FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
ORSERDU is indicated for the treatment of postmenopausal women or adult men with estrogen receptor (ER)-positive, human epidermal growth factor receptor 2 (HER2)‑negative, ESR1-mutated advanced or metastatic breast cancer with disease progression following at least one line of endocrine therapy.
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
Select patients for treatment of ER-positive, HER2-negative advanced or metastatic breast cancer with ORSERDU based on the presence of ESR1 mutation(s) in plasma specimen using an FDA-approved test [see Indications and Usage (1) and Clinical Studies (14)].
Information on FDA-approved tests for detection of ESR1 mutations in breast cancer is available at: http://www.fda.gov/CompanionDiagnostics.
2.2 Recommended Dosage
The recommended dosage of ORSERDU is 345 mg taken orally with food once daily until disease progression or unacceptable toxicity occurs.
Take ORSERDU at approximately the same time each day. Take with food to reduce nausea and vomiting [see Adverse Reactions (6.1)].
Swallow ORSERDU tablet(s) whole. Do not chew, crush, or split prior to swallowing. Do not take any ORSERDU tablets that are broken, cracked, or that look damaged.
If a dose is missed for more than 6 hours or vomiting occurs, skip the dose and take the next dose the following day at its regularly scheduled time.
2.3 Dosage Modifications for Adverse Reactions
The recommended dose reduction levels for adverse reactions are listed in Table 1:
Table 1: ORSERDU Dose Reduction Levels for Adverse Reactions
| 1 If further dose reduction below 172 mg once daily is required, permanently discontinue ORSERDU. | ||
| Dose Reduction | Dosage | Number and Strength of Tablets |
| First-dose reduction | 258 mg once daily | Three 86 mg tablets |
| Second-dose reduction | 172 mg once daily1 | Two 86 mg tablets |
Recommended dosage modifications of ORSERDU for adverse reactions are provided in Table 2 [see Adverse Reactions (6.1)].
Table 2: ORSERDU Dosage Modification Guidelines for Adverse Reactions
| Severity | Dosage Modification |
| Grade 1 | Continue ORSERDU at current dose level. |
| Grade 2 | Consider interruption of ORSERDU until recovery to Grade ≤ 1 or baseline. Then resume ORSERDU at the same dose level. |
| Grade 3 | Interrupt ORSERDU until recovery to Grade ≤ 1 or baseline. Then resume ORSERDU at the next lower dose level. If the Grade 3 toxicity recurs, interrupt ORSERDU until recovery to Grade ≤ 1 or baseline. Then resume ORSERDU reduced by another dose level. |
| Grade 4 | Interrupt ORSERDU until recovery to Grade ≤ 1 or baseline. Then resume ORSERDU reduced by one dose level. If a Grade 4 or intolerable adverse reaction recurs, permanently discontinue ORSERDU. |
2.4 Dosage Modifications for Use with Concomitant CYP3A4 Inducers and Inhibitors
Avoid concomitant use of ORSERDU with strong or moderate CYP3A4 inducers and inhibitors [see Drug Interactions (7.1)].
2.5 Dosage Modifications for Hepatic Impairment
Avoid use of ORSERDU in patients with severe hepatic impairment (Child-Pugh C). Reduce the ORSERDU dosage to 258 mg once daily for patients with moderate hepatic impairment (Child-Pugh B). No dosage adjustment is recommended for patients with mild hepatic impairment (Child-Pugh A) [see Clinical Pharmacology (12.3)].
3 DOSAGE FORMS AND STRENGTHS
Tablets: Elacestrant 345 mg (equivalent to 400 mg elacestrant hydrochloride) and 86 mg (equivalent to 100 mg elacestrant hydrochloride):
- 345 mg: light blue, unscored, oval film-coated biconvex tablet, imprinted with “MH” on one side and plain on the other side.
- 86 mg: light blue, unscored, round film-coated biconvex tablet, imprinted with “ME” on one side and plain on the other side.
5 WARNINGS AND PRECAUTIONS
5.1 Dyslipidemia
Hypercholesterolemia and hypertriglyceridemia occurred in patients taking ORSERDU at an incidence of 30% and 27%, respectively. The incidence of Grade 3 and 4 hypercholesterolemia and hypertriglyceridemia were 0.9% and 2.2%, respectively [see Adverse Reactions (6.1)].
Monitor lipid profile prior to starting and periodically while taking ORSERDU.
5.2 Embryo-Fetal Toxicity
Based on findings in animals and its mechanism of action, ORSERDU can cause fetal harm when administered to a pregnant woman. Administration of elacestrant to pregnant rats resulted in adverse developmental outcomes, including embryo-fetal mortality and structural abnormalities, at maternal exposures below the recommended dose based on area under the curve (AUC).
Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with ORSERDU and for 1 week after the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with ORSERDU and for 1 week after the last dose [see Use in Specific Populations (8.1, 8.3) and Clinical Pharmacology (12.1)].
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Dyslipidemia [see Warnings and Precautions (5.1)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of ORSERDU was evaluated in 467 patients with ER+/HER2- advanced breast cancer following CDK4/6 inhibitor therapy in EMERALD, a randomized, open-label, multicenter study [see Clinical Studies (14)]. Patients received ORSERDU 345 mg orally once daily (n=237) or standard of care (SOC) consisting of fulvestrant or an aromatase inhibitor (n=230). Among patients who received ORSERDU, 22% were exposed for 6 months or longer and 9% were exposed for greater than one year.
Serious adverse reactions occurred in 12% of patients who received ORSERDU. Serious adverse reactions in >1% of patients who received ORSERDU were musculoskeletal pain (1.7%) and nausea (1.3%). Fatal adverse reactions occurred in 1.7% of patients who received ORSERDU, including cardiac arrest, septic shock, diverticulitis, and unknown cause (one patient each).
Permanent discontinuation of ORSERDU due to an adverse reaction occurred in 6% of patients. Adverse reactions which resulted in permanent discontinuation of ORSERDU in >1% of patients were musculoskeletal pain (1.7%) and nausea (1.3%).
Dosage interruptions of ORSERDU due to an adverse reaction occurred in 15% of patients. Adverse reactions which resulted in dosage interruption of ORSERDU in >1% of patients were nausea (3.4%), musculoskeletal pain (1.7%), and increased ALT (1.3%).
Dosage reductions of ORSERDU due to an adverse reaction occurred in 3% of patients. Adverse reactions which required dosage reductions of ORSERDU in >1% of patients were nausea (1.7%).
The most common (≥10%) adverse reactions, including laboratory abnormalities, of ORSERDU were musculoskeletal pain, nausea, increased cholesterol, increased AST, increased triglycerides, fatigue, decreased hemoglobin, vomiting, increased ALT, decreased sodium, increased creatinine, decreased appetite, diarrhea, headache, constipation, abdominal pain, hot flush, and dyspepsia.
Table 3 summarizes the adverse reactions in EMERALD.
Table 3: Adverse Reactions (>10%) in Patients with ER-positive, HER2-negative, Advanced or Metastatic Breast Cancer Who Received ORSERDU in EMERALDa
| aAdverse reactions were graded using NCI CTCAE version 5.0. | ||||
| bIncludes other related terms | ||||
| cOnly includes Grade 3 adverse reactions. | ||||
| Adverse Reaction | ORSERDU (n=237) | Fulvestrant or an Aromatase Inhibitor (n=230) |
||
| All Grades (%) | Grade 3 or 4 c
(%) | All Grades (%) | Grade 3 or 4 c
(%) |
|
| Musculoskeletal and connective tissue disorders | ||||
| Musculoskeletal painb | 41 | 7 | 39 | 1 |
| Gastrointestinal disorders | ||||
| Nausea | 35 | 2.5 | 19 | 0.9 |
| Vomitingb | 19 | 0.8 | 9 | 0 |
| Diarrhea | 13 | 0 | 10 | 1 |
| Constipation | 12 | 0 | 6 | 0 |
| Abdominal painb | 11 | 1 | 10 | 0.9 |
| Dyspepsia | 10 | 0 | 2.6 | 0 |
| General disorders | ||||
| Fatigueb | 26 | 2 | 27 | 1 |
| Metabolism and nutrition disorders | ||||
| Decreased appetite | 15 | 0.8 | 10 | 0.4 |
| Nervous system | ||||
| Headache | 12 | 2 | 12 | 0 |
| Vascular disorders | ||||
| Hot flush | 11 | 0 | 8 | 0 |
Clinically relevant adverse reactions in < 10% of patients who received ORSERDU included rash, insomnia, dyspnea, cough, dizziness, stomatitis and gastroesophageal reflux disease.
Table 4 summarizes the laboratory abnormalities in EMERALD.
Table 4: Select Laboratory Abnormalities (>10%) That Worsened from Baseline in Patients with ER-positive, HER2-negative, Advanced or Metastatic Breast Cancer Who Received ORSERDU in EMERALDa
| aThe denominator used to calculate the rate varied from 29 to 236 for ORSERDU and from 37 to 225 for fulvestrant or an aromatase inhibitor based on the number of patients with a baseline value and at least one post-treatment value. | ||||
| Laboratory Abnormality | ORSERDUa | Fulvestrant or an Aromatase Inhibitora | ||
| All Grades (%) | Grade 3 or 4 (%) | All Grades (%) | Grade 3 or 4 (%) | |
| Chemistry | ||||
| Cholesterol increased | 30 | 1 | 17 | 0 |
| Aspartate aminotransferase increased | 29 | 0 | 34 | 1 |
| Triglycerides increased | 27 | 2 | 15 | 1 |
| Alanine aminotransferase increased | 17 | 0 | 24 | 1 |
| Sodium decreased | 16 | 1 | 15 | 0 |
| Creatinine increased | 16 | 0 | 6 | 0 |
| Hematology | ||||
| Hemoglobin decreased | 26 | 1 | 20 | 2 |
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on ORSERDU
Strong and Moderate CYP3A4 Inhibitors
Avoid concomitant use of strong or moderate CYP3A inhibitors with ORSERDU.
Elacestrant is a CYP3A4 substrate. Concomitant use of a strong or moderate CYP3A4 inhibitor increase elacestrant exposure [see Clinical Pharmacology (12.3)], which may increase the risk of adverse reactions of ORSERDU.
Strong and Moderate CYP3A4 Inducers
Avoid concomitant use of strong or moderate CYP3A inducers with ORSERDU.
Elacestrant is a CYP3A4 substrate. Concomitant use of a strong or moderate CYP3A4 inducer decreases elacestrant exposure [see Clinical Pharmacology (12.3)], which may decrease effectiveness of ORSERDU.
7.2 Effect of ORSERDU on Other Drugs
P-gp Substrates
Reduce the dosage of P-gp substrates per their Prescribing Information when minimal concentration changes may lead to serious or life-threatening adverse reactions.
Elacestrant is a P-gp inhibitor. Concomitant use of ORSERDU with a P-gp substrate increased the concentrations of P-gp substrate [see Clinical Pharmacology (12.3)], which may increase the adverse reactions associated with a P-gp substrate.
BCRP Substrates
Reduce the dosage of BCRP substrates per their Prescribing Information when minimal concentration changes may lead to serious or life-threatening adverse reactions.
Elacestrant is a BCRP inhibitor. Concomitant use of ORSERDU with a BCRP substrate increased the plasma concentrations of BCRP substrate [see Clinical Pharmacology (12.3)], which may increase the adverse reactions associated with a BCRP substrate.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings in animals and its mechanism of action, ORSERDU can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. There are no available human data on ORSERDU use in pregnant women to inform the drug-associated risk. In an animal reproduction study, oral administration of elacestrant to pregnant rats during organogenesis caused embryo-fetal mortality and structural abnormalities at maternal exposures below the recommended dose based on AUC (see Data). Advise pregnant women and females of reproductive potential of the potential risk to a fetus.
The background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2%-4% and 15%-20%, respectively.
Animal Data
In an embryo-fetal development study in pregnant rats, administration of oral doses of elacestrant up to 30 mg/kg/day during the period of organogenesis resulted in maternal toxicity (reduced body weight gain, low food consumption, red vulvar discharge) and embryo-fetal mortality (increased resorptions, post-implantation loss, and reduced number of live fetuses) at ≥ 3 mg/kg/day (approximately 0.1 times the human AUC at the recommended dose). Additional adverse effects included reduced fetal weight and external malformations of the limbs (hyperflexion, malrotation) and head (domed, misshapen, flattened) with corresponding skeletal malformations of the skull at doses ≥ 10 mg/kg/day (approximately 0.5 times the human AUC at the recommended dose).
8.2 Lactation
Risk Summary
There are no data on the presence of elacestrant in human milk, its effects on milk production, or the breastfed child. Because of the potential for serious adverse reactions in the breastfed child, advise lactating women to not breastfeed during treatment with ORSERDU and for 1 week after the last dose.
8.3 Females and Males of Reproductive Potential
ORSERDU can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify the pregnancy status in females of reproductive potential prior to initiating ORSERDU treatment.
Contraception
Females
Advise females of reproductive potential to use effective contraception during treatment with ORSERDU and for 1 week after the last dose.
Males
Advise male patients with female partners of reproductive potential to use effective contraception during treatment with ORSERDU and for 1 week after the last dose.
Infertility
Based on findings from animal studies, ORSERDU may impair fertility in females and males of reproductive potential [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of ORSERDU in pediatric patients have not been established.
8.5 Geriatric Use
Of 237 patients who received ORSERDU in the EMERALD trial, 43% were 65 years of age or older and 17% were 75 years of age or older. No overall differences in safety or effectiveness of ORSERDU were observed between patients 65 years or older of age compared to younger patients. There are an insufficient number of patients 75 years of age or older to assess whether there are differences in safety or effectiveness.
8.6 Hepatic Impairment
Avoid use of ORSERDU in patients with severe hepatic impairment (Child-Pugh C). Reduce the dose of ORSERDU in patients with moderate hepatic impairment (Child-Pugh B). No dosage adjustment is recommended for patients with mild hepatic impairment (Child-Pugh A) [see Dosage and Administration (2.5) and Clinical Pharmacology (12.3)].
11 DESCRIPTION
Elacestrant hydrochloride is the salt form of elacestrant, an estrogen receptor antagonist, that has the chemical name: (6R)-6-(2-(N-(4-(2-(ethylamino)ethyl)benzyl)-N-ethylamino)-4-methoxyphenyl)-5,6,7,8-tetrahydronaphthalen-2-ol dihydrochloride. Elacestrant hydrochloride is the dihydrochloride salt and the molecular formula is C30H38N2O2.2HCL. The relative molecular mass is 531.56 g/mol. The chemical structure of elacestrant hydrochloride is shown below:
Elacestrant hydrochloride is a white to off-white to grey solid and is freely soluble in 0.01N HCI.
ORSERDU (elacestrant) 345 mg film-coated tablet contains 400 mg of elacestrant hydrochloride (approximately 345 mg of elacestrant free base).
ORSERDU (elacestrant) 86 mg film-coated tablet contains 100 mg of elacestrant hydrochloride (approximately 86 mg of elacestrant free base).
Both tablet strengths contain the following inactive ingredients: colloidal silicon dioxide, crospovidone, magnesium stearate (non-bovine), microcrystalline cellulose, and silicified microcrystalline cellulose. The tablets also contain Opadry II Blue (polyvinyl alcohol, titanium dioxide, polyethylene glycol, FD&C Blue #1 and talc).
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Elacestrant is an estrogen receptor antagonist that binds to estrogen receptor-alpha (ERα). In ER-positive (ER+) HER2-negative (HER2-) breast cancer cells, elacestrant inhibited 17β-estradiol mediated cell proliferation at concentrations inducing degradation of ERα protein mediated through proteasomal pathway. Elacestrant demonstrated in vitro and in vivo antitumor activity including in ER+ HER2- breast cancer models resistant to fulvestrant and cyclin-dependent kinase 4/6 inhibitors and those harboring estrogen receptor 1 gene (ESR1) mutations.
12.2 Pharmacodynamics
Elacestrant exposure-response relationships and the time course of pharmacodynamics have not been fully characterized.
Cardiac Electrophysiology
ORSERDU does not cause a mean increase in QTc interval > 20 msec at the approved recommended dose.
12.3 Pharmacokinetics
The steady-state mean (%CV) maximum concentration (Cmax) of elacestrant is 119 ng/mL (43.6%) and the area under the concentration-time curve (AUC0-24h) is 2440 ng*h/mL (44.3%) after administration of the recommended dosage of 345 mg once daily. The Cmax and AUC of elacestrant increase more than proportionally over a dosage range from 43 mg to 862 mg once daily (0.125 to 2.5 times the approved recommended dosage). Steady state is reached by Day 6 and the mean accumulation ratio based on AUC0-24h is 2-fold.
Absorption
The time to achieve peak plasma concentration (tmax) ranges from 1 to 4 hours. The elacestrant oral bioavailability is approximately 10%.
Effect of Food
Administration of ORSERDU 345 mg with a high-fat meal (800 to 1000 calories, 50% fat) increased Cmax by 42% and AUC by 22% compared to fasted administration.
Distribution
The estimated apparent volume of distribution is 5800L. Plasma protein binding of elacestrant is >99% and independent of concentration.
Elimination
The elimination half-life of elacestrant is 30 to 50 hours. The estimated mean (% CV) clearance of elacestrant is 186 L/hr (43.5%) and renal clearance is ≤ 0.14 L/hr.
Metabolism
Elacestrant is primarily metabolized by CYP3A4 and to a lesser extent by CYP2A6 and CYP2C9.
Excretion
Following a single radiolabeled oral dose of 345 mg, 82% was recovered in feces (34% unchanged) and 7.5% was recovered in urine (< 1% unchanged).
Specific Populations
There were no clinically significant differences in the pharmacokinetics of elacestrant based on age (24 to 89 years), sex, and body weight (41 to 143 kg).
Patients with Hepatic Impairment
There were no clinically significant differences in the Cmax and AUC of elacestrant in subjects with mild hepatic impairment (Child-Pugh A). The AUC of elacestrant increased in subjects with moderate hepatic impairment (Child-Pugh B) by 83%.
Elacestrant has not been studied in subjects with severe hepatic impairment (Child-Pugh C).
Drug Interaction Studies
Clinical Studies
There were no clinically significant differences in the pharmacokinetics of elacestrant when used concomitantly with cimetidine (weak CYP3A inhibitor), omeprazole (gastric acid-reducing agent), or warfarin (highly protein-bound drug).
Table 5 describes the effect of other drugs on the pharmacokinetics of elacestrant and Table 6 describes the effect of elacestrant on the pharmacokinetics of other drugs.
Table 5: Effect of Other Drugs on Elacestrant
| aPredicted changes in Cmax and AUC of elacestrant. | |||
| Concomitant Drug | Elacestrant Dose | Fold Increased or Percent Decrease of Elacestrant With Concomitant Drug | |
| Cmax | AUC | ||
| CYP3A Inhibitors | |||
| Strong Inhibitor
Itraconazole | 172 mg once daily | 4.4 | 5.3 |
| Moderate Inhibitor
Fluconazolea | 345 mg single dose | 1.6 | 2.3 |
| CYP3A Inducers | |||
| Strong Inducer
Rifampin | 345 mg single dose | 73% | 86% |
| Moderate Inducer
Efavirenza | 345 mg single dose | 44-63% | 55-73% |
Table 6: Effect of Elacestrant on Other Drugs
| Concomitant Drug | Elacestrant Dose | Fold Increase of Concomitant Drug With Elacestrant | |
| Cmax | AUC | ||
| Substrate of P-gp | |||
| Digoxin | 345 mg single dose | 1.3 | 1.1 |
| Substrate of BCRP | |||
| Rosuvastatin | 345 mg single dose | 1.5 | 1.2 |
In Vitro Studies
Cytochrome P450 (CYP) Enzymes: Elacestrant is not an inhibitor of CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, or CYP3A.
Elacestrant is not an inducer of CYP1A2, CYP2A6, CYP2B6, CYP2C9, CYP2C19, or CYP3A.
Transporter Systems: Elacestrant is a substrate for OATP2B1, but not P-gp.
Elacestrant is not an inhibitor of OAT1, OAT3, OCT2, MATE1, MATE2-K, OCT1, OATP1B1, OATP1B3 or OATP2B1.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with elacestrant.
Elacestrant was not mutagenic in an in vitro bacterial reverse mutation (Ames) assay or clastogenic in either in vitro chromosome aberration assays or an in vivo rat bone marrow micronucleus assay.
Fertility studies with elacestrant in animals have not been conducted. In repeated-dose toxicity studies up to 26 weeks duration in rats and 39 weeks duration in cynomolgus monkeys, adverse reactions were observed in female reproductive organs including atrophy of the vagina, cervix, and uterus and follicular cysts in the ovary at doses ≥ 10 mg/kg/day in rats and cynomolgus monkeys (≥ 0.3 times the human AUC at the recommended dose). Decreased cellularity of Leydig cells and degeneration/atrophy of the seminiferous epithelium in the testis were observed in male rats at a dose of 50 mg/kg/day (approximately 2.6 times the human AUC at the recommended dose).
14 CLINICAL STUDIES
The efficacy of ORSERDU was evaluated in EMERALD (NCT03778931), a randomized, open-label, active-controlled, multicenter trial that enrolled 478 postmenopausal women and men with ER+/HER2- advanced or metastatic breast cancer of which 228 patients had ESR1 mutations. Patients were required to have disease progression on one or two prior lines of endocrine therapy, including one line containing a CDK4/6 inhibitor. Eligible patients could have received up to one prior line of chemotherapy in the advanced or metastatic setting.
Patients were randomized (1:1) to receive ORSERDU 345 mg orally once daily (n=239), or investigator’s choice of endocrine therapy (n=239), which included fulvestrant (n=166), or an aromatase inhibitor (n=73; anastrozole, letrozole or exemestane). Randomization was stratified by ESR1 mutation status (detected vs not detected), prior treatment with fulvestrant (yes vs no), and visceral metastasis (yes vs no). ESR1 mutational status was determined by blood circulating tumor deoxyribonucleic acid (ctDNA) using the Guardant360 CDx assay and was limited to ESR1 missense mutations in the ligand binding domain (between codons 310 to 547). Patients were treated until disease progression or unacceptable toxicity.
The major efficacy outcome was progression-free survival (PFS), assessed by a blinded imaging review committee (BIRC). An additional efficacy outcome measure was overall survival (OS).
A statistically significant difference in PFS was observed in the intention to treat (ITT) population and in the subgroup of patients with ESR1 mutations. An exploratory analysis of PFS in the 250 (52%) patients without ESR1 mutations showed a HR 0.86 (95% CI: 0.63, 1.19) indicating that the improvement in the ITT population was primarily attributed to the results seen in the ESR1 mutated population.
Among the patients with ESR1 mutations (n=228), the median age was 63 years (range: 28-89); 100% were female; 72% were White, 5.7% Asian, 3.5% Black, 0.4% Other, 18.4% unknown/not reported; 8.8% were Hispanic/Latino; and baseline ECOG performance status was 0 (57%) or 1 (43%). Most patients had visceral disease (71%); 62% had received 1 line of endocrine therapy and 39% had received 2 lines of endocrine therapy in the advanced or metastatic setting. All patients had received prior treatment with a CDK4/6 inhibitor, 24% had received prior fulvestrant, and 25% had received prior chemotherapy in the advanced or metastatic setting.
Efficacy results are presented in Table 7 and Figure 1 for patients with ESR1 mutations.
Table 7: Efficacy Results for EMERALD (Patients with ESR1 Mutations)
| CI=confidence interval; ESR1=estrogen receptor 1 | ||
| aPFS results based on blinded imaging review committee. | ||
| bKaplan-Meier estimate; 95% CI based on the Brookmeyer-Crowley method using a linear transformation. | ||
| cCox proportional hazards model stratified by prior treatment with fulvestrant (yes vs no) and visceral metastasis (yes vs no). | ||
| dStratified log-rank test two-sided p-value. | ||
| eNS – Not statistically significant. | ||
| ORSERDU (N = 115) | Fulvestrant or an Aromatase Inhibitor (N=113) |
|
| Progression-free Survival (PFS)a | ||
| Number of PFS Events, n (%) | 62 (54) | 78 (69) |
| Median PFS monthsb (95% CI) | 3.8 (2.2, 7.3) | 1.9 (1.9, 2.1) |
| Hazard ratioc (95% CI) | 0.55 (0.39, 0.77) | |
| p-valued | 0.0005 | |
| Overall Survival (OS) | ||
| Number of OS Events, n (%) | 61 (53) | 60 (53) |
| Hazard ratioc (95% CI) | 0.90 (0.63, 1.30) | |
| p-valued | NSe | |
Figure 1: Kaplan-Meier Curve for PFS in EMERALD (Patients with ESR1 Mutations, BIRC Assessment)
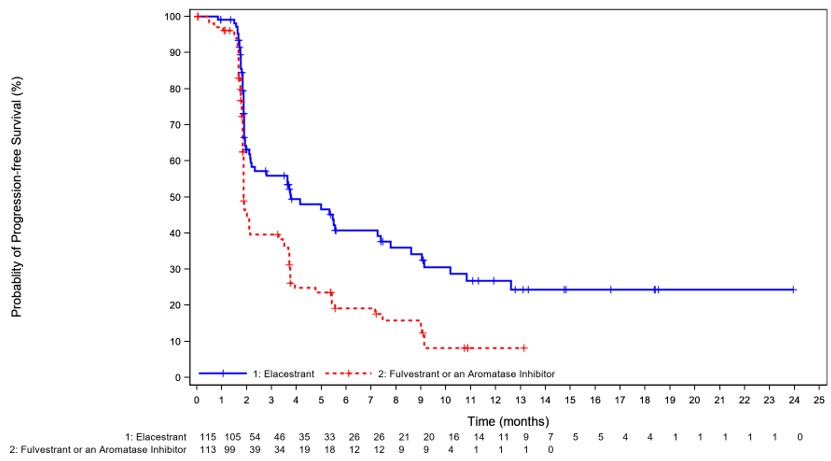
+ Censored times
16 HOW SUPPLIED/STORAGE AND HANDLING
ORSERDU (elacestrant) film-coated tablets for oral use are supplied as follows:
| Tablet Strength | Tablet Color and Shape | Tablet Markings | Pack Size | NDC Code |
| Elacestrant 345 mg (equivalent to 400 mg elacestrant hydrochloride) | Light blue; Oval | “MH” | Bottle of 30 Tablets with Child Resistant Closure (CRC). | NDC 72187-0102-3 |
| Elacestrant 86 mg (equivalent to 100 mg elacestrant hydrochloride) | Light blue; Round | “ME” | Bottle of 30 Tablets with Child Resistant Closure (CRC). | NDC 72187-0101-3 |
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Dyslipidemia
Advise patients that hypercholesterolemia and hypertriglyceridemia may occur while taking ORSERDU. Inform patients that lipid profile monitoring will be performed prior to starting and periodically while taking ORSERDU [see Warnings and Precautions (5.1) and Adverse Reactions (6.1)].
Embryo-Fetal Toxicity
Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.2) and Use in Specific Populations (8.1)].
Advise females of reproductive potential to use effective contraception during treatment with ORSERDU and for 1 week after the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with ORSERDU and for 1 week after the last dose [see Use in Specific Populations (8.3)].
Lactation
Advise women not to breastfeed during treatment with ORSERDU and for 1 week after the last dose [see Use in Specific Populations (8.2)].
Infertility
Advise males and females of reproductive potential that ORSERDU may impair fertility [see Use in Specific Populations (8.3)].
Drug Interactions
Advise patients to inform their healthcare providers of all concomitant medications, including prescription medicines, over-the-counter drugs, and herbal products [see Drug Interactions (7.1, 7.2)].
Other Common Adverse Reactions
Advise patients that other adverse reactions with ORSERDU treatment may include musculoskeletal pain, nausea, fatigue, vomiting, decreased appetite, diarrhea, headache, constipation, abdominal pain, hot flush, and dyspepsia, [see Adverse Reactions (6.1)].
Dosing Instructions
Instruct patients to take ORSERDU at approximately the same time each day and to swallow the tablet(s) whole. Tablets should not be chewed, crushed, or split prior to swallowing [see Dosage and Administration (2.2)].
Advise patients to take ORSERDU with food to reduce nausea and vomiting [see Dosage and Administration (2.2) and Adverse Reactions (6.1)].
Instruct patients that if a dose of ORSERDU is missed for more than 6 hours or vomiting occurs, skip the dose and take the next dose the following day at its regularly scheduled time [see Dosage and Administration (2.2)].
ORSERDU is a trademark of the Menarini Group.
©Stemline Therapeutics, Inc., a Menarini Group company
Manufactured for: Stemline Therapeutics, Inc., New York, NY 10022
| This Patient Information has been approved by the U.S. Food and Drug Administration. | Issued: 01/2023 |
|
PATIENT INFORMATION
ORSERDU™ (or-SER-doo)
|
|
|
What is ORSERDU? ORSERDU is a prescription medicine to treat women who have gone through menopause and adult men with estrogen receptor (ER)-positive, human epidermal growth factor receptor 2 (HER2)-negative, ESR1-mutated advanced breast cancer or breast cancer that has spread to other parts of the body (metastatic), and whose disease has progressed after endocrine therapy.
|
|
|
Before taking ORSERDU, tell your healthcare provider about all of your medical conditions, including if you:
Tell your healthcare provider about all of the medicines you take, including prescription and over-the counter medicines, vitamins, and herbal supplements. ORSERDU and other medicines may affect each other causing side effects. Know the medicines you take. Keep a list of them to show your healthcare provider or pharmacist when you get a new medicine. |
|
|
How should I take ORSERDU?
|
|
|
What are the possible side effects of ORSERDU? ORSERDU may cause serious side effects, including:
The most common side effects of ORSERDU include:
Your healthcare provider may decrease your dose, temporarily stop, or completely stop treatment with ODSERDU, if you develop certain side effects.
|
|
|
How should I store ORSERDU?
Keep ORSERDU and all medicines out of the reach of children. |
|
|
General information about the safe and effective use of ORSERDU. Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use ORSERDU for a condition for which it was not prescribed. Do not give ORSERDU to other people, even if they have the same symptoms you have. It may harm them. You can ask your healthcare provider or pharmacist for more information about ORSERDU that is written for health professionals. |
|
|
What are the ingredients in ORSERDU? Active ingredient: elacestrant Inactive ingredients: colloidal silicon dioxide, crospovidone, magnesium stearate (non-bovine), microcrystalline cellulose, silicified microcrystalline cellulose and Opadry II Blue (polyvinyl alcohol, titanium dioxide, polyethylene glycol, FD&C Blue #1 and talc). ORSERDU is a trademark of the Menarini Group. ©Stemline Therapeutics, Inc., a Menarini Group Company Manufactured for: Stemline Therapeutics, Inc., New York, NY 10022 For more information, go to www.ORSERDU.com or call 1-877-332-7961 |
|

