FULL PRESCRIBING INFORMATION
WARNING: EXCESSIVE SEDATION AND SUDDEN LOSS OF CONSCIOUSNESS
Patients treated with ZULRESSO are at risk of excessive sedation or sudden loss of consciousness during administration [see Warnings and Precautions (5.1)].
Because of the risk of serious harm, patients must be monitored for excessive sedation and sudden loss of consciousness and have continuous pulse oximetry monitoring. Patients must be accompanied during interactions with their child(ren) [see Warnings and Precautions (5.1)].
Because of these risks, ZULRESSO is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the ZULRESSO REMS [see Warnings and Precautions (5.2)].
1 INDICATIONS AND USAGE
ZULRESSO is indicated for the treatment of postpartum depression (PPD) in patients 15 years and older [see Clinical Studies (14)].
2 DOSAGE AND ADMINISTRATION
2.1 Important Considerations Prior to Initiating and During Therapy
A healthcare provider must be available on site to continuously monitor the patient, and intervene as necessary, for the duration of the ZULRESSO infusion.
Monitor patients for hypoxia using continuous pulse oximetry equipped with an alarm. Assess for excessive sedation every 2 hours during planned, non-sleep periods [see Warnings and Precautions (5.1)].
Initiate ZULRESSO treatment early enough during the day to allow for recognition of excessive sedation [see Warnings and Precautions (5.1)].
2.2 Recommended Dosage
Administer ZULRESSO as a continuous intravenous (IV) infusion over a total of 60 hours (2.5 days) as follows:
- 0 to 4 hours: Initiate with a dosage of 30 mcg/kg/hour
- 4 to 24 hours: Increase dosage to 60 mcg/kg/hour
- 24 to 52 hours: Increase dosage to 90 mcg/kg/hour (a reduction in dosage to 60 mcg/kg/hour may be considered during this time period for patients who do not tolerate 90 mcg/kg/hour)
- 52 to 56 hours: Decrease dosage to 60 mcg/kg/hour
- 56 to 60 hours: Decrease dosage to 30 mcg/kg/hour
If excessive sedation occurs at any time during the infusion, stop the infusion until the symptoms resolve. The infusion may be resumed at the same or lower dose as clinically appropriate.
2.3 Preparation and Storage Instructions
ZULRESSO is supplied in vials as a concentrated solution that requires dilution prior to administration. After dilution, the product can be stored in infusion bags under refrigerated conditions for up to 96 hours. However, given that the diluted product can be used for only 12 hours at room temperature, each 60-hour infusion will require the preparation of at least five infusion bags.
Prepare according to the following steps using aseptic technique:
- Visually inspect the vials of ZULRESSO for particulate matter and discoloration prior to administration. ZULRESSO is a clear, colorless solution. Do not use if the solution is discolored or particulate matter is present.
- The 60-hour infusion will generally require the preparation of five infusion bags. Additional bags will be needed for patients weighing ≥ 90 kg.
- For each infusion bag:
- Prepare and store in a polyolefin, non-DEHP, nonlatex bag, only. Dilute in the infusion bag immediately after the initial puncture of the drug product vial.
- Withdraw 20 mL of ZULRESSO from the vial and place in the infusion bag. Dilute with 40 mL of Sterile Water for Injection, and further dilute with 40 mL of 0.9% Sodium Chloride Injection (total volume of 100 mL) to achieve a target concentration of 1 mg/mL.
- Immediately place the infusion bag under refrigerated conditions until use.
Diluted ZULRESSO storage instructions:
- If not used immediately after dilution, store under refrigerated conditions for up to 96 hours. Prolonged storage at room temperature may support adventitious microbial growth.
- Each prepared bag of diluted ZULRESSO may be used for up to 12 hours of infusion time at room temperature. Discard any unused ZULRESSO after 12 hours of infusion.
2.4 Administration Instructions
ZULRESSO must be diluted before administration [see Dosage and Administration (2.3)]. The following are important administration instructions:
- Use a programmable peristaltic infusion pump to ensure accurate delivery of ZULRESSO.
- Administer ZULRESSO via a dedicated line. Do not inject other medications into the infusion bag or mix with ZULRESSO.
- Fully prime infusion administration sets with admixture before inserting into the pump and connecting to the venous catheter.
- Use a PVC, non-DEHP, nonlatex infusion set. Do not use in-line filter infusion sets.
2.5 Recommendations in Patients with End Stage Renal Disease
Avoid use of ZULRESSO in patients with end stage renal disease (ESRD) with eGFR of < 15 mL/minute/1.73 m2 because of the potential accumulation of the solubilizing agent, betadex sulfobutyl ether sodium [see Clinical Pharmacology (12.3, 12.6)].
3 DOSAGE FORMS AND STRENGTHS
Injection: 100 mg/20 mL (5 mg/mL) clear, colorless solution in a single-dose vial.
5 WARNINGS AND PRECAUTIONS
5.1 Excessive Sedation and Sudden Loss of Consciousness
In clinical studies in adults, ZULRESSO caused sedation and somnolence that required dose interruption or reduction in some patients during the infusion (5% of ZULRESSO-treated patients compared to 0% of placebo-treated patients). Some adult patients were also reported to have loss of consciousness or altered state of consciousness during the ZULRESSO infusion (4% of the ZULRESSO-treated patients compared with 0% of the placebo-treated patients). Time to full recovery from loss or altered state of consciousness, after dose interruption, ranged from 15 to 60 minutes in clinical studies in adults. A healthy 55-year-old man participating in a cardiac repolarization study experienced severe somnolence and <1 minute of apnea while receiving two times the maximum recommended dosage of ZULRESSO (180 mcg/kg/hour).
In an open-label clinical study in 20 patients ages 15 to 17 years, one patient experienced dizziness and loss of consciousness.
All patients with loss of or altered state of consciousness recovered with dose interruption. There was no clear association between loss or alteration of consciousness and pattern or timing of dose. Not all patients who experienced a loss or alteration of consciousness reported sedation or somnolence before the episode.
During the infusion, monitor patients for sedative effects every 2 hours during planned, non-sleep periods. Immediately stop the infusion if there are signs or symptoms of excessive sedation.
After symptoms resolve, the infusion may be resumed at the same or lower dose as clinically appropriate [see Dosage and Administration (2.2)].
Immediately stop the infusion if pulse oximetry reveals hypoxia. After hypoxia, the infusion should not be resumed.
Patients should be cautioned against engaging in potentially hazardous activities requiring mental alertness, such as driving after infusion until any sedative effects of ZULRESSO have dissipated. Patients must be accompanied during interactions with their child(ren) while receiving the infusion because of the potential for excessive sedation and sudden loss of consciousness.
Concomitant use of opioids, antidepressants, or other CNS depressants such as benzodiazepines or alcohol may increase the likelihood or severity of adverse reactions related to sedation [see Drug Interactions (7.1, 7.2)].
Because of the risk of serious harm resulting from excessive sedation or sudden loss of consciousness, ZULRESSO is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the ZULRESSO REMS [see Warnings and Precautions (5.2)].
5.2 ZULRESSO Risk Evaluation and Mitigation Strategy (REMS)
ZULRESSO is available only through a restricted program under a REMS called the ZULRESSO REMS because excessive sedation or sudden loss of consciousness can result in serious harm [see Warnings and Precautions (5.1)].
Notable requirements of the ZULRESSO REMS include the following:
- Healthcare facilities must enroll in the program and ensure that ZULRESSO is only administered to patients who are enrolled in the ZULRESSO REMS.
- Pharmacies must be certified with the program and must only dispense ZULRESSO to healthcare facilities who are certified in the ZULRESSO REMS.
- Patients must be enrolled in the ZULRESSO REMS prior to administration of ZULRESSO.
- Wholesalers and distributors must be registered with the program and must only distribute to certified healthcare facilities and pharmacies.
Further information, including a list of certified healthcare facilities, is available at www.zulressorems.com or 1-844-472-4379.
5.3 Suicidal Thoughts and Behaviors
In pooled analyses of placebo-controlled trials of chronically administered antidepressant drugs (SSRIs and other antidepressant classes) that included approximately 77,000 adult patients and 4,500 pediatric patients, the incidence of suicidal thoughts and behaviors in antidepressant-treated patients age 24 years and younger was greater than in placebo-treated patients. There was considerable variation in risk of suicidal thoughts and behaviors among drugs, but there was an increased risk identified in young patients for most drugs studied. There were differences in absolute risk of suicidal thoughts and behaviors across the different indications, with the highest incidence in patients with major depressive disorder (MDD). The drug-placebo differences in the number of cases of suicidal thoughts and behaviors per 1000 patients treated are provided in Table 1.
|
|
| Age Range (years) | Drug-Placebo Difference in Number of Patients with Suicidal Thoughts or Behaviors per 1000 Patients Treated |
| Increases Compared to Placebo | |
| <18 | 14 additional patients |
| 18-24 | 5 additional patients |
| Decreases Compared to Placebo | |
| 25-64 | 1 fewer patient |
ZULRESSO does not directly affect monoaminergic systems. Because of this and the comparatively low number of exposures to ZULRESSO, the risk of developing suicidal thoughts and behaviors with ZULRESSO is unknown. Consider changing the therapeutic regimen, including discontinuing ZULRESSO, in patients whose depression becomes worse or who experience emergent suicidal thoughts and behaviors.
6 ADVERSE REACTIONS
The following adverse reactions are discussed in more detail in other sections of the labeling:
- Excessive Sedation and Sudden Loss of Consciousness [see Boxed Warning, Warnings and Precautions (5.1)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The safety of ZULRESSO was evaluated in 140 adult patients with postpartum depression (PPD). A titration to a target dosage of 90 mcg/kg/hour was evaluated in 102 adult patients and a titration to a target dose of 60 mcg/kg/hour was evaluated in 38 adult patients [see Clinical Studies (14)]. Patients were then followed for 4 weeks.
The most common adverse reactions (incidence ≥5% and at least twice the rate of placebo) were sedation/somnolence, dry mouth, loss of consciousness, and flushing/hot flush (Table 2).
Adverse Reactions Leading to Discontinuation, Dosage Interruption, or Dosage Reduction
In the pooled placebo controlled-studies in adults, the incidence of patients who discontinued due to any adverse reaction was 2% of ZULRESSO-treated patients compared to 1% of placebo-treated patients. The adverse reactions leading to treatment discontinuation in ZULRESSO-treated patients were sedation-related (loss of consciousness, vertigo, syncope, and presyncope) or infusion site pain.
In the pooled placebo controlled-studies in adults, the incidence of patients who had an interruption or reduction of the dosage due to any adverse reaction was 7% of ZULRESSO-treated patients compared to 3% of placebo-treated patients. The adverse reactions leading to dose reduction or interruption in ZULRESSO-treated patients were sedation-related (loss of consciousness, syncope, somnolence, dizziness, fatigue), infusion site events, changes in blood pressure, or medication error due to infusion pump malfunction. Three ZULRESSO-treated patients who had a dosage interruption because of loss of consciousness subsequently resumed and completed treatment after resolution of symptoms; two patients who had dosage interruption because of loss of consciousness did not resume the infusion.
Table 2 presents the adverse reactions that occurred in ZULRESSO-treated adult PPD patients at a rate of at least 2% and at a higher rate than in the placebo-treated patients during the 60-hour treatment period.
| Placebo (n=107) | Maximum dosage 60 mcg/kg/hour (n=38) | Maximum dosage 90 mcg/kg/hour (Recommended dosage) (n=102) |
|
| Cardiac Disorders | |||
| Tachycardia | - | - | 3% |
| Gastrointestinal Disorders | |||
| Diarrhea | 1% | 3% | 2% |
| Dry mouth | 1% | 11% | 3% |
| Dyspepsia | - | - | 2% |
| Oropharyngeal pain | - | 3% | 2% |
| Nervous System Disorders | |||
| Dizziness, presyncope, vertigo | 7% | 13% | 12% |
| Loss of consciousness | - | 5% | 3% |
| Sedation, somnolence | 6% | 21% | 13% |
| Vascular Disorders | |||
| Flushing, hot flush | - | 5% | 2% |
Patients 15 to 17 Years
The safety of ZULRESSO was evaluated in an open-label study in patients 15 to 17 years. A titration to a target dosage of 90 mcg/kg/hour was evaluated in 20 patients with PPD. Patients were then followed for 4 weeks. Adverse reactions reported in the clinical study were generally similar to those observed in clinical studies of ZULRESSO in adults with PPD.
7 DRUG INTERACTIONS
7.1 CNS Depressants
Concomitant use of ZULRESSO with CNS depressants (e.g., opioids, benzodiazepines) may increase the likelihood or severity of adverse reactions related to sedation [see Warnings and Precautions (5.1)].
7.2 Antidepressants
In the placebo-controlled studies, a higher percentage of ZULRESSO-treated patients who used concomitant antidepressants reported sedation-related events [see Warnings and Precautions (5.1)].
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to antidepressants during pregnancy. Healthcare providers are encouraged to register patients by calling the National Pregnancy Registry for Antidepressants at 1-844-405-6185 or visiting online at https://womensmentalhealth.org/clinical-and-research-programs/pregnancyregistry/antidepressants/ .
Risk Summary
Based on findings from animal studies of other drugs that enhance GABAergic inhibition, ZULRESSO may cause fetal harm. Available data from case reports with ZULRESSO use in pregnant women are insufficient to establish a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. In animal reproduction studies, malformations were not seen in rats or rabbits at plasma levels up to 5 and 6 times the maximum recommended human dose (MRHD), respectively. Developmental toxicities were seen in the fetuses of rats and rabbits at 5 and ≥3 times the plasma levels at the MRHD, respectively. Reproductive toxicities were seen in rabbits at ≥3 times the plasma levels at the MRHD. These effects were not seen in rats and rabbits at 2 and 1.2 times the plasma levels at the MRHD. Brexanolone administered to pregnant rats during pregnancy and lactation resulted in lower pup survival at doses which were associated with ≥2 times the plasma levels at the MRHD and a neurobehavioral deficit in female offspring at 5 times the plasma levels at the MRHD. These effects were not seen at 0.8 times and 2 times the plasma levels at the MRHD, respectively (see Data).
In published animal studies, administration of other drugs that enhance GABAergic inhibition to neonatal rats caused widespread apoptotic neurodegeneration in the developing brain. The window of vulnerability to these changes in rats (postnatal days 0-14) corresponds to the period of brain development that takes place during the third trimester of pregnancy in humans.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Animal Data
In pregnant rats and rabbits, no malformations were seen when brexanolone was given during the period of organogenesis at continuous intravenous doses up to 60 and 30 mg/kg/day, respectively. These doses were associated with maternal plasma levels 5 and 6 times the plasma levels at the MRHD of 90 mcg/kg/hour, in rats and rabbits, respectively. In rats, a decrease in fetal body weights was seen at 60 mg/kg/day (5 times the plasma level at the MRHD). In rabbits, increased numbers of late resorptions and a decrease in fetal body weights were seen at doses equal to and greater than 15 mg/kg/day (3 times the plasma levels at the MRHD) with fewer live fetuses and a higher post implantation loss seen at 30 mg/kg/day (6 times the plasma levels at the MRHD) in the presence of maternal toxicity (decreased food consumption and decreased body weight gain and/or body weight loss). Effects in rats and rabbits were not seen at 2 and 1.2 times the plasma levels at the MRHD, respectively.
When brexanolone was administered to pregnant rats by continuous intravenous administration at 30 and 60 mg/kg/day (2 and 5 times plasma levels at the MRHD, respectively) during the period of organogenesis and throughout pregnancy and lactation, increased numbers of dead pups and fewer live pups at birth were seen. This effect was not seen at 0.8 times the plasma levels at the MRHD. Decreased pup viability between postnatal day 0 and 4 in the presence of maternal toxicity (decreased body weight gain and food consumption during lactation) was seen at 5 times the plasma levels at the MRHD. These effects were not seen at 2 times the plasma levels at the MRHD. A neurobehavioral deficit, characterized by slower habituation in the maximal startle response in the auditory startle test, was seen in female offspring of dams dosed at 5 times the plasma levels at the MRHD. This effect was not seen at 2 times the plasma levels at the MRHD.
8.2 Lactation
Risk Summary
Available data from a lactation study in 12 adult women indicate that brexanolone is transferred to breastmilk in nursing mothers. However, the relative infant dose (RID) is low, 1% to 2% of the maternal weight-adjusted dosage (see Data). Also, as ZULRESSO has low oral bioavailability (<5%) in adults, infant exposure is expected to be low. There were no reports of effects of ZULRESSO on milk production. There are no data on the effects of ZULRESSO on a breastfed infant. Available data on the use of ZULRESSO during lactation do not suggest a significant risk of adverse reactions to breastfed infants from exposure to ZULRESSO. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for ZULRESSO and any potential adverse effects on the breastfed child from ZULRESSO or from the underlying maternal condition.
A study was conducted in twelve healthy adult lactating women treated with intravenous ZULRESSO according to the recommended 60-hour dosing regimen (maximum dosage was 90 mcg/kg/hour). Concentrations of ZULRESSO in breast milk were at low levels (<10 ng/mL) in >95% of women by 36 hours after the end of the infusion of ZULRESSO. The calculated maximum relative infant dose for ZULRESSO during the infusion was 1% to 2%.
8.4 Pediatric Use
Safety and effectiveness of ZULRESSO for the treatment of PPD have been established in patients 15 to 17 years.
Use of ZULRESSO in this population is supported by evidence from adequate and well-controlled studies in adults with PPD, pharmacokinetic data in adults and patients 15 to 17 years, and safety data in patients 15 to 17 years [see Warnings and Precautions (5.3), Adverse Reactions (6.1), and Clinical Pharmacology (12.3)].
The safety and effectiveness of ZULRESSO in patients less than 15 years of age have not been established.
8.5 Geriatric Use
PPD is a condition associated with pregnancy; there is no geriatric experience with ZULRESSO.
8.6 Hepatic Impairment
Dosage adjustment in patients with hepatic impairment is not necessary. Modest increases in exposure to unbound brexanolone and modest decreases in exposure to total brexanolone were observed in patients with moderate to severe hepatic impairment (Child-Pugh≥7) with no associated change in tolerability [see Clinical Pharmacology (12.3)].
8.7 Renal Impairment
No dosage adjustment is recommended in patients with mild (eGFR 60 to 89 mL/minute/1.73 m2), moderate (eGFR 30 to 59 mL/minute/1.73 m2) or severe (eGFR 15 to 29 mL/minute/1.73 m2) renal impairment [see Clinical Pharmacology (12.3)].
Avoid use of ZULRESSO in patients with end stage renal disease (ESRD) with eGFR of < 15 mL/minute/1.73 m2 because of the potential accumulation of the solubilizing agent, betadex sulfobutyl ether sodium [see Clinical Pharmacology (12.3, 12.6)].
9 DRUG ABUSE AND DEPENDENCE
9.1 Controlled Substance
ZULRESSO contains brexanolone, a Schedule IV controlled substance under the Controlled Substances Act.
9.2 Abuse
In a human abuse potential study, 90 mcg/kg, 180 mcg/kg (two times the maximum recommended infusion rate), and 270 mcg/kg (three times the maximum recommended infusion rate) ZULRESSO infusions over a one-hour period were compared to oral alprazolam administration (1.5 mg and 3 mg). On positive subjective measures of "drug liking", "overall drug liking", "high" and "good drug effects", the 90 mcg/kg dosage produced scores that were similar to placebo. Scores on these positive subjective measures for both dosages of ZULRESSO 90 mcg/kg and 180 mcg/kg were lower than both alprazolam doses. However, the scores on the positive subjective measures for ZULRESSO 270 mcg/kg dosage were similar to those produced by both doses of alprazolam. In this study, 3% of subjects administered ZULRESSO 90 mcg/kg and 13% administered ZULRESSO 270 mcg/kg reported euphoric mood, compared to none administered placebo during the one-hour administration.
9.3 Dependence
In the PPD clinical studies conducted with ZULRESSO, end of treatment occurred through tapering. Thus, in these studies it was not possible to assess whether abrupt discontinuation of ZULRESSO produced withdrawal symptoms indicative of physical dependence. It is recommended that ZULRESSO be tapered according to the dosage recommendations, unless symptoms warrant immediate discontinuation [see Dosage and Administration (2.2), Warnings and Precautions (5.1)].
10 OVERDOSAGE
Human Experience
There is limited clinical trial experience regarding human overdosage with ZULRESSO. In premarketing clinical studies, two cases of accidental overdosage due to infusion pump malfunction resulted in transient loss of consciousness. Both patients regained consciousness approximately 15 minutes after discontinuation of the infusion without supportive measures. After full resolution of symptoms, both patients subsequently resumed and completed treatment. Overdosage may result in excessive sedation, including loss of consciousness [see Warnings and Precautions (5.1)] and the potential for accompanying respiratory changes.
Management of Overdose
In case of overdosage, stop the infusion immediately and initiate supportive measures as necessary. Brexanolone is rapidly cleared from plasma [see Clinical Pharmacology (12.3)]. Consult a Certified Poison Control Center at 1-800-222-1222 for latest recommendations.
11 DESCRIPTION
ZULRESSO contains brexanolone, a neuroactive steroid gamma-aminobutyric acid (GABA) A receptor positive modulator, that is chemically identical to endogenous allopregnanolone.
The molecular formula of brexanolone is C21H34O2. The relative molecular mass is 318.5 Da. The chemical structure is:
ZULRESSO (brexanolone) injection is a sterile, clear, colorless, and preservative-free solution. ZULRESSO 5 mg/mL is hypertonic and must be diluted prior to administration as an intravenous infusion [see Dosage and Administration (2.3)]. Each mL of solution contains 5 mg of brexanolone, 250 mg of betadex sulfobutyl ether sodium, 0.265 mg of citric acid monohydrate, 2.57 mg of sodium citrate dihydrate, and water for injection. Hydrochloric acid or sodium hydroxide may be used during manufacturing to adjust pH.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The mechanism of action of brexanolone in the treatment of PPD is not fully understood, but is thought to be related to its positive allosteric modulation of GABAA receptors.
12.2 Pharmacodynamics
Brexanolone potentiated GABA-mediated currents from recombinant human GABAA receptors in mammalian cells expressing α1β2γ2 receptor subunits, α4β3δ receptor subunits, and α6β3δ receptor subunits.
Brexanolone exposure-response relationships and the time course of pharmacodynamics response are unknown.
Cardiac Electrophysiology
The effect of brexanolone on the QT interval was evaluated in a Phase 1 randomized, placebo and positive controlled, double-blind, three-period crossover thorough QT study in 30 healthy adult subjects. At 1.9-times the exposure occurring at the highest recommended infusion rate (90 mcg/kg/hour), brexanolone did not prolong the QT interval to a clinically relevant extent.
12.3 Pharmacokinetics
Brexanolone exhibited dose proportional pharmacokinetics over a dosage range of 30 mcg/kg/hour to 270 mcg/kg/hour (three times the maximum recommended dosage). Mean steady state exposure at 60 mcg/kg/hour and 90 mcg/kg/hour was around 52 ng/mL and 79 ng/mL, respectively.
Distribution
The volume of distribution of brexanolone was approximately 3 L/kg, suggesting extensive distribution into tissues. Plasma protein binding was greater than 99% and is independent of plasma concentrations.
Elimination
The terminal half-life of brexanolone is approximately 9 hours. The total plasma clearance of brexanolone is approximately 1 L/h/kg.
Metabolism
Brexanolone is extensively metabolized by non-CYP based pathways via three main routes - keto-reduction (AKRs), glucuronidation (UGTs), and sulfation (SULTs). There are three major circulating metabolites that are pharmacologically inactive and do not contribute to the overall efficacy of ZULRESSO.
Excretion
Following administration of radiolabeled brexanolone, 47% was recovered in feces (primarily as metabolites) and 42% in urine (with less than 1% as unchanged brexanolone).
Specific Populations
Patients 15 to 17 years
Brexanolone pharmacokinetics were evaluated in 20 patients with PPD (15 to 17 years) and were comparable to those in adult patients with PPD.
Patients with Renal or Hepatic Impairment
No clinically significant differences in the pharmacokinetics of brexanolone were observed based on renal impairment (severe) study or hepatic impairment (mild, moderate, severe) study. The effect of end stage renal disease (ESRD, eGFR < 15 mL/minute/1.73 m2) on brexanolone pharmacokinetics is unknown. However, avoid use of ZULRESSO in patients with ESRD [see Use in Specific Populations (8.7)].
Drug Interaction Studies
No studies were conducted to evaluate the effects of other drugs on ZULRESSO.
No clinically significant differences in the pharmacokinetics of phenytoin (CYP2C9 substrate) were observed when it was used concomitantly with brexanolone.
12.6 Betadex Sulfobutyl Ether Sodium Pharmacokinetics
Betadex sulfobutyl ether sodium is a solubilizing agent in ZULRESSO. In patients with severe renal impairment (eGFR 15-29 mL/minute/1.73 m2), betadex sulfobutyl ether sodium AUCinf increased 5.5-fold and Cmax increased 1.7-fold. Avoid use of ZULRESSO in patients with ESRD [see Use in Specific Populations (8.7)].
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Carcinogenicity studies of brexanolone have not been performed.
Mutagenesis
Brexanolone was not genotoxic when tested in an in vitro microbial mutagenicity (Ames) assay, an in vitro micronucleus assay in human peripheral blood lymphocytes, and an in vivo rat bone marrow micronucleus assay.
Impairment of Fertility
Treatment of female and male rats with brexanolone at doses equal to and greater than 30 mg/kg/day, which is associated with 2 times the plasma levels at the maximum recommended human dose (MRHD) of 90 mcg/kg/hour, caused impairment of female and male fertility and reproduction. In female rats, brexanolone was associated with decreased mating and fertility indices, an increase in number of days to mating, prolonged/irregular estrous cycles, an increase in the number of early resorptions, and post implantation loss. Reversal of effects in females was observed following a 28-day recovery period. In male rats, brexanolone was associated with decreased mating and fertility indices, decreased conception rate, lower prostate, seminal vesicle, and epididymis weight, as well as decreased sperm numbers. Impaired female and male fertility and reproduction were not observed at 0.8 times the MRHD.
14 CLINICAL STUDIES
The efficacy of ZULRESSO in the treatment of postpartum depression (PPD) was demonstrated in two multicenter, randomized, double-blind, placebo-controlled studies (referred to as Studies 1 and 2) in women (18 to 45 years) with PPD who met the Diagnostic and Statistical Manual of Mental Disorders criteria for a major depressive episode (DSM-IV) with onset of symptoms in the third trimester or within 4 weeks of delivery. In these studies, patients received a 60-hour continuous intravenous infusion of ZULRESSO or placebo and were then followed for 4 weeks. Study 1 (NCT02942004) included patients with severe PPD (Hamilton Depression Rating Scale (HAM-D) score ≥ 26), and Study 2 (NCT02942017) included patients with moderate PPD (HAM-D score of 20 to 25). A titration to the recommended target dosage of 90 mcg/kg/hour was evaluated in both studies (patients received 30 mcg/kg/hour for 4 hours, 60 mcg/kg/hour for 20 hours, 90 mcg/kg/hour for 28 hours, followed by a taper to 60 mcg/kg/hour for 4 hours and then 30 mcg/kg/hour for 4 hours). A titration to a target dosage of 60 mcg/kg/hour (patients received 30 mcg/kg/hour for 4 hours, 60 mcg/kg/hour for 52 hours, then 30 mcg/kg/hour for 4 hours) was also evaluated in Study 1.
Demographic and baseline disease characteristics were generally similar across treatment groups in the pooled Studies 1 and 2. Most patients were White (63%) or Black (34%); 18% of patients identified as Hispanic or Latina; the average age of women receiving ZULRESSO was 28 years. Most patients (76%) had onset of PPD symptoms within 4 weeks after delivery, with the remainder having onset during the third trimester. Baseline oral antidepressant use was reported for 23% of patients.
The primary endpoint was the mean change from baseline in depressive symptoms as measured by the HAM-D total score at the end of the infusion (Hour 60). A pre-specified secondary efficacy endpoint was the mean change from baseline in HAM-D total score at Day 30. In both placebo-controlled studies, titration to a target dose of ZULRESSO 90 mcg/kg/hour was superior to placebo in improvement of depressive symptoms. In a group of 38 patients in Study 1, a ZULRESSO titration to a target dose of 60 mcg/kg/hour was also superior to placebo in improvement of depressive symptoms.
|
HAM-D: Hamilton depression rating scale; ITT: intention to treat; SD: standard deviation; LS: least squares; SE: standard error; CI: confidence interval; *: statistically significant after multiplicity adjustments |
||||
| Study Number | Treatment Group (# ITT subject) | Primary Endpoint: Change from Baseline in HAM-D Total Score at Hour 60 | ||
| Mean Baseline Score (SD) | LS Mean Change from Baseline (SE) | Placebo-subtracted Difference (95% CI) Unadjusted p-value | ||
| 1 | ZULRESSO target dosage 90 mcg/kg/hour (n=41)* | 28.4 (2.5) | -17.7 (1.2) | -3.7 (-6.9, -0.5) P=0.0252 |
| Placebo (n=43) | 28.6 (2.5) | -14.0 (1.1) | ||
| ZULRESSO target dosage 60 mcg/kg/hour (n=38)* | 29.0 (2.7) | -19.5 (1.2) | -5.5 (-8.8, -2.2) P=0.0013 |
|
| Placebo (n=43) | 28.6 (2.5) | -14.0 (1.1) | ||
| 2 | ZULRESSO target dosage 90 mcg/kg/hour (n=51)* | 22.6 (1.6) | -14.6 (0.8) | -2.5 (-4.5, -0.5) P=0.0160 |
| Placebo (n=53) | 22.7 (1.6) | -12.1 (0.8) | ||
Examination of subgroups by race did not suggest differences in response.
Time Course of Treatment Response
Figure 1 shows the time course of response for the ZULRESSO 90 mcg/kg/hour-target and 60 mcg/kg/hour-target groups compared to the placebo group for Study 1.
Figure 1: Change from Baseline in HAM-D Total Score Over Time (Days) in Study 1
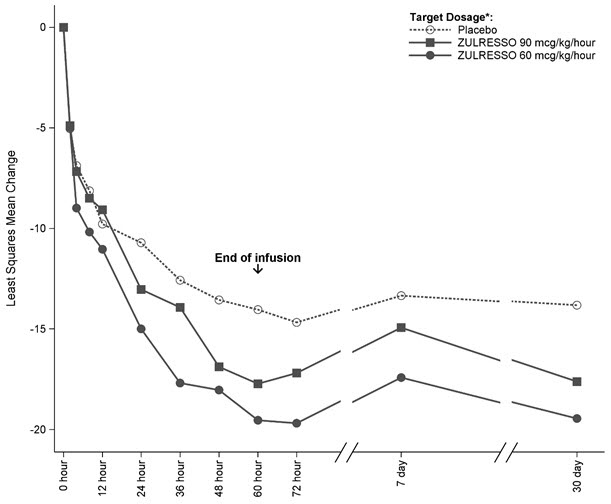
*ZULRESSO was administered via a 60-hour intravenous infusion as follows:
90 mcg/kg/hour-target dosage: 30 mcg/kg/hour for 4 hours, 60 mcg/kg/hour for 20 hours, 90 mcg/kg/hour for 28 hours,
60 mcg/kg/hour for 4 hours, 30 mcg/kg/hour for 4 hours
60 mcg/kg/hour-target dosage: 30 mcg/kg/hour for 4 hours, 60 mcg/kg/hour for 52 hours, 30 mcg/kg/hour for 4 hours
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
ZULRESSO injection is supplied as 100 mg brexanolone in 20 mL (5 mg/mL) single-dose vials containing a sterile, preservative-free, clear, colorless solution. NDC 72152-547-20
Storage and Handling
Store the undiluted ZULRESSO product at 2°C to 8°C (36°F to 46°F). Do not freeze. Store protected from light.
The diluted product in the infusion bag can be used at room temperature for up to 12 hours. If the diluted product is not used immediately after dilution, store under refrigerated conditions for up to 96 hours [see Dosage and Administration (2.3)].
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Excessive Sedation and Sudden Loss of Consciousness
Patients may experience loss of consciousness or altered state of consciousness during the ZULRESSO infusion. Advise patients to report signs of excessive sedation that may occur during the infusion. Patients must not be the primary caregiver of dependents and must be accompanied during interactions with their child(ren) [see Warnings and Precautions (5.1)].
ZULRESSO Risk Evaluation and Mitigation Strategy (REMS)
ZULRESSO is available only through a restricted program called the ZULRESSO REMS [see Warnings and Precautions (5.2)].
Inform the patient of the following notable requirements:
- Patients must be enrolled in the ZULRESSO REMS Program prior to administration.
- Patients must be monitored during administration of ZULRESSO and report any signs and symptoms of excessive sedation to a healthcare provider.
Potential for Abuse
Advise patients that ZULRESSO can be abused or lead to dependence [see Drug Abuse and Dependence (9)].
Concomitant Medications
Caution patients that opioids or other CNS depressants, such as benzodiazepines, taken in combination with ZULRESSO may increase the severity of sedative effects [see Warnings and Precautions (5.1, 5.2), Drug Interactions (7.1)].
Suicide Thoughts and Behaviors
Advise patients and caregivers to look for the emergence of suicidal thoughts and behavior and instruct them to report such symptoms to the healthcare provider [see Warnings and Precautions (5.3)].
Pregnancy
Advise women to notify their healthcare provider if they could possibly be pregnant prior to therapy with ZULRESSO. Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise patients that there is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to ZULRESSO during pregnancy [see Use in Specific Populations (8.1)].
Manufactured for:
Sage Therapeutics, Inc.,
Cambridge, MA 02142 USA
ZULRESSO, the ZULRESSO logo, SAGE THERAPEUTICS, and the SAGE THERAPEUTICS logo are registered trademarks of Sage Therapeutics, Inc. All other trademarks referenced herein are the properties of their respective owners.
v3.0
| This Medication Guide has been approved by the U.S. Food and Drug Administration. v3.0 |
Revised: 6/2022 |
|
MEDICATION GUIDE
ZULRESSO® (zul reh' soe) |
|
|
What is the most important information I should know about ZULRESSO? ZULRESSO can cause serious side effects, including:
|
|
|
What is ZULRESSO? ZULRESSO is a prescription medicine used to treat a certain type of depression called Postpartum Depression in individuals 15 years and older. It is not known if ZULRESSO is safe and effective in individuals less than 15 years of age. |
|
|
Before receiving ZULRESSO, tell your healthcare provider about all your medical conditions, including if you:
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. ZULRESSO and some medicines may interact with each other and cause serious side effects. Especially tell your healthcare provider if you take:
Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine. Your healthcare provider will decide if other medicines can be taken with ZULRESSO. |
|
|
How will I receive ZULRESSO?
|
|
|
What should I avoid while receiving ZULRESSO?
|
|
|
What are the possible side effects of ZULRESSO? ZULRESSO can cause serious side effects, including:
The most common side effects of ZULRESSO include:
These are not all the side effects of ZULRESSO. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|
|
General information about ZULRESSO. Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. You can ask your healthcare provider for information about ZULRESSO that is written for health professionals. |
|
|
What are the ingredients in ZULRESSO? Active ingredient: brexanolone Inactive ingredients: betadex sulfobutyl ether sodium, citric acid monohydrate, sodium citrate dihydrate, and water for injection. Hydrochloric acid or sodium hydroxide may be added during manufacturing to adjust pH. Manufactured for: Sage Therapeutics, Inc., Cambridge, MA 02142 ZULRESSO, the ZULRESSO logo, SAGE THERAPEUTICS, and the SAGE THERAPEUTICS logo are registered trademarks of Sage Therapeutics, Inc. All other trademarks referenced herein are the properties of their respective owners. For more information about ZULRESSO go to www.zulresso.com or call 844-472-4379. |
|

