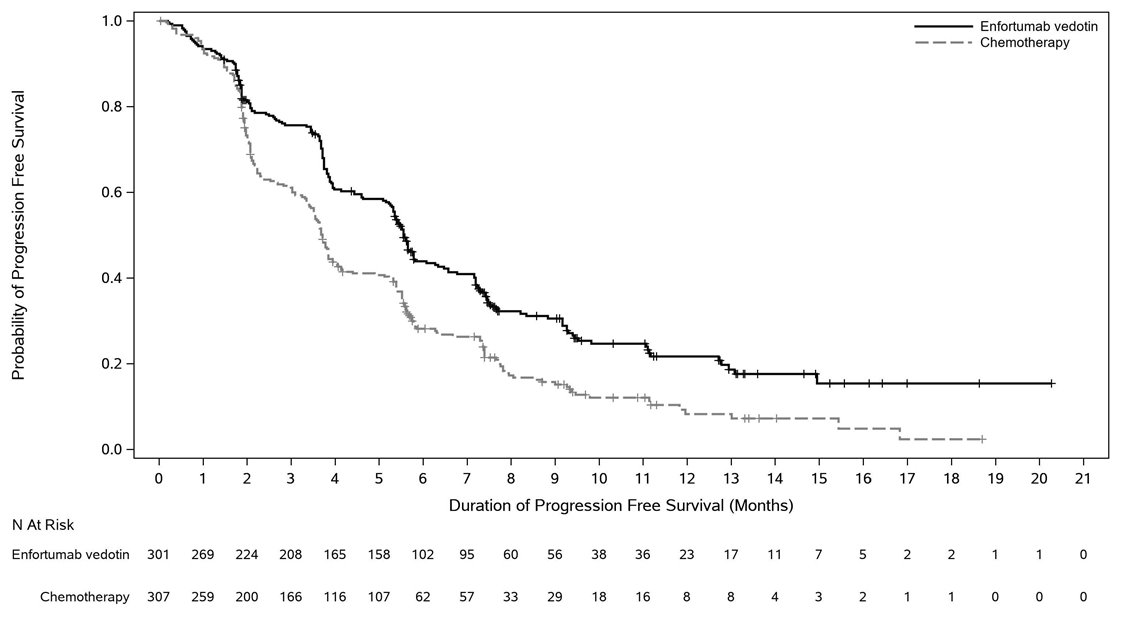
Table 16 and Figures 4-5 summarize the efficacy results for EV-301.
| Endpoint | PADCEV
n=301 | Chemotherapy
n=307 |
|---|---|---|
|
Overall Survival* |
||
|
Number (%) of patients with events |
134 (44.5) |
167 (54.4) |
|
Median in months (95% CI) |
12.9 (10.6, 15.2) |
9.0 (8.1, 10.7) |
|
Hazard ratio (95% CI) |
0.70 (0.56, 0.89) |
|
|
p-value |
0.0014 |
|
|
Progression Free Survival* |
||
|
Number (%) of patients with events |
201 (66.8) |
231 (75.2) |
|
Median in months (95% CI) |
5.6 (5.3, 5.8) |
3.7 (3.5, 3.9) |
|
Hazard ratio (95% CI) |
0.62 (0.51, 0.75) |
|
|
p-value |
<0.0001 |
|
|
Overall Response Rate (CR + PR)† |
||
|
ORR (%) (95% CI) |
40.6 (34.9, 46.5) |
17.9 (13.7, 22.8) |
|
p-value |
<0.0001 |
|
|
Complete response rate (%) |
4.9 |
2.7 |
|
Partial response rate (%) |
35.8 |
15.2 |
EV-201, Cohort 1
The efficacy of PADCEV as a single agent was also investigated in Cohort 1 of EV-201 (NCT03219333), a single-arm, multi-cohort, multicenter trial that enrolled 125 patients with locally advanced or metastatic urothelial cancer who received prior treatment with a PD-1 or PD-L1 inhibitor and a platinum-based chemotherapy. Patients were excluded if they had active central nervous system (CNS) metastases, ongoing sensory or motor neuropathy ≥Grade 2, heart failure, or uncontrolled diabetes defined as hemoglobin A1C (HbA1c) ≥8% or HbA1c ≥7% with associated diabetes symptoms.
PADCEV was administered at a dose of 1.25 mg/kg, as an intravenous (IV) infusion on days 1, 8, and 15 of each 28-day cycle.
The median age was 69 years (range: 40 to 84 years) and 70% were male. Racial demographics were reported as White (85%), Asian (9%), Black (2%), Other (0.8%) or not reported (4%). Four percent of patients were Hispanic or Latino. All patients had a baseline Eastern Cooperative Oncology Group (ECOG) performance status of 0 (32%) or 1 (68%). Ninety percent of patients had visceral metastases including 40% with liver metastases. Approximately two-thirds (67%) of patients had pure transitional cell carcinoma (TCC) histology; 33% had TCC with other histologic variants. The median number of prior systemic therapies was 3 (range: 1 to 6). Sixty-six percent of patients received prior cisplatin-based regimens, 26% received prior carboplatin-based regimens, and an additional 8% received both cisplatin and carboplatin-based regimens.
The major efficacy outcome measures were confirmed objective response rate (ORR) and duration of response (DOR) assessed by BICR using RECIST v1.1.
Efficacy results are presented in Table 17.
| Endpoint | PADCEV
n=125 |
|---|---|
| NE = not estimable | |
|
|
|
Confirmed ORR (95% CI) |
44% (35.1, 53.2) |
|
Complete Response Rate (CR) |
12% |
|
Partial Response Rate (PR) |
32% |
|
Median* Duration of Response, months (95% CI) |
7.6 (6.3, NE) |
Previously Treated Cisplatin Ineligible Patients with Locally Advanced or Metastatic Urothelial Cancer
EV-201, Cohort 2
The efficacy of PADCEV as a single agent was also evaluated in Cohort 2 of EV-201, a single-arm, multi-cohort, multicenter trial in 89 patients with locally advanced or metastatic urothelial cancer who received prior treatment with a PD-1 or PD-L1 inhibitor and were cisplatin ineligible and did not receive platinum in the locally advanced or metastatic setting. Patients were excluded if they had active CNS metastases, ongoing sensory or motor neuropathy ≥Grade 2, heart failure, or uncontrolled diabetes defined as hemoglobin A1C (HbA1c) ≥8% or HbA1c ≥7% with associated diabetes symptoms.
PADCEV was administered at a dose of 1.25 mg/kg, as an intravenous (IV) infusion on days 1, 8, and 15 of each 28-day cycle.
The median age was 75 years (range: 49 to 90 years), 74% were male. Racial demographics were reported as White (70%), Asian (22%) or not reported (8%). One percent of patients were Hispanic or Latino. Patients had a baseline ECOG performance status of 0 (42%), 1 (46%) and 2 (12%). Forty-three percent of patients had tumors located in the upper tract that included the renal pelvis and ureter. Seventy-nine percent of patients had visceral metastases and 24% had liver metastases.
Reasons for cisplatin ineligibility included: 66% with baseline creatinine clearance of 30 – 59 mL/min, 7% with ECOG PS of 2, 15% with Grade 2 or greater hearing loss, and 12% with more than one cisplatin-ineligibility criteria. Seventy percent of patients had TCC histology; 13% had TCC with squamous differentiation and 17% had TCC with other histologic variants.
The median number of prior systemic therapies was 1 (range: 1 to 4).
Efficacy results are presented in Table 18 below.
| NE = not estimable | |
|
|
|
Endpoint |
PADCEV n=89 |
|
Confirmed ORR (95% CI) |
51% (39.8, 61.3) |
|
Complete Response Rate (CR) |
22% |
|
Partial Response Rate (PR) |
28% |
|
Median* Duration of Response, months (95% CI) |
13.8 (6.4, NE) |
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
PADCEV (enfortumab vedotin-ejfv) 20 mg and 30 mg are supplied as a sterile, preservative-free, white to off-white lyophilized powder in single-dose vials. PADCEV vials are available in the following packages:
- •
- Carton of one 20 mg single-dose vial (NDC 51144-020-01)
- •
- Carton of one 30 mg single-dose vial (NDC 51144-030-01)
Storage
Store PADCEV vials refrigerated at 2ºC to 8ºC (36ºF to 46ºF) in the original carton. Do not freeze. Do not shake.
Special Handling
PADCEV is a hazardous drug. Follow applicable special handling and disposal procedures.1
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Skin Reactions
Inform patients that severe skin reactions including SJS and TEN with fatal outcomes have occurred after administration of PADCEV, predominantly during the first cycle of treatment but may occur later.
Advise patients to contact their healthcare provider immediately if they develop new target lesions, progressively worsening skin reactions, severe blistering or peeling of the skin [see Boxed Warning and Warnings and Precautions (5.1)].
Hyperglycemia
Inform patients about the risk of hyperglycemia and how to recognize associated symptoms [see Warnings and Precautions (5.2)].
Pneumonitis/ILD
Advise patients to immediately report new or worsening respiratory symptoms [see Warnings and Precautions (5.3)].
Peripheral Neuropathy
Inform patients to report to their healthcare provider any numbness and tingling of the hands or feet or muscle weakness [see Warnings and Precautions (5.4)].
Ocular disorders
Advise patients to contact their healthcare provider if they experience any visual changes [see Warnings and Precautions (5.5)]. In order to prevent or treat dry eyes, advise patients to use artificial tear substitutes.
Infusion Site Extravasation
Inform patients that infusion site reactions have occurred after administration of PADCEV. These reactions generally occurred immediately after administration but, in some instances, had a delayed onset (e.g., 24 hours). Instruct patients to contact their healthcare provider immediately if they experience an infusion site reaction [see Warnings and Precautions (5.6)].
Embryo-Fetal Toxicity
Advise pregnant women and females of reproductive potential of the potential risk to the fetus. Advise females to inform their healthcare providers of a known or suspected pregnancy [see Warnings and Precautions (5.7) and Use in Specific Population (8.1)].
Females and Males of Reproductive Potential
Advise female patients of reproductive potential to use effective contraception during treatment with PADCEV and for 2 months after the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with PADCEV and for 4 months after the last dose [see Use in Specific Populations (8.3)].
Lactation
Advise women not to breastfeed during treatment with PADCEV and for 3 weeks after the last dose [see Use in Specific Populations (8.2)].
Infertility
Advise females and males of reproductive potential that PADCEV may impair fertility [see Use in Specific Populations (8.3)].
Manufactured and Marketed by:
Astellas Pharma US, Inc.
Northbrook, Illinois 60062
Distributed and Marketed by:
Seagen Inc.
Bothell, WA 98021
1-855-4SEAGEN
U.S. License 2124
PADCEV is a registered trademark jointly owned by Agensys, Inc. and Seagen Inc.
©2023 Agensys, Inc. and Seagen Inc.
397124-EV-USA
|
PATIENT INFORMATION PADCEV® (PAD-sev) (enfortumab vedotin-ejfv) for injection |
|||
|
If your healthcare provider prescribes PADCEV in combination with pembrolizumab for you, also read the Medication Guide that comes with pembrolizumab for important information about pembrolizumab. |
|||
|
What is the most important information I should know about PADCEV? PADCEV may cause serious side effects, including: Skin reactions. Skin reactions including severe skin reactions have happened in people treated with PADCEV and may be more common when PADCEV is given with pembrolizumab. In some cases, these severe skin reactions have caused death. Most severe skin reactions occurred during the first cycle of treatment but may happen later. Your healthcare provider will monitor you, may stop your treatment with PADCEV completely or for a period of time (temporarily), may change your dose, and may prescribe medicines if you get skin reactions. Tell your healthcare provider right away if you develop any of these signs of a new or worsening skin reaction: |
|||
|
|
||
|
See “What are the possible side effects of PADCEV?” for more information about side effects. |
|||
|
What is PADCEV? PADCEV is a prescription medicine used to treat adults with bladder cancer and cancers of the urinary tract (renal pelvis, ureter or urethra) that has spread or cannot be removed by surgery.
It is not known if PADCEV is safe and effective in children. |
|||
|
Before receiving PADCEV, tell your healthcare provider about all of your medical conditions, including if you:
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Taking PADCEV with certain other medicines may cause side effects. |
|||
|
How will I receive PADCEV?
|
|||
|
What are the possible side effects of PADCEV? PADCEV may cause serious side effects, including:
|
|||
|
Your healthcare provider may decrease your dose of PADCEV, or temporarily or completely stop your treatment with PADCEV if you have severe side effects. The most common side effects of PADCEV when used in combination with pembrolizumab include: |
|||
|
|
|
|
|
The most common side effects of PADCEV when used alone include: |
|||
|
|
|
|
|
PADCEV may cause fertility problems in females and males, which may affect the ability to have children. Talk to your healthcare provider if you have concerns about fertility. These are not all of the possible side effects of PADCEV. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|||
|
General information about the safe and effective use of PADCEV. Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. You can ask your pharmacist or healthcare provider for information about PADCEV that is written for healthcare professionals. |
|||
|
What are the ingredients in PADCEV? Active ingredient: enfortumab vedotin-ejfv Inactive ingredients: histidine, histidine hydrochloride monohydrate, polysorbate 20, and trehalose dihydrate. Manufactured and Marketed by: Astellas Pharma US, Inc., Northbrook, Illinois 60062 Distributed and Marketed by: Seagen Inc., Bothell, WA 98021 U.S. License 2124 PADCEV is a registered trademark jointly owned by Agensys, Inc. and Seagen Inc. ©2023 Agensys, Inc. and Seagen Inc. For more information, go to www.padcev.com or call 1-888-4-PADCEV 397124-EV-USA |
|||
This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: 12/2023