FULL PRESCRIBING INFORMATION
WARNING: SERIOUS INFECTIONS AND MALIGNANCY
SERIOUS INFECTIONS
Patients treated with CIMZIA are at increased risk for developing serious infections that may lead to hospitalization or death [see Warnings and Precautions (5.1) and Adverse Reactions (6.1)] . Most patients who developed these infections were taking concomitant immunosuppressants such as methotrexate or corticosteroids.
CIMZIA should be discontinued if a patient develops a serious infection or sepsis.
Reported infections include:
- Active tuberculosis, including reactivation of latent tuberculosis. Patients with tuberculosis have frequently presented with disseminated or extrapulmonary disease. Patients should be tested for latent tuberculosis before CIMZIA use and during therapy. Treatment for latent infection should be initiated prior to CIMZIA use.
- Invasive fungal infections, including histoplasmosis, coccidioidomycosis, candidiasis, aspergillosis, blastomycosis, and pneumocystosis. Patients with histoplasmosis or other invasive fungal infections may present with disseminated, rather than localized disease. Antigen and antibody testing for histoplasmosis may be negative in some patients with active infection. Empiric anti-fungal therapy should be considered in patients at risk for invasive fungal infections who develop severe systemic illness.
- Bacterial, viral and other infections due to opportunistic pathogens, including Legionella and Listeria.
The risks and benefits of treatment with CIMZIA should be carefully considered prior to initiating therapy in patients with chronic or recurrent infection.
Patients should be closely monitored for the development of signs and symptoms of infection during and after treatment with CIMZIA, including the possible development of tuberculosis in patients who tested negative for latent tuberculosis infection prior to initiating therapy. [see Warnings and Precautions (5.1) and Adverse Reactions (6.1)].
MALIGNANCY
Lymphoma and other malignancies, some fatal, have been reported in children and adolescent patients treated with TNF blockers, of which CIMZIA is a member [see Warnings and Precautions (5.2)]. CIMZIA is not indicated for use in pediatric patients.
1 INDICATIONS AND USAGE
1.1 Crohn's Disease
CIMZIA is indicated for reducing signs and symptoms of Crohn's disease and maintaining clinical response in adult patients with moderately to severely active disease who have had an inadequate response to conventional therapy.
1.2 Rheumatoid Arthritis
CIMZIA is indicated for the treatment of adults with moderately to severely active rheumatoid arthritis (RA).
1.3 Psoriatic Arthritis
CIMZIA is indicated for the treatment of adult patients with active psoriatic arthritis (PsA).
1.4 Ankylosing Spondylitis
CIMZIA is indicated for the treatment of adults with active ankylosing spondylitis (AS). [see Clinical Studies (14.4)]
1.5 Non-radiographic Axial Spondyloarthritis
CIMZIA is indicated for the treatment of adults with active non-radiographic axial spondyloarthritis (nr-axSpA) with objective signs of inflammation [see Clinical Studies (14.5)].
1.6 Plaque Psoriasis
CIMZIA is indicated for the treatment of adults with moderate-to-severe plaque psoriasis (PsO) who are candidates for systemic therapy or phototherapy [see Clinical Studies (14.6)]
2 DOSAGE AND ADMINISTRATION
CIMZIA is administered by subcutaneous injection . Injection sites should be rotated and injections should not be given into areas where the skin is tender, bruised, red or hard. When a 400 mg dose is needed (given as two subcutaneous injections of 200 mg), injections should occur at separate sites in the thigh or abdomen.
The solution should be carefully inspected visually for particulate matter and discoloration prior to administration. The solution should be a clear colorless to yellow liquid, essentially free from particulates and should not be used if cloudy or if foreign particulate matter is present. CIMZIA does not contain preservatives; therefore, unused portions of drug remaining in the syringe or vial should be discarded.
2.1 Crohn's Disease
The recommended initial adult dose of CIMZIA is 400 mg (given as two subcutaneous injections of 200 mg) initially, and at Weeks 2 and 4. In patients who obtain a clinical response, the recommended maintenance regimen is 400 mg every four weeks.
2.2 Rheumatoid Arthritis
The recommended dose of CIMZIA for adult patients with rheumatoid arthritis is 400 mg (given as two subcutaneous injections of 200 mg) initially and at Weeks 2 and 4, followed by 200 mg every other week. For maintenance dosing, CIMZIA 400 mg every 4 weeks can be considered [see Clinical Studies (14.2)] .
2.3 Psoriatic Arthritis
The recommended dose of CIMZIA for adult patients with psoriatic arthritis is 400 mg (given as 2 subcutaneous injections of 200 mg each) initially and at week 2 and 4, followed by 200 mg every other week. For maintenance dosing, CIMZIA 400 mg every 4 weeks can be considered [see Clinical Studies (14.3)] .
2.4 Ankylosing Spondylitis
The recommended dose of CIMZIA for adult patients with ankylosing spondylitis is 400 mg (given as 2 subcutaneous injections of 200 mg each) initially and at weeks 2 and 4, followed by 200 mg every 2 weeks or 400 mg every 4 weeks.
2.5 Non-radiographic Axial Spondyloarthritis
The recommended dose of CIMZIA for adult patients with non-radiographic axial spondyloarthritis is 400 mg (given as 2 subcutaneous injections of 200 mg each) initially and at weeks 2 and 4, followed by 200 mg every 2 weeks or 400 mg every 4 weeks.
2.6 Plaque Psoriasis
The recommended dose of CIMZIA for adults with moderate-to-severe plaque psoriasis is 400 mg (given as 2 subcutaneous injections of 200 mg each) every other week.
For some patients (with body weight ≤ 90 kg), CIMZIA 400 mg (given as 2 subcutaneous injections of 200 mg each) initially and at Weeks 2 and 4, followed by 200 mg every other week can be considered [see Clinical Studies (14.6)] .
2.7 Preparation and Administration of CIMZIA Using the Lyophilized Powder for Injection
CIMZIA Lyophilized powder should be prepared and administered by a health care professional. CIMZIA is provided in a package that contains everything required to reconstitute and inject the drug [see How Supplied/Storage and Handling (16)] . Step-by-step preparation and administration instructions are provided below.
Preparation and Storage
- If refrigerated, remove CIMZIA from the refrigerator and allow the vial(s) to sit at room temperature for 30 minutes before reconstituting. Do not warm the vial in any other way. Use appropriate aseptic technique when preparing and administering CIMZIA.
- Reconstitute the vial(s) of CIMZIA with 1 mL of Sterile Water for Injection, USP using the 20-gauge needle provided. The sterile water for injection should be directed at the vial wall rather than directly on CIMZIA.
- Gently swirl each vial of CIMZIA for about one minute without shaking, assuring that all of the powder comes in contact with the Sterile Water for Injection. The swirling should be as gentle as possible in order to avoid creating a foaming effect.
- Continue swirling every 5 minutes as long as non-dissolved particles are observed. Full reconstitution may take as long as 30 minutes. The final reconstituted solution contains 200 mg/mL and should be clear to opalescent, colorless to pale yellow liquid essentially free from particulates.
- Once reconstituted, CIMZIA can be stored in the vials for up to 24 hours between 2° to 8° C (36° to 46° F) prior to injection. Do not freeze.
Administration
- Prior to injecting, reconstituted CIMZIA should be at room temperature but do not leave reconstituted CIMZIA at room temperature for more than two hours prior to administration.
- Withdraw the reconstituted solution into a separate syringe for each vial using a new 20-gauge needle for each vial so that each syringe contains 1 mL of CIMZIA (200 mg of certolizumab pegol).
- Replace the 20-gauge needle(s) on the syringes with a 23-gauge(s) for administration.
- Inject the full contents of the syringe(s) subcutaneously, by pinching the skin of the thigh or abdomen. Where a 400 mg dose is required, two injections are required, therefore, separate sites should be used for each 200 mg injection.
2.8 Preparation and Administration of CIMZIA Using the Prefilled Syringe
After proper training in subcutaneous injection technique, a patient may self-inject with the CIMZIA Prefilled Syringe if a physician determines that it is appropriate.
- If refrigerated, remove the prefilled syringe from the carton and let it warm to room temperature.
- Inspect the liquid in the prefilled syringe. It should be clear and colorless to yellow and free from particulates. Discard the syringe if cloudy, discolored or contains particulates.
- Suitable sites for injection include the thigh or abdomen at least 2 inches away from the navel. Inject at least 1 inch from the previous site.
- Do not inject into areas where the skin is tender, bruised, red or hard, or where there are scars or stretch marks.
The needle shield inside the removable cap of the CIMZIA prefilled syringe contains a derivative of natural rubber latex which may cause allergic reactions and should be handled with caution by latex-sensitive individuals [see Warnings and Precautions (5.4)] .
2.9 Monitoring to Assess Safety
Before initiation of therapy with CIMZIA, all patients must be evaluated for both active and inactive (latent) tuberculosis infection. The possibility of undetected latent tuberculosis should be considered in patients who have immigrated from or traveled to countries with a high prevalence of tuberculosis or had close contact with a person with active tuberculosis. Appropriate screening tests (e.g. tuberculin skin test and chest x-ray) should be performed in all patients.
4 CONTRAINDICATIONS
CIMZIA is contraindicated in patients with a history of hypersensitivity reaction to certolizumab pegol or to any of the excipients. Reactions have included angioedema, anaphylaxis, serum sickness, and urticaria [see Warnings and Precautions (5.4)] .
5 WARNINGS AND PRECAUTIONS
5.1 Risk of Serious Infections
Patients treated with CIMZIA are at an increased risk for developing serious infections involving various organ systems and sites that may lead to hospitalization or death.
Opportunistic infections due to bacterial, mycobacterial, invasive fungal, viral, parasitic, or other opportunistic pathogens including aspergillosis, blastomycosis, candidiasis, coccidioidomycosis, histoplasmosis, legionellosis, listeriosis, pneumocystosis and tuberculosis have been reported with TNF blockers. Patients have frequently presented with disseminated rather than localized disease.
Treatment with CIMZIA should not be initiated in patients with an active infection, including clinically important localized infections. Patients greater than 65 years of age, patients with co-morbid conditions, and/or patients taking concomitant immunosuppressants (e.g. corticosteroids or methotrexate) may be at a greater risk of infection. The risks and benefits of treatment should be considered prior to initiating therapy in patients:
- with chronic or recurrent infection
- who have been exposed to tuberculosis
- with a history of an opportunistic infection
- who have resided or traveled in areas of endemic tuberculosis or endemic mycoses, such as histoplasmosis, coccidioidomycosis, or blastomycosis
- with underlying conditions that may predispose them to infection
Tuberculosis
Cases of reactivation of tuberculosis or new tuberculosis infections have been observed in patients receiving CIMZIA, including patients who have previously or concomitantly received treatment for latent or active tuberculosis. Reports included cases of pulmonary and extrapulmonary (i.e., disseminated) tuberculosis. Evaluate patients for tuberculosis risk factors and test for latent infection prior to initiating CIMZIA and periodically during therapy.
Treatment of latent tuberculosis infection prior to therapy with TNF-blocking agents has been shown to reduce the risk of tuberculosis reactivation during therapy. Prior to initiating CIMZIA, assess if treatment for latent tuberculosis is needed; and consider an induration of 5 mm or greater a positive tuberculin skin test result, even for patients previously vaccinated with Bacille Calmette-Guerin (BCG).
Consider anti-tuberculosis therapy prior to initiation of CIMZIA in patients with a past history of latent or active tuberculosis in whom an adequate course of treatment cannot be confirmed, and for patients with a negative test for latent tuberculosis but having risk factors for tuberculosis infection. Despite previous or concomitant treatment for latent tuberculosis, cases of active tuberculosis have occurred in patients treated with CIMZIA. Some patients who have been successfully treated for active tuberculosis have redeveloped tuberculosis while being treated with CIMZIA. Consultation with a physician with expertise in the treatment of tuberculosis is recommended to aid in the decision of whether initiating anti-tuberculosis therapy is appropriate for an individual patient.
Strongly consider tuberculosis in patients who develop a new infection during CIMZIA treatment, especially in patients who have previously or recently traveled to countries with a high prevalence of tuberculosis, or who have had close contact with a person with active tuberculosis.
Monitoring
Patients should be closely monitored for the development of signs and symptoms of infection during and after treatment with CIMZIA, including the development of tuberculosis in patients who tested negative for latent tuberculosis infection prior to initiating therapy. Tests for latent tuberculosis infection may also be falsely negative while on therapy with CIMZIA.
CIMZIA should be discontinued if a patient develops a serious infection or sepsis. A patient who develops a new infection during treatment with CIMZIA should be closely monitored, undergo a prompt and complete diagnostic workup appropriate for an immunocompromised patient, and appropriate antimicrobial therapy should be initiated.
Invasive Fungal Infections
For patients who reside or travel in regions where mycoses are endemic, invasive fungal infection should be suspected if they develop a serious systemic illness. Appropriate empiric antifungal therapy should be considered while a diagnostic workup is being performed. Antigen and antibody testing for histoplasmosis may be negative in some patients with active infection. When feasible, the decision to administer empiric antifungal therapy in these patients should be made in consultation with a physician with expertise in the diagnosis and treatment of invasive fungal infections and should take into account both the risk for severe fungal infection and risks of antifungal therapy.
5.2 Malignancies
In the controlled portions of clinical studies of some TNF blockers, more cases of malignancies have been observed among patients receiving TNF blockers compared to control patients. During controlled and open-labeled portions of CIMZIA studies of Crohn's disease and other diseases, malignancies (excluding non-melanoma skin cancer) were observed at a rate (95% confidence interval) of 0.5 (0.4, 0.7) per 100 patient-years among 4,650 CIMZIA-treated patients versus a rate of 0.6 (0.1, 1.7) per 100 patient-years among 1,319 placebo-treated patients. During CIMZIA studies of psoriasis, malignancies (excluding non-melanoma skin cancer) were observed corresponding to an incidence rate of 0.5 (0.2, 1.0) per 100 subject-years among a total of 995 subjects who received CIMZIA. The size of the control group and limited duration of the controlled portions of the studies precludes the ability to draw firm conclusions.
Malignancies, some fatal, have been reported among children, adolescents, and young adults who received treatment with TNF-blocking agents (initiation of therapy ≤ 18 years of age), of which CIMZIA is a member. Approximately half the cases were lymphomas, including Hodgkin's and non-Hodgkin's lymphoma. The other cases represented a variety of different malignancies and included rare malignancies usually associated with immunosuppression and malignancies that are not usually observed in children and adolescents. The malignancies occurred after a median of 30 months of therapy (range 1 to 84 months). Most of the patients were receiving concomitant immunosuppressants. These cases were reported post-marketing and are derived from a variety of sources including registries and spontaneous post-marketing reports. CIMZIA is not indicated for use in pediatric patients.
In the controlled portions of clinical trials of all the TNF blockers, more cases of lymphoma have been observed among patients receiving TNF blockers compared to control patients. In controlled studies of CIMZIA for Crohn's disease and other investigational uses, there was one case of lymphoma among 2,657 Cimzia-treated patients and one case of Hodgkin's lymphoma among 1,319 placebo-treated patients.
In the CIMZIA RA clinical trials (placebo-controlled and open label) a total of three cases of lymphoma were observed among 2,367 patients. This is approximately 2-fold higher than expected in the general population. Patients with RA, particularly those with highly active disease, are at a higher risk for the development of lymphoma. In the CIMZIA PsO clinical trials (placebo-controlled and open label) there was one case of Hodgkin's lymphoma.
Rates in clinical studies for CIMZIA cannot be compared to the rates of clinical trials of other TNF blockers and may not predict the rates observed when CIMZIA is used in a broader patient population. Patients with Crohn's disease that require chronic exposure to immunosuppressant therapies may be at higher risk than the general population for the development of lymphoma, even in the absence of TNF blocker therapy [see Adverse Reactions (6.1)] . The potential role of TNF blocker therapy in the development of malignancies in adults is not known.
Postmarketing cases of hepatosplenic T-cell lymphoma (HSTCL), a rare type of T-cell lymphoma that has a very aggressive disease course and is usually fatal, have been reported in patients treated with TNF blockers, including CIMZIA. The majority of reported TNF blocker cases occurred in adolescent and young adult males with Crohn's disease or ulcerative colitis. Almost all of these patients had received treatment with the immunosuppressants azathioprine and/or 6-mercaptopurine (6-MP) concomitantly with a TNF blocker at or prior to diagnosis. It is uncertain whether the occurrence of HSTCL is related to use of a TNF blocker or a TNF blocker in combination with these other immunosuppressants. The potential risk of using a TNF blocker in combination with azathioprine or 6-MP should be carefully considered.
Cases of acute and chronic leukemia have been reported in association with post-marketing TNF-blocker use in RA and other indications. Even in the absence of TNF-blocker therapy, patients with RA may be at a higher risk (approximately 2-fold) than the general population for the development of leukemia.
Melanoma and Merkel cell carcinoma have been reported in patients treated with TNF blockers, including CIMZIA. Periodic skin examinations are recommended for all patients, particularly those with risk factors for skin cancer.
5.3 Heart Failure
Cases of worsening congestive heart failure (CHF) and new onset CHF have been reported with TNF blockers, including CIMZIA. CIMZIA has not been formally studied in patients with CHF; however, in clinical studies in patients with CHF with another TNF blocker, worsening congestive heart failure (CHF) and increased mortality due to CHF were observed. Exercise caution in patients with heart failure and monitor them carefully [see Adverse Reactions (6.1)].
5.4 Hypersensitivity Reactions
The following symptoms that could be compatible with hypersensitivity reactions have been reported rarely following CIMZIA administration to patients: angioedema, anaphylaxis, dyspnea, hypotension, rash, serum sickness, and urticaria. Some of these reactions occurred after the first administration of CIMZIA. If such reactions occur, discontinue further administration of CIMZIA and institute appropriate therapy. There are no data on the risks of using CIMZIA in patients who have experienced a severe hypersensitivity reaction towards another TNF blocker; in these patients caution is needed [see Adverse Reactions (6.1)] .
The needle shield inside the removable cap of the CIMZIA prefilled syringe contains a derivative of natural rubber latex which may cause an allergic reaction in individuals sensitive to latex.
5.5 Hepatitis B Virus Reactivation
Use of TNF blockers, including CIMZIA, has been associated with reactivation of hepatitis B virus (HBV) in patients who are chronic carriers of this virus. In some instances, HBV reactivation occurring in conjunction with TNF blocker therapy has been fatal. The majority of reports have occurred in patients concomitantly receiving other medications that suppress the immune system, which may also contribute to HBV reactivation.
Test patients for HBV infection before initiating treatment with CIMZIA. For patients who test positive for HBV infection, consultation with a physician with expertise in the treatment of hepatitis B is recommended. Adequate data are not available on the safety or efficacy of treating patients who are carriers of HBV with anti-viral therapy in conjunction with TNF blocker therapy to prevent HBV reactivation. Patients who are carriers of HBV and require treatment with CIMZIA should be closely monitored for clinical and laboratory signs of active HBV infection throughout therapy and for several months following termination of therapy.
In patients who develop HBV reactivation, discontinue CIMZIA and initiate effective anti-viral therapy with appropriate supportive treatment. The safety of resuming TNF blocker therapy after HBV reactivation is controlled is not known. Therefore, exercise caution when considering resumption of CIMZIA therapy in this situation and monitor patients closely.
5.6 Neurologic Reactions
Use of TNF blockers, of which CIMZIA is a member, has been associated with rare cases of new onset or exacerbation of clinical symptoms and/or radiographic evidence of central nervous system demyelinating disease, including multiple sclerosis, and with peripheral demyelinating disease, including Guillain-Barré syndrome. Exercise caution in considering the use of CIMZIA in patients with pre-existing or recent-onset central or peripheral nervous system demyelinating disorders. Rare cases of neurological disorders, including seizure disorder, optic neuritis, and peripheral neuropathy have been reported in patients treated with CIMZIA [see Adverse Reactions (6.1)] .
5.7 Hematological Reactions
Rare reports of pancytopenia, including aplastic anemia, have been reported with TNF blockers. Adverse reactions of the hematologic system, including medically significant cytopenia (e.g., leukopenia, pancytopenia, thrombocytopenia) have been infrequently reported with CIMZIA [see Adverse Reactions (6.1)] . The causal relationship of these events to CIMZIA remains unclear.
Although no high risk group has been identified, exercise caution in patients being treated with CIMZIA who have ongoing, or a history of, significant hematologic abnormalities. Advise all patients to seek immediate medical attention if they develop signs and symptoms suggestive of blood dyscrasias or infection (e.g., persistent fever, bruising, bleeding, pallor) while on CIMZIA. Consider discontinuation of CIMZIA therapy in patients with confirmed significant hematologic abnormalities.
5.8 Use with Biological Disease-Modifying Antirheumatic Drugs (Biological DMARDs)
Serious infections were seen in clinical studies with concurrent use of anakinra (an interleukin-1 antagonist) and another TNF blocker, etanercept, with no added benefit compared to etanercept alone. A higher risk of serious infections was also observed in combination use of TNF blockers with abatacept and rituximab. Because of the nature of the adverse events seen with this combination therapy, similar toxicities may also result from the use of CIMZIA in this combination. Therefore, the use of CIMZIA in combination with other biological DMARDs is not recommended [see Drug Interactions (7.1)].
5.9 Autoimmunity
Treatment with CIMZIA may result in the formation of autoantibodies and rarely, in the development of a lupus-like syndrome. If a patient develops symptoms suggestive of a lupus-like syndrome following treatment with CIMZIA, discontinue treatment [see Adverse Reactions (6.1)] .
5.10 Immunizations
Patients treated with CIMZIA may receive vaccinations, except for live or live attenuated vaccines. No data are available on the response to live vaccinations or the secondary transmission of infection by live vaccines in patients receiving CIMZIA.
In a placebo-controlled clinical trial of patients with rheumatoid arthritis, no difference was detected in antibody response to vaccine between CIMZIA and placebo treatment groups when the pneumococcal polysaccharide vaccine and influenza vaccine were administered concurrently with CIMZIA. Similar proportions of patients developed protective levels of anti-vaccine antibodies between CIMZIA and placebo treatment groups; however patients receiving CIMZIA and concomitant methotrexate had a lower humoral response compared with patients receiving CIMZIA alone. The clinical significance of this is unknown.
5.11 Immunosuppression
Since TNF mediates inflammation and modulates cellular immune responses, the possibility exists for TNF blockers, including CIMZIA, to affect host defenses against infections and malignancies. The impact of treatment with CIMZIA on the development and course of malignancies, as well as active and/or chronic infections, is not fully understood [see Warnings and Precautions (5.1, 5.2, 5.5) and Adverse Reactions (6.1)] . The safety and efficacy of CIMZIA in patients with immunosuppression has not been formally evaluated.
6 ADVERSE REACTIONS
The most serious adverse reactions were:
- Serious Infections [see Warnings and Precautions (5.1)]
- Malignancies [see Warnings and Precautions (5.2)]
- Heart Failure [see Warnings and Precautions (5.3)]
- Hypersensitivity Reactions [see Warnings and Precautions (5.4)]
- Hepatitis B Virus Reactivation [see Warnings and Precautions (5.5)]
- Neurologic Reactions [see Warnings and Precautions (5.6)]
- Hematologic Reactions [see Warnings and Precautions (5.7)]
- Autoimmunity [see Warnings and Precautions (5.9)]
- Immunosuppression [see Warnings and Precautions (5.11)]
6.1 Clinical Trials Experience
Because clinical studies are conducted under widely varying and controlled conditions, adverse reaction rates observed in clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug, and may not predict the rates observed in a broader patient population in clinical practice.
In premarketing controlled trials of all patient populations combined the most common adverse reactions (≥ 8%) were upper respiratory infections (18%), rash (9%) and urinary tract infections (8%).
Adverse Reactions Most Commonly Leading to Discontinuation of Treatment in Premarketing Controlled Trials
The proportion of patients with Crohn's disease who discontinued treatment due to adverse reactions in the controlled clinical studies was 8% for CIMZIA and 7% for placebo. The most common adverse reactions leading to the discontinuation of CIMZIA (for at least 2 patients and with a higher incidence than placebo) were abdominal pain (0.4% CIMZIA, 0.2% placebo), diarrhea (0.4% CIMZIA, 0% placebo), and intestinal obstruction (0.4% CIMZIA, 0% placebo).
The proportion of patients with rheumatoid arthritis who discontinued treatment due to adverse reactions in the controlled clinical studies was 5% for CIMZIA and 2.5% for placebo. The most common adverse reactions leading to discontinuation of CIMZIA were tuberculosis infections (0.5%); and pyrexia, urticaria, pneumonia, and rash (0.3%).
Controlled Studies with Crohn's Disease
The data described below reflect exposure to CIMZIA at 400 mg subcutaneous dosing in studies of patients with Crohn's disease. In the safety population in controlled studies, a total of 620 patients with Crohn's disease received CIMZIA at a dose of 400 mg, and 614 subjects received placebo (including subjects randomized to placebo in Study CD2 following open label dosing of CIMZIA at Weeks 0, 2, 4). In controlled and uncontrolled studies, 1,564 patients received CIMZIA at some dose level, of whom 1,350 patients received 400 mg CIMZIA. Approximately 55% of subjects were female, 45% were male, and 94% were Caucasian. The majority of patients in the active group were between the ages of 18 and 64.
During controlled clinical studies, the proportion of patients with serious adverse reactions was 10% for CIMZIA and 9% for placebo. The most common adverse reactions (occurring in ≥ 5% of CIMZIA-treated patients, and with a higher incidence compared to placebo) in controlled clinical studies with CIMZIA were upper respiratory infections (e.g. nasopharyngitis, laryngitis, viral infection) in 20% of CIMZIA-treated patients and 13% of placebo-treated patients, urinary tract infections (e.g. bladder infection, bacteriuria, cystitis) in 7% of CIMZIA-treated patients and in 6% of placebo-treated patients, and arthralgia (6% CIMZIA, 4% placebo).
Other Adverse Reactions
The most commonly occurring adverse reactions in controlled trials of Crohn's disease were described above. Other serious or significant adverse reactions reported in controlled and uncontrolled studies in Crohn's disease and other diseases, occurring in patients receiving CIMZIA at doses of 400 mg or other doses include:
Blood and lymphatic system disorders: Anemia, leukopenia, lymphadenopathy, pancytopenia, and thrombophilia.
Cardiac disorders: Angina pectoris, arrhythmias, atrial fibrillation, cardiac failure, hypertensive heart disease, myocardial infarction, myocardial ischemia, pericardial effusion, pericarditis, stroke and transient ischemic attack.
Eye disorders: Optic neuritis, retinal hemorrhage, and uveitis.
General disorders and administration site conditions: Bleeding and injection site reactions.
Hepatobiliary disorders: Elevated liver enzymes and hepatitis.
Immune system disorders: Alopecia totalis.
Psychiatric disorders: Anxiety, bipolar disorder, and suicide attempt.
Renal and urinary disorders: Nephrotic syndrome and renal failure.
Reproductive system and breast disorders: Menstrual disorder.
Skin and subcutaneous tissue disorders: Dermatitis, erythema nodosum, and urticaria.
Vascular disorders: Thrombophlebitis, vasculitis.
Controlled Studies with Rheumatoid Arthritis
CIMZIA was studied primarily in placebo-controlled trials and in long-term follow-up studies. The data described below reflect the exposure to CIMZIA in 2,367 RA patients, including 2,030 exposed for at least 6 months, 1,663 exposed for at least one year and 282 for at least 2 years; and 1,774 in adequate and well-controlled studies. In placebo-controlled studies, the population had a median age of 53 years at entry; approximately 80% were females, 93% were Caucasian and all patients were suffering from active rheumatoid arthritis, with a median disease duration of 6.2 years. Most patients received the recommended dose of CIMZIA or higher.
Table 1 summarizes the reactions reported at a rate of at least 3% in patients treated with CIMZIA 200 mg every other week compared to placebo (saline formulation), given concomitantly with methotrexate.
| Adverse Reaction
(Preferred Term) | Placebo+ MTX
* (%)
N =324 | CIMZIA 200 mg EOW + MTX(%)
N =640 |
|---|---|---|
|
||
| Upper respiratory tract infection | 2 | 6 |
| Headache | 4 | 5 |
| Hypertension | 2 | 5 |
| Nasopharyngitis | 1 | 5 |
| Back pain | 1 | 4 |
| Pyrexia | 2 | 3 |
| Pharyngitis | 1 | 3 |
| Rash | 1 | 3 |
| Acute bronchitis | 1 | 3 |
| Fatigue | 2 | 3 |
Hypertensive adverse reactions were observed more frequently in patients receiving CIMZIA than in controls. These adverse reactions occurred more frequently among patients with a baseline history of hypertension and among patients receiving concomitant corticosteroids and non-steroidal anti-inflammatory drugs.
Patients receiving CIMZIA 400 mg as monotherapy every 4 weeks in rheumatoid arthritis controlled clinical trials had similar adverse reactions to those patients receiving CIMZIA 200 mg every other week.
Other Adverse Reactions
Other infrequent adverse reactions (occurring in less than 3% of RA patients) were similar to those seen in Crohn's disease patients.
Psoriatic Arthritis Clinical Study
CIMZIA has been studied in 409 patients with psoriatic arthritis (PsA) in a placebo-controlled trial. The safety profile for patients with PsA treated with CIMZIA was similar to the safety profile seen in patients with RA and previous experience with CIMZIA .
Ankylosing Spondylitis Clinical Study
CIMZIA has been studied in 325 patients with axial spondyloarthritis of whom the majority had ankylosing spondylitis (AS) in a placebo-controlled study (AS-1). The safety profile for patients in study AS-1 treated with CIMZIA was similar to the safety profile seen in patients with RA.
Non-radiographic Axial Spondyloarthritis Clinical Study
CIMZIA has been studied in 317 patients with non-radiographic axial spondyloarthritis (nr-axSpA-1). The safety profile for patients with nr-axSpA treated with CIMZIA was similar to the safety profile seen in patients with RA and previous experience with CIMZIA.
Plaque Psoriasis Clinical Studies
In clinical studies, a total of 1112 subjects with plaque psoriasis were treated with CIMZIA. Of these, 779 subjects were exposed for at least 12 months, 551 for 18 months, and 66 for 24 months.
Data from three placebo-controlled studies (Studies PS-1, PS-2, and PS-3) in 1020 subjects (mean age 46 years, 66% males, 94% white) were pooled to evaluate the safety of CIMZIA [see Clinical Studies (14)].
Placebo-Controlled Period (Week 0-16)
In the placebo-controlled period of Studies PS-1, PS-2 and PS-3 in the 400 mg group, adverse events occurred in 63.5% of subjects in the CIMZIA group compared to 61.8% of subjects in the placebo group. The rates of serious adverse events were 4.7% in the CIMZIA group and 4.5% in the placebo group. Table 2 summarizes the adverse reactions that occurred at a rate of at least 1% and at a higher rate in the CIMZIA group than in the placebo group.
| Adverse Reactions | Cimzia 400 mg every other week
n (%) N=342 | Cimzia 200 mg
* every other week
n (%) N=350 | Placebo
n (%) N=157 |
|---|---|---|---|
|
|||
| Upper respiratory tract infections † | 75 (21.9) | 68 (19.4) | 33 (21.0) |
| Headache ‡ | 13 (3.8) | 10 (2.9) | 4 (2.5) |
| Injection site reactions § | 11 (3.2) | 6 (1.7) | 1 (0.6) |
| Cough | 11 (3.2) | 4 (1.1) | 3 (1.9) |
| Herpes infections ¶ | 5 (1.5) | 5 (1.4) | 2 (1.3) |
Elevated Liver Enzymes
Elevated liver enzymes were reported more frequently in the CIMZIA-treated subjects (4.3% in the 200 mg group and 2.3% in the 400 mg group) than in the placebo-treated subjects (2.5%). Of CIMZIA-treated subjects who had elevation of liver enzymes, two subjects were discontinued from the trial. In controlled Phase 3 studies of CIMZIA in adults with PsO with a controlled period duration ranging from 0 to 16 weeks, AST and/or ALT elevations ≥5 × ULN occurred in 0.9% of CIMZIA 200 mg or CIMZIA 400 mg arms and none in placebo arm.
Adverse Reactions of Special Interest Across Indications
Infections
The incidence of infections in controlled studies in Crohn's disease was 38% for CIMZIA-treated patients and 30% for placebo-treated patients. The infections consisted primarily of upper respiratory infections (20% for CIMZIA, 13% for placebo). The incidence of serious infections during the controlled clinical studies was 3% per patient-year for CIMZIA-treated patients and 1% for placebo-treated patients. Serious infections observed included bacterial and viral infections, pneumonia, and pyelonephritis.
The incidence of new cases of infections in controlled clinical studies in rheumatoid arthritis was 0.91 per patient-year for all CIMZIA-treated patients and 0.72 per patient-year for placebo-treated patients. The infections consisted primarily of upper respiratory tract infections, herpes infections, urinary tract infections, and lower respiratory tract infections. In the controlled rheumatoid arthritis studies, there were more new cases of serious infection adverse reactions in the CIMZIA treatment groups, compared to the placebo groups (0.06 per patient-year for all CIMZIA doses vs. 0.02 per patient-year for placebo). Rates of serious infections in the 200 mg every other week dose group were 0.06 per patient-year and in the 400 mg every 4 weeks dose group were 0.04 per patient-year. Serious infections included tuberculosis, pneumonia, cellulitis, and pyelonephritis. In the placebo group, no serious infection occurred in more than one subject. There is no evidence of increased risk of infections with continued exposure over time [see Warnings and Precautions (5.1)] .
In controlled clinical studies in psoriasis, the incidence rates of infections were similar in the CIMZIA and placebo groups. The infections consisted primarily of upper respiratory tract infections and viral infections (including herpes infections). Serious adverse events of infection occurred in CIMZIA-treated patients during the placebo-controlled periods of the pivotal studies (pneumonia, abdominal abscess, and hematoma infection) and Phase 2 study (urinary tract infection, gastroenteritis, and disseminated tuberculosis).
Tuberculosis and Opportunistic Infections
In completed and ongoing global clinical studies in all indications including 5,118 CIMZIA-treated patients, the overall rate of tuberculosis is approximately 0.61 per 100 patient-years across all indications.
The majority of cases occurred in countries with high endemic rates of TB. Reports include cases of disseminated (miliary, lymphatic, and peritoneal) as well as pulmonary TB. The median time to onset of TB for all patients exposed to CIMZIA across all indications was 345 days. In the studies with CIMZIA in RA, there were 36 cases of TB among 2,367 exposed patients, including some fatal cases. Rare cases of opportunistic infections have also been reported in these clinical trials. In Phase 2 and Phase 3 studies with CIMZIA in plaque psoriasis, there were 2 cases of TB among 1112 exposed patients [see Warnings and Precautions (5.1)].
Malignancies
In clinical studies of CIMZIA, the overall incidence rate of malignancies was similar for CIMZIA-treated and control patients. For some TNF blockers, more cases of malignancies have been observed among patients receiving those TNF blockers compared to control patients [see Warnings and Precautions (5.2)].
Heart Failure
In placebo-controlled and open-label studies, cases of new or worsening heart failure have been reported for CIMZIA-treated patients. The majority of these cases were mild to moderate and occurred during the first year of exposure [see Warnings and Precautions (5.3)].
Hypersensitivity Reactions
The following symptoms that could be compatible with hypersensitivity reactions have been reported rarely following CIMZIA administration to patients: angioedema, allergic dermatitis, dizziness (postural), dyspnea, hot flush, hypotension, injection site reactions, malaise, pyrexia, rash, serum sickness, and (vasovagal) syncope [see Warnings and Precautions (5.4)] .
Autoantibodies
In clinical studies in Crohn's disease, 4% of patients treated with CIMZIA and 2% of patients treated with placebo that had negative baseline ANA titers developed positive titers during the studies. One of the 1,564 Crohn's disease patients treated with CIMZIA developed symptoms of a lupus-like syndrome.
In clinical trials of TNF blockers, including CIMZIA, in patients with RA, some patients have developed ANA. Four patients out of 2,367 patients treated with CIMZIA in RA clinical studies developed clinical signs suggestive of a lupus-like syndrome. The impact of long-term treatment with CIMZIA on the development of autoimmune diseases is unknown [see Warnings and Precautions (5.9)].
6.2 Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to certolizumab pegol in the studies described below with the incidence of antibodies in other studies or to other products may be misleading.
Patients with Crohn's disease were tested at multiple time points for antibodies to certolizumab pegol during Studies CD1 and CD2. In patients continuously exposed to CIMZIA, the overall percentage of patients who were antibody positive to CIMZIA on at least one occasion was 8%; approximately 6% were neutralizing in vitro. No apparent correlation of antibody development to adverse events or efficacy was observed. Patients treated with concomitant immunosuppressants had a lower rate of antibody development than patients not taking immunosuppressants at baseline (3% and 11%, respectively). The following adverse events were reported in Crohn's disease patients who were antibody-positive (N = 100) at an incidence at least 3% higher compared to antibody-negative patients (N = 1,242): abdominal pain, arthralgia, edema peripheral, erythema nodosum, injection site erythema, injection site pain, pain in extremity, and upper respiratory tract infection.
In two long-term (up to 7 years of exposure), open-label Crohn's disease studies, overall 23% (207/903) of patients developed antibodies against certolizumab pegol on at least one occasion. Of the 207 patients who were antibody positive, 152 (73%) had a persistent reduction of drug plasma concentration, which represents 17% (152/903) of the study population. The data from these two studies do not suggest an association between the development of antibodies and adverse events.
The overall percentage of patients with antibodies to certolizumab pegol detectable on at least one occasion was 7% (105 of 1,509) in the rheumatoid arthritis placebo-controlled trials. Approximately one third (3%, 39 of 1,509) of these patients had antibodies with neutralizing activity in vitro. Patients treated with concomitant immunosuppressants (MTX) had a lower rate of antibody development than patients not taking immunosuppressants at baseline. Patients treated with concomitant immunosuppressant therapy (MTX) in RA-I, RA-II, RA-III had a lower rate of neutralizing antibody formation overall than patients treated with CIMZIA monotherapy in RA-IV (2% vs. 8%). Both the loading dose of 400 mg every other week at Weeks 0, 2 and 4 and concomitant use of MTX were associated with reduced immunogenicity.
Antibody formation was associated with lowered drug plasma concentration and reduced efficacy. In patients receiving the recommended CIMZIA dosage of 200 mg every other week with concomitant MTX, the ACR20 response was lower among antibody positive patients than among antibody-negative patients (Study RA-I, 48% versus 60%; Study RA-II 35% versus 59%, respectively). In Study RA-III, too few patients developed antibodies to allow for meaningful analysis of ACR20 response by antibody status. In Study RA-IV (monotherapy), the ACR20 response was 33% versus 56%, antibody-positive versus antibody-negative status, respectively [see Clinical Pharmacology (12.3)]. No association was seen between antibody development and the development of adverse events.
Approximately 8 % (22/265) and 19% (54/281) of subjects with psoriasis who received CIMZIA 400 mg every 2 weeks and CIMZIA 200 mg every 2 weeks for 48 weeks, respectively, developed antibodies to certolizumab pegol. Of the subjects who developed antibodies to certolizumab pegol, 45% (27/60) had antibodies that were classified as neutralizing. Antibody formation was associated with lowered drug plasma concentration and reduced efficacy.
A more sensitive and drug tolerant electrochemiluminesence (ECL)-based bridging assay was used for the first time in the nr-axSpA-1 study, resulting in a greater proportion of samples having measurable antibodies to certolizumab pegol and thus a greater incidence of patients being classed as antibody positive. In the placebo-controlled trial in patients with non-radiographic axial spondyloarthritis, after up to 52 weeks of treatment, the overall incidence of patients who were antibody positive to certolizumab pegol was 97% (248/255 patients). Of these antibody positive patients, higher titers were associated with reduced certolizumab pegol plasma levels.
The data above reflect the percentage of patients whose test results were considered positive for antibodies to certolizumab pegol in an ELISA or ECL-based bridging assay, and are highly dependent on the sensitivity and specificity of the assay.
6.3 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of CIMZIA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to estimate reliably their frequency or establish a causal relationship to drug exposure.
Vascular disorder: systemic vasculitis has been identified during post-approval use of TNF blockers.
Skin: case of severe skin reactions, including Stevens-Johnson syndrome, toxic epidermal necrolysis, erythema multiforme, new or worsening psoriasis (all sub-types including pustular and palmoplantar), and lichenoid skin reaction have been identified during post-approval use of TNF blockers.
Immune System Disorders: sarcoidosis
Neoplasms benign, malignant and unspecified (including cysts and polyps): Melanoma, Merkel cell carcinoma (neuroendocrine carcinoma of the skin) [see Warnings and Precautions (5.2)].
7 DRUG INTERACTIONS
7.1 Use with Anakinra, Abatacept, Rituximab, and Natalizumab
An increased risk of serious infections has been seen in clinical studies of other TNF-blocking agents used in combination with anakinra or abatacept, with no added benefit. Formal drug interaction studies have not been performed with rituximab or natalizumab. Because of the nature of the adverse events seen with these combinations with TNF blocker therapy, similar toxicities may also result from the use of CIMZIA in these combinations. There is not enough information to assess the safety and efficacy of such combination therapy. Therefore, the use of CIMZIA in combination with anakinra, abatacept, rituximab, or natalizumab is not recommended [see Warnings and Precautions (5.8)] .
7.2 Live Vaccines
Avoid use of live (including attenuated) vaccines concurrently with CIMZIA [see Warnings and Precautions (5.10)] .
7.3 Laboratory Tests
Interference with certain coagulation assays has been detected in patients treated with CIMZIA. Certolizumab pegol may cause erroneously elevated activated partial thromboplastin time (aPTT) assay results in patients without coagulation abnormalities. This effect has been observed with the PTT-Lupus Anticoagulant (LA) test and Standard Target Activated Partial Thromboplastin time (STA-PTT) Automate tests from Diagnostica Stago, and the HemosIL APTT-SP liquid and HemosIL lyophilized silica tests from Instrumentation Laboratories. Other aPTT assays may be affected as well. Interference with thrombin time (TT) and prothrombin time (PT) assays has not been observed. There is no evidence that CIMZIA therapy has an effect on in vivo coagulation.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to CIMZIA during pregnancy. For more information, healthcare providers or patients can contact:
MotherToBaby Pregnancy Studies conducted by the Organization of Teratology Information Specialists (OTIS). The OTIS AutoImmune Diseases Study at 1-877-311-8972 or visit http://mothertobaby.org/pregnancy-studies/
Risk Summary
Limited data from the ongoing pregnancy registry on use of CIMZIA in pregnant women are not sufficient to inform a risk of major birth defects or other adverse pregnancy outcomes. However, certolizumab pegol plasma concentrations obtained from two studies of CIMZIA use during the third trimester of pregnancy demonstrated that placental transfer of certolizumab pegol was negligible in most infants at birth, and low in other infants at birth (see Data) . There are risks to the mother and fetus associated with active rheumatoid arthritis or Crohn's disease. The theoretical risks of administration of live or live-attenuated vaccines to the infants exposed in utero to CIMZIA should be weighed against the benefits of vaccinations (see Clinical Considerations) . No adverse developmental effects were observed in animal reproduction studies during which pregnant rats were administered intravenously a rodent anti-murine TNFα pegylated Fab' fragment (cTN3 PF) similar to certolizumab pegol during organogenesis at up to 2.4 times the recommended human dose of 400 mg every four weeks.
The estimated background risk of major birth defects and miscarriage for the indicated population(s) are unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risks of major birth defects and miscarriage in clinically recognized pregnancies are 2 to 4% and 15 to 20%, respectively.
Clinical Considerations
Disease-Associated Maternal and/or Embryo/Fetal Risk
Published data suggest that the risk of adverse pregnancy outcomes in women with rheumatoid arthritis or Crohn's disease is correlated with maternal disease activity and that active disease increases the risk of adverse pregnancy outcomes, including fetal loss, preterm delivery (before 37 weeks of gestation), low birth weight (less than 2500 g) and small for gestational age birth.
Fetal/Neonatal Adverse Reactions
Due to its inhibition of TNFα, CIMZIA administered during pregnancy could affect immune responses in the in utero-exposed newborn and infant. The clinical significance of BLQ or low levels is unknown for in utero-exposed infants. Additional data available from one exposed infant suggest that CIMZIA may be eliminated at a slower rate in infants than in adults (see Data). The safety of administering live or live-attenuated vaccines in exposed infants is unknown.
Data
Human Data
A limited number of pregnancies have been reported in the ongoing pregnancy exposure registry. Due to the small number of CIMZIA-exposed pregnancies with known outcomes (n=54), no meaningful comparisons between the exposed group and control groups may be conducted to determine an association with CIMZIA and major birth defects or adverse pregnancy outcomes.
A multicenter clinical study was conducted in 16 women treated with CIMZIA at a maintenance dose of 200 mg every 2 weeks or 400 mg every 4 weeks during the third trimester of pregnancy for rheumatological diseases or Crohn's disease. The last dose of CIMZIA was given on average 11 days prior to delivery (range 1 to 27 days). Certolizumab pegol plasma concentrations were measured in samples from mothers and infants using an assay that can measure certolizumab pegol concentrations at or above 0.032 mcg/mL. Certolizumab pegol plasma concentrations measured in the mothers at delivery (range: 4.96 to 49.4 mcg/mL) were consistent with non-pregnant women's plasma concentrations in Study RA-I [see Clinical Studies (14.2)] . Certolizumab pegol plasma concentrations were not measurable in 13 out of 15 infants at birth. The concentration of certolizumab pegol in one infant was 0.0422 mcg/mL at birth (infant/mother plasma ratio of 0.09%). In a second infant, delivered by emergency Caesarean section, the concentration was 0.485 mcg/mL (infant/mother plasma ratio of 4.49%). At Week 4 and Week 8, all 15 infants had no measurable concentrations. Among 16 exposed infants, one serious adverse reaction was reported in a neonate who was treated empirically with intravenous antibiotics due to an increased white blood cell count; blood cultures were negative. The certolizumab pegol plasma concentrations for this infant were not measurable at birth, Week 4, or Week 8.
In another clinical study conducted in 10 pregnant women with Crohn´s disease treated with CIMZIA (400 mg every 4 weeks for every mother), certolizumab pegol concentrations were measured in maternal blood as well as in cord and infant blood at the day of birth with an assay that can measure concentrations at or above 0.41 mcg/mL. The last dose of CIMZIA was given on average 19 days prior to delivery (range 5 to 42 days). Plasma certolizumab pegol concentrations ranged from not measurable to 1.66 mcg/mL in cord blood and 1.58 mcg/mL in infant blood; and ranged from 1.87 to 59.57 mcg/mL in maternal blood. Plasma certolizumab pegol concentrations were lower (by at least 75%) in the infants than in mothers suggesting low placental transfer of certolizumab pegol. In one infant, the plasma certolizumab pegol concentration declined from 1.02 to 0.84 mcg/mL over 4 weeks suggesting that certolizumab pegol may be eliminated at a slower rate in infants than adults.
Animal Data
Because certolizumab pegol does not cross-react with mouse or rat TNFα, reproduction studies were performed in rats using a rodent anti-murine TNFα pegylated Fab' fragment (cTN3 PF) similar to certolizumab pegol. Animal reproduction studies have been performed in rats during organogenesis at intravenous doses up to 100 mg/kg (about 2.4 times the recommended human dose of 400 mg, based on the surface area) and have revealed no evidence of harm to the fetus due to cTN3 PF.
8.2 Lactation
Risk Summary
In a multicenter clinical study of 17 lactating women treated with CIMZIA at 200 mg every 2 weeks or 400 mg every 4 weeks, minimal certolizumab pegol concentrations were observed in breast milk. No serious adverse reactions were noted in the 17 infants in the study. There are no data on the effects on milk production. In a separate study, certolizumab pegol concentrations were not detected in the plasma of 9 breastfed infants at 4 weeks post-partum (see Data) . The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for CIMZIA and any potential adverse effects on the breastfed infant from CIMZIA or from the underlying maternal condition.
Data
A multicenter clinical study designed to evaluate breast milk was conducted in 17 lactating women who were at least 6 weeks post-partum and had received at least 3 consecutive doses of CIMZIA 200 mg every 2 weeks or 400 mg every 4 weeks for rheumatological disease or Crohn's disease. The effects of certolizumab pegol on milk production were not studied. The concentration of certolizumab pegol in breast milk was not measurable in 77 (56 %) of the 137 samples taken over the dosing periods using an assay that can measure certolizumab pegol concentrations at or above 0.032 mcg/mL. The median of the estimated average daily infant doses was 0.0035 mg/kg/day (range: 0 to 0.01 mg/kg/day). The percentage of the maternal dose (200 mg CIMZIA dosed once every 2 weeks), that reaches an infant ranged from 0.56% to 4.25% based on samples with measurable certolizumab pegol concentration. No serious adverse reactions were noted in the 17 breastfed infants in the study.
In a separate study, plasma certolizumab pegol concentrations were collected 4 weeks after birth in 9 breastfed infants whose mothers had been currently taking CIMZIA (regardless of being exclusively breastfed or not). Certolizumab pegol in infant plasma was not measurable i.e., below 0.032 mcg/mL.
8.4 Pediatric Use
Safety and effectiveness in pediatric patients have not been established.
CIMZIA was evaluated for the treatment of pediatric patients with moderately to severely active Crohn's disease. Efficacy was not demonstrated in an open-label, randomized, parallel-group, multiple dose study for a period of up to 62 weeks in 99 subjects aged 6 to 17 years. The study was ended prematurely because of a high number of patient discontinuations.
Due to its inhibition of TNFα, CIMZIA administered during pregnancy could affect immune responses in the in utero-exposed newborn and infant [see Use in Specific Populations (8.1)].
8.5 Geriatric Use
Clinical studies of CIMZIA did not include sufficient numbers of patients aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. Population pharmacokinetic analyses of patients enrolled in CIMZIA clinical studies concluded that there was no apparent difference in drug concentration regardless of age. Because there is a higher incidence of infections in the elderly population in general, use caution when treating the elderly with CIMZIA [see Warnings and Precautions (5.1)] .
11 DESCRIPTION
Certolizumab pegol is a TNF blocker. CIMZIA is a recombinant, humanized antibody Fab' fragment, with specificity for human tumor necrosis factor alpha (TNFα), conjugated to an approximately 40kDa polyethylene glycol (PEG2MAL40K). The Fab' fragment is manufactured in E. coli and is subsequently subjected to purification and conjugation to PEG2MAL40K, to generate certolizumab pegol. The Fab' fragment is composed of a light chain with 214 amino acids and a heavy chain with 229 amino acids. The molecular weight of certolizumab pegol is approximately 91 kiloDaltons.
CIMZIA (certolizumab pegol) for injection is supplied as a sterile white, lyophilized powder in a single-dose vial for subcutaneous use. After reconstitution of the lyophilized powder with 1 mL Sterile Water for Injection, USP, the final concentration is 200 mg/mL with a deliverable volume of 1 mL (200 mg) and a pH of approximately 5.2. Each single-dose vial provides 200 mg certolizumab pegol, lactic acid (0.9 mg), polysorbate (0.1 mg), and sucrose (100 mg).
CIMZIA (certolizumab pegol) injection is supplied as a sterile, clear to opalescent, colorless to pale yellow solution that may contain particulates in a single-dose prefilled syringe for subcutaneous use. Each prefilled syringe delivers 1 mL of solution containing 200 mg certolizumab pegol, sodium acetate (1.36 mg), sodium chloride (7.31 mg), and Water for Injection, USP.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Certolizumab pegol binds to human TNFα with a KD of 90pM. TNFα is a key pro-inflammatory cytokine with a central role in inflammatory processes. Certolizumab pegol selectively neutralizes TNFα (IC 90 of 4 ng/mL for inhibition of human TNFα in the in vitro L929 murine fibrosarcoma cytotoxicity assay) but does not neutralize lymphotoxin α (TNFβ). Certolizumab pegol cross-reacts poorly with TNF from rodents and rabbits, therefore in vivo efficacy was evaluated using animal models in which human TNFα was the physiologically active molecule.
Certolizumab pegol was shown to neutralize membrane-associated and soluble human TNFα in a dose-dependent manner. Incubation of monocytes with certolizumab pegol resulted in a dose-dependent inhibition of LPS-induced TNFα and IL-1β production in human monocytes.
Certolizumab pegol does not contain a fragment crystallizable (Fc) region, which is normally present in a complete antibody, and therefore does not fix complement or cause antibody-dependent cell-mediated cytotoxicity in vitro. It does not induce apoptosis in vitro in human peripheral blood-derived monocytes or lymphocytes, nor does certolizumab pegol induce neutrophil degranulation.
A tissue reactivity study was carried out ex vivo to evaluate potential cross-reactivity of certolizumab pegol with cryosections of normal human tissues. Certolizumab pegol showed no reactivity with a designated standard panel of normal human tissues.
12.2 Pharmacodynamics
Biological activities ascribed to TNFα include the upregulation of cellular adhesion molecules and chemokines, upregulation of major histocompatibility complex (MHC) class I and class II molecules, and direct leukocyte activation. TNFα stimulates the production of downstream inflammatory mediators, including interleukin-1, prostaglandins, platelet activating factor, and nitric oxide. Elevated levels of TNFα have been implicated in the pathology of Crohn's disease and rheumatoid arthritis. Certolizumab pegol binds to TNFα, inhibiting its role as a key mediator of inflammation. TNFα is strongly expressed in the bowel wall in areas involved by Crohn's disease and fecal concentrations of TNFα in patients with Crohn's disease have been shown to reflect clinical severity of the disease. After treatment with certolizumab pegol, patients with Crohn's disease demonstrated a decrease in the levels of C-reactive protein (CRP). Increased TNFα levels are found in the synovial fluid of rheumatoid arthritis patients and play an important role in the joint destruction that is a hallmark of this disease.
12.3 Pharmacokinetics
Absorption
A total of 126 healthy subjects received doses of up to 800 mg certolizumab pegol subcutaneously (sc) and up to 10 mg/kg intravenously (IV) in four pharmacokinetic studies. Data from these studies demonstrate that single intravenous and subcutaneous doses of certolizumab pegol have predictable dose-related plasma concentrations with a linear relationship between the dose administered and the maximum plasma concentration (C max), and the Area Under the certolizumab pegol plasma concentration versus time Curve (AUC). A mean C max of approximately 43 to 49 mcg/mL occurred at Week 5 during the initial loading dose period using the recommended dose regimen for the treatment of patients with rheumatoid arthritis (400 mg sc at Weeks 0, 2 and 4 followed by 200 mg every other week).
Certolizumab pegol plasma concentrations were broadly dose-proportional and pharmacokinetics observed in patients with rheumatoid arthritis, Crohn's disease, and plaque psoriasis were consistent with those seen in healthy subjects.
Following subcutaneous administration, peak plasma concentrations of certolizumab pegol were attained between 54 and 171 hours post-injection. Certolizumab pegol has bioavailability (F) of approximately 80% (ranging from 76% to 88%) following subcutaneous administration compared to intravenous administration.
Distribution
The steady state volume of distribution (Vss) was estimated as 4.7 to 8 L in the population pharmacokinetic analysis for patients with Crohn's disease, patients with rheumatoid arthritis, and adult patients with plaque psoriasis.
Metabolism
The metabolism of certolizumab pegol has not been studied in human subjects. Data from animals indicate that once cleaved from the Fab' fragment the PEG moiety is mainly excreted in urine without further metabolism.
Elimination
PEGylation, the covalent attachment of PEG polymers to peptides, delays the metabolism and elimination of these entities from the circulation by a variety of mechanisms, including decreased renal clearance, proteolysis, and immunogenicity. Accordingly, certolizumab pegol is an antibody Fab' fragment conjugated with PEG in order to extend the terminal plasma elimination half-life (t 1/2) of the Fab'. The terminal elimination phase half-life (t 1/2) was approximately 14 days for all doses tested. The clearance following IV administration to healthy subjects ranged from 9.21 mL/h to 14.38 mL/h. The clearance following sc dosing was estimated 17 mL/h in the Crohn's disease population PK analysis with an inter-subject variability of 38% (CV) and an inter-occasion variability of 16%. Similarly, the clearance following sc dosing was estimated as 21.0 mL/h in the RA population PK analysis, with an inter-subject variability of 30.8% (%CV) and inter-occasion variability 22.0%. The clearance following subcutaneous dosing in patients with plaque psoriasis was 14 mL/h with an inter-subject variability of 22.2% (CV). The route of elimination of certolizumab pegol has not been studied in human subjects. Studies in animals indicate that the major route of elimination of the PEG component is via urinary excretion.
Specific Populations
Population pharmacokinetic analysis was conducted on data from patients with rheumatoid arthritis and patients with Crohn's disease, to evaluate the effect of age, race, gender, methotrexate use, concomitant medication, creatinine clearance and presence of anti-certolizumab antibodies on pharmacokinetics of certolizumab pegol. A population pharmacokinetic analysis was also conducted on data from patients with plaque psoriasis to evaluate the effect of age, gender, body weight, and presence of anti-certolizumab pegol antibodies. Only bodyweight and presence of anti-certolizumab antibodies significantly affected certolizumab pegol pharmacokinetics. Pharmacokinetic exposure was inversely related to body weight but pharmacodynamic exposure-response analysis showed that no additional therapeutic benefit would be expected from a weight-adjusted dose regimen. When assessed using the previous ELISA method, the presence of anti-certolizumab antibodies was associated with a ≥ 3 to 4 fold increase in clearance.
Geriatric Patients: Pharmacokinetics of certolizumab pegol was not different in elderly compared to young adults.
Racial or Ethnic Groups: A specific clinical study showed no difference in pharmacokinetics between Caucasian and Japanese subjects.
Male and Female Patients: Pharmacokinetics of certolizumab pegol was similar in male and female subjects.
Patients with Renal Impairment: Specific clinical studies have not been performed to assess the effect of renal impairment on the pharmacokinetics of CIMZIA. The pharmacokinetics of the PEG (polyethylene glycol) fraction of certolizumab pegol is expected to be dependent on renal function but has not been assessed in renal impairment. There are insufficient data to provide a dosing recommendation in moderate and severe renal impairment.
Drug Interaction Studies
Methotrexate pharmacokinetics is not altered by concomitant administration with CIMZIA in patients with rheumatoid arthritis. The effect of methotrexate on CIMZIA pharmacokinetics was not studied. However, methotrexate-treated patients have lower incidence of antibodies to CIMZIA. Thus, therapeutic plasma levels are more likely to be sustained when CIMZIA is administered with methotrexate in patients with rheumatoid arthritis.
Formal drug-drug interaction studies have not been conducted with CIMZIA upon concomitant administration with corticosteroids, nonsteroidal anti-inflammatory drugs, analgesics or immunosuppressants.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, and Impairment of Fertility
Long-term animal studies of CIMZIA have not been conducted to assess its carcinogenic potential. Certolizumab pegol was not genotoxic in the Ames test, the human peripheral blood lymphocytes chromosomal aberration assay, or the mouse bone marrow micronucleus assay.
Since certolizumab pegol does not cross-react with mouse or rat TNFα, reproduction studies were performed in rats using a rodent anti-murine TNFα pegylated Fab fragment (cTN3 PF), similar to certolizumab pegol. The cTN3 PF had no effects on the fertility and general reproductive performance of male and female rats at intravenous doses up 100 mg/kg, administered twice weekly.
14 CLINICAL STUDIES
14.1 Crohn's Disease
The efficacy and safety of CIMZIA were assessed in two double-blind, randomized, placebo-controlled studies in patients aged 18 years and older with moderately to severely active Crohn's disease, as defined by a Crohn's Disease Activity Index (CDAI) of 220 to 450 points, inclusive. CIMZIA was administered subcutaneously at a dose of 400 mg in both studies. Stable concomitant medications for Crohn's disease were permitted.
Study CD1
Study CD1 was a randomized placebo-controlled study in 662 patients with active Crohn's disease. CIMZIA or placebo was administered at Weeks 0, 2, and 4 and then every four weeks to Week 24. Assessments were done at Weeks 6 and 26. Clinical response was defined as at least a 100-point reduction in CDAI score compared to baseline, and clinical remission was defined as an absolute CDAI score of 150 points or lower.
The results for Study CD1 are provided in Table 3. At Week 6, the proportion of clinical responders was statistically significantly greater for CIMZIA-treated patients compared to controls. The difference in clinical remission rates was not statistically significant at Week 6. The difference in the proportion of patients who were in clinical response at both Weeks 6 and 26 was also statistically significant, demonstrating maintenance of clinical response.
| Timepoint | % Response or Remission (95% CI) | |
|---|---|---|
| Placebo
(N = 328) | CIMZIA 400 mg
(N = 331) |
|
| Week 6 | ||
| Clinical Response * | 27% (22%, 32%) | 35% (30%, 40%) † |
| Clinical Remission * | 17% (13%, 22%) | 22% (17%, 26%) |
| Week 26 | ||
| Clinical Response | 27% (22%, 31%) | 37% (32%, 42%) † |
| Clinical Remission | 18% (14%, 22%) | 29% (25%, 34%) † |
| Both Weeks 6 & 26 | ||
| Clinical Response | 16% (12%, 20%) | 23% (18%, 28%) † |
| Clinical Remission | 10% (7%, 13%) | 14% (11%, 18%) |
Study CD2
Study CD2 was a randomized treatment-withdrawal study in patients with active Crohn's disease. All patients who entered the study were dosed initially with CIMZIA 400 mg at Weeks 0, 2, and 4 and then assessed for clinical response at Week 6 (as defined by at least a 100-point reduction in CDAI score). At Week 6, a group of 428 clinical responders was randomized to receive either CIMZIA 400 mg or placebo, every four weeks starting at Week 8, as maintenance therapy through Week 24. Non-responders at Week 6 were withdrawn from the study. Final evaluation was based on the CDAI score at Week 26. Patients who withdrew or who received rescue therapy were considered not to be in clinical response. Three randomized responders received no study injections, and were excluded from the ITT analysis.
The results for clinical response and remission are shown in Table 4. At Week 26, a statistically significantly greater proportion of Week 6 responders were in clinical response and in clinical remission in the CIMZIA-treated group compared to the group treated with placebo.
| % Response or Remission (95% CI) | ||
|---|---|---|
| CIMZIA 400 mg ×3 + Placebo
N = 210 | CIMZIA
400 mg N = 215 |
|
| Week 26 | ||
| Clinical Response * | 36% (30%, 43%) | 63% (56%, 69%) † |
| Clinical Remission * | 29% (22%, 35%) | 48% (41%, 55%) † |
Baseline use of immunosuppressants or corticosteroids had no impact on the clinical response to CIMZIA.
14.2 Rheumatoid Arthritis
The efficacy and safety of CIMZIA were assessed in four randomized, placebo-controlled, double-blind studies (RA-I, RA-II, RA-III, and RA-IV) in patients ≥ 18 years of age with moderately to severely active rheumatoid arthritis diagnosed according to the American College of Rheumatology (ACR) criteria. Patients had ≥ 9 swollen and tender joints and had active RA for at least 6 months prior to baseline. CIMZIA was administered subcutaneously in combination with MTX at stable doses of at least 10 mg weekly in Studies RA-I, RA-II, and RA-III. CIMZIA was administered as monotherapy in Study RA-IV.
Study RA-I and Study RA-II evaluated patients who had received MTX for at least 6 months prior to study medication, but had an incomplete response to MTX alone. Patients were treated with a loading dose of 400 mg at Weeks 0, 2 and 4 (for both treatment arms) or placebo followed by either 200 mg or 400 mg of CIMZIA or placebo every other week, in combination with MTX for 52 weeks in Study RA-I and for 24 weeks in Study RA-II. Patients were evaluated for signs and symptoms and structural damage using the ACR20 response at Week 24 (RA-I and RA-II) and modified Total Sharp Score (mTSS) at Week 52 (RA-I). The open-label extension follow-up study enrolled 846 patients who received 400 mg of CIMZIA every other week.
Study RA-III evaluated 247 patients who had active disease despite receiving MTX for at least 6 months prior to study enrollment. Patients received 400 mg of CIMZIA every four weeks for 24 weeks without a prior loading dose. Patients were evaluated for signs and symptoms of RA using the ACR20 at Week 24.
Study RA-IV (monotherapy) evaluated 220 patients who had failed at least one DMARD use prior to receiving CIMZIA. Patients were treated with CIMZIA 400 mg or placebo every 4 weeks for 24 weeks. Patients were evaluated for signs and symptoms of active RA using the ACR20 at Week 24.
Clinical Response
The percent of CIMZIA-treated patients achieving ACR20, 50, and 70 responses in Studies RA-I and RA-IV are shown in Table 5. CIMZIA-treated patients had higher ACR20, 50 and 70 response rates at 6 months compared to placebo-treated patients. The results in study RA-II (619 patients) were similar to the results in RA-I at Week 24. The results in study RA-III (247 patients) were similar to those seen in study RA-IV. Over the one-year Study RA-I, 13% of CIMZIA-treated patients achieved a major clinical response, defined as achieving an ACR70 response over a continuous 6-month period, compared to 1% of placebo-treated patients.
| Study RA-I
Methotrexate Combination (24 and 52 weeks) | Study RA-IV
Monotherapy (24 weeks) |
|||||
|---|---|---|---|---|---|---|
| Response | Placebo + MTX | CIMZIA
* 200 mg + MTX
q 2 weeks | CIMZIA * 200 mg + MTX - Placebo + MTX | Placebo | CIMZIA
† 400 mg
q 4 weeks | CIMZIA † 400 mg - Placebo |
| N=199 | N=393 | (95% CI) ‡ | N=109 | N=111 | (95% CI) ‡ | |
|
||||||
| ACR20 | ||||||
| Week 24 | 14% | 59% | 45% (38%, 52%) | 9% | 46% | 36% (25%, 47%) |
| Week 52 | 13% | 53% | 40% (33%, 47%) | N/A | N/A | |
| ACR50 | ||||||
| Week 24 | 8% | 37% | 30% (24%, 36%) | 4% | 23% | 19% (10%, 28%) |
| Week 52 | 8% | 38% | 30% (24%, 37%) | N/A | N/A | |
| ACR70 | ||||||
| Week 24 | 3% | 21% | 18% (14%, 23%) | 0% | 6% | 6% (1%, 10%) |
| Week 52 | 4% | 21% | 18% (13%, 22%) | N/A | N/A | |
| Major Clinical Response § | 1% | 13% | 12% (8%, 15%) | |||
| Study RA-I | Study RA-IV | |||||||
|---|---|---|---|---|---|---|---|---|
| Parameter * | Placebo +
MTX N=199 | CIMZIA
† 200 mg + MTX q 2 weeks
N=393 | Placebo
N=109 | CIMZIA
‡ 400 mg q 4 weeks
Monotherapy N=111 |
||||
| Baseline | Week 24 | Baseline | Week 24 | Baseline | Week 24 | Baseline | Week 24 | |
|
||||||||
| Number of tender joints (0-68) | 28 | 27 | 29 | 9 | 28 (12.5) | 24 (15.4) | 30 (13.7) | 16 (15.8) |
| Number of swollen joints (0-66) | 20 | 19 | 20 | 4 | 20 (9.3) | 16 (12.5) | 21 (10.1) | 12 (11.2) |
| Physician global assessment § | 66 | 56 | 65 | 25 | 4 (0.6) | 3 (1.0) | 4 (0.7) | 3 (1.1) |
| Patient global assessment § | 67 | 60 | 64 | 32 | 3 (0.8) | 3 (1.0) | 3 (0.8) | 3 (1.0) |
| Pain §¶ | 65 | 60 | 65 | 32 | 55 (20.8) | 60 (26.7) | 58 (21.9) | 39 (29.6) |
| Disability index (HAQ) # | 1.75 | 1.63 | 1.75 | 1.00 | 1.55 (0.65) | 1.62 (0.68) | 1.43 (0.63) | 1.04 (0.74) |
| CRP (mg/L)
| 16.0 | 14.0 | 16.0 | 4.0 | 11.3 | 13.5 | 11.6 | 6.4 |
The percent of patients achieving ACR20 responses by visit for Study RA-I is shown in Figure 1. Among patients receiving CIMZIA, clinical responses were seen in some patients within one to two weeks after initiation of therapy.
|
| Figure 1 Study RA-I ACR20 Response Over 52 Weeks * |
|
|
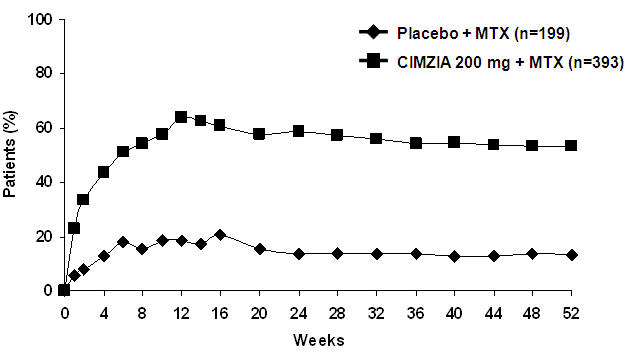
Radiographic Response
In Study RA-I, inhibition of progression of structural damage was assessed radiographically and expressed as the change in modified Total Sharp Score (mTSS) and its components, the Erosion Score (ES) and Joint Space Narrowing (JSN) score, at Week 52, compared to baseline. CIMZIA inhibited the progression of structural damage compared to placebo plus MTX after 12 months of treatment as shown in Table 7. In the placebo group, 52% of patients experienced no radiographic progression (mTSS ≤0.0) at Week 52 compared to 69% in the CIMZIA 200 mg every other week treatment group. Study RA-II showed similar results at Week 24.
| Placebo + MTX
N=199 Mean (SD) | CIMZIA 200 mg + MTX
N=393 Mean (SD) | CIMZIA 200 mg + MTX –
Placebo + MTX Mean Difference |
|
|---|---|---|---|
| An ANCOVA was fitted to the ranked change from baseline for each measure with region and treatment as factors and rank baseline as a covariate. | |||
| mTSS | |||
| Baseline | 40 (45) | 38 (49) | -- |
| Week 24 | 1.3 (3.8) | 0.2 (3.2) | -1.1 |
| Week 52 | 2.8 (7.8) | 0.4 (5.7) | -2.4 |
| Erosion Score | |||
| Baseline | 14 (21) | 15 (24) | -- |
| Week 24 | 0.7 (2.1) | 0.0 (1.5) | -0.7 |
| Week 52 | 1.5 (4.3) | 0.1 (2.5) | -1.4 |
| JSN Score | |||
| Baseline | 25 (27) | 24 (28) | -- |
| Week 24 | 0.7 (2.4) | 0.2 (2.5) | -0.5 |
| Week 52 | 1.4 (5.0) | 0.4 (4.2) | -1.0 |
Physical Function Response
In studies RA-I, RA-II, RA-III, and RA-IV, CIMZIA-treated patients achieved greater improvements from baseline than placebo-treated patients in physical function as assessed by the Health Assessment Questionnaire – Disability Index (HAQ-DI) at Week 24 (RA-II, RA-III and RA-IV) and at Week 52 (RA-I).
14.3 Psoriatic Arthritis
The efficacy and safety of CIMZIA were assessed in a multi-center, randomized, double-blind, placebo controlled trial (PsA001) in 409 patients aged 18 years and older with active psoriatic arthritis despite DMARD therapy. Patients in this study had ≥ 3 swollen and tender joints and adult-onset PsA of at least 6 months' duration as defined by the Classification Criteria for Psoriatic Arthritis (CASPAR) criteria, and increased acute phase reactants. Patients had failed one or more DMARDs. Previous treatment with one anti-TNF biologic therapy was allowed, and 20% of patients had prior anti-TNF biologic exposure. Patients receiving concomitant NSAIDs and conventional DMARDs were 73% and 70 % respectively.
Patients received a loading dose of CIMZIA 400 mg at Weeks 0, 2 and 4 (for both treatment arms) or placebo followed by either CIMZIA 200 mg every other week or CIMZIA 400 mg every 4 weeks or placebo every other week. Patients were evaluated for signs and symptoms and structural damage using the ACR20 response at Week 12 and modified Total Sharp Score (mTSS) at Week 24.
Clinical Response
The percentage of CIMZIA-treated patients achieving ACR20, 50 and 70 responses in study PsA001 are shown in Table 8. ACR20 response rates at weeks 12 and 24 were higher for each CIMZIA dose group relative to placebo (95% confidence intervals for CIMZIA 200 mg minus placebo at weeks 12 and 24 of (23%, 45%) and (30%, 51%), respectively and 95% confidence intervals for CIMZIA 400 mg minus placebo at weeks 12 and 24 of (17%, 39%) and (22%, 44%), respectively). The results of the components of the ACR response criteria are shown in Table 9.
Patients with enthesitis at baseline were evaluated for mean improvement in Leeds Enthesitis Index (LEI). CIMZIA-treated patients receiving either 200 mg every 2 weeks or 400 mg every 4 weeks showed a reduction in enthesitis of 1.8 and 1.7, respectively as compared with a reduction in placebo-treated patients of 0.9 at week 12. Similar results were observed for this endpoint at week 24. Treatment with CIMZIA resulted in improvement in skin manifestations in patients with PsA.
| Response * | Placebo | CIMZIA
†200 mg
Q2W | CIMZIA
‡ 400 mg
Q4W |
|---|---|---|---|
| N=136 | N=138 | N=135 | |
|
|||
| ACR20 | |||
| Week 12 | 24% | 58% | 52% |
| Week 24 | 24% | 64% | 56% |
| ACR50 | |||
| Week 12 | 11% | 36% | 33% |
| Week 24 | 13% | 44% | 40% |
| ACR70 | |||
| Week 12 | 3% | 25% | 13% |
| Week 24 | 4% | 28% | 24% |
| Parameter | Placebo
*
N=136 | CIMZIA
† 200 mg Q2W
N=138 | CIMZIA
‡ 400 mg Q4W
N=135 |
|||
|---|---|---|---|---|---|---|
| Baseline | Week 12 | Baseline | Week 12 | Baseline | Week 12 | |
| All values presented represent the mean | ||||||
| Results are from the randomized set (either with imputation or observed case) | ||||||
|
||||||
| Number of tender joints (0-68) § | 20 | 17 | 22 | 11 | 20 | 11 |
| Number of swollen joints (0-66) § | 10 | 9 | 11 | 4 | 11 | 5 |
| Physician global assessment §, ¶ | 59 | 44 | 57 | 25 | 58 | 29 |
| Patient global assessment §, ¶ | 57 | 50 | 60 | 33 | 60 | 40 |
| Pain §, # | 60 | 50 | 60 | 33 | 61 | 39 |
| Disability index (HAQ) §, Þ | 1.30 | 1.15 | 1.33 | 0.87 | 1.29 | 0.90 |
| CRP (mg/L) | 18.56 | 14.75 | 15.36 | 5.67 | 13.71 | 6.34 |
The percent of patients achieving ACR20 responses by visit for PsA001 is shown in Figure 2.
| Randomized Set. Non-responder imputation used for patients with missing data or those who escaped therapy. |
|
| Figure 2: Study PsA001-ACR20 Response Over 24 Weeks * |
|
|
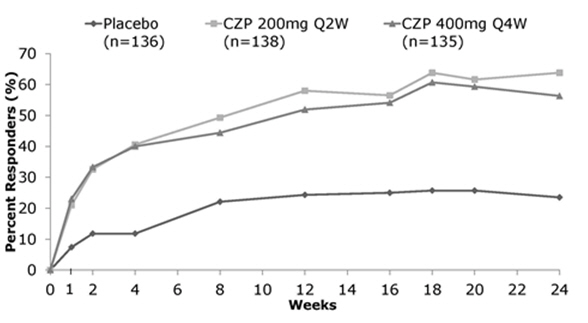
Radiographic Response
In study PsA001, inhibition of progression of structural damage was assessed radiographically and expressed as the change in modified total Sharp score (mTSS) and its components, the Erosion Score (ES) and Joint Space Narrowing score (JSN) at week 24, compared to baseline. The mTSS score was modified for psoriatic arthritis by addition of hand distal interphalangeal (DIP) joints.
Patients treated with CIMZIA 200 mg every other week demonstrated greater reduction in radiographic progression compared with placebo-treated patients at Week 24 as measured by change from baseline in total modified mTSS Score (estimated mean score was 0.18 in the placebo group compared with -0.02 in the CIMZIA 200 mg group; 95% CI for the difference was (-0.38, -0.04)). Patients treated with CIMZIA 400 mg every four weeks did not demonstrate greater inhibition of radiographic progression compared with placebo-treated patients at Week 24.
Physical Function Response
In Study PsA001, CIMZIA-treated patients showed improvement in physical function as assessed by the Health Assessment Questionnaire – Disability Index (HAQ-DI) at Week 24 as compared to placebo (estimated mean change from baseline was 0.19 in the placebo group compared with 0.54 in the CIMZIA 200 mg group; 95% CI for the difference was (-0.47, -0.22) and 0.46 in the CIMZIA 400 mg group; 95% CI for the difference was (-0.39, -0.14)).
14.4 Ankylosing Spondylitis
The efficacy and safety of CIMZIA were assessed in one multicenter, randomized, double-blind, placebo-controlled study (AS-1) in 325 patients ≥18 years of age with adult-onset active axial spondyloarthritis for at least 3 months. The majority of patients in the study had active AS.
Patients had active disease as defined by the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) ≥4, and spinal pain ≥4 on a 0 to 10 Numerical Rating Scale (NRS). Patients must have been intolerant to or had an inadequate response to at least one NSAID. Patients were treated with a loading dose of CIMZIA 400 mg at Weeks 0, 2 and 4 (for both treatment arms) or placebo followed by either 200 mg of CIMZIA every 2 weeks or 400 mg of CIMZIA every 4 weeks or placebo. Concomitant NSAIDs were received by 91% of the AS patients. The primary efficacy variable was the proportion of patients achieving an ASAS20 response at Week 12.
Clinical Response
In study AS-1, at Week 12, a greater proportion of AS patients treated with CIMZIA 200 mg every 2 weeks or 400 mg every 4 weeks achieved ASAS 20 response compared to AS patients treated with placebo (Table 10). Responses were similar in patients receiving CIMZIA 200 mg every 2 weeks and CIMZIA 400 mg every 4 weeks. The results of the components of the ASAS response criteria and other measures of disease activity are shown in Table 11.
| Parameters | Placebo
N=57 | CIMZIA
* 200 mg every 2 weeks
N=65 | CIMZIA
† 400 mg every 4 weeks
N=56 |
|---|---|---|---|
| All percents reflect the proportion of patients who responded in the full analysis set | |||
| ASAS20 | |||
| Week 12 | 37% | 57% | 64% |
| Week 24 | 33% | 68% | 70% |
| ASAS40 | |||
| Week 12 | 19% | 40% | 50% |
| Week 24 | 16% | 48% | 59% |
| Placebo
N=57 | CIMZIA
* 200 mg every 2 weeks
N=65 | CIMZIA
† 400 mg every 4 weeks
N=56 |
||||
|---|---|---|---|---|---|---|
| Baseline | Week 12 | Baseline | Week 12 | Baseline | Week 12 | |
| All values presented represent the mean in the full analysis set | ||||||
|
||||||
| ASAS20 response criteria | ||||||
| -Patient Global Assessment (0-10) | 6.9 | 5.6 | 7.3 | 4.2 | 6.8 | 3.8 |
| -Total spinal pain (0-10) | 7.3 | 5.8 | 7.0 | 4.3 | 6.9 | 4.0 |
| -BASFI (0-10) ‡ | 6.0 | 5.2 | 5.6 | 3.8 | 5.7 | 3.8 |
| -Inflammation (0-10) | 6.7 | 5.5 | 6.7 | 3.8 | 6.4 | 3.4 |
| BASDAI (0-10) § | 6.4 | 5.4 | 6.5 | 4.0 | 6.2 | 3.7 |
| BASMI ¶ | 4.8 | 4.4 | 4.2 | 3.6 | 4.3 | 3.9 |
The percent of AS patients achieving ASAS20 responses by visit for Study AS001 is shown in Figure 3. Among patients receiving CIMZIA, clinical responses were seen in some AS patients within one to two weeks after initiation of therapy.
Figure 3: Study AS-1: ASAS20 response over 24 weeks in AS patients *
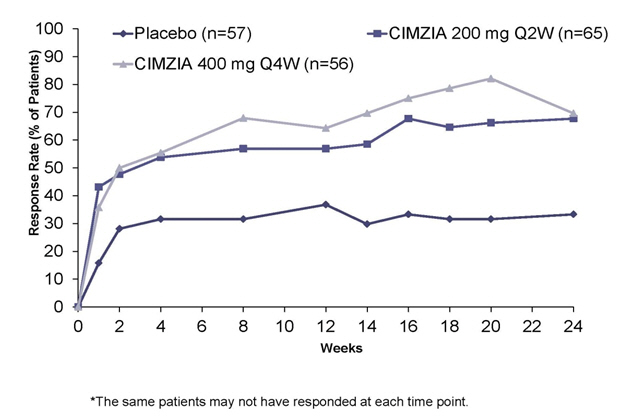
14.5 Non-radiographic Axial Spondyloarthritis
The efficacy and safety of CIMZIA were assessed in a multicenter, randomized, double-blind, placebo-controlled study (nr-axSpA-1) (NCT02552212) in 317 subjects ≥18 years of age with adult-onset active axial spondyloarthritis for at least 12 months. Patients must have had objective signs of inflammation indicated by C-reactive protein (CRP) levels above the upper limit of normal and/or sacroiliitis on magnetic resonance imaging (MRI), indicative of inflammatory disease [positive CRP (> ULN) and/or positive MRI], but without definitive radiographic evidence of structural damage on sacroiliac joints. Patients had active disease as defined by the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) ≥4, and spinal pain ≥4 on a 0 to 10 Numerical Rating Scale (NRS). Patients must have been intolerant to or had an inadequate response to at least two NSAIDs. Patients were treated with a loading dose of CIMZIA 400 mg at Weeks 0, 2 and 4 or placebo followed by 200 mg of CIMZIA every 2 weeks or placebo. Utilization and dose adjustment of concomitant medications (including NSAIDs, DMARDs, corticosteroids, opioids) were permitted at any time. Patients were allowed to transition to use of open-label CIMZIA at any time at the discretion of the investigator. However, no patients transitioned before Week 12. The primary endpoint was the proportion of patients achieving an Ankylosing Spondylitis Disease Activity Score-Major Improvement (ASDAS-MI) response at Week 52. The ASDAS is a composite weighted scoring system that assesses disease activity, including patient-reported outcomes and CRP levels. A response in ASDAS-Major Improvement (MI) is indicated by a change from baseline of ≥2.0 in the ASDAS and/or reaching the lowest possible ASDAS value.
Clinical Response
In study nr-axSpA-1, at Week 52, a greater proportion of nr-axSpA patients treated with CIMZIA had ASDAS-MI response compared to patients treated with placebo. At both Weeks 12 and 52, ASAS40 responses were greater for patients treated with CIMZIA compared to patients treated with placebo (Table 12). The components of the ASDAS-MI and ASAS response criteria are shown in Table 13.
| Parameters | Placebo | CIMZIA * 200 mg every 2 weeks | CIMZIA 200 mg versus Placebo Odds ratio
(95% CI) |
|---|---|---|---|
| N=158 | N=159 | ||
|
|||
| ASDAS-MI | |||
| Week 52 | 7% | 47% | 15.2
(7.3 , 31.6) |
| ASAS-40 | |||
| Week 12 | 11% | 48% | 7.4
(4.1, 13.4) |
| Week 52 | 16% | 57% | 7.4
(4.3, 12.6) |
| Placebo | CIMZIA * 200 mg every 2 weeks | |||
|---|---|---|---|---|
| N=158 | N=159 | |||
| Baseline
(SD) | Week 12
(SD) | Baseline
(SD) | Week 12
(SD) |
|
| Mean and standard deviation in parenthesis were presented based on full analysis set. | ||||
|
||||
| Total Spinal Pain (0-10) | 6.9 (1.8) | 6.0 (2.3) | 7.0 (1.9) | 3.9 (2.6) |
| Patient Global Assessment of Disease Activity (0-10) | 6.7 (2.0) | 5.9 (2.4) | 6.8 (1.9) | 3.8 (2.6) |
| C-Reactive Protein (mg/L) | 15.8 (17.7) | 13.2 (17.2) | 15.8 (17.8) | 6.7 (15.1) |
| BASDAI (0-10) † | 6.8 (1.3) | 5.7 (2.1) | 6.9 (1.4) | 3.9 (2.2) |
| - Back Pain | 7.4 (1.3) | 6.2 (2.1) | 7.4 (1.4) | 4.1 (2.5) |
| - Peripheral pain and swelling (0-10) | 6.2 (2.2) | 5.3 (2.5) | 6.3 (2.3) | 3.7 (2.4) |
| - Inflammation ‡ | 6.7 (1.8) | 5.5 (2.4) | 6.9 (1.8) | 3.6 (2.4) |
| BASFI (0-10) § | 5.4 (2.2) | 4.9 (2.4) | 5.4 (2.1) | 3.2 (2.3) |
| BASMI ¶ | 2.8 (1.4) | 2.7 (1.4) | 3.0 (1.3) | 2.6 (1.4) |
The percentage of nr-axSpA patients achieving ASDAS-MI response by visit for study nr-axSpA-1 is shown in Figure 4.
|
|
Figure 4: Study nr-axSpA-1: ASDAS-MI response over 12 weeks * |
|
|
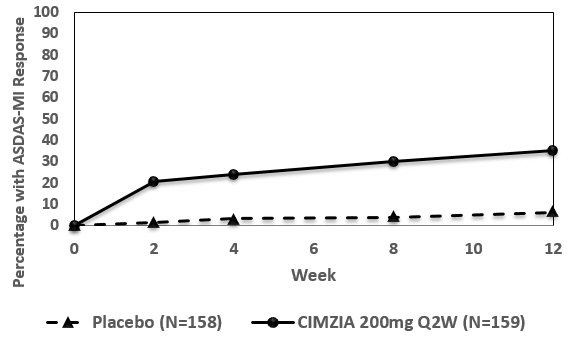
In study AS-1, at Week 12, patients with nr-axSpA treated with CIMZIA 200 mg every 2 weeks and CIMZIA 400 mg every 4 weeks had an ASAS 20 response of 42% and 47%, respectively, compared to 20% of patients treated with placebo. The ASAS 40 response in patients treated with CIMZIA 200 mg every 2 weeks and 400 mg every 4 weeks was 30% and 37%, respectively, compared to 11% of patients treated with placebo at Week 12 (see Section 14.4).
14.6 Plaque Psoriasis
Three multicenter, randomized, double-blind studies (Study PS-1 [NCT02326298], Study PS-2 [NCT02326272], and Study PS-3 [NCT02346240]) enrolled subjects 18 years of age or older with moderate-to-severe plaque psoriasis who were eligible for systemic therapy or phototherapy. Subjects had a Physician Global Assessment (PGA) of ≥ 3 ("moderate") on a 5-category scale of overall disease severity, a Psoriasis Area and Severity Index (PASI) score ≥ 12, and body surface area (BSA) involvement of ≥ 10%.
Studies PS-1 (234 subjects) and PS-2 (227 subjects) randomized subjects to placebo, CIMZIA 200 mg every other week (following a loading dose of CIMZIA 400 mg at Weeks 0, 2, and 4), or CIMZIA 400 mg every other week. Studies PS-1 and PS-2 assessed the co-primary endpoints of the proportion of patients who achieved a PASI 75 and PGA of "clear" or "almost clear" with at least a 2-point improvement at Week 16. Other evaluated outcomes were PASI 90 at Week 16 and maintenance of efficacy to Week 48.
Study PS-3 randomized 559 subjects to receive placebo, CIMZIA 200 mg every other week (following a loading dose of CIMZIA 400 mg at Weeks 0, 2, and 4), CIMZIA 400 mg every other week up to Week 16, or a biologic comparator (up to Week 12). Study PS-3 assessed the proportion of patients who achieved a PASI 75 at Week 12 as the primary endpoint. Other evaluated outcomes were PGA of "clear" or "almost clear" at Week 16, PASI 75 at Week 16, PASI 90 at Week 16, and maintenance of efficacy to Week 48.
Of the 850 subjects randomized to receive placebo or CIMZIA in these placebo-controlled studies, 29% of patients were naïve to prior systemic therapy for the treatment of psoriasis, 47% had received prior phototherapy or chemophototherapy, and 30% had received prior biologic therapy for the treatment of psoriasis. Of the 850 subjects, 14% had received at least one TNF alpha agent and 16% had received an anti-IL agent. Eighteen percent of subjects reported a history of psoriatic arthritis at baseline.
Across all studies and treatment groups, the mean PASI score at baseline was 20 and ranged from 12 to 69. The baseline PGA score ranged from moderate (70%) to severe (30%). Mean baseline BSA was 25% and ranged from 10% to 96%. Subjects were predominantly men (64%) and White (94%), with a mean age of 46 years.
Clinical Response
Table 15 presents the efficacy results of PS-1, PS-2, and PS-3 at Week 16.
| Study PS-1 | Study PS-2 | Study PS-3 † | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Placebo
(N=51) | CIMZIA 200mg
‡
Q2W (N=95) | CIMZIA 400mg
Q2W (N=88) | Placebo
(N=49) | CIMZIA 200mg
Q2W (N=91) | CIMZIA 400mg
Q2W (N=87) | Placebo
(N=57) | CIMZIA 200mg
Q2W (N=165) | CIMZIA 400mg
Q2W (N=167) |
|
|
|||||||||
| PGA of 0 or 1 §, ¶ | 4% | 45% | 55% | 3% | 61% | 65% | 4% | 52% | 62% |
| PASI 75 § | 7% | 65% | 75% | 13% | 81% | 82% | 4% | 69% | 75% |
| PASI 90 | 0% | 36% | 44% | 5% | 50% | 52% | 0% | 40% | 49% |
Examination of age, gender, prior use of biologics, and prior use of systemic therapies did not identify difference in response to CIMZIA among these subgroups.
Based on a post-hoc subgroup analysis in subjects with moderate-to-severe psoriasis, stratified by ≤90 kg or >90 kg, subjects with both lower body weight and lower disease severity may achieve an acceptable response with CIMZIA 200 mg.
Maintenance of Response
In PS-1 and PS-2, among subjects who were PASI 75 responders at Week 16 and received CIMZIA 400 mg every other week, the PASI 75 response rates at Week 48 were 94% and 81%, respectively. In subjects who were PASI 75 responders at Week 16 and received CIMZIA 200 mg every other week, the PASI 75 response rates at Week 48 were 81% and 74%, respectively.
In PS-1 and PS-2, among subjects who were PGA clear or almost clear responders at Week 16 and received CIMZIA 400 mg every other week, the PGA response rates at Week 48 were 79% and 73%, respectively. In subjects who were PGA clear or almost clear responders at Week 16 and received CIMZIA 200 mg every other week, the PGA response rates at Week 48 were 79% and 76%, respectively.
In PS-3 study, subjects who achieved a PASI 75 response at Week 16 were re-randomized to either continue treatment with CIMZIA or be withdrawn from therapy (i.e., receive placebo). At Week 48, 98% of subjects who continued on CIMZIA 400 mg every other week were PASI 75 responders as compared to 36% of subjects who were re-randomized to placebo. Among PASI 75 responders at Week 16 who received CIMZIA 200 mg every other week and were re-randomized to either CIMZIA 200 mg every other week or placebo, there was also a higher percentage of PASI 75 responders at Week 48 in the CIMZIA group as compared to placebo (80% and 46%, respectively).
16 HOW SUPPLIED/STORAGE AND HANDLING
Storage and Stability
Refrigerate carton between 2 to 8 °C (36 to 46 °F). Do not freeze. Do not separate contents of carton prior to use. Do not use beyond expiration date, which is located on the drug label and carton. Protect solution from light.
Unopened CIMZIA vials may also be stored at room temperature up to a maximum of 25°C (77°F) for 6 months, but not exceeding the original expiration date. If stored at room temperature, do not place back in refrigerator and write the new expiration date on the carton in the space provided.
Lyophilized Powder for Reconstitution:
NDC 50474-700-62
CIMZIA (certolizumab pegol) for injection is supplied as a sterile white, lyophilized powder in a single-dose vial for subcutaneous use.
| Qty. | Item |
|---|---|
| 2 | Type I glass vials with rubber stopper and overseals each containing 200 mg of lyophilized CIMZIA for reconstitution. |
| 2 | 2 mL Type I glass vials containing 1 mL sterile Water for Injection |
| 2 | 3 mL plastic syringes |
| 4 | 20 gauge needles (1 inch) |
| 2 | 23 gauge needles (1 inch) |
| 8 | Alcohol swabs |
Prefilled Syringe
NDC 50474-710-79
CIMZIA (certolizumab pegol) injection is supplied as a sterile, clear to opalescent, colorless to pale yellow solution in a single-dose prefilled syringe for subcutaneous use.
2 alcohol swabs and 2 single-dose prefilled glass syringes with a fixed 25 ½ gauge thin-wall needle, each containing 200 mg (1 mL) of CIMZIA. The needle shield inside the removable cap of the CIMZIA prefilled syringe contains a derivative of natural rubber latex which may cause allergic reactions and should be handled with caution by latex-sensitive individuals [see Warnings and Precautions (5.4)].
Prefilled Syringe Starter Kit
NDC 50474-710-81
6 alcohol swabs and 6 single dose prefilled glass syringes with a fixed 25 ½ gauge thin-wall needle. The Starter Kit contains 3 sets of 2 prefilled syringes to provide sufficient drug supply for the initial 3 induction doses at the start of treatment. Each prefilled syringe contains 200 mg (1 mL) of CIMZIA.
When necessary, CIMZIA prefilled syringes may be stored at room temperature up to 77 °F (25 °C) in the original carton to protect from light for a single period of up to 7 days. Once a CIMZIA prefilled syringe has been stored at room temperature, do not place back in refrigerator. Write the date removed from the refrigerator in the space provided on the carton and discard if not used within the 7-day period.
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide and Instructions for Use)
Risk of Serious Infections
Inform patients that CIMZIA may lower the ability of the immune system to fight infections. Instruct patients of the importance of contacting their doctor if they develop any symptoms of infection, including tuberculosis and reactivation of hepatitis B virus infections.
Because caution should be exercised in prescribing CIMZIA to patients with clinically important active infections, advise patients of the importance of informing their health care providers about all aspects of their health [see Warnings and Precautions (5.1, 5.5)] .
Malignancies
Counsel patients about the possible risk of lymphoma and other malignancies while receiving CIMZIA [see Warnings and Precautions (5.2)] .
Other Medical Conditions
Advise patients to report any signs of new or worsening medical conditions such as heart disease, neurological disease, or autoimmune disorders [see Warnings and Precautions (5.3, 5.6, 5.9). Advise patients to report promptly any symptoms suggestive of a cytopenia such as bruising, bleeding, or persistent fever [see Warnings and Precautions (5.7)] .
Hypersensitivity Reactions
Advise patients to seek immediate medical attention if they experience any symptoms of severe hypersensitivity reactions. Advise latex-sensitive patients that the needle shield inside the removable cap of the CIMZIA prefilled syringe contains a derivative of natural rubber latex [see Warnings and Precautions (5.4)] .
Pregnancy
Advise patients that there is a pregnancy registry that monitors pregnancy outcomes in women exposed to CIMZIA during pregnancy, patients can call 1-877-311-8972 [see Use in Specific Populations 8.1] .
Preparation and Administration of CIMZIA Using the Prefilled Syringe
Instruct patients and caregivers on how to inject the Prefilled Syringe. Complete instructions are provided in the Instructions for Use packaged in each CIMZIA Prefilled Syringe kit.
- If refrigerated, remove the prefilled syringe from the carton and let it warm to room temperature.
- Inspect the liquid in the prefilled syringe. It should be clear and colorless to yellow and free from particulates. Discard the syringe if cloudy, discolored or contains particulates.
- Suitable sites for injection include the thigh or abdomen. Inject at least 1 inch from the previous site.
- Do not inject into areas where the skin is tender, bruised, red or hard, or where there are scars or stretch marks.
Instruct patients and caregivers in proper syringe and needle disposal technique.
- To avoid needle-stick injury, do not to place the needle cap back on the syringe or otherwise recap the needle.
- Properly dispose of needles and syringes in a puncture-proof container.
- Do not reuse the injection materials.
| This Medication Guide has been approved by the U.S. Food and Drug Administration. | Revised: April 2020 | |||
| MEDICATION GUIDE
CIMZIA ® (CIM-zee-uh) (certolizumab pegol) injection for subcutaneous use |
||||
| What is the most important information I should know about CIMZIA?
CIMZIA may cause serious side effects, including:
|
||||
|
|
|||
|
||||
| Stop using CIMZIA, and tell your healthcare provider right away if you have any of the symptoms of an infection listed above. | ||||
|
||||
| What is CIMZIA?
CIMZIA is a prescription medicine called a Tumor Necrosis Factor (TNF) blocker used in adults to:
|
||||
Before receiving CIMZIA, tell your healthcare provider about all of your medical conditions, including if you:
Know the medicines you take. Keep a list of them to show to your healthcare provider and pharmacist when you get a new medicine. |
||||
How will I receive CIMZIA?
|
||||
| What are the possible side effects of CIMZIA?
CIMZIA can cause serious side effects, including:
|
||||
|
|
|||
|
||||
|
|
|||
The most common side effects of CIMZIA include upper respiratory infections (flu, cold), rash, urinary tract infections (bladder infections). These are not all of the possible side effects of CIMZIA. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
||||
How should I store CIMZIA?
|
||||
| General information about the safe and effective use of CIMZIA.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use CIMZIA for a condition for which it was not prescribed. Do not give CIMZIA to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about CIMZIA that is written for health professionals. |
||||
| What are the ingredients in CIMZIA?
CIMZIA lyophilized powder: Active ingredient: certolizumab pegol Inactive ingredients: lactic acid, polysorbate, sucrose CIMZIA lyophilized powder is mixed with sterile Water for Injection. CIMZIA prefilled syringe: Active ingredient: certolizumab pegol Inactive ingredients: sodium acetate, sodium chloride, Water for Injection CIMZIA has no preservatives. Product manufactured by: UCB, Inc. 1950 Lake Park Drive Smyrna, GA 30080 US License No. 1736 For more information, go to www.CIMZIA.com or call 1-866-424-6942. |
||||
Instructions for Use
CIMZIA
® (CIM-zee-uh)
(certolizumab pegol)
injection for subcutaneous use
Prefilled Syringes
Read this Instructions for Use booklet that comes with CIMZIA before you start receiving it, and before each injection of CIMZIA. This Instructions for Use booklet does not take the place of talking with your healthcare provider about your medical condition or treatment. These instructions are for 1 injection only. You may need more than 1 injection at a time depending on your prescribed dose of CIMZIA.
| Do not share your CIMZIA Prefilled Syringe with needle attached with another person. You may give another person an infection or get an infection from them.
Important: The plastic needle shield inside the removable cap contains natural rubber. Tell your healthcare provider if you are allergic to latex. Supplies you will need to give your CIMZIA injection: See Figure A and Figure B.
CIMZIA comes in a tray containing 2 prefilled glass syringes. Use a new CIMZIA syringe for each injection. |
|
Storage information:
|
|
| Setting up for your CIMZIA injection:
Step 1. If refrigerated, take the carton containing the prefilled syringes of CIMZIA out of the refrigerator. Check the expiration date on the syringe carton and label. See Figure C. |
|
| If the expiration date has passed,
do not use the syringe. Call your pharmacist for questions about the expiration date. Do not use CIMZIA if the carton has been opened or tampered with.
|
|
| Step 2.
Remove the prefilled syringe from the carton and let it warm to room temperature, this will take about 30 minutes. Do not warm the syringe in any other way. If you are not using the second syringe, put the carton containing the remaining prefilled syringe back in the refrigerator. |
|
| Step 3.
Find a clean, flat work surface, such as a table. |
|
| Step 4.
Make sure the liquid medicine in the prefilled syringe is clear and colorless to yellow and free from particles. Do not inject the medicine if it is cloudy, discolored, or contains particles. Call your healthcare provider or pharmacist if you have any questions about your CIMZIA prefilled syringe. |
|
| Step 5.
Gather all the supplies you will need for your injection. |
|
| Step 6.
Wash your hands with soap and warm water and dry with a clean towel. |
|
| Selecting and preparing your injection site:
|
|
| Step 7.
Choose your injection site(s) on your stomach or upper thighs. See Figure D. 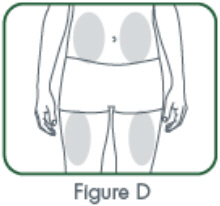
|
|
| Step 8.
Clean your injection site with an alcohol swab. Let the area dry completely. |
|
| Giving your CIMZIA injection:
Step 9. Pick up the prefilled syringe with 1 hand and hold it with the needle pointing up. You may see air bubbles. This is normal. There is no need to remove air bubbles before giving your injection. Injecting the solution with air bubbles will not harm you. With your other hand, remove the plastic ring needle cap by pulling straight up on the plastic ring. See Figure E. |
|
| Do not touch the needle and do not let the needle touch any surface.
Do not bend the needle. Place the plastic ring needle cap to the side. |
|
| Step 10.
With your other hand, gently pinch a fold of skin at the cleaned injection site. See Figure F. |
|
| Step 11.
With a quick, "dart-like" motion, insert the needle into your skin at about a 45 degree angle. Release the pinched skin, keeping the syringe in position. Slowly push on the plunger all the way down until the syringe is empty. See Figure G. |
|
| Step 12.
When the syringe is empty, pull the needle out of your skin while carefully keeping the needle at the same angle as inserted. See Figure H. |
|
| Step 13.
Place a dry cotton ball or gauze pad over the injection site for several seconds. See Figure I. |
|
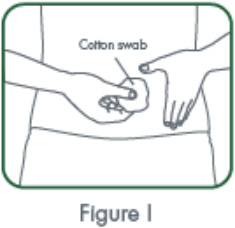 |
|
| Do not rub the injection site. Do not use an alcohol swab as it may cause stinging. If there is a little bleeding, cover the injection site with a small bandage.
To avoid a needle-stick injury, do not try to recap the needle. Do not reuse any of your injection supplies. |
|
|
Disposal of your syringes with needles attached:
|
|
| Do not throw away (dispose of) loose syringes and needles in your household trash. |
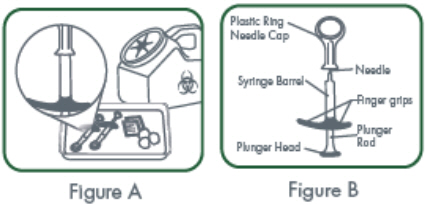
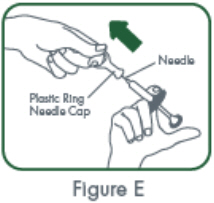
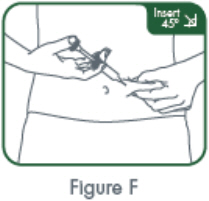
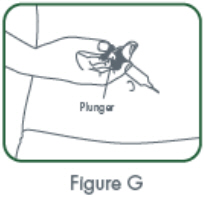
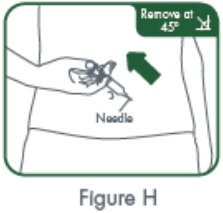
- If you do not have a FDA-cleared sharps disposal container, you may use a household container that is:
- made of a heavy-duty plastic
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out
- upright and stable during use
- leak-resistant
- properly labeled to warn of hazardous waste inside the container
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: http://www.fda.gov/safesharpsdisposal.
Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Do not recycle your used sharps disposal container.
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Product manufactured by:
UCB, Inc.
1950 Lake Park Drive
Smyrna, GA 30080
Revised: 04/2020
CIMZIA®
(certolizumab pegol)
Preparation and Administration of Lyophilized
Powder for Injection
(200 mg/vial)
CIMZIA Lyophilized powder should be prepared and administered by a health care professional.
Contents:
2 inner kits, each containing 1 vial of CIMZIA 200 mg, 1 vial of sterile water for injection (USP 1 mL), 1 single-dose plastic syringe, 4 alcohol swabs, two reconstitution needles, and one dosing needle.
Refrigerate carton at 2 to 8°C (36 to 46°F). DO NOT FREEZE. Do not separate contents of carton prior to use. Do not use beyond expiration date on container.
Unopened CIMZIA vials may also be stored at room temperature up to a maximum of 25°C (77°F) for 6 months, but not exceeding the original expiration date. If stored at room temperature, do not place back in refrigerator and write the new expiration date on the carton in the space provided.
If refrigerated, remove CIMZIA from the refrigerator and allow the vial(s) to sit at room temperature for 30 minutes before reconstituting. Do not warm the vial in any other way. Using appropriate aseptic technique, reconstitute each lyophilized vial of CIMZIA with 1 mL of sterile water for injection using a syringe with a fresh 20-gauge needle. The sterile water for injection should be directed at the vial wall rather than directly on CIMZIA. (See Figure 1.)
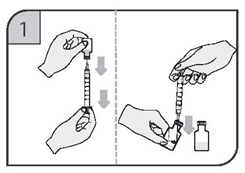
The resultant solution will contain 200 mg/mL.
Gently swirl each vial of CIMZIA for about one minute without shaking, ensuring that all of the powder comes into contact with the Sterile Water for Injection. The swirling should be as gentle as possible in order to avoid creating a foaming effect. (See Figure 2.)
Continue swirling every 5 minutes as long as non-dissolved particles are observed. Full reconstitution may take as long as 30 minutes. The final reconstituted solution contains 200 mg/mL and should be clear to opalescent, colorless to pale yellow liquid and essentially free from particulates.
Once reconstituted, CIMZIA can be stored in the vials for up to 24 hours between 2° to 8° C (36° to 46° F) prior to injection. Do not freeze.
When ready to inject, reconstituted CIMZIA should be at room temperature. Do not leave reconstituted CIMZIA at room temperature for more than 2 hours prior to administration.
Withdraw the reconstituted solution into a separate syringe for each vial using a new 20-gauge needle for each vial so that each syringe contains 1 mL of CIMZIA (200 mg of certolizumab pegol). (See Figure 3.)
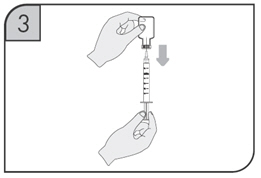
Replace the 20-gauge needle(s) on the syringes with a 23-gauge needle(s) for administration. (See Figure 4.)
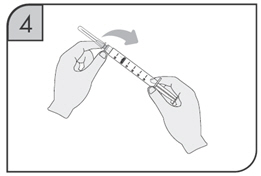
Inject the full contents of the syringe(s) subcutaneously by pinching the skin of the abdomen (See Figure 5) or thigh (See Figure 6). Where a 400 mg dose is required, two injections are required. Therefore, separate sites should be used for each 200 mg injection.
| OR |
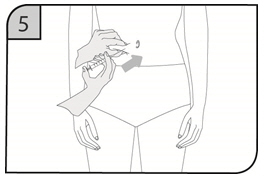
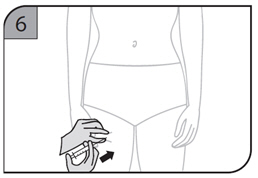
You can also visit the CIMZIA Web site at www.cimziahcp.com
06/2018
U.S. License No. 1736
Manufactured by UCB Inc. Smyrna GA 30080
UCB Pharma S.A.
PRINCIPAL DISPLAY PANEL - Kit Carton
NDC 50474-700-62
Rx ONLY
OPEN HERE
cimzia
®
(certolizumab pegol)
200 mg/vial
FOR SUBCUTANEOUS USE ONLY
Single-dose vial. Discard unused portion.
No US standard of potency
MUST BE RECONSTITUTED
Dispense the enclosed Medication Guide to each patient.
See package insert for reconstitution and dosage information.
After reconstitution with 1 mL of Sterile Water for Injection, USP, each mL
contains 200 mg of certolizumab pegol.
Storage
Refrigerate carton at 2° to 8° C (36° to 46° F) in carton to protect from
light. Do Not Freeze.
Unopened vials may be stored at room temperature up to a maximum of
25°C (77°F) for 6 months. If stored at room temperature, do not place back
in refrigerator and discard after:
___/___/___
Contains 2 vials of certolizumab pegol, each containing 200 mg
ucb
DO NOT SEPARATE CONTENTS
OF CARTON PRIOR TO USE

PRINCIPAL DISPLAY PANEL - 1 mL Syringe Carton Box
NDC 50474-710-81
Rx ONLY
Dispense the enclosed Medication Guide to each patient.
cimzia
®
(certolizumab pegol)
STARTER KIT
3 cartons of 2 x 200 mg/mL
PREFILLED SYRINGES
FOR SUBCUTANEOUS INJECTION USE ONLY
The entire carton is to be dispensed as one unit.
Each unit carton contains three individual cartons (doses).
Each individual carton (dose) contains:
2 single-dose prefilled syringes (200 mg/1mL) and 2 alcohol swabs.
