USE IN PREGNANCY
When used in pregnancy during the second and third trimesters, drugs that act directly on the renin-angiotensin system can cause injury and even death to the developing fetus. When pregnancy is detected, TEVETEN® HCT Tablets should be discontinued as soon as possible. See WARNINGS: Fetal/Neonatal Morbidity and Mortality.
DESCRIPTION
TEVETEN® HCT 600/12.5 mg and TEVETEN® HCT 600/25 mg (eprosartan mesylate-hydrochlorothiazide) combine an angiotensin II receptor (AT1 subtype) antagonist and a diuretic, hydrochlorothiazide. TEVETEN® (eprosartan mesylate) is a non-biphenyl non-tetrazole angiotensin II receptor (AT1) antagonist. A selective non-peptide molecule, TEVETEN® is chemically described as the monomethanesulfonate of (E)-2-butyl-1-(p-carboxybenzyl)-α-2-thienylmethylimidazole-5-acrylic acid. Its empirical formula is C23H24N2O4S•CH4O3S and molecular weight is 520.625. Its structural formula is:
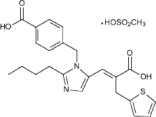
Eprosartan mesylate is a white to off-white free-flowing crystalline powder that is insoluble in water, freely soluble in ethanol, and melts between 248°C and 250°C. Hydrochlorothiazide is 6-chloro-3,4-dihydro-2 H 1,2,4-benzothiadiazine-7-sulfonamide 1,1-dioxide. Its empirical formula is C7H8ClN3O4S2 and its structural formula is:
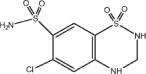
Hydrochlorothiazide is a white, or practically white, crystalline powder with a molecular weight of 297.74, which is slightly soluble in water, but freely soluble in sodium hydroxide solution. TEVETEN® HCT is available for oral administration in film-coated, non-scored, capsule-shaped tablet combinations of eprosartan mesylate and hydrochlorothiazide. TEVETEN® HCT 600/12.5 mg contains 735.8 mg of eprosartan mesylate (equivalent to 600 mg eprosartan) and 12.5 mg hydrochlorothiazide in a butterscotch-colored tablet. TEVETEN® HCT 600/25 mg contains 735.8 mg of eprosartan mesylate (equivalent to 600 mg eprosartan) and 25 mg hydrochlorothiazide in a brick-red tablet. Inactive ingredients of both tablets: microcrystalline cellulose, lactose monohydrate, pregelatinized starch, crospovidone, magnesium stearate, and purified water. Ingredients of the OPADRY® 85F27320 butterscotch film coating: polyethylene glycol 3350, talc, polyvinyl alcohol, titanium dioxide, iron oxide black, and iron oxide yellow. Ingredients of the OPADRY® II 85F24297 pink film coating: polyethylene glycol 3350, titanium dioxide, talc, polyvinyl alcohol, iron oxide red, and iron oxide yellow.
CLINICAL PHARMACOLOGY
Mechanism of Action
Eprosartan: Angiotensin II (formed from angiotensin I in a reaction catalyzed by angiotensin-converting enzyme [kininase II]), a potent vasoconstrictor, is the principal pressor agent of the renin-angiotensin system. Angiotensin II also stimulates aldosterone synthesis and secretion by the adrenal cortex, cardiac contraction, renal resorption of sodium, activity of the sympathetic nervous system, and smooth muscle cell growth. Eprosartan blocks the vasoconstrictor and aldosterone-secreting effects of angiotensin II by selectively blocking the binding of angiotensin II to the AT1 receptor found in many tissues (e.g., vascular smooth muscle, adrenal gland). There is also an AT2 receptor found in many tissues but it is not known to be associated with cardiovascular homeostasis. Eprosartan does not exhibit any partial agonist activity at the AT1 receptor. Its affinity for the AT1 receptor is 1,000 times greater than for the AT2 receptor. In vitro binding studies indicate that eprosartan is a reversible, competitive inhibitor of the AT1 receptor. Blockade of the AT1 receptor removes the negative feedback of angiotensin II on renin secretion, but the resulting increased plasma renin activity and circulating angiotensin II do not overcome the effect of eprosartan on blood pressure. TEVETEN® HCT does not inhibit kininase II, the enzyme that converts angiotensin I to angiotensin II and degrades bradykinin; whether this has clinical relevance is not known. It does not bind to or block other hormone receptors or ion channels known to be important in cardiovascular regulation.
Hydrochlorothiazide: Hydrochlorothiazide is a thiazide diuretic. Thiazides affect the renal tubular mechanisms of electrolyte reabsorption, directly increasing excretion of sodium and chloride in approximately equivalent amounts. Indirectly, the diuretic action of hydrochlorothiazide reduces plasma volume, with consequent increases in plasma renin activity, increases in aldosterone secretion, increases in urinary potassium loss, and decreases in serum potassium. The renin-aldosterone link is mediated by angiotensin II, so coadministration of an angiotensin II receptor antagonist tends to reverse the potassium loss associated with these diuretics. The mechanism of the antihypertensive effect of thiazides is unknown.
Pharmacokinetics
General
Eprosartan: Absolute bioavailability following a single 300-mg oral dose of eprosartan is approximately 13%. Eprosartan plasma concentrations peak at 1 to 2 hours after an oral dose in the fasted state. Administering eprosartan with food delays absorption, and causes variable changes (<25%) in Cmax and AUC values which do not appear clinically important. Plasma concentrations of eprosartan increase in a slightly less than dose-proportional manner over the 100 mg to 800 mg dose range. The mean terminal elimination half-life of eprosartan following multiple oral doses of 600 mg was approximately 20 hours. Eprosartan does not significantly accumulate with chronic use.
Metabolism and Excretion:
Eprosartan: Eprosartan is eliminated by biliary and renal excretion, primarily as unchanged compound. Less than 2% of an oral dose is excreted in the urine as a glucuronide. There are no active metabolites following oral and intravenous dosing with [14C] eprosartan in human subjects. Eprosartan was the only drug-related compound found in the plasma and feces. Following intravenous [14C] eprosartan, about 61% of the material is recovered in the feces and about 37% in the urine. Following an oral dose of [14C] eprosartan, about 90% is recovered in the feces and about 7% in the urine. Approximately 20% of the radioactivity excreted in the urine was an acyl glucuronide of eprosartan with the remaining 80% being unchanged eprosartan. Eprosartan is not metabolized by cytochrome P450 enzymes.
Distribution
Eprosartan: Plasma protein binding of eprosartan is high (approximately 98%) and constant over the concentration range achieved with therapeutic doses. The pooled population pharmacokinetic analysis from two Phase 3 trials of 299 men and 172 women with mild to moderate hypertension (aged 20 to 93 years) showed that eprosartan exhibited a population mean oral clearance (CL/F) for an average 60-year-old patient of 48.5 L/hr. The population mean steady-state volume of distribution (Vss/F) was 308 L. Eprosartan pharmacokinetics were not influenced by weight, race, gender or severity of hypertension at baseline. Oral clearance was shown to be a linear function of age with CL/F decreasing 0.62 L/hr for every year increase.
Special Populations
Pediatric: Eprosartan pharmacokinetics have not been investigated in patients younger than 18 years of age.
Geriatric: Following single oral dose administration of eprosartan to healthy elderly men, (aged 68 to 78 years), AUC, Cmax, and Tmax eprosartan values increased, on average, by approximately twofold, compared to healthy young men (aged 20 to 38 years) who received the same dose. The extent of plasma protein binding is not influenced by age.
Gender: There was no difference in the pharmacokinetics and plasma protein binding between men and women following single oral dose administration of eprosartan.
Race: A pooled population pharmacokinetic analysis of 442 Caucasian and 29 non-Caucasian hypertensive patients showed that oral clearance and steady-state volume of distribution were not influenced by race.
Renal Insufficiency: Following administration of 600 mg once daily, there was a 70-90% increase in AUC, and a 30-50% increase in Cmax in moderate or severe renal impairment. The unbound eprosartan fractions increased by 35% and 59% in patients with moderate and severe renal impairment, respectively. No initial dosing adjustment is generally necessary in patients with moderate or severe renal impairment, with maximum dose not exceeding 600 mg daily. Eprosartan was poorly removed by hemodialysis (CLHD<1L/hr) (see DOSAGE AND ADMINISTRATION).
Hepatic Insufficiency: Eprosartan AUC (but not Cmax) values increased, on average, by approximately 40% in men with decreased hepatic function compared to healthy men after a single 100 mg oral dose of eprosartan. The extent of eprosartan plasma protein binding was not influenced by hepatic dysfunction. No dosage adjustment is necessary for patients with hepatic impairment.
Drug Interactions
Eprosartan: Concomitant administration of eprosartan with digoxin had no effect on a single oral-dose digoxin pharmacokinetics. Concomitant administration of eprosartan and warfarin had no effect on steady-state prothrombin time ratios (INR) in healthy volunteers. Concomitant administration of eprosartan and glyburide in diabetic patients did not affect 24-hour plasma glucose profiles. Eprosartan pharmacokinetics were not affected by concomitant administration of ranitidine. Eprosartan did not inhibit human cytochrome P450 enzymes CYP1A, 2A6, 2C9/8, 2C19, 2D6, 2E, and 3A in vitro. Eprosartan steady-state plasma concentrations were not affected by concomitant administration of ketoconazole or fluconazole, potent inhibitors of CYP3A and 2C9, respectively.
Pharmacodynamics and Clinical Effects
Eprosartan: Eprosartan inhibits the pharmacologic effects of angiotensin II infusions in healthy adult men. Single oral doses of eprosartan from 10 mg to 400 mg have been shown to inhibit the vasopressor, renal vasoconstrictive and aldosterone secretory effects of infused angiotensin II with complete inhibition evident at doses of 350 mg and above. Eprosartan inhibits the pressor effects of angiotensin II infusions. A single oral dose of 350 mg of eprosartan inhibits pressor effects by approximately 100% at peak, with approximately 30% inhibition persisting for 24 hours. The absence of angiotensin II AT1 agonist activity has been demonstrated in healthy adult men. In hypertensive patients treated chronically with eprosartan, there was a twofold rise in angiotensin II plasma concentration and a twofold rise in plasma renin activity, while plasma aldosterone levels remained unchanged. Serum potassium levels also remained unchanged in these patients. Achievement of maximal blood pressure response to a given dose in most patients may take 2 to 3 weeks of treatment. Onset of blood pressure reduction is seen within 1 to 2 hours of dosing with few instances of orthostatic hypotension. Blood pressure control is maintained with once- or twice-daily dosing over a 24-hour period. Discontinuing treatment with eprosartan does not lead to a rapid rebound increase in blood pressure. There was no change in mean heart rate in patients treated with eprosartan in controlled clinical trials. Eprosartan increases mean effective renal plasma flow (ERPF) in salt-replete and salt-restricted normal subjects. A dose-related increase in ERPF of 25% to 30% occurred in salt-restricted normal subjects, with the effect plateauing between 200 mg and 400 mg doses. There was no change in ERPF in hypertensive patients and patients with renal insufficiency on normal salt diets. Eprosartan did not reduce glomerular filtration rate in patients with renal insufficiency or in patients with hypertension, after 7 days and 28 days of dosing, respectively. In hypertensive patients and patients with chronic renal insufficiency, eprosartan did not change fractional excretion of sodium and potassium. Eprosartan (1200 mg once daily for 7 days or 300 mg twice daily for 28 days) had no effect on the excretion of uric acid in healthy men, patients with essential hypertension or those with varying degrees of renal insufficiency. There were no effects on mean levels of fasting triglycerides, total cholesterol, HDL cholesterol, LDL cholesterol or fasting glucose.
Clinical Trials
Eprosartan Mesylate: The safety and efficacy of TEVETEN® has been evaluated in controlled clinical trials worldwide that enrolled predominantly hypertensive patients with sitting DBP ranging from 95 mmHg to ≤115 mmHg. There is also some experience with use of eprosartan together with other antihypertensive drugs in more severe hypertension. The antihypertensive effects of TEVETEN® were demonstrated principally in five placebo-controlled trials (4 to 13 weeks' duration) including dosages of 400 mg to 1200 mg given once daily (two studies), 25 mg to 400 mg twice daily (two studies), and one study comparing total daily doses of 400 mg to 800 mg given once daily or twice daily. The five studies included 1,111 patients randomized to eprosartan and 395 patients randomized to placebo. The studies showed dose-related antihypertensive responses. At study endpoint, patients treated with TEVETEN® at doses of 600 mg to 1200 mg given once daily experienced significant decreases in sitting systolic and diastolic blood pressure at trough, with differences from placebo of approximately 5-10/3-6 mmHg. Limited experience is available with the dose of 1200 mg administered once daily. In a direct comparison of 200 mg to 400 mg b.i.d. with 400 mg to 800 mg q.d. of TEVETEN®, effects at trough were similar. Patients treated with TEVETEN® at doses of 200 mg to 400 mg given twice daily experienced significant decreases in sitting systolic and diastolic blood pressure at trough, with differences from placebo of approximately 7-10/4-6 mmHg. Peak (1 to 3 hours) effects were uniformly, but moderately, larger than trough effects with b.i.d. dosing, with the trough-to-peak ratio for diastolic blood pressure 65% to 80%. In the once-daily dose-response study, trough-to-peak responses of ≤50% were observed at some doses (including 1200 mg), suggesting attenuation of effect at the end of the dosing interval. The antihypertensive effect of TEVETEN® was similar in men and women, but was somewhat smaller in patients over 65. There were too few black subjects to determine whether their response was similar to Caucasians. In general, blacks (usually a low renin population) have had smaller responses to ACE inhibitors and angiotensin II inhibitors than Caucasian populations. Angiotensin-converting enzyme (ACE) inhibitor-induced cough (a dry, persistent cough) can lead to discontinuation of ACE inhibitor therapy. In one study, patients who had previously coughed while taking an ACE inhibitor were treated with eprosartan, an ACE inhibitor (enalapril) or placebo for six weeks. The incidence of dry, persistent cough was 2.2% on eprosartan, 4.4% on placebo, and 20.5% on the ACE inhibitor; p=0.008 for the comparison of eprosartan with enalapril. In a second study comparing the incidence of cough in 259 patients treated with eprosartan to 261 patients treated with the ACE inhibitor enalapril, the incidence of dry, persistent cough in eprosartan-treated patients (1.5%) was significantly lower (p=0.018) than that observed in patients treated with the ACE inhibitor (5.4%). In addition, analysis of overall data from six double-blind clinical trials involving 1,554 patients showed an incidence of spontaneously reported cough in patients treated with eprosartan of 3.5%, similar to placebo (2.6%).
Hydrochlorothiazide: After oral administration of hydrochlorothiazide, diuresis begins within 2 hours, peaks in about 4 hours, and lasts about 6 to 12 hours.
Eprosartan Mesylate – Hydrochlorothiazide: Four adequate and well-controlled studies were conducted to assess the antihypertensive effectiveness of TEVETEN HCT®/(Eprosartan Mesylate/ hydrochlorothiazide) in 1457 patients with mild-to-moderate essential hypertension. In a 2x2 factorial study with 112-119 hypertensive patients per arm, the mean baseline- and placebo-subtracted reductions in blood pressure at 8 weeks were 3.6/2.1 mmHg on eprosartan 600 mg, 5.6/1.9 mmHg on hydrochlorothiazide 12.5 mg, and 10.0/5.0 mmHg on the combination.
INDICATIONS AND USAGE
TEVETEN® HCT is indicated for the treatment of hypertension. It may be used alone or in combination with other antihypertensives such as calcium channel blockers. This fixed dose combination is not indicated for initial therapy (see DOSAGE AND ADMINISTRATION).
CONTRAINDICATIONS
TEVETEN® HCT is contraindicated in patients who are hypersensitive to this product or any of its components. Because of the hydrochlorothiazide component, this product is contraindicated in patients with anuria or hypersensitivity to other sulfonamide-derived drugs.
WARNINGS
Fetal/Neonatal Morbidity and Mortality
Drugs that act directly on the renin-angiotensin system can cause fetal and neonatal morbidity and death when administered to pregnant women. Several dozen cases have been reported in the world literature in patients who were taking angiotensin-converting enzyme inhibitors. When pregnancy is detected, TEVETEN® HCT should be discontinued as soon as possible. The use of drugs that act directly on the renin-angiotensin system during the second and third trimesters of pregnancy has been associated with fetal and neonatal injury, including hypotension, neonatal skull hypoplasia, anuria, reversible or irreversible renal failure, and death. Oligohydramnios has also been reported, presumably resulting from decreased fetal renal function; oligohydramnios in this setting has been associated with fetal limb contractures, craniofacial deformation, and hypoplastic lung development. Prematurity, intrauterine growth retardation, and patent ductus arteriosus have also been reported, although it is not clear whether these occurrences were due to exposure to the drug. These adverse effects do not appear to have resulted from intrauterine drug exposure that has been limited to the first trimester. Mothers whose embryos and fetuses are exposed to an angiotensin II receptor antagonist only during the first trimester should be so informed. Nonetheless, when patients become pregnant, physicians should advise the patient to discontinue the use of eprosartan as soon as possible. Rarely (probably less often than once in every thousand pregnancies), no alternative to a drug acting on the renin-angiotensin system will be found. In these rare cases, the mothers should be apprised of the potential hazards to their fetuses, and serial ultrasound examinations should be performed to assess the intra-amniotic environment. If oligohydramnios is observed, TEVETEN® HCT should be discontinued unless it is considered life-saving for the mother. Contraction stress testing (CST), a nonstress test (NST) or biophysical profiling (BPP) may be appropriate, depending upon the week of pregnancy. Patients and physicians should be aware, however, that oligohydramnios may not appear until after the fetus has sustained irreversible injury. Infants with histories of in utero exposure to an angiotensin II receptor antagonist should be closely observed for hypotension, oliguria, and hyperkalemia. If oliguria occurs, attention should be directed toward support of blood pressure and renal perfusion. Exchange transfusion or dialysis may be required as means of reversing hypotension and/or substituting for disordered renal function. Eprosartan mesylate, alone or in combination with hydrochlorothiazide, has been shown to produce maternal and fetal toxicities (maternal and fetal mortality, low maternal body weight and food consumption, resorptions, abortions and litter loss) in pregnant rabbits given oral doses as low as 10 mg eprosartan/kg/day and 3 mg hydrochlorothiazide/kg/day. No maternal or fetal adverse effects were observed in rabbits at 3 mg eprosartan/kg/day alone or in combination with 1 mg/kg/day of hydrochlorothiazide; this oral dose yielded a systemic exposure (AUC) to unbound eprosartan approximately equal to the human systemic exposure achieved with the dose of eprosartan mesylate contained in the maximum recommended human dose of TEVETEN® HCT (600 mg eprosartan/day). No adverse effects on in utero or postnatal development and maturation of offspring were observed when eprosartan mesylate was administered to pregnant rats at oral doses up to 1000 mg eprosartan/kg/day (the 1000 mg eprosartan/kg/day dose in non-pregnant rats yielded systemic exposure to unbound eprosartan approximately 0.8 times the exposure achieved in humans given 600 mg/day). Thiazides cross the placental barrier and appear in cord blood. There is a risk of fetal or neonatal jaundice, thrombocytopenia, and possibly other adverse reactions that have occurred in adults.
Hypotension in Volume- and/or Salt-Depleted Patients
In patients with an activated renin-angiotensin system, such as volume- and/or salt-depleted patients (e.g., those being treated with diuretics), symptomatic hypotension may occur. These conditions should be corrected prior to administration of TEVETEN® HCT, or the treatment should start under close medical supervision. If hypotension occurs, the patient should be placed in the supine position and, if necessary, given an intravenous infusion of normal saline. A transient hypotensive response is not a contraindication to further treatment, which usually can be continued without difficulty once the blood pressure has stabilized.
Hydrochlorothiazide
Impaired Hepatic Function: Thiazides should be used with caution in patients with impaired hepatic function or progressive liver disease, since minor alterations of fluid and electrolyte balance may precipitate hepatic coma.
Hypersensitivity Reactions: Hypersensitivity reactions to hydrochlorothiazide may occur in patients with or without a history of allergy or bronchial asthma, but are more likely in patients with such a history.
Acute Myopia and Secondary Angle-Closure Glaucoma
Hydrochlorothiazide, a sulfonamide, can cause an idiosyncratic reaction, resulting in acute transient myopia and acute angle-closure glaucoma. Symptoms include acute onset of decreased visual acuity or ocular pain and typically occur within hours to weeks of drug initiation. Untreated acute angle-closure glaucoma can lead to permanent vision loss. The primary treatment is to discontinue hydrochlorothiazide as rapidly as possible. Prompt medical or surgical treatments may need to be considered if the intraocular pressure remains uncontrolled. Risk factors for developing acute angle-closure glaucoma may include a history of sulfonamide or penicillin allergy.
Systemic Lupus Erythematosus: Thiazide diuretics have been reported to cause exacerbation or activation of systemic lupus erythematosus. Lithium Interaction: Lithium generally should not be given with thiazides (see PRECAUTIONS, Drug Interactions, Hydrochlorothiazide, Lithium).
PRECAUTIONS
General
Hyperuricemia may occur or frank gout may be precipitated in certain patients receiving thiazide therapy. Thiazides have been shown to increase the urinary excretion of magnesium; this may result in hypomagnesemia. Thiazides may decrease urinary calcium excretion. Thiazides may cause intermittent and slight elevation of serum calcium in the absence of known disorders of calcium metabolism. Marked hypercalcemia may be evidence of hidden hyperparathyroidism. Thiazides should be discontinued before carrying out tests for parathyroid function. In diabetic patients, dosage adjustment of insulin or oral hypoglycemic agents may be required. Hyperglycemia may occur with thiazide diuretics. Thus, latent diabetes mellitus may become manifest during thiazide therapy. The antihypertensive effects of hydrochlorothiazide may be enhanced in postsympathectomy patients.
Electrolyte Imbalance
Periodic determination of serum electrolytes to detect possible electrolyte imbalance should be performed at appropriate intervals. All patients receiving thiazide therapy should be observed for clinical signs of fluid or electrolyte imbalance: hyponatremia, hypochloremic alkalosis, and hypokalemia. Serum and urine electrolyte determinations are particularly important when the patient is vomiting excessively or receiving parenteral fluids. Warning signs or symptoms of fluid and electrolyte imbalance, irrespective of cause, include: dryness of mouth, thirst, weakness, lethargy, drowsiness, restlessness, confusion, seizures, muscle pains or cramps, muscular fatigue, hypotension, oliguria, tachycardia, and gastrointestinal disturbances such as nausea and vomiting. Hypokalemia may develop, especially with brisk diuresis, when severe cirrhosis is present, or after prolonged therapy. Interference with adequate oral electrolyte intake will also contribute to hypokalemia. Hypokalemia may cause cardiac arrhythmia and may also sensitize or exaggerate the response of the heart to the toxic effects of digitalis (e.g., increased ventricular irritability). Although any chloride deficit is generally mild and usually does not require specific treatment except under extraordinary circumstances (as in liver disease or renal disease), chloride replacement may be required in the treatment of metabolic alkalosis. Dilutional hyponatremia may occur in edematous patients in hot weather; appropriate therapy is water restriction, rather than administration of salt except in rare instances when the hyponatremia is life-threatening. In actual salt depletion, appropriate replacement is the therapy of choice.
Risk of Renal Impairment
As a consequence of inhibiting the renin-angiotensin-aldosterone system, changes in renal function have been reported in susceptible individuals treated with angiotensin II antagonists; in some patients, these changes in renal function were reversible upon discontinuation of therapy. In patients whose renal function may depend on the activity of the renin-angiotensin-aldosterone system (e.g., patients with severe congestive heart failure), treatment with angiotensin-converting enzyme inhibitors and angiotensin II receptor antagonists has been associated with oliguria and/or progressive azotemia and (rarely) with acute renal failure and/or death. TEVETEN® HCT would be expected to behave similarly. In studies of ACE inhibitors in patients with unilateral or bilateral renal artery stenosis, increases in serum creatinine or BUN have been reported. Similar effects have been reported with angiotensin II antagonists; in some patients, these effects were reversible upon discontinuation of therapy. Thiazides should be used with caution in severe renal disease. In patients with renal disease, thiazides may precipitate azotemia. Cumulative effects of the drug may develop in patients with impaired renal function. If progressive renal impairment becomes evident, consider withholding or discontinuing diuretic therapy.
Information for Patients
Pregnancy: Female patients of childbearing age should be told about the consequences of second- and third-trimester exposure to drugs that act on the renin-angiotensin system, and they should also be told that these consequences do not appear to have resulted from intrauterine drug exposure that has been limited to the first trimester. These patients should be asked to report pregnancies to their physicians as soon as possible so that treatment may be discontinued under medical supervision.
Symptomatic Hypotension: A patient receiving TEVETEN® HCT should be cautioned that lightheadedness can occur, especially during the first days of therapy, and that it should be reported to the prescribing physician. The patient should be told that if syncope occurs, TEVETEN® HCT should be discontinued until the physician has been consulted. All patients should be cautioned that inadequate fluid intake, excessive perspiration, diarrhea, or vomiting can lead to an excessive fall in blood pressure, with the same consequences of lightheadedness and possible syncope.
Potassium Supplements: A patient receiving TEVETEN® HCT should be told not to use potassium supplements or salt substitutes containing potassium without consulting the prescribing physician (see PRECAUTIONS, Drug Interactions, Eprosartan Mesylate).
Drug Interactions
Eprosartan Mesylate: Eprosartan has been shown to have no effect on the pharmacokinetics of digoxin and the pharmacodynamics of warfarin and glyburide. Thus, no dosing adjustments are necessary during concomitant use with these agents. Because eprosartan is not metabolized by the cytochrome P450 system, inhibitors of CYP450 enzyme would not be expected to affect its metabolism, and ketoconazole and fluconazole, potent inhibitors of CYP3A and 2C9, respectively, have been shown to have no effect on eprosartan pharmacokinetics. Ranitidine also has no effect on eprosartan pharmacokinetics. Eprosartan (up to 400 mg b.i.d. or 800 mg q.d.) doses have been safely used concomitantly with a thiazide diuretic (hydrochlorothiazide). Eprosartan doses of up to 300 mg b.i.d. have been safely used concomitantly with sustained-release calcium channel blockers (sustained-release nifedipine) with no clinically significant adverse interactions. As with other drugs that block angiotensin II or its effects, concomitant use of potassium-sparing diuretics (e.g., spironolactone, triamterene, amiloride), potassium supplements or salt substitutes containing potassium may lead to increases in serum potassium (see PRECAUTIONS, Information for Patients, Potassium Supplements).
Non-Steroidal Anti-Inflammatory Agents including Selective Cyclooxygenase-2 Inhibitors (COX-2 Inhibitors)
In patients who are elderly, volume-depleted (including those on diuretic therapy), or with compromised renal function, co-administration of NSAIDs, including selective COX-2 inhibitors, with angiotensin II receptor antagonists, including eprosartan, may result in deterioration of renal function, including possible acute renal failure. These effects are usually reversible. Monitor renal function periodically in patients receiving eprosartan and NSAID therapy.
The antihypertensive effect of angiotensin II receptor antagonists, including eprosartan may be attenuated by NSAIDs including selective COX-2 inhibitors.
Hydrochlorothiazide: When administered concurrently the following drugs may interact with thiazide diuretics: Alcohol, barbiturates, or narcotics – potentiation of orthostatic hypotension may occur. Antidiabetic drug (oral agents and insulin) – dosage adjustment of the antidiabetic drug may be required. Other antihypertensive drugs – additive effect or potentiation. Cholestyramine and colestipol resins – Absorption of hydrochlorothiazide is impaired in the presence of anionic exchange resins. Single doses of either cholestyramine or colestipol resins bind the hydrochlorothiazide and reduce its absorption from the gastrointestinal tract by up to 85% and 43%, respectively. Corticosteroids, ACTH – intensified electrolyte depletion, particularly hypokalemia. Pressor amines (e.g., norepinephrine) – possible decreased response to pressor amines but not sufficient to preclude their use. Skeletal muscle relaxants, nondepolarizing (e.g., tubocurarine) – possible increased responsiveness to the muscle relaxant. Lithium – should not generally be given with diuretics. Diuretic agents reduce the renal clearance of lithium and add a high risk of lithium toxicity. Refer to the package insert for lithium preparations before use of such preparations with TEVETEN® HCT. Nonsteroidal Anti-Inflammatory Drugs – in some patients, the administration of a nonsteroidal anti-inflammatory agent can reduce the diuretic, natriuretic, and antihypertensive effects of loop, potassium-sparing and thiazide diuretics. Therefore, when TEVETEN® HCT and nonsteroidal anti-inflammatory agents are used concomitantly, the patient should be observed closely to determine if the desired effect of the diuretic is obtained.
Carcinogenesis, Mutagenesis, Impairment of Fertility
No carcinogenicity studies have been conducted with eprosartan mesylate in combination with hydrochlorothiazide. Eprosartan mesylate was not carcinogenic in dietary restricted rats or ad libitum fed mice dosed at 600 mg and 2000 mg eprosartan/kg/day, respectively, for up to 2 years. In male and female rats, the systemic exposure (AUC) to unbound eprosartan at the dose evaluated was only approximately 25% of the exposure achieved in humans given TEVETEN® HCT. In mice, the systemic exposure (AUC) to unbound eprosartan was approximately 35 times the exposure achieved in humans given TEVETEN® HCT. Two-year feeding studies in mice and rats conducted under the auspices of the National Toxicology Program (NTP) uncovered no evidence of a carcinogenic potential of hydrochlorothiazide in female mice (at doses of up to approximately 600 mg/kg/day) or in male and female rats (at doses of up to approximately 100 mg/kg/day). The NTP, however, found equivocal evidence for hepatocarcinogenicity in male mice. Eprosartan mesylate was not mutagenic in vitro in mammalian cells (mouse lymphoma assay). Eprosartan mesylate alone or in combination with hydrochlorothiazide was not mutagenic in vitro in bacteria (Ames test) and did not cause structural chromosomal damage in vivo (mouse micronucleus assay). In human peripheral lymphocytes in vitro, eprosartan mesylate in combination with hydrochlorothiazide was positive for clastogenicity with and without metabolic activation. In the same assay, eprosartan mesylate alone was associated with polyploidy but there was only equivocal evidence of structural chromosomal damage. Hydrochlorothiazide was not genotoxic in vitro in the Ames test and in the Chinese Hamster Ovary (CHO) test for chromosomal aberrations, or in vivo in assays using mouse germinal cell chromosomes, Chinese hamster bone marrow chromosomes, and the Drosophila sex-linked recessive lethal trait gene. Positive test results were obtained in the in vitro CHO Sister Chromatid Exchange (clastogenicity) and Mouse Lymphoma Cell (mutagenicity) assays and in the Aspergillus nidulans non-disjunction assay. No fertility studies have been conducted with eprosartan mesylate in combination with hydrochlorothiazide. Eprosartan mesylate had no adverse effects on the reproductive performance of male or female rats at oral doses up to 1000 mg eprosartan/kg/day. Hydrochlorothiazide had no adverse effects on the fertility of mice and rats of either sex in studies wherein these species were exposed, via their diet, to doses of up to 100 and 4 mg/kg/day, respectively, prior to conception and throughout gestation.
Pregnancy
Pregnancy Category C (first trimester) and D (second and third trimesters): See WARNINGS: Fetal/Neonatal Morbidity and Mortality.
Nursing Mothers
Eprosartan is excreted in animal milk; it is not known whether eprosartan is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from eprosartan, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother. Thiazides appear in human milk. Because of the potential for adverse effects on the nursing infant, a decision should be made whether to discontinue nursing or discontinue the drug, taking into account the importance of the drug to the mother.
Geriatric Use
In the controlled clinical trials where patients received eprosartan/hydrochlorothiazide combination therapy, 15% to 33% of the patients were 65 years of age or greater. There was no difference in the effect of TEVETEN® HCT 600/12.5 mg treatment according to age. However, following single oral dose administration of eprosartan to healthy elderly men, (aged 68 to 78 years), AUC, Cmax, and Tmax eprosartan values increased, on average, by approximately twofold, compared to healthy young men (aged 20 to 38 years) who received the same dose. (See Pharmacokinetics, Special Populations).
ADVERSE REACTIONS
TEVETEN® HCT 600/12.5 mg has been evaluated for safety in 268 patients in double-blind, controlled clinical trials. Most of these patients were treated with TEVETEN® HCT 600/12.5 mg for 29 to 60 days. Eprosartan/hydrochlorothiazide combination therapy has been evaluated for safety in 890 patients in open-label, long-term clinical trials. Approximately 50% of these patients were treated with eprosartan/hydrochlorothiazide for over 2 years. Eprosartan/hydrochlorothiazide combination therapy was well tolerated. Most adverse events were of mild or moderate severity and did not require discontinuation of therapy. Adverse experiences were similar in patients regardless of age, gender, or race. In the controlled clinical trials, about 3% of the 268 patients treated with TEVETEN® HCT 600/12.5 mg discontinued therapy due to clinical adverse experiences.
Adverse Events Occurring at an Incidence of Greater Than 3% Among TEVETEN® HCT Treated Patients
The following table lists adverse events that occurred at an incidence of >3% among TEVETEN® HCT 600/12.5 mg- or monotherapy-treated patients who participated in the controlled clinical trials. Of the 268 patients who received TEVETEN® HCT 600/12.5 mg during the double-blind treatment period in the controlled trials, 110 patients were reported to have adverse events.
|
Placebo (N=246) |
Eprosartan 600 mg (N=275) |
HCTZ 12.5 mg (N=117) |
HCTZ 25 mg (N=52) | Eprosartan
600 mg/HCTZ 12.5 mg (N=268) |
|
| Preferred Term | n (%) | n (%) | n (%) | n (%) | n (%) |
| Dizziness | 4 (1.6) | 5 (1.8) | 2 (1.7) | 2 (3.8) | 11 (4.1) |
| Headache | 22 (8.9) | 10 (3.6) | 4 (3.4) | 3 (5.8) | 9 (3.4) |
| Back pain | 6 (2.4) | 7 (2.5) | 2 (1.7) | 2 (3.8) | 7 (2.6) |
| Fatigue | 6 (2.4) | 5 (1.8) | 1 (0.9) | 2 (3.8) | 5 (1.9) |
| Myalgia | 8 (3.3) | 2 (0.7) | 3 (2.6) | 0 (0.0) | 1 (0.4) |
| Upper Respiratory Tract Infection | 8 (3.3) | 2 (0.7) | 0 (0.0) | 2 (3.8) | 1 (0.4) |
| Sinusitis | 4 (1.6) | 1 (0.4) | 0 (0.0) | 2 (3.8) | 0 (0.0) |
| Viral Infection | 4 (1.6) | 0 (0.0) | 2 (1.7) | 2 (3.8) | 0 (0.0) |
The adverse events reported in over 600 patients that received TEVETEN®/hydrochlorothiazide combination therapy for at least 1 year in the open-label, long-term clinical trials were comparable to those reported in the controlled trials.
Eprosartan Mesylate: In addition to the adverse events above, potentially important adverse events that are included in the current labeling for TEVETEN® monotherapy are listed below. Most of these adverse events occurred in <1% of patients, or were as frequent or more frequent in the placebo group. It is not known if these events were related to eprosartan usage: Body as a Whole: alcohol intolerance, asthenia, substernal chest pain, dependent edema, peripheral edema, facial edema, fatigue, fever, hot flushes, influenza-like symptoms, injury, malaise, pain, rigors, viral infection; Cardiovascular: angina pectoris, bradycardia, abnormal ECG, specific abnormal ECG, extrasystoles, atrial fibrillation, hypotension (including orthostatic hypotension), tachycardia, palpitations; Gastrointestinal: abdominal pain, anorexia, constipation, diarrhea, dry mouth, dyspepsia, esophagitis, flatulence, gastritis, gastroenteritis, gingivitis, nausea, periodontitis, toothache, vomiting; Hematologic: anemia, purpura; Liver and Biliary: increased SGOT, increased SGPT; Metabolic and Nutritional: increased creatine phosphokinase, diabetes mellitus, glycosuria, gout, hypercholesterolemia, hyperglycemia, hyperkalemia, hypokalemia, hyponatremia, hypertriglyceridemia; Musculoskeletal: arthralgia, arthritis, aggravated arthritis, arthrosis, skeletal pain, tendinitis; Nervous System/Psychiatric: anxiety, ataxia, depression, dizziness, insomnia, migraine, neuritis, nervousness, paresthesia, somnolence, tremor, vertigo; Resistance Mechanism: herpes simplex, otitis externa, otitis media, upper respiratory tract infection; Respiratory: asthma, bronchitis, coughing, epistaxis, pharyngitis, rhinitis; Skin and Appendages: eczema, furunculosis, pruritus, rash, maculopapular rash, increased sweating; Special Senses: conjunctivitis, abnormal vision, xerophthalmia, tinnitus; Urinary: albuminuria, cystitis, hematuria, micturition frequency, polyuria, renal calculus, urinary incontinence, urinary tract infection; Vascular: leg cramps, peripheral ischemia.
Hydrochlorothiazide: Other adverse events that have been reported for hydrochlorothiazide, without regard to causality, are listed below: Body as a Whole: weakness; Cardiovascular: hypotension (including orthostatic hypotension); Digestive: pancreatitis, jaundice (intrahepatic cholestatic jaundice), diarrhea, vomiting, sialadenitis, cramping, constipation, gastric irritation, nausea, anorexia; Hematologic: aplastic anemia, agranulocytosis, leukopenia, hemolytic anemia, thrombocytopenia; Hypersensitivity: anaphylactic reactions, necrotizing angiitis (vasculitis and cutaneous vasculitis), respiratory distress including pneumonitis, and pulmonary edema, photosensitivity, fever, urticaria, rash, purpura; Metabolic: electrolyte imbalance including hyponatremia, hypokalemia, and hypochloremic alkalosis, hyperglycemia, glycosuria, hyperuricemia; Musculoskeletal: muscle spasm; Nervous System/Psychiatric: vertigo, paresthesias, restlessness; Renal: renal failure, renal dysfunction, interstitial nephritis, azotemia; Skin: erythema multiform, including Stevens-Johnson syndrome, exfoliative dermatitis, including toxic epidermal necrolysis, alopecia; Special Senses: transient blurred vision, xanthopsia; Urogenital: impotence.
Laboratory Test Findings:
In placebo-controlled studies, clinically important changes in standard laboratory parameters were rarely associated with administration of TEVETEN®. Patients were rarely withdrawn from TEVETEN® because of laboratory test results. Laboratory test findings that have been reported for TEVETEN® are listed below: Creatinine, Blood Urea Nitrogen: Minor elevations in creatinine and in BUN occurred in 0.6% and 1.3%, respectively, of patients taking TEVETEN® and 0.9% and 0.3%, respectively, of patients given placebo in controlled clinical trials. Two patients were withdrawn from clinical trials for elevations in serum creatinine and BUN, and three additional patients were withdrawn for increases in serum creatinine. Liver Function Tests: Minor elevations of ALAT, ASAT, and alkaline phosphatase occurred for comparable percentages of patients taking TEVETEN® or placebo in controlled clinical trials. An elevated ALAT of >3.5 x ULN occurred in 0.1% of patients taking TEVETEN® (one patient) and in no patient given placebo in controlled clinical trials. Four patients were withdrawn from clinical trials for an elevation in liver function tests. Hemoglobin: A greater than 20% decrease in hemoglobin was observed in 0.1% of patients taking TEVETEN® (one patient) and in no patient given placebo in controlled clinical trials. Two patients were withdrawn from clinical trials for anemia. Leukopenia: A WBC count of ≤3.0 x 103/mm3 occurred in 0.3% of patients taking TEVETEN® and in 0.3% of patients given placebo in controlled clinical trials. One patient was withdrawn from clinical trials for leukopenia. Neutropenia: A neutrophil count of ≤1.5 x 103/mm3 occurred in 1.3% of patients taking TEVETEN® and in 1.4% of patients given placebo in controlled clinical trials. No patient was withdrawn from any clinical trials for neutropenia. Thrombocytopenia: A platelet count of ≤100 x 109/L occurred in 0.3% of patients taking TEVETEN® (one patient) and in no patient given placebo in controlled clinical trials. Four patients receiving TEVETEN® in clinical trials were withdrawn for thrombocytopenia. In one case, thrombocytopenia was present prior to dosing with TEVETEN®. Serum Potassium: A potassium value of ≥5.6 mmol/L occurred in 0.9% of patients taking TEVETEN® and 0.3% of patients given placebo in controlled clinical trials. One patient was withdrawn from clinical trials for hyperkalemia and three for hypokalemia.
Additional Information:
Among the adverse events reported for patients receiving either TEVETEN® monotherapy or TEVETEN®/hydrochlorothiazide combination therapy in the TEVETEN® HCT clinical trials, some adverse events are not included in the current labeling for either TEVETEN® or hydrochlorothiazide monotherapy. The adverse events which are not currently included in the labeling for TEVETEN® or hydrochlorothiazide monotherapy include the following: angioedema, bilirubinemia, blood urea nitrogen increased, edema periorbital, eosinophilia, and NPN increased. The majority of these adverse events were reported in the open-label, long-term trials and were reported in small numbers of patients receiving TEVETEN® alone or TEVETEN® in combination with hydrochlorothiazide. All of these adverse events were either not reported in patients receiving TEVETEN® monotherapy or combination therapy with hydrochlorothiazide during the double-blind period of the controlled trials, or were reported at an incidence of ≤1% or in only one patient per treatment group in the controlled trials. The overall safety profile of the TEVETEN®/hydrochlorothiazide combination treatment is as expected based on the safety profile of each of the components and what is generally known about the patient population.
OVERDOSAGE
Eprosartan Mesylate: Limited data are available regarding overdosage. Appropriate symptomatic and supportive therapy should be given if overdosage should occur. There was no mortality in rats and mice receiving oral doses of up to 3000 mg eprosartan/kg and in dogs receiving oral doses of up to 1000 mg eprosartan/kg.
Hydrochlorothiazide: The most common signs and symptoms observed are those caused by electrolyte depletion (hypokalemia, hypochloremia, and hyponatremia) and dehydration resulting from excessive diuresis. If digitalis has also been administered, hypokalemia may accentuate cardiac arrhythmias. The degree to which hydrochlorothiazide is removed by hemodialysis has not been established. The oral LD50 of hydrochlorothiazide is greater than 10 g/kg in both mice and rats.
DOSAGE AND ADMINISTRATION
The usual recommended starting dose of eprosartan is 600 mg once daily when used as monotherapy in patients who are not volume-depleted (see WARNINGS, Hypotension in Volume- and/or Salt-Depleted Patients). Eprosartan can be administered once or twice daily and total daily doses ranging from 400 mg to 800 mg. There is limited experience with doses beyond 800 mg/day. If the antihypertensive effect measured at trough using once-daily monotherapy dosing is inadequate, a twice-a-day regimen at the same total daily dose or an increase in dose may give a more satisfactory response. Achievement of maximum blood pressure reduction in most patients may take 2 to 3 weeks. Hydrochlorothiazide is effective in doses of 12.5 mg to 50 mg once daily. To minimize dose-independent side effects, it is usually appropriate to begin combination therapy only after a patient has failed to achieve the desired effect with monotherapy. The side effects (see WARNINGS) of eprosartan are generally rare and apparently independent of dose; those of hydrochlorothiazide are a mixture of dose-dependent (primarily hypokalemia) and dose-independent (e.g., pancreatitis) phenomena, the former much more common than the latter. Therapy with any combination of eprosartan and hydrochlorothiazide will be associated with both sets of dose-independent side effects.
Replacement Therapy
TEVETEN® HCT may be substituted for the individual components. The usual recommended dose of TEVETEN® HCT is 600 mg/12.5 mg once daily when used as combination therapy in patients who are not volume-depleted (see WARNINGS, Hypotension in Volume-and/or Salt-Depleted Patients). If the antihypertensive effect measured at trough using TEVETEN® HCT 600/12.5 mg is inadequate, patients may be titrated to TEVETEN® HCT 600/25 mg once daily. Higher doses have not been studied in combination. Achievement of maximum blood pressure reduction in most patients may take 2 to 3 weeks. If the patient under treatment with TEVETEN® HCT requires additional blood pressure control at trough, or to maintain a twice a day dosing schedule of monotherapy, 300 mg TEVETEN® may be added as evening dose. TEVETEN® HCT may be used in combination with other antihypertensive agents such as calcium channel blockers if additional blood-pressure-lowering effect is required. Discontinuation of treatment with eprosartan does not lead to a rapid rebound increase in blood pressure.
Elderly, Hepatically Impaired or Renally Impaired Patients: No initial dosing adjustment is generally necessary for elderly or hepatically impaired patients or those with renal impairment. No initial dosing adjustment is generally necessary in patients with moderate and severe renal impairment with maximum dose not exceeding 600 mg daily. TEVETEN® HCT may be taken with or without food.
HOW SUPPLIED
TEVETEN® HCT is available as film-coated, capsule-shaped tablets, debossed with “SOLVAY” on one side and “5147” or “5150” on the other, supplied as bottles of 30 tablets as follows:
| Eprosartan (mg) | HCTZ (mg) | Color | NDC |
| 600 | 12.5 | Butterscotch | 54868-5281-1 |
