FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
1.1 Patients with Cancer Receiving Myelosuppressive Chemotherapy
UDENYCA is indicated to decrease the incidence of infection, as manifested by febrile neutropenia, in patients with non-myeloid malignancies receiving myelosuppressive anti-cancer drugs associated with a clinically significant incidence of febrile neutropenia [see Clinical Studies (14.1)].
Limitations of Use
UDENYCA is not indicated for the mobilization of peripheral blood progenitor cells for hematopoietic stem cell transplantation.
2 DOSAGE AND ADMINISTRATION
2.1 Patients with Cancer Receiving Myelosuppressive Chemotherapy
The recommended dosage of UDENYCA is a single subcutaneous injection of 6 mg administered once per chemotherapy cycle. For dosing in pediatric patients weighing less than 45 kg, refer to Table 1. Do not administer UDENYCA between 14 days before and 24 hours after administration of cytotoxic chemotherapy.
2.2 Patients with Hematopoietic Subsyndrome of Acute Radiation Syndrome
The recommended dose of UDENYCA is two doses, 6 mg each, administered subcutaneously one week apart. For dosing in pediatric patients weighing less than 45 kg, refer to Table 1. Administer the first dose as soon as possible after suspected or confirmed exposure to radiation levels greater than 2 gray (Gy). Administer the second dose one week after the first dose.
Obtain a baseline complete blood count (CBC). Do not delay administration of UDENYCA if a CBC is not readily available. Estimate a patient’s absorbed radiation dose (i.e., level of radiation exposure) based on information from public health authorities, biodosimetry if available, or clinical findings such as time to onset of vomiting or lymphocyte depletion kinetics.
2.3 Administration
UDENYCA is administered subcutaneously via a single-dose prefilled autoinjector, a single-dose prefilled syringe for manual use or for use with the on-body injector (OBI) for UDENYCA, which is co-packaged with a single dose prefilled syringe for OBI. Use of the OBI for UDENYCA is not recommended for patients with Hematopoietic Subsyndrome of Acute Radiation Syndrome. Use of the OBI has not been studied in pediatric patients.
Prior to use‚ remove the carton from the refrigerator and allow UDENYCA to reach room temperature for a minimum of 30 minutes. Discard any UDENYCA left at room temperature for greater than 48 hours.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Do not administer UDENYCA if discoloration or particulates are observed.
The needle cap on the prefilled syringe and prefilled autoinjector is not made with natural rubber latex.
Pediatric Patients Weighing Less than 45 kg
The UDENYCA prefilled syringe is not designed to allow for direct administration of doses less than 0.6 mL (6 mg). The syringe does not bear graduation marks which are necessary to accurately measure doses of UDENYCA less than 0.6 mL (6 mg) for direct administration to patients. Thus, the direct administration to patients requiring dosing of less than 0.6 mL (6 mg) is not recommended due to the potential for dosing errors. Refer to Table 1.
|
||
| Body Weight | UDENYCA Dose | Volume to Administer |
|---|---|---|
| Less than 10 kg* | See below* | See below* |
| 10 - 20 kg | 1.5 mg | 0.15 mL |
| 21 - 30 kg | 2.5 mg | 0.25 mL |
| 31 - 44 kg | 4 mg | 0.4 mL |
The UDENYCA prefilled autoinjector is not suitable for use in pediatric patients weighing less than 45 kg. The UDENYCA prefilled autoinjector delivers the entire contents (6 mg in 0.6 mL) in a single injection and is not adjustable.
2.4 Advice to Give to Patients or Caregivers Regarding Administration via the Prefilled Autoinjector
Only adults can self-administer UDENYCA with the prefilled autoinjector. If subcutaneous injections can be given at home, refer the patient or caregiver to the dose delivery information provided in the Instructions for Use. Provide training to patients or caregivers to ensure they understand how to administer UDENYCA via the prefilled autoinjector. Ensure patients or caregivers understand how to identify that a full dose has been administered by listening for the second 'click' and checking that the 'Orange Indicator' completely blocks the viewing window. Instruct patients or caregivers using the prefilled autoinjector to notify their healthcare provider immediately in order to determine the need for a replacement dose of UDENYCA if they suspect that the full dose may not have been administered.
2.5 Special Healthcare Provider Instructions for the On-body Injector for UDENYCA
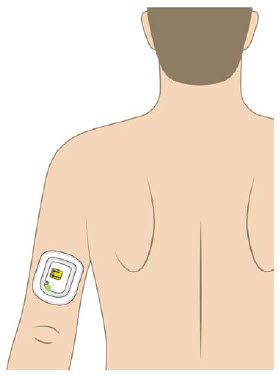
A healthcare provider must fill the on-body injector (OBI) with UDENYCA using the prefilled syringe and then apply the OBI for UDENYCA to the patient's skin (abdomen or back of arm). The back of the arm may only be used if there is a caregiver available to monitor the status of the OBI for UDENYCA. Approximately 27 hours after the OBI for UDENYCA is applied to the patient's skin, UDENYCA will be delivered over approximately 5 minutes. A healthcare provider may initiate administration with the OBI for UDENYCA on the same day as the administration of cytotoxic chemotherapy, as long as the OBI for UDENYCA delivers UDENYCA no less than 24 hours after administration of cytotoxic chemotherapy.
The prefilled syringe co-packaged in UDENYCA ONBODY must only be used with the OBI for UDENYCA. The prefilled syringe contains additional solution to compensate for liquid loss during filling of the OBI and delivery through the OBI for UDENYCA. If the prefilled syringe co-packaged in UDENYCA ONBODY is used for manual subcutaneous injection, the patient will receive an overdose. If the single-dose prefilled syringe for manual use is used with the OBI for UDENYCA, the patient may receive less than the recommended dose.
Do not use the OBI for UDENYCA to deliver any other drug product except the UDENYCA prefilled syringe co-packaged with the OBI for UDENYCA.
The OBI for UDENYCA should be applied to intact, non-irritated skin on the arm or abdomen.
A missed dose could occur due to an OBI for UDENYCA failure or leakage. If the patient misses a dose, a new dose should be administered by single-dose prefilled syringe for manual use, as soon as possible after detection.
Refer to the Healthcare Provider Instructions for Use for the OBI for UDENYCA for full administration information.
2.6 Advice to give to Patients Regarding Administration via the On-body Injector for UDENYCA
Advise patients to avoid activities such as traveling, driving, or operating heavy machinery during hours 26-29 following application of the on-body injector (OBI) for UDENYCA (this includes the 5-minute delivery period plus an hour post-delivery). Patients should have a caregiver nearby for the first use.
Refer the patient to the dose delivery information written on the Patient Instructions for Use. Provide training to patients to ensure they understand when the dose delivery of UDENYCA will begin and how to monitor the OBI for UDENYCA for completed delivery. Ensure patients understand how to identify signs of malfunction of OBI for UDENYCA [see Warnings and Precautions (5.12) and Patient Counseling Information (17)]. Instruct patients using the OBI to notify their healthcare professional immediately in order to determine the need for a replacement dose of UDENYCA if they suspect that the device may not have performed as intended [see Warnings and Precautions (5.12)].
3 DOSAGE FORMS AND STRENGTHS
UDENYCA is a clear, colorless, preservative free solution available as:
- Injection: 6 mg/0.6 mL in a single-dose prefilled syringe for manual use only.
- Injection: 6 mg/0.6 mL in a single-dose prefilled autoinjector.
- Injection: 6mg/0.6 mL in a single-dose prefilled syringe co-packaged with the on-body injector (OBI) for UDENYCA (UDENYCA ONBODY).
4 CONTRAINDICATIONS
UDENYCA is contraindicated in patients with a history of serious allergic reactions to pegfilgrastim products or filgrastim products. Reactions have included anaphylaxis [see Warnings and Precautions (5.3)].
5 WARNINGS AND PRECAUTIONS
5.1 Splenic Rupture
Splenic rupture, including fatal cases, can occur following the administration of pegfilgrastim products. Evaluate for an enlarged spleen or splenic rupture in patients who report left upper abdominal or shoulder pain after receiving UDENYCA.
5.2 Acute Respiratory Distress Syndrome
Acute respiratory distress syndrome (ARDS) can occur in patients receiving pegfilgrastim products. Evaluate patients who develop fever and lung infiltrates or respiratory distress after receiving UDENYCA for ARDS. Discontinue UDENYCA in patients with ARDS.
5.3 Serious Allergic Reactions
Serious allergic reactions, including anaphylaxis, can occur in patients receiving pegfilgrastim products. The majority of reported events occurred upon initial exposure. Allergic reactions, including anaphylaxis, can recur within days after the discontinuation of initial anti-allergic treatment. Permanently discontinue UDENYCA in patients with serious allergic reactions. Do not administer UDENYCA to patients with a history of serious allergic reactions to pegfilgrastim products or filgrastim products.
5.4 Allergies to Acrylics
The on-body injector (OBI) for UDENYCA uses acrylic adhesive. For patients who have reactions to acrylic adhesives, use of this product may result in a significant reaction.
5.5 Use in Patients with Sickle Cell Disorders
Severe and sometimes fatal sickle cell crises can occur in patients with sickle cell disorders receiving pegfilgrastim products. Discontinue UDENYCA if sickle cell crisis occurs.
5.6 Glomerulonephritis
Glomerulonephritis has occurred in patients receiving pegfilgrastim products. The diagnoses were based upon azotemia, hematuria (microscopic and macroscopic), proteinuria, and renal biopsy. Generally, events of glomerulonephritis resolved after dose reduction or discontinuation of pegfilgrastim products. If glomerulonephritis is suspected, evaluate for cause. If causality is likely, consider dose-reduction or interruption of UDENYCA.
5.7 Leukocytosis
White blood cell (WBC) counts of 100 x 109/L or greater have been observed in patients receiving pegfilgrastim products. Monitoring of complete blood count (CBC) during UDENYCA therapy is recommended.
5.8 Thrombocytopenia
Thrombocytopenia has been reported in patients receiving pegfilgrastim products. Monitor platelet counts.
5.9 Capillary Leak Syndrome
Capillary leak syndrome has been reported after G-CSF administration, including pegfilgrastim products, and is characterized by hypotension, hypoalbuminemia, edema, and hemoconcentration. Episodes vary in frequency, severity and may be life-threatening if treatment is delayed. Patients who develop symptoms of capillary leak syndrome should be closely monitored and receive standard symptomatic treatment, which may include a need for intensive care.
5.10 Potential for Tumor Growth Stimulatory Effects on Malignant Cells
The granulocyte colony-stimulating factor (G-CSF) receptor through which pegfilgrastim products and filgrastim products act has been found on tumor cell lines. The possibility that pegfilgrastim products act as a growth factor for any tumor type, including myeloid malignancies and myelodysplasia, diseases for which pegfilgrastim products are not approved, cannot be excluded.
5.11 Myelodysplastic Syndrome (MDS) and Acute Myeloid Leukemia (AML) in Patients with Breast and Lung Cancer
MDS and AML have been associated with the use of pegfilgrastim products in conjunction with chemotherapy and/or radiotherapy in patients with breast and lung cancer. Monitor patients for signs and symptoms of MDS/AML in these settings.
5.12 Potential Device failures
Missed or partial doses have been reported in patients receiving UDENYCA via the on-body injector (OBI) due to the device not performing as intended. In the event of a missed or partial dose, patients may be at increased risk of events such as neutropenia, febrile neutropenia and/or infection than if the dose had been correctly delivered.
Instruct patients using the OBI to notify their healthcare professional immediately in order to determine the need for a replacement dose of UDENYCA if they suspect that the device may not have performed as intended.
5.13 Aortitis
Aortitis has been reported in patients receiving pegfilgrastim products. It may occur as early as the first week after start of therapy. Manifestations may include generalized signs and symptoms such as fever, abdominal pain, malaise, back pain, and increased inflammatory markers (e.g., c-reactive protein and white blood cell count). Consider aortitis in patients who develop these signs and symptoms without known etiology. Discontinue UDENYCA if aortitis is suspected.
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are discussed in greater detail in other sections of the labeling:
- Splenic Rupture [see Warnings and Precautions (5.1)]
- Acute Respiratory Distress Syndrome [see Warnings and Precautions (5.2)]
- Serious Allergic Reactions [see Warnings and Precautions (5.3)]
- Allergies to Acrylics [see Warnings and Precautions (5.4)]
- Use in Patients with Sickle Cell Disorders [see Warnings and Precautions (5.5)]
- Glomerulonephritis [see Warnings and Precautions (5.6)]
- Leukocytosis [see Warnings and Precautions (5.7)]
- Thrombocytopenia [see Warnings and Precautions (5.8)]
- Capillary Leak Syndrome [see Warnings and Precautions (5.9)]
- Potential for Tumor Growth Stimulatory Effects on Malignant Cells [see Warnings and Precautions (5.10)]
- Myelodysplastic syndrome [see Warnings and Precautions (5.11)]
- Acute myeloid leukemia [see Warnings and Precautions (5.11)]
- Aortitis [see Warnings and Precautions (5.13)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Pegfilgrastim clinical trials safety data are based upon 932 patients receiving pegfilgrastim in seven randomized clinical trials. The population was 21 to 88 years of age and 92% female. The ethnicity was 75% Caucasian, 18% Hispanic, 5% Black, and 1% Asian. Patients with breast (n = 823), lung and thoracic tumors (n = 53) and lymphoma (n = 56) received pegfilgrastim after nonmyeloablative cytotoxic chemotherapy. Most patients received a single 100 mcg/kg (n = 259) or a single 6 mg (n = 546) dose per chemotherapy cycle over 4 cycles.
The following adverse reaction data in Table 2 are from a randomized, double-blind, placebo-controlled study in patients with metastatic or non-metastatic breast cancer receiving docetaxel 100 mg/m2 every 21 days (Study 3). A total of 928 patients were randomized to receive either 6 mg pegfilgrastim (n = 467) or placebo (n = 461). The patients were 21 to 88 years of age and 99% female. The ethnicity was 66% Caucasian, 31% Hispanic, 2% Black, and <1% Asian, Native American or other.
The most common adverse reactions occurring in ≥ 5% of patients and with a between-group difference of ≥ 5% higher in the pegfilgrastim arm in placebo controlled clinical trials are bone pain and pain in extremity.
| Body System Adverse Reaction | Placebo (N = 461) | pegfilgrastim 6 mg SC on Day 2 (N = 467) |
|---|---|---|
| Musculoskeletal and connective tissue disorders | ||
| Bone pain | 26% | 31% |
| Pain in extremity | 4% | 9% |
Leukocytosis
In clinical studies, leukocytosis (WBC counts > 100 x 109/L was observed in less than 1% of 932 patients with non-myeloid malignancies receiving pegfilgrastim. No complications attributable to leukocytosis were reported in clinical studies.
6.2 Immunogenicity
The observed incidence of anti-drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-drug antibodies in the studies described below with the incidence of anti-drug antibodies in other studies, including those of pegfilgrastim or of other pegfilgrastim products.
Binding antibodies to pegfilgrastim were detected using a BIAcore assay. The approximate limit of detection for this assay is 500 ng/mL. Pre-existing binding antibodies were detected in approximately 6% (51/849) of patients with metastatic breast cancer. Four of 521 pegfilgrastim-treated subjects who were negative at baseline developed binding antibodies to pegfilgrastim following treatment. None of these 4 patients had evidence of neutralizing antibodies detected using a cell-based bioassay.
6.3 Postmarketing Experience
The following adverse reactions have been identified during post approval use of pegfilgrastim products. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Splenic rupture and splenomegaly (enlarged spleen) [see Warnings and Precautions (5.1)]
- Acute respiratory distress syndrome (ARDS) [see Warnings and Precautions (5.2)]
- Allergic reactions/hypersensitivity, including anaphylaxis, skin rash, urticaria, generalized erythema and flushing [see Warnings and Precautions (5.3)]
- Sickle cell crisis [see Warnings and Precautions (5.5)]
- Glomerulonephritis [see Warnings and Precautions (5.6)]
- Leukocytosis [see Warnings and Precautions (5.7)]
- Thrombocytopenia [see Warnings and Precautions (5.8)]
- Capillary leak syndrome [see Warnings and Precautions (5.9)]
- Injection site reactions
- Sweet’s syndrome (acute febrile neutrophilic dermatosis), cutaneous vasculitis
- Application site reactions (including events such as application site hemorrhage, application site pain, application site discomfort, application site bruise, and application site erythema) have been reported with the use of the on-body-injector for UDENYCA.
- Contact dermatitis and local skin reactions such as rash, pruritus, and urticaria have been reported with the use of the on-body-injector for UDENYCA, possibly indicating a hypersensitivity reaction to the adhesive.
- Myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) in patients with breast and lung cancer receiving chemotherapy and/or radiotherapy [see Warnings and Precautions (5.11)]
- Aortitis [see Warnings and Precautions (5.13)]
- Alveolar hemorrhage
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Although available data with UDENYCA or pegfilgrastim product use in pregnant women are insufficient to establish whether there is a drug associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes, there are available data from published studies in pregnant women exposed to filgrastim products. These studies have not established an association of filgrastim product use during pregnancy with major birth defects, miscarriage, or adverse maternal or fetal outcomes.
In animal studies, no evidence of reproductive/developmental toxicity occurred in the offspring of pregnant rats that received cumulative doses of pegfilgrastim approximately 10 times the recommended human dose (based on body surface area). In pregnant rabbits, increased embryo lethality and spontaneous abortions occurred at 4 times the maximum recommended human dose simultaneously with signs of maternal toxicity (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risks of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Animal Data
Pregnant rabbits were dosed with pegfilgrastim subcutaneously every other day during the period of organogenesis. At cumulative doses ranging from the approximate human dose to approximately 4 times the recommended human dose (based on body surface area), the treated rabbits exhibited decreased maternal food consumption, maternal weight loss, as well as reduced fetal body weights and delayed ossification of the fetal skull; however, no structural anomalies were observed in the offspring from either study. Increased incidences of post-implantation losses and spontaneous abortions (more than half the pregnancies) were observed at cumulative doses approximately 4 times the recommended human dose, which were not seen when pregnant rabbits were exposed to the recommended human dose.
Three studies were conducted in pregnant rats dosed with pegfilgrastim at cumulative doses up to approximately 10 times the recommended human dose at the following stages of gestation: during the period of organogenesis, from mating through the first half of pregnancy, and from the first trimester through delivery and lactation. No evidence of fetal loss or structural malformations was observed in any study. Cumulative doses equivalent to approximately 3 and 10 times the recommended human dose resulted in transient evidence of wavy ribs in fetuses of treated mothers (detected at the end of gestation but no longer present in pups evaluated at the end of lactation).
8.2 Lactation
Risk Summary
There are no data on the presence of pegfilgrastim products in human milk, the effects on the breastfed child, or the effects on milk production. Other filgrastim products are secreted poorly into breast milk, and filgrastim products are not absorbed orally by neonates. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for UDENYCA and any potential adverse effects on the breastfed child from UDENYCA or from the underlying maternal condition.
8.4 Pediatric Use
The safety and effectiveness of UDENYCA have been established in pediatric patients. No overall differences in safety were identified between adult and pediatric patients based on postmarketing surveillance and review of the scientific literature.
Use of UDENYCA in pediatric patients for chemotherapy-induced neutropenia is based on UDENYCA's approval as a biosimilar to pegfilgrastim and evidence from adequate and well controlled studies of pegfilgrastim in adults with additional pharmacokinetic and safety data of pegfilgrastim in pediatric patients with sarcoma [see Clinical Pharmacology (12.3) and Clinical Studies (14.1)].
The use of UDENYCA to increase survival in pediatric patients acutely exposed to myelosuppressive doses of radiation is based on UDENYCA's approval as a biosimilar to pegfilgrastim and evidence from efficacy studies of pegfilgrastim conducted in animals and clinical data supporting the use of pegfilgrastim in patients with cancer receiving myelosuppressive chemotherapy. Efficacy studies of pegfilgrastim products could not be conducted in humans with acute radiation syndrome for ethical and feasibility reasons. Results from population modeling and simulation indicate that two doses of pegfilgrastim (Table 1), administered one week apart provide pediatric patients with exposures comparable to that in adults receiving two 6 mg doses one week apart [see Dosage and Administration (2.3), Clinical Pharmacology (12.3) and Clinical Studies (14.2)].
10 OVERDOSAGE
Overdosage of pegfilgrastim products may result in leukocytosis and bone pain. Events of edema, dyspnea, and pleural effusion have been reported in a single patient who administered pegfilgrastim on 8 consecutive days in error. In the event of overdose, the patient should be monitored for adverse reactions [see Adverse Reactions (6)].
11 DESCRIPTION
Pegfilgrastim-cbqv is a covalent conjugate of recombinant methionyl human G-CSF and monomethoxypolyethylene glycol. Recombinant methionyl human G-CSF is a water-soluble, 175 amino acid protein with a molecular weight of approximately 19 kilodaltons (kDa). Recombinant methionyl human G-CSF is obtained from the bacterial fermentation of a strain of E coli transformed with a genetically engineered plasmid containing the human G-CSF gene. During the pegfilgrastim-cbqv manufacturing process, fermentation is carried out in nutrient medium containing the antibiotic kanamycin. However, kanamycin is cleared in the manufacturing process and is not detectable in the final product. To produce pegfilgrastim-cbqv, a 20 kDa monomethoxypolyethylene glycol molecule is covalently bound to the N-terminal methionyl residue of recombinant methionyl human G-CSF. The average molecular weight of pegfilgrastim-cbqv is approximately 39 kDa.
UDENYCA (pegfilgrastim-cbqv) injection is provided in three presentations:
- UDENYCA for manual subcutaneous injection is supplied in 0.6 mL prefilled syringes. The prefilled syringe does not bear graduation marks and is designed to deliver the entire contents of the syringe (6 mg/0.6 mL).
- UDENYCA for subcutaneous injection is supplied in a 0.6 mL prefilled single-dose autoinjector. The prefilled autoinjector delivers the entire contents (6 mg in 0.6mL) in a single injection and is not adjustable.
- On-body injector (OBI) for UDENYCA is supplied with a prefilled syringe containing 0.67 mL of UDENYCA in solution that delivers 0.6 mL of UDENYCA in solution when used with the OBI for UDENYCA. The syringe does not bear graduation marks and is only to be used with the OBI for UDENYCA.
The delivered 0.6 mL dose from either the prefilled syringe for manual subcutaneous injection, the prefilled autoinjector or the OBI for UDENYCA contains 6 mg pegfilgrastim-cbqv (based on protein weight) in a sterile, clear, colorless, preservative-free solution (pH 4.0) containing acetate (0.35 mg), polysorbate 20 (0.02 mg), sodium (0.02 mg), and sorbitol (30 mg) in Water for Injection, USP.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Pegfilgrastim products are colony-stimulating factors that act on hematopoietic cells by binding to specific cell surface receptors, thereby stimulating proliferation, differentiation, commitment, and end cell functional activation.
12.2 Pharmacodynamics
Animal data and clinical data in humans suggest a correlation between pegfilgrastim products exposure and the duration of severe neutropenia as a predictor of efficacy. Selection of the dosing regimen of UDENYCA is based on reducing the duration of severe neutropenia.
12.3 Pharmacokinetics
The pharmacokinetics of pegfilgrastim were studied in 379 patients with cancer. The pharmacokinetics of pegfilgrastim were nonlinear, and clearance decreased with increases in dose. Neutrophil receptor binding is an important component of the clearance of pegfilgrastim, and serum clearance is directly related to the number of neutrophils. In addition to numbers of neutrophils, body weight appeared to be a factor. Patients with higher body weights experienced higher systemic exposure to pegfilgrastim after receiving a dose normalized for body weight. A large variability in the pharmacokinetics of pegfilgrastim was observed. The half-life of pegfilgrastim ranged from 15 to 80 hours after subcutaneous injection. In healthy volunteers, the pharmacokinetics of pegfilgrastim were comparable when delivered subcutaneously via a manual prefilled syringe versus via the on-body-injector (OBI) for UDENYCA.
Specific Populations
No gender-related differences were observed in the pharmacokinetics of pegfilgrastim, and no differences were observed in the pharmacokinetics of geriatric patients (≥ 65 years of age) compared with younger patients (< 65 years of age) [see Use in Specific Populations (8.5)].
Renal Impairment
In a study of 30 subjects with varying degrees of renal dysfunction, including end stage renal disease, renal dysfunction had no effect on the pharmacokinetics of pegfilgrastim.
Pediatric Patients with Cancer Receiving Myelosuppressive Chemotherapy
The pharmacokinetics and safety of pegfilgrastim were studied in 37 pediatric patients with sarcoma in Study 4 [see Clinical Studies 14.1]. The mean (± standard deviation [SD]) systemic exposure (AUC0-inf) of pegfilgrastim after subcutaneous administration at 100 mcg/kg was 47.9 (± 22.5) mcg·hr/mL in the youngest age group (0 to 5 years, n = 11), 22.0 (± 13.1) mcg·hr/mL in the 6 to 11 years age group (n = 10), and 29.3 (± 23.2) mcg·hr/mL in the 12 to 21 years age group (n = 13). The terminal elimination half-lives of the corresponding age groups were 30.1 (± 38.2) hours, 20.2 (± 11.3) hours, and 21.2 (± 16.0) hours, respectively.
Patients Acutely Exposed to Myelosuppressive Doses of Radiation
The pharmacokinetics of pegfilgrastim is not available in patients acutely exposed to myelosuppressive doses of radiation. Based on limited pharmacokinetic data in irradiated non-human primates, the area under the concentration-time curve (AUC), reflecting the exposure to pegfilgrastim in non-human primates following a 300 mcg/kg dose of pegfilgrastim, appears to be greater than in humans receiving a 6 mg dose. Results from population modeling and simulation indicate that two 6 mg doses of pegfilgrastim administered one week apart in adults result in clinically relevant effects on duration of grade 3 and 4 neutropenia. In addition, weight-based dosing in pediatric patients weighing less than 45 kg [see Dosage and Administration, Section 2.3, Table 1] provides exposures comparable to those in adults receiving two 6 mg doses one week apart.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No carcinogenicity or mutagenesis studies have been performed with pegfilgrastim products.
Pegfilgrastim did not affect reproductive performance or fertility in male or female rats at cumulative weekly doses approximately 6 to 9 times higher than the recommended human dose (based on body surface area).
14 CLINICAL STUDIES
14.1 Patients with Cancer Receiving Myelosuppressive Chemotherapy
Pegfilgrastim was evaluated in three randomized, double-blind, controlled studies. Studies 1 and 2 were active- controlled studies that employed doxorubicin 60 mg/m2 and docetaxel 75 mg/m2 administered every 21 days for up to 4 cycles for the treatment of metastatic breast cancer. Study 1 investigated the utility of a fixed dose of pegfilgrastim. Study 2 employed a weight-adjusted dose. In the absence of growth factor support, similar chemotherapy regimens have been reported to result in a 100% incidence of severe neutropenia (ANC < 0.5 x 109/L) with a mean duration of 5 to 7 days and a 30% to 40% incidence of febrile neutropenia. Based on the correlation between the duration of severe neutropenia and the incidence of febrile neutropenia found in studies with filgrastim, duration of severe neutropenia was chosen as the primary endpoint in both studies, and the efficacy of pegfilgrastim was demonstrated by establishing comparability to filgrastim-treated patients in the mean days of severe neutropenia.
In Study 1, 157 patients were randomized to receive a single subcutaneous injection of pegfilgrastim (6 mg) on day 2 of each chemotherapy cycle or daily subcutaneous filgrastim (5 mcg/kg/day) beginning on day 2 of each chemotherapy cycle. In Study 2, 310 patients were randomized to receive a single subcutaneous injection of pegfilgrastim (100 mcg/kg) on day 2 or daily subcutaneous filgrastim (5 mcg/kg/day) beginning on day 2 of each chemotherapy cycle.
Both studies met the major efficacy outcome measure of demonstrating that the mean days of severe neutropenia of pegfilgrastim-treated patients did not exceed that of filgrastim-treated patients by more than 1 day in cycle 1 of chemotherapy. The mean days of cycle 1 severe neutropenia in Study 1 were 1.8 days in the pegfilgrastim arm compared to 1.6 days in the filgrastim arm [difference in means 0.2 (95% CI -0.2, 0.6)] and in Study 2 were 1.7 days in the pegfilgrastim arm compared to 1.6 days in the filgrastim arm [difference in means 0.1 (95% CI -0.2, 0.4)].
A secondary endpoint in both studies was days of severe neutropenia in cycles 2 through 4 with results similar to those for cycle 1.
Study 3 was a randomized, double-blind, placebo-controlled study that employed docetaxel 100 mg/m2 administered every 21 days for up to 4 cycles for the treatment of metastatic or non-metastatic breast cancer. In this study, 928 patients were randomized to receive a single subcutaneous injection of pegfilgrastim (6 mg) or placebo on day 2 of each chemotherapy cycle. Study 3 met the major trial outcome measure of demonstrating that the incidence of febrile neutropenia (defined as temperature ≥ 38.2°C and ANC ≤ 0.5 x109/L) was lower for pegfilgrastim-treated patients as compared to placebo-treated patients (1% versus 17%, respectively, p < 0.001). The incidence of hospitalizations (1% versus 14%) and IV anti-infective use (2% versus 10%) for the treatment of febrile neutropenia was also lower in the pegfilgrastim-treated patients compared to the placebo-treated patients.
Study 4 was a multicenter, randomized, open-label study to evaluate the efficacy, safety, and pharmacokinetics [see Clinical Pharmacology (12.3)] of pegfilgrastim in pediatric and young adult patients with sarcoma. Patients with sarcoma receiving chemotherapy age 0 to 21 years were eligible. Patients were randomized to receive subcutaneous pegfilgrastim as a single dose of 100 mcg/kg (n= 37) or subcutaneous filgrastim at a dose 5 mcg/kg/day (n=6) following myelosuppressive chemotherapy. Recovery of neutrophil counts was similar in the pegfilgrastim and filgrastim groups. The most common adverse reaction reported was bone pain.
14.2 Patients with Hematopoietic Subsyndrome of Acute Radiation Syndrome
Efficacy studies of pegfilgrastim products could not be conducted in humans with acute radiation syndrome for ethical and feasibility reasons. Approval of this indication was based on efficacy studies conducted in animals and data supporting pegfilgrastim's effect on severe neutropenia in patients with cancer receiving myelosuppressive chemotherapy [see Dosage and Administration (2.1)].
The recommended dose of UDENYCA is two doses, 6 mg each, administered one week apart for humans exposed to myelosuppressive doses of radiation. For pediatric patients weighing less than 45 kg, dosing of UDENYCA is weight based and is provided in Table 1 [see Dosage and Administration (2.3)]. This dosing regimen is based on population modeling and simulation analyses. The exposure associated with this dosing regimen is expected to provide sufficient pharmacodynamic activity to treat humans exposed to myelosuppressive doses of radiation [see Clinical Pharmacology (12.3)]. The safety of pegfilgrastim at a dose of 6 mg has been assessed on the basis of clinical experience in patients with cancer receiving myelosuppressive chemotherapy.
The efficacy of pegfilgrastim for the acute radiation syndrome setting was studied in a randomized, placebo- controlled non-human primate model of radiation injury. Rhesus macaques were randomized to either a control (n = 23) or treated (n = 23) cohort. On study day 0, animals (n = 6 to 8 per irradiation day) were exposed to total body irradiation (TBI) of 7.50 ± 0.15 Gy delivered at 0.8 ± 0.03 Gy/min, representing a dose that would be lethal in 50% of animals by 60 days of follow-up (LD50/60). Animals were administered subcutaneous injections of a blinded treatment (control article [5% dextrose in water] or pegfilgrastim [300-319 mcg/kg/day]) on study day 1 and on study day 8. The primary endpoint was survival. Animals received medical management consisting of intravenous fluids, antibiotics, blood transfusions, and other support as required.
Pegfilgrastim significantly (at 0.0014 level of significance) increased 60-day survival in irradiated non-human primates: 91% survival (21/23) in the pegfilgrastim group compared to 48% survival (11/23) in the control group.
16 HOW SUPPLIED/STORAGE AND HANDLING
UDENYCA single-dose prefilled syringe for manual use
UDENYCA (pegfilgrastim-cbqv) injection is a clear, colorless, preservative-free solution supplied in a prefilled single-dose syringe containing 6 mg pegfilgrastim-cbqv, supplied with a 29-gauge, 1⁄2-inch needle with an UltraSafe Passive™ Needle Guard.
The needle cap of the prefilled syringe is not made with natural rubber latex.
UDENYCA is provided in a dispensing pack containing one sterile 6 mg/0.6 mL prefilled syringe (NDC 70114-101-01).
UDENYCA prefilled syringe does not bear graduation marks and is intended only to deliver the entire contents of the syringe (6 mg/0.6 mL) for direct administration. Use of the prefilled syringe is not recommended for direct administration for pediatric patients weighing less than 45 kg who require doses that are less than the full contents of the syringe.
Store refrigerated between 2° to 8°C (36° to 46°F) in the carton to protect from light. Do not shake. Discard UDENYCA stored at room temperature for more than 48 hours. Avoid freezing; if frozen, thaw in the refrigerator before administration. Discard UDENYCA if frozen more than once.
UDENYCA single-dose prefilled autoinjector
UDENYCA (pegfilgrastim-cbqv) injection is a clear, colorless, preservative-free solution supplied in a prefilled single-dose autoinjector containing 6 mg pegfilgrastim-cbqv.
The needle cap of the prefilled autoinjector is not made with natural rubber latex.
UDENYCA is provided in a dispensing pack containing one 6 mg/0.6 mL prefilled autoinjector (NDC 70114-120-01).
The UDENYCA prefilled autoinjector is not suitable for use in pediatric patients weighing less than 45 kg. The UDENYCA prefilled autoinjector delivers the entire contents (6 mg in 0.6 mL) in a single injection and is not adjustable.
Store refrigerated between 2° to 8°C (36° to 46°F) in the carton to protect from light. Do not shake. Discard UDENYCA stored at room temperature for more than 48 hours. Avoid freezing; if frozen, thaw in the refrigerator before administration. Discard UDENYCA if frozen more than once.
UDENYCA ONBODY
UDENYCA ONBODY is provided in a carton containing one sterile prefilled syringe and one sterile on-body injector (OBI) for UDENYCA (NDC 70114-130-01).
The UDENYCA injection single-dose prefilled syringe contains 0.67 mL of a clear, colorless solution that delivers 6 mg/0.6 mL of pegfilgrastim-cbqv when used with the OBI for UDENYCA. The prefilled syringe is supplied with a 29-gauge, 1/2-inch needle. The syringe does not bear graduation marks and is only to be used with the OBI for UDENYCA.
The needle cap of the prefilled syringe is not made with natural rubber latex.
Store UDENYCA ONBODY in the refrigerator at 2°C to 8°C (36°F to 46°F) until 30 minutes prior to use. Because the OBI for UDENYCA is at room temperature during the period of use, UDENYCA ONBODY should not be held at room temperature longer than 12 hours prior to use. Discard UDENYCA ONBODY if stored at room temperature for more than 12 hours.
Do not use the OBI for UDENYCA if its packaging has been previously opened.
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information and Instructions for Use)
Advise patients of the following risks and potential risks with UDENYCA:
- Splenic rupture and splenomegaly
- Acute Respiratory Distress Syndrome
- Serious allergic reactions
- Sickle cell crisis
- Glomerulonephritis
- Increased risk of Myelodysplastic Syndrome and/or Acute Myeloid Leukemia in patients with breast and lung cancer who receive UDENYCA in conjunction with chemotherapy and/or radiation therapy
- Capillary Leak Syndrome
- Aortitis
Advise patients acutely exposed to myelosuppressive doses of radiation (Hematopoietic Subsyndrome of Acute Radiation Syndrome) that efficacy studies of pegfilgrastim products for this indication could not be conducted in humans for ethical and feasibility reasons and that, therefore, approval of this use was based on efficacy studies conducted in animals [see Clinical Studies (14.2)].
Instruct patients who self-administer UDENYCA using the single-dose prefilled syringe of the:
- Importance of following the Instructions for Use (see Instructions for Use).
- Dangers of reusing syringes.
- Importance of following local requirements for proper disposal of used syringes.
For patients who will use the UDENYCA prefilled autoinjector, tell them that they:
- Will hear two "clicks" during the UDENYCA injection. The first 'click' means the start of injection and second 'click' means the end of injection.
- To start injection, push the prefilled autoinjector body down. Continue holding down after hearing the first 'click'.
- In the viewing window, the orange indicator will advance to show the progress of the injection.
When injection has finished, there will be a second 'click' and the 'Orange Indicator' will completely block the viewing window.
Advise patients on the use of the on-body injector (OBI) for UDENYCA:
- Review the Patient Information and Patient Instructions for Use with the patient and provide the instructions to the patient.
- Refer the patient to the dose delivery information written on the Patient Instructions for Use.
- Tell the patient when their dose delivery of UDENYCA will begin and when their dose delivery should be completed.
- Advise the patient that serious allergic reactions can happen with UDENYCA. Patients should have a caregiver nearby for the first use. Patients should plan to be in a place where they can appropriately monitor the OBI for UDENYCA during the approximately 5 minutes UDENYCA delivery and for an hour after the delivery. Advise the patient to avoid traveling, driving, or operating heavy machinery during hours 26-29 following application of the OBI for UDENYCA.
- If the OBI for UDENYCA is placed on the back of the arm, remind the patient that a caregiver must be available to monitor the OBI for UDENYCA.
- If a patient calls the healthcare provider regarding any OBI for UDENYCA problems, the healthcare provider is advised to call Coherus at 1-800-483-3692.
- Advise the patient:
- to call their healthcare provider immediately if the status light on the OBI for UDENYCA is flashing red (see the Patient Instructions for Use).
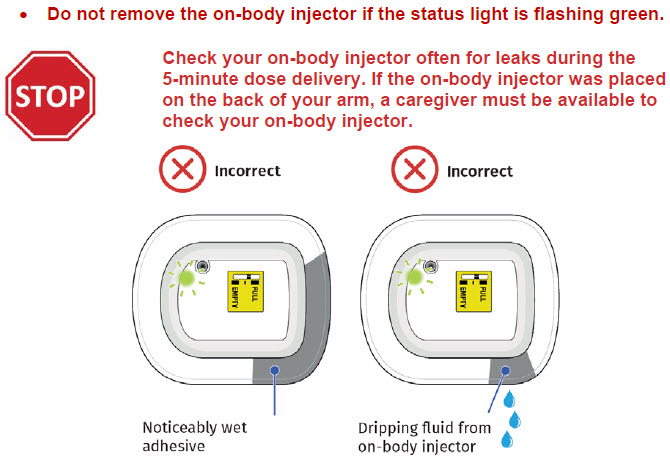
- to inform their healthcare provider if the adhesive on the OBI for UDENYCA becomes saturated with fluid, or there is dripping, as this may be evidence of significant product leakage, resulting in inadequate or missed dose (see the Patient Instructions for Use).
- to keep the OBI for UDENYCA dry for approximately the last 3 hours prior to the dose delivery start to better enable potential leak detection.
- that the OBI for UDENYCA should only be exposed to temperatures between 41°F and 104°F (5°C to 40°C).
- to keep the OBI for UDENYCA at least 4 inches away from electrical equipment such as cell phones, cordless telephones, microwaves, and other common appliances. Failure to keep the OBI for UDENYCA at least this recommended distance may interfere with operation and can lead to a missed or incomplete dose of UDENYCA.
- that if the needle is exposed after OBI for UDENYCA removal, place the used OBI for UDENYCA in a sharps disposal container to avoid accidental needle stick and call their healthcare provider immediately.
- to remove the OBI for UDENYCA after the green light shines continuously and to place the used OBI for UDENYCA in a sharps disposal container (see the Patient Instructions for Use).
- Advise the patient:
- do not reapply the OBI for UDENYCA if the OBI for UDENYCA comes off before full dose is delivered and instead call their healthcare provider immediately, as they may need a replacement dose.
- avoid bumping the OBI for UDENYCA or knocking the OBI for UDENYCA off the body.
- do not use other materials to hold the on-body injector in place that could cover audio/visual indicators or compress the on-body injector against the patient’s skin, as this could dislodge the cannula and lead to a missed dose or incomplete dose of UDENYCA.
- do not expose the OBI for UDENYCA to medical imaging studies (e.g., X-ray scan, MRI, CT scan and ultrasound), radiation treatment, and oxygen rich environments such as hyperbaric chambers to avoid OBI for UDENYCA damage and patient injury.
- Advise the patient to avoid:
- airport X-ray scans and request a manual pat down instead; remind patients who elect to request a manual pat down to exercise care to avoid having the OBI for UDENYCA dislodged during the pat down process.
- sleeping on the OBI for UDENYCA or applying pressure on the OBI for UDENYCA as this may affect OBI for UDENYCA performance.
- getting body lotions, creams, oils, and cleaning agents near the OBI for UDENYCA as these products may loosen the adhesive.
- use of lotions, creams, or oils on their arms and abdomen prior to their next scheduled OBI for UDENYCA dose (to help with device adherence to the skin).
- using bath tubs, hot tubs, whirlpools, or saunas and avoid exposing the OBI for UDENYCA to direct sunlight as these may affect the drug.
- peeling off or disturbing the OBI for UDENYCA adhesive before delivery of full dose is complete.
Coherus
BioSciences
UDENYCA® (pegfilgrastim-cbqv)
Manufactured by: Coherus BioSciences, Inc., Redwood City, California 94065-1442
U.S. License No. 2023
© 2021 Coherus BioSciences Inc. All rights reserved.
For more information, go to www.UDENYCA.com or call 1-800-4UDENYCA (1-800-483-3692)
PMD-0212, Rev. 00
| Patient Information
UDENYCA® (yoo-den-i-kah) (pegfilgrastim-cbqv) Injection |
|
| What is UDENYCA?
UDENYCA is a man-made form of granulocyte colony-stimulating factor (G-CSF). G-CSF is a substance produced by the body. It stimulates the growth of neutrophils, a type of white blood cell important in the body’s fight against infection. Acute Radiation Syndrome: The effectiveness of pegfilgrastim for this use was only studied in animals, because it could not be studied in people. |
|
| Do not take UDENYCAif you have had a serious allergic reaction to pegfilgrastim products or filgrastim products. | |
Before you receive UDENYCA, tell your healthcare provider about all of your medical conditions, including if you:
|
|
How will I receive UDENYCA?
|
|
| What are possible side effects of UDENYCA? UDENYCA may cause serious side effects, including:
These are not all the possible side effects of UDENYCA. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|
How should I store UDENYCA?
|
|
| General information about the safe and effective use of UDENYCA.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use UDENYCA for a condition for which it was not prescribed. Do not give UDENYCA to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about UDENYCA that is written for health professionals. |
|
| What are the ingredients in UDENYCA?
Active ingredient: pegfilgrastim-cbqv Inactive ingredients: acetate, polysorbate 20, sodium, and sorbitol in Water for Injection. |
|
| |
|
| Manufactured by: Coherus BioSciences, Inc., Redwood City, California 94065-1442 U.S. License No. 2023 © 2023 Coherus BioSciences Inc. All rights reserved. For more Information go to www.UDENYCA.com or call 1-800-4UDENYCA (1-800-483-3692) PMD-0006 Rev. 06 | Coherus BioSciences |
| This Patient Information has been approved by the U.S. Food and Drug Administration | Revised: 03/2023 |
| Instructions for Use UDENYCA® (yoo-den-i-kah) (pegfilgrastim-cbqv) Injection Single-Dose Prefilled Syringe |
|
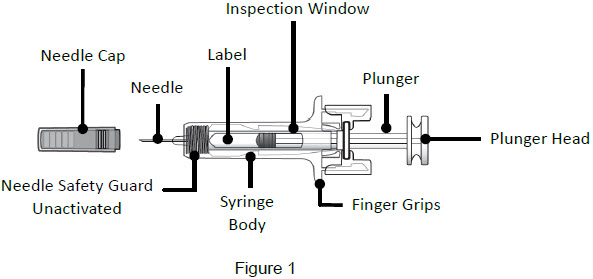
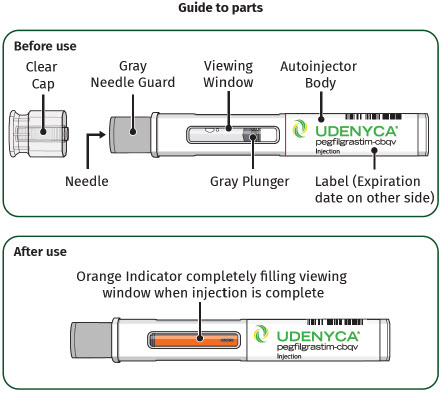
| Guide to Parts | |
| Before Use | |
 |
|
| Important: The needle is covered by a needle cap before use | |
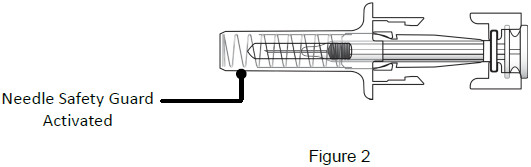
| After Use | |
 |
|
| Important Read the Patient Information for important information you need to know about UDENYCA before using these Instructions for Use. |
|
| Storing the UDENYCA prefilled syringe | |
|  |
| Using the prefilled syringe | |
| |
| Call your healthcare provider if you have any questions. | |
| Prepare the injection | |
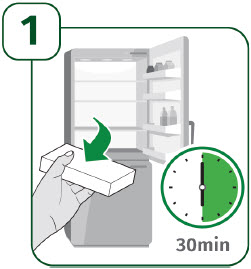
| 1 - Remove carton from refrigerator and check expiration date | |
| 1A: Remove the carton from the refrigerator and check the expiration date printed on the carton. (See Figure 3) Do not use if the expiration date has passed. | 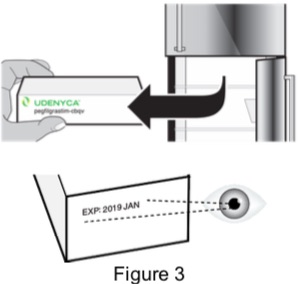 |
| 1B: Open the carton and remove the sealed syringe tray. (See Figure 4) |  |
| 2 - Allow UDENYCA to reach room temperature and gather supplies | |
| 2A: Place the sealed syringe tray on a flat, clean work surface and allow it to sit at room temperature for at least 30 minutes. (See Figure 5) Do not attempt to warm up the syringe in any other way, such as a microwave, hot water, or direct sunlight. |  |
2B: Gather the following supplies: (See Figure 6)
| 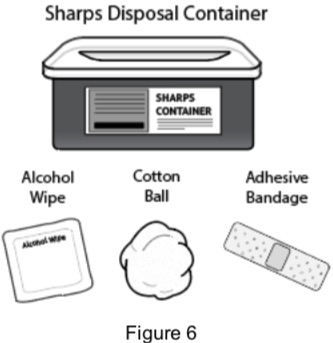 |
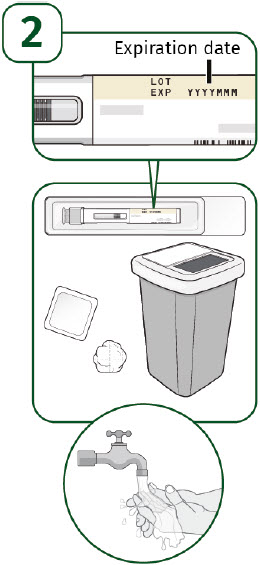
| 3 - Wash your hands and remove syringe from tray | |
| 3A: Wash your hands well with soap and warm water. (See Figure 7) |  |
| 3B: Open the tray by peeling away the cover. Remove the prefilled syringe from the tray by grasping the middle of the syringe body and carefully pulling it out of the tray. (See Figure 8) For Safety reasons:
| 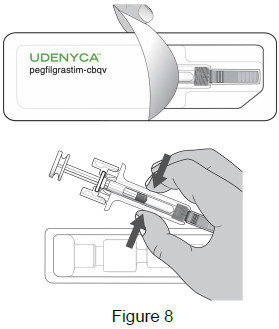 |
| 4 - Inspect the syringe and medicine | |
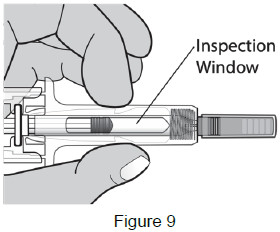
| Check the medicine through the Inspection Window. The medicine should be clear and colorless. It is normal to see 1 or more air bubbles in the syringe. Removal of the air is not needed. (See Figure 9) Do not use the prefilled syringe if:
|  |
| Select and clean injection site | |
| 5 - Select and clean the injection site | |
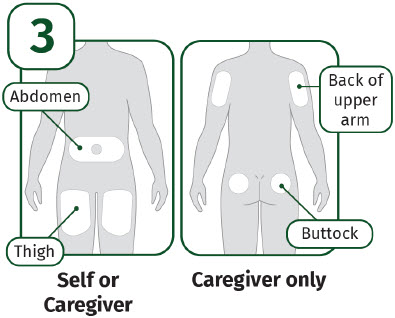
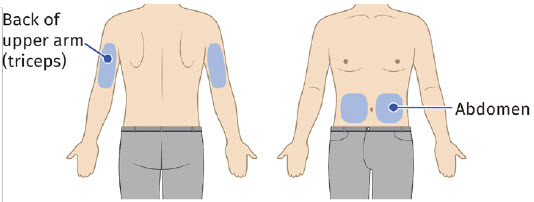
5A: Select the injection site. The recommended injection sites for a subcutaneous injection are the: (See Figure 10)
If you want to use the same injection site, make sure it is not the same spot on the injection site you used for a previous injection. | 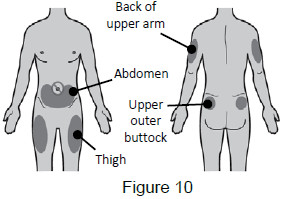 |
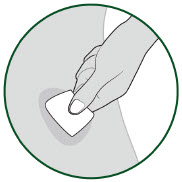
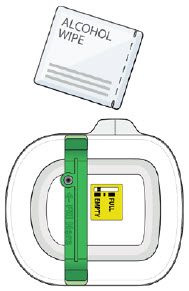
| 5B: Clean the injection site with an alcohol wipe.
(See Figure 11) Do not touch this area again before injection. | 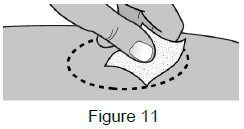 |
| Inject the dose | |
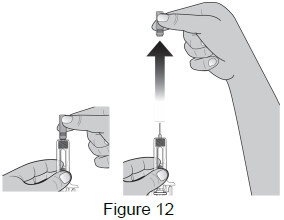
| 6 - Remove needle cap | |
Remove the needle cap by pulling it straight off.
(See Figure 12)
|  |
| 7 – Position fingers | |
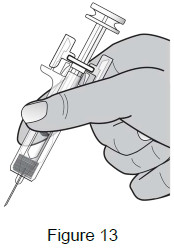
Grasp the body of the syringe like a dart (just under the finger grips) with your thumb and index fingers. (See Figure 13)
|  |
| 8 – Pinch the skin and insert the needle | |
| 8A: Use your free hand to gently pinch the cleaned injection site to create a firm surface. (See Figure 14) | 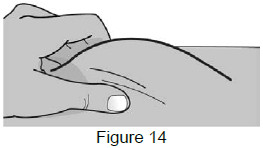 |
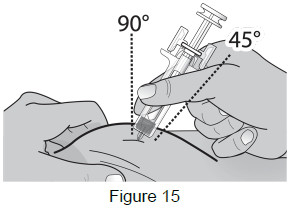
8B: Hold the pinch. Insert the needle into the skin at a 45 to 90-degree angle. (See Figure 15)
|  |
| 8C: After fully inserting the needle, release the
pinched skin and use your free hand to stabilize the bottom of the syringe. Then move your other hand into injection position with your thumb on the plunger head. (See Figure 16) | 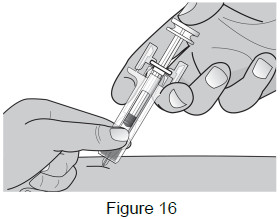 |
| 9 – Push plunger head down to deliver dose | |
| 9A: Using slow and constant pressure, push the plunger head down until it reaches the bottom. This will help to ensure that you receive the full dose. (See Figure 17) | 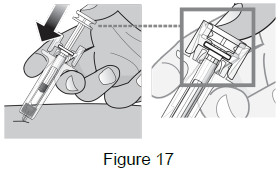 |
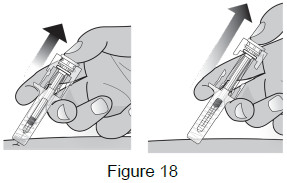
| 9B: While the needle is still inserted, slowly move your thumb back, allowing the plunger to rise. This will release the needle safety guard to safely cover the needle. Then remove the syringe from the injection site. (See Figure 18) Important: When you remove the syringe, if it looks like the medicine is still in the syringe, this means you have not received a full dose. Call your healthcare provider right away.
|  |
| Dispose | |
| 10 – Dispose of used prefilled syringes | |
| Put the used prefilled syringe into an FDA-cleared sharps disposal container right away after use. Do not throw away the syringe in the household trash. (See Figure 19) | 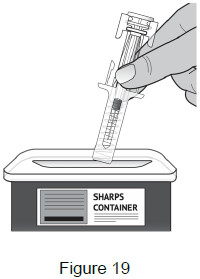 |
|
|
| This Instructions for Use has been approved by the U.S. Food and Drug Administration. | Issued: 09/2019 |
Coherus
BioSciences
UDENYCA® (pegfilgrastim-cbqv)
Manufactured by: Coherus BioSciences, Inc., Redwood City, California 94065-1442
U.S. License No. 2023
© 2019 Coherus BioSciences Inc. All rights reserved.
For more Information go to www.UDENYCA.com or call 1-800-4UDENYCA (1-800-483-3692)
PMD-0005, Rev. 02
| INSTRUCTIONS FOR USE UDENYCA™ (yoo-den-i-kah) (pegfilgrastim-cbqv) injection, for subcutaneous use Single-Dose Prefilled Autoinjector |
|
 | Storing the UDENYCA prefilled autoinjector
|
 | Take the UDENYCA carton out of the refrigerator.
|
 | Check the expiration date on the prefilled autoinjector label. Do not use the prefilled autoinjector if the expiration date has passed. Place the following supplies on a clean, flat surface:
|
  | Choose an injection site.
|
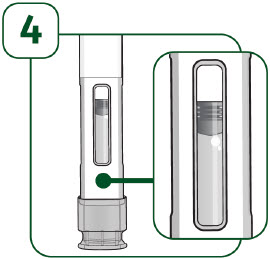 | Check the viewing window:
|
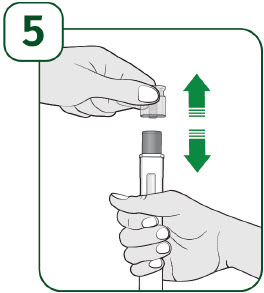 | Hold the prefilled autoinjector with the clear cap facing up and pull the clear cap straight off.
|
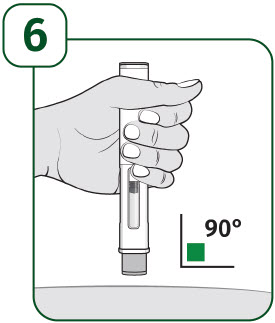 | Hold the body of the prefilled autoinjector in one hand so you can see the viewing window. Place the gray needle guard against your skin at a 90-degree angle. |
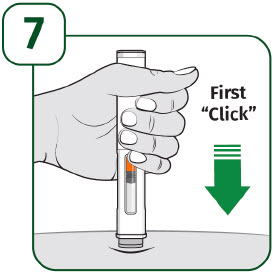 | Press the prefilled autoinjector firmly against the skin and you will hear the first "click". The orange indicator will start to move along the viewing window. |
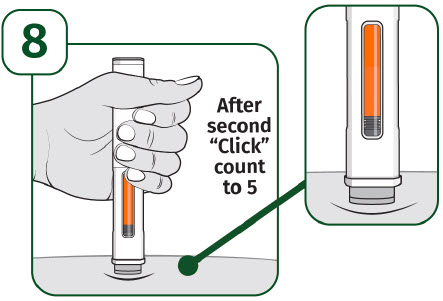 | Keep holding the prefilled autoinjector firmly against the skin until the orange indicator stops moving and you hear the second "click". After you hear the second "click", continue to hold the prefilled autoinjector in place and count to 5. When the injection is complete, make sure the orange indicator completely fills the viewing window before slowly lifting the prefilled autoinjector away from the skin. |
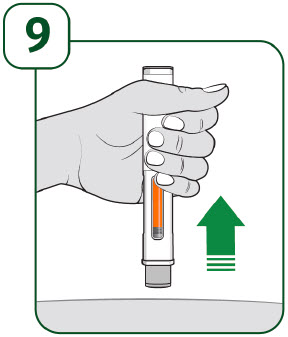 | The gray needle guard will cover the needle tip. If the viewing window is not completely filled by the orange indicator or if there are more than a few drops of liquid on the injection site, contact your healthcare provider right away. You may not have received your full dose. After completing the injection, place a cotton ball or gauze pad over the injection site.
|
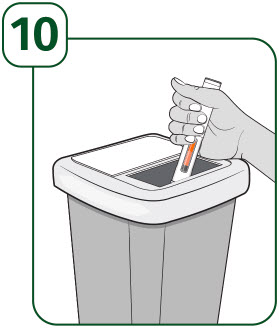 | How should I throw away (dispose of) the used UDENYCA prefilled autoinjector?
|
| This Instructions for Use has been approved by the U.S. Food and Drug Administration. | Issued: 03/2023 |
| |
|
| Manufactured by: Coherus BioSciences, Inc., Redwood City California 94065-1442 U.S. License No. 2023 ©2023 Coherus BioSciences, Inc. All rights reserved. For more information, go to www.UDENYCA.com, or call 1-800-4UDENYCA (1-800-483-3692) PMD-0175 Rev. 00 | Coherus BIOSCIENCES |
| Patient Information
UDENYCA® (yoo-den-i-kah) (pegfilgrastim-cbqv) injection on-body injector for UDENYCA |
|
What is the most important information I need to know about receiving UDENYCA with the on-body injector for UDENYCA?
|
|
| What is UDENYCA?
UDENYCA is a prescription medicine used to help reduce the chance of infection due to a low white blood cell count, in people with certain types of cancer (non-myeloid), who receive anti-cancer medicines (chemotherapy) that can cause fever and low blood cell count. |
|
| Do not take UDENYCA if you have had a serious allergic reaction to pegfilgrastim products or filgrastim products. | |
Before you receive UDENYCA, tell your healthcare provider about all of your medical conditions, including if you:
|
|
| How will I receive UDENYCA? See the Instructions for Use for detailed information about how you will receive a dose of UDENYCA with the on-body injector for UDENYCA, and how to remove and dispose of the on-body injector for UDENYCA.
|
|
While the on-body injector for UDENYCA is in place you should avoid:
|
|
| What are the possible side effects of UDENYCA? UDENYCA may cause serious side effects, including:
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|
| General information about the safe and effective use of UDENYCA.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. If you would like more information about UDENYCA, talk with your healthcare provider or pharmacist. You can ask your pharmacist for information about UDENYCA that is written for health professionals. |
|
| What are the ingredients in UDENYCA?
Active ingredient: pegfilgrastim-cbqv Inactive ingredients: acetate, polysorbate 20, sodium, and sorbitol in Water for Injection. |
|
| |
|
| Manufactured by: Coherus BioSciences, Inc., Redwood City California 94065-1442 U.S. License No. 2023 ©2021 Coherus BioSciences Inc. All rights reserved. For more information go to www.UDENYCA.com, or call 1-800-4UDENYCA (1-800-483-3692) PMD-0213, Rev.00 | Coherus BioSciences |
| This Patient Information has been approved by the U.S. Food and Drug Administration. | Issued: 12/2023 |
| Healthcare Provider INSTRUCTIONS FOR USE UDENYCA (yoo-den-i-kah) ONBODY™ (pegfilgrastim-cbqv) injection, for subcutaneous use |
|
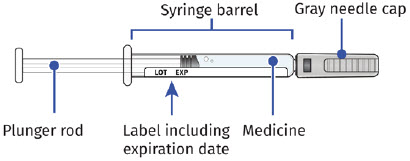
| Guide to Parts UDENYCA Prefilled Syringe |
|
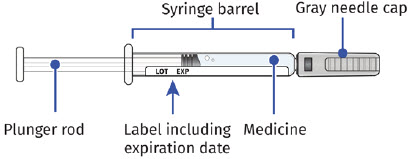 |
|
| On-body Injector for UDENYCA | |
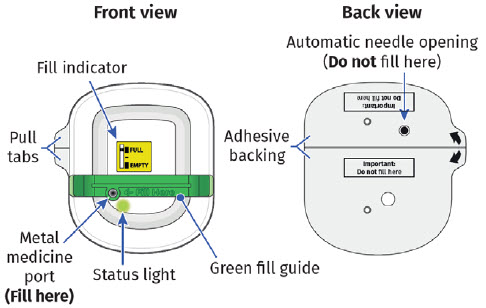 |
|
| Important READ THE FOLLOWING INSTRUCTIONS BEFORE USING UDENYCA ONBODY Prescribing Information
|
|
| Step 1: Prepare A| Remove ONBODY from the refrigerator and open carton. Place the syringe tray and on-body injector tray on a clean, flat, well-lit work surface. Allow to come to room temperature for 30 minutes prior to use. Do not warm the UDENYCA ONBODY components using a heat source. Check to make sure it contains:
Do not use the on-body injector:
 Choose the flattest site for the on-body injector application. Ask the patient about their ability to monitor and remove the on-body injector.
|
|
|  |
| Step 2: Get ready A| Open on-body injector tray and remove plastic lid. |
|
|
Wash your hands before use. Open the on-body injector tray by fully removing the tray cover and removing the plastic lid.
| 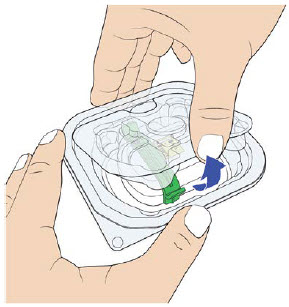 |
B| Remove UDENYCA prefilled syringe from tray.
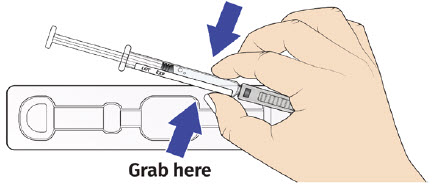 For safety reasons:
For safety reasons:
D| Remove air bubbles in prefilled syringe.  E| Orient the syringe with the needle facing downwards. Center the needle directly over the metal medicine port at a 90-degree angle. Insert the needle all the way into the port, avoiding sides. E| Orient the syringe with the needle facing downwards. Center the needle directly over the metal medicine port at a 90-degree angle. Insert the needle all the way into the port, avoiding sides. |
|
Insert needle into metal medicine port at a 90-degree angle only.
| 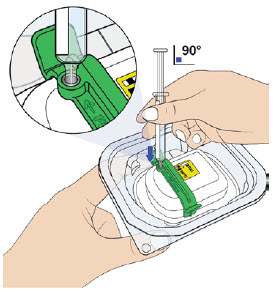 |
| F| Fully depress the plunger rod to expel entire contents of the prefilled syringe into the on-body injector. | |
|
After filling completely, you will hear two beeps. The status light will start to flash green. Remove the needle straight from the metal medicine port and dispose the empty syringe right away into a puncture proof container.
| 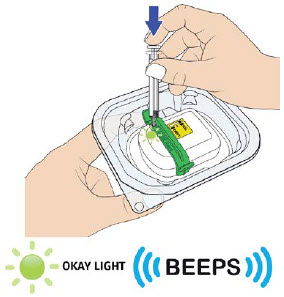 |
| G| Check to see if the on-body injector is full. | |
|
You should see the green status light flashing and a black line next to FULL on the fill indicator. If this is not the case, do not use. Start again with a new UDENYCA ONBODY, and call Coherus BioSciences at 1-800-4UDENYCA (1-800-483-3692). | 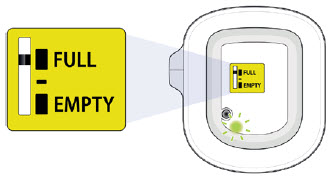 |
H| Remove on-body injector from tray, then firmly squeeze and lift the green fill guide away from the on-body injector.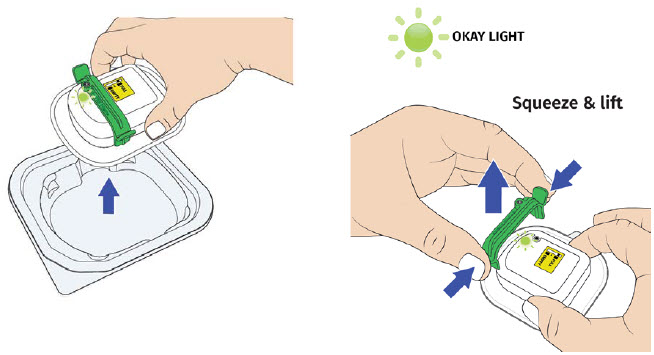 Dispose of green fill guide.
Dispose of green fill guide.A drop of medicine may be visible on the metal medicine port when the green fill guide is removed. |
|
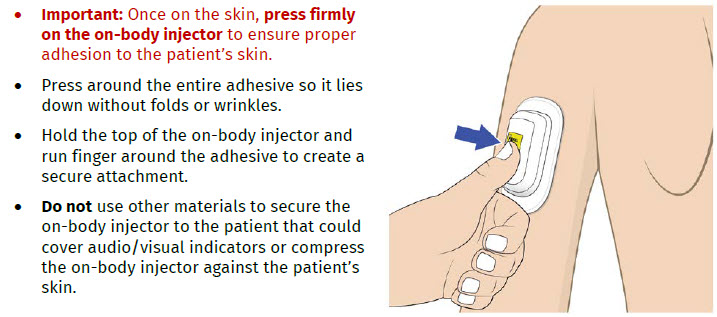
| Step 3: Apply A| Peel away both pull tabs to show the adhesive. Never touch hands or gloves to the adhesive. Make sure skin is dry prior to applying the on-body injector. 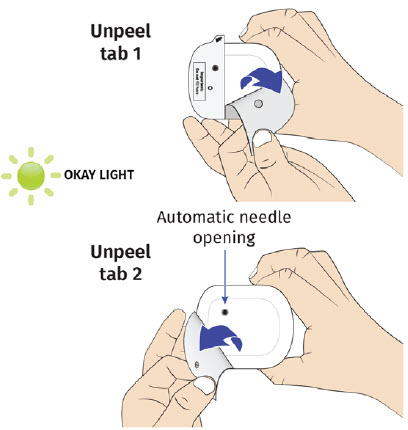
Call Coherus BioSciences at 1-800-4UDENYCA (1-800-483-3692). B| Securely apply the on-body injector without folding and without wrinkling to the cleaned injection site so it is visible and can be monitored by the patient or caregiver. |
|
|  |
 |
|
| Back of upper arm (triceps)
Vertical with the light facing down toward the elbow | 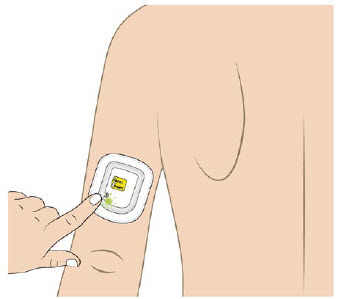 |
| Abdomen
Horizontal with the light facing up and visible to the patient | 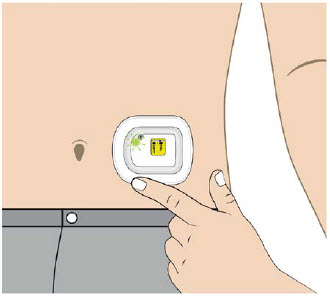 |
 If the adhesive is wrinkled or has folds anywhere that prevent the on-body injector from securely adhering, remove the on-body injector. Start again with a new ONBODY and call Coherus BioSciences at 1-800-4UDENYCA (1-800-483-3692). If the adhesive is wrinkled or has folds anywhere that prevent the on-body injector from securely adhering, remove the on-body injector. Start again with a new ONBODY and call Coherus BioSciences at 1-800-4UDENYCA (1-800-483-3692).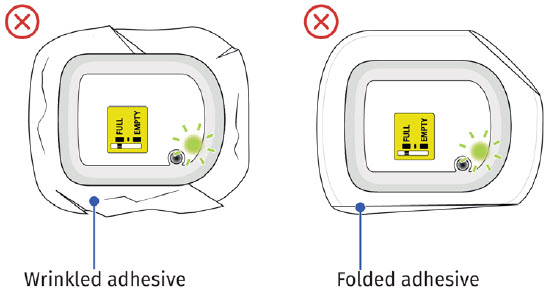 |
|
| Step 4: Finish A| Provide the Patient Instructions for Use Booklet for the patient to take home. Fill in the Dose Delivery information on the Patient Instructions for Use Booklet, and review the following instructions with your patient:
 If at any time the on-body injector beeps continuously for five minutes, and the status light is flashing red, take the on-body injector off of the patient.
If the patient reports the red status light is on, they may not have received the full dose. Schedule a follow-up appointment and report the incident to Coherus BioSciences at 1-800-4UDENYCA (1-800-483-3692). 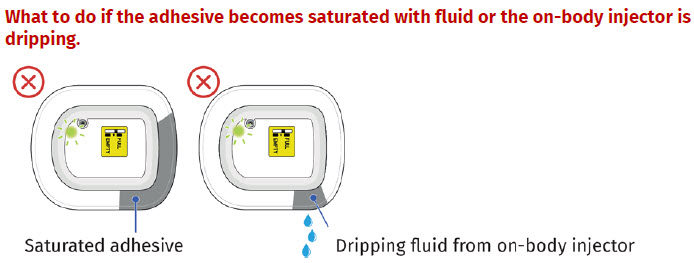 If the patient reports an on-body injector leak, they may not have received the full dose. Schedule a follow-up appointment and report the incident to Coherus BioSciences at 1-800-4UDENYCA (1-800-483-3692). |
|
| |
|
| Manufactured by:
Coherus BioSciences, Inc. Redwood City, CA 94065-1442 US License No. 2023 https://udenyca.com 1-800-4UDENYCA (1-800-483-3692) Issued: 12/2023 PMD-0214 Rev. 00 |
|
Electromagnetic Compatibility
The delivery system is designed to conform to the electromagnetic compatibility (EMC) standard IEC 60601-1-2:2020 and to operate accurately in conjunction with other medical equipment which also meets the requirements of this standard under home and hospital use environments.
To avoid electromagnetic interference (EMI) that may affect the performance of the delivery system [(i) Dose accuracy, (ii) treatment duration, (iii) Injection Depth, (iv) Visual and audible feedback], do not use the delivery system near sources of strong electric and magnetic interference (EMI), such as MRI, ionizing radiation, CT, diathermy, electromagnetic security systems (e.g., metal detectors), and large electric motors. In addition, portable and mobile RF communication equipment, such as RF emitters, cellular telephones, 2- way radios, BluetoothTM devices, and microwave ovens in close proximity to this device may affect the operation of the delivery system. Some of these EMI sources (mostly RF emitters) may not be visible and the device can potentially be exposed to fields from these EMI sources without the user’s awareness.
If you identify or suspect that external RF sources or other equipment are influencing delivery system operation (from known or unknown sources), try to (as applicable) increase the delivery system distance from the EMI source.
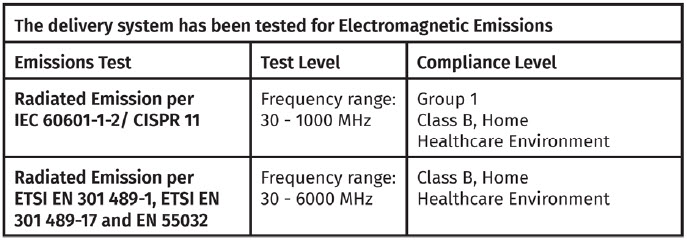
Electromagnetic Emissions
The delivery system has been tested and found to comply with the limits for a Class B digital device, pursuant to Part 15 of the FCC rules. These limits are designed to provide reasonable protection against harmful interference in a residential installation.
If this delivery system does cause harmful interference to radio or television reception, which can be determined by turning the radio or television off and on, the user is encouraged to try to correct the interference by one or more of the following measures: Reorient or relocate the receiving antenna. Increase the separation between the equipment and receiver. Connect the radio or television to an outlet on a circuit different from that to which the receiver is connected. Consult the dealer or an experienced radio/TV technician.
Electromagnetic Immunity
The delivery system has been tested to comply in either a Home Healthcare Environment or Professional Healthcare Environment.

Proximity fields from RF wireless communications equipment Immunity
The delivery system is tested per IEC 61000-4-3 at Frequencies and Levels as specified below to ensure Enclosure Port Immunity to RF wireless communications equipment.
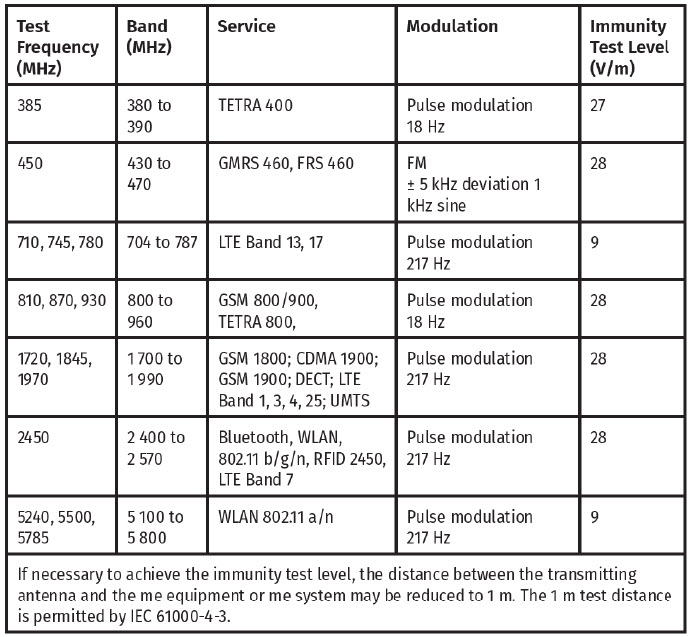
WARNING: Portable RF communications equipment (including peripherals such as antenna cables and external antennas) should be used no closer than 30 cm (12 inches) to any part of the Delivery System. Otherwise, degradation of the performance of this equipment could result.
WARNING: Use of this delivery system adjacent to other equipment should be avoided because it could result in improper operation. If such use is necessary, this equipment and the other equipment should be observed to verify that they are operating normally.
Symbols Glossary
| Description | Symbol/Sub clause |
|---|---|
| Name or trade mark of the product |  |
| Part number |  |
| Name or trade name and address of the manufacturer |  |
| Serial number or batch code, preceded by the word 'LOT', or the serial number/NDC number; |  |
| Warnings and/or precautions to take (in text format) |  |
| Storage temperature and any other special storage and/or handling conditions (for the combination) | 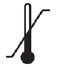 |
| Humidity limitation (for the combination) | 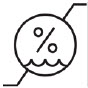 |
| Pressure limitation (for the combination) | 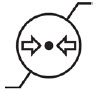 |
| Do not use if package is damaged | 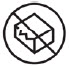 |
| Keep away from sunlight |  |
| Keep dry |  |
| For electrical devices - Water and dust ingress rating |  |
| For electrical devices – refer to instruction for use |  |
| "Rx only" labeling of prescription devices |  |
| Sterile & Sterilization by EO |  |
| Expiration date (Use by) |  |
| The device is for single use |  |
| Should not enter the MRI scanner room |  |
| Type BF applied parts |  |
| CSA Certificate |  |
| Battery specification Li-MnO2 Battery 3V/850mAH (CR14250) | N/A |
| WEEE directive compliance |  |
| UDENYCA® (pegfilgrastim-cbqv) prefilled syringe |  |
| On-body injector for UDENYCA® (pegfilgrastim-cbqv) |  |
| Patient INSTRUCTIONS FOR USE UDENYCA (yoo-den-i-kah) ONBODY™ (pegfilgrastim-cbqv) injection, for subcutaneous use |
|
| Dose Delivery Information | |
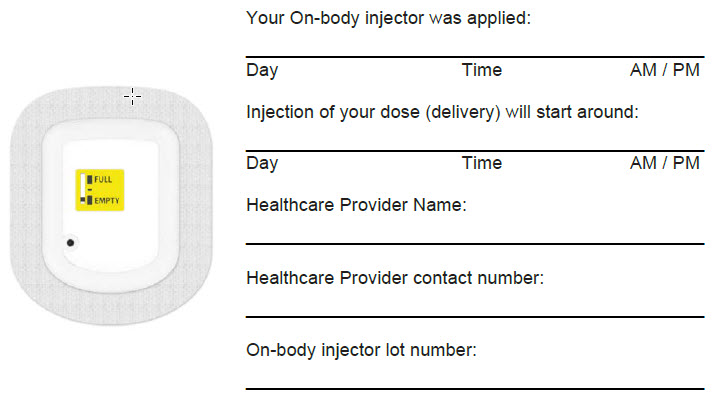 |
|
| Get to Know Your On-body Injector | |
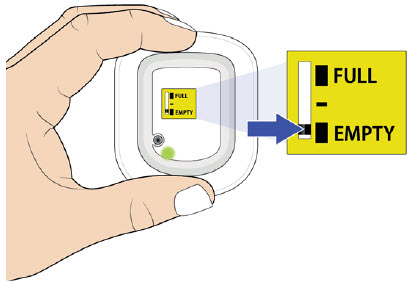
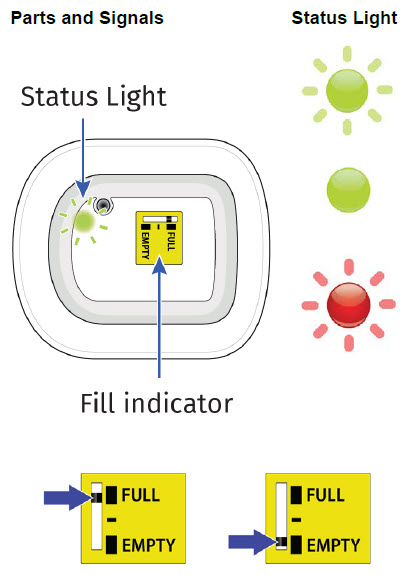 | Flashing Green: The on-body injector is working properly. Do not remove the on-body injector if the status light is flashing green. Solid Green (or off): Signals dose delivery is complete. Check to see if fill indicator reads empty. Flashing Red: On-body injector error. If you hear beeping at any time, check the status light. If it is flashing red, call your healthcare provider right away as you may need a replacement dose. Fill indicator: Black line shows how much UDENYCA is in the on-body injector. |
| Important Information On-body Injector for UDENYCA Description
|
|
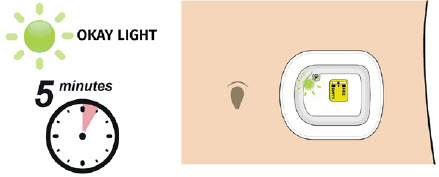
| Step 1: Monitor On-body Injector A| For the next 27 hours, occasionally check the status light for at least 10 seconds. If the status light is flashing green, it is okay. 
|
|
| Step 2: Observe Dose Delivery A| After about 27 hours, your on-body injector will begin to deliver your dose of UDENYCA.
|
|
| Step 3: Verify Dose Complete A| After the beep, check the color of the status light.  B| Grab the edge of the adhesive pad. Slowly peel off the on-body injector. B| Grab the edge of the adhesive pad. Slowly peel off the on-body injector.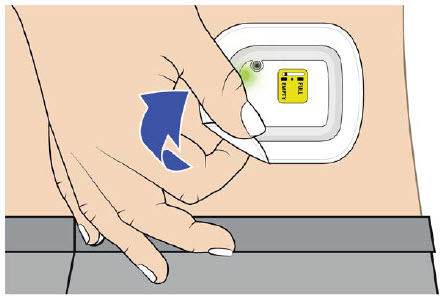
|
|
Step 4: Finish
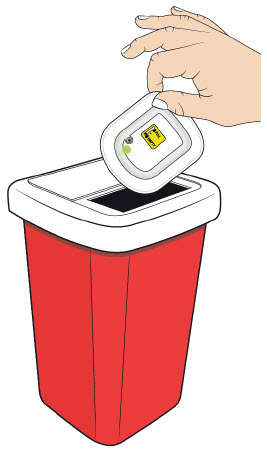
 B| Properly dispose of the on-body injector. B| Properly dispose of the on-body injector.
|
|
|  |
| Frequently Asked Questions How do I know it is safe to remove the on-body injector? 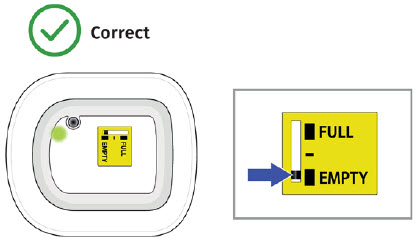 It is safe to remove the on-body injector after checking the following: It is safe to remove the on-body injector after checking the following:

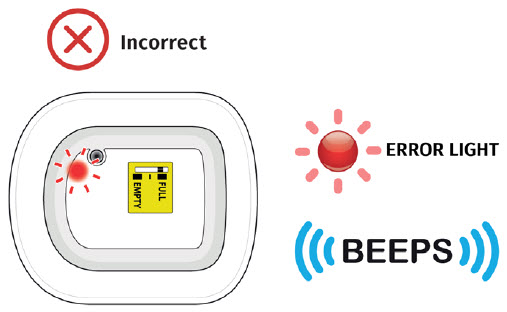  Call your healthcare provider right away if the on-body injector at any time comes away from your skin before your full dose delivery, as you may need a replacement dose.
Call your healthcare provider right away if the on-body injector at any time comes away from your skin before your full dose delivery, as you may need a replacement dose.Do not reapply it. Caution the needle may be exposed. Dispose of the on-body injector into a sharps disposal container right away.  If there is blood, press a clean cotton ball or gauze pad on the application site. Apply an adhesive bandage if needed.
If there is blood, press a clean cotton ball or gauze pad on the application site. Apply an adhesive bandage if needed.
 Call your healthcare provider right away if you experience persistent or worsening redness or tenderness at the application site, as this can be a sign of infection.
Call your healthcare provider right away if you experience persistent or worsening redness or tenderness at the application site, as this can be a sign of infection.
|
|
| |
|
| Manufactured by:
Coherus BioSciences, Inc. Redwood City, CA 94065-1442 US License No. 2023 https://udenyca.com 1-800-4UDENYCA (1-800-483-3692) Issued: 12/2023 PMD-0215 Rev. 00 |
|
Electromagnetic Compatibility
The delivery system is designed to conform to the electromagnetic compatibility (EMC) standard IEC 60601-1-2:2020 and to operate accurately in conjunction with other medical equipment which also meets the requirements of this standard under home and hospital use environments.
To avoid electromagnetic interference (EMI) that may affect the performance of the delivery system [(i) Dose accuracy, (ii) treatment duration, (iii) Injection Depth, (iv) Visual and audible feedback], do not use the delivery system near sources of strong electric and magnetic interference (EMI), such as MRI, ionizing radiation, CT, diathermy, electromagnetic security systems (e.g., metal detectors), and large electric motors. In addition, portable and mobile RF communication equipment, such as RF emitters, cellular telephones, 2- way radios, BluetoothTM devices, and microwave ovens in close proximity to this device may affect the operation of the delivery system. Some of these EMI sources (mostly RF emitters) may not be visible and the device can potentially be exposed to fields from these EMI sources without the user’s awareness.
If you identify or suspect that external RF sources or other equipment are influencing delivery system operation (from known or unknown sources), try to (as applicable) increase the delivery system distance from the EMI source.
Electromagnetic Emissions
The delivery system has been tested and found to comply with the limits for a Class B digital device, pursuant to Part 15 of the FCC rules. These limits are designed to provide reasonable protection against harmful interference in a residential installation.
If this delivery system does cause harmful interference to radio or television reception, which can be determined by turning the radio or television off and on, the user is encouraged to try to correct the interference by one or more of the following measures: Reorient or relocate the receiving antenna. Increase the separation between the equipment and receiver. Connect the radio or television to an outlet on a circuit different from that to which the receiver is connected. Consult the dealer or an experienced radio/TV technician.
Electromagnetic Immunity
The delivery system has been tested to comply in either a Home Healthcare Environment or Professional Healthcare Environment.
Proximity fields from RF wireless communications equipment Immunity
The delivery system is tested per IEC 61000-4-3 at Frequencies and Levels as specified below to ensure Enclosure Port Immunity to RF wireless communications equipment.
WARNING: Portable RF communications equipment (including peripherals such as antenna cables and external antennas) should be used no closer than 30 cm (12 inches) to any part of the Delivery System. Otherwise, degradation of the performance of this equipment could result.
WARNING: Use of this delivery system adjacent to other equipment should be avoided because it could result in improper operation. If such use is necessary, this equipment and the other equipment should be observed to verify that they are operating normally.
Symbols Glossary
| Description | Symbol/Sub clause |
|---|---|
| Name or trade mark of the product | |
| Part number | |
| Name or trade name and address of the manufacturer | |
| Serial number or batch code, preceded by the word ‘LOT’, or the serial number/NDC number; | |
| Warnings and/or precautions to take (in text format) | |
| Storage temperature and any other special storage and/or handling conditions (for the combination) | |
| Humidity limitation (for the combination) | |
| Pressure limitation (for the combination) | |
| Do not use if package is damaged | |
| Keep away from sunlight | |
| Keep dry | |
| For electrical devices - Water and dust ingress rating | |
| For electrical devices – refer to instruction for use | |
| "Rx only" labeling of prescription devices | |
| Sterile & Sterilization by EO | |
| Expiration date (Use by) | |
| The device is for single use | |
| Should not enter the MRI scanner room | |
| Type BF applied parts | |
| CSA Certificate | |
| Battery specification Li-MnO2 Battery 3V/850mAH (CR14250) | N/A |
| WEEE directive compliance | |
| UDENYCA® (pegfilgrastim-cbqv) prefilled syringe | |
| On-body injector for UDENYCA® (pegfilgrastim-cbqv) | |
Carton Label - One 6 mg/0.6 mL Single-Dose Prefilled Syringe - UDENYCA
PRINCIPAL DISPLAY PANEL
Coherus
BioSciences
6 mg/
0.6 mL
NDC 70114-101-01
UDENYCA®
pegfilgrastim-cbqv
Injection
Rx Only
6 mg in 0.6 mL Single-Dose Prefilled
Syringe
For Subcutaneous Use
Sterile Solution - No Preservative
Pegylated Recombinant
Methionyl Human Granulocyte
Colony-Stimulating Factor
(PEG-r-metHuG-CSF) derived
from E. coli
One 6 mg/0.6 mL single-dose
Prefilled Syringe

Carton Label - One 6 mg/0.6 mL Single-Dose Prefilled AUTOINJECTOR - UDENYCA
PRINCIPAL DISPLAY PANEL
NDC 70114-120-01
UDENYCA®
pegfilgrastim-cbqv
Injection
Rx Only
6 mg in 0.6 mL Single-Dose Prefilled AUTOINJECTOR
For Subcutaneous Use Sterile Solution - No Preservative
One 6 mg/0.6 mL Single-Dose Prefilled Autoinjector
Pegylated Recombinant Methionyl Human
Granulocyte Colony-Stimulating Factor
(PEG-r-metHuG-CSF) derived from E. coli
Coherus™
6 mg/0.6 mL

Carton Label - One 6 mg/0.6 mL Single-Dose Prefilled On-body Injector (OBI) - UDENYCA
PRINCIPAL DISPLAY PANEL
Coherus®
6 mg/
0.6 mL*
See package insert for full
Prescribing Information
and Instructions for Use.
Rx
Only
1 - Single-Dose UDENYCA Prefilled Syringe
1 - Sterile On-body Injector for UDENYCA
NDC 70114-130-01
UDENYCA® ONBODY™
(pegfilgrastim-cbqv)
Injection
Pegylated Recombinant Methionyl Human Granulocyte
Colony-Stimulating Factor (PEG-r-metHuG-CSF) derived from E. Coli
*Prefilled syringe contains 0.67 mL to deliver 6 mg/0.6 mL
when used with On-bodv Injector.
For Subcutaneous Use Only
Carton Contents (intended to be dispensed as a unit):
- 1 Sterile On-body Injector for UDENYCA
- 1 UDENYCA Prefilled Syringe Labeled for Use With
On-body Injector Only - 1 Prescribing Information and 1 Patient information
- 1 Patient Instructions for Use
- 1 Healthcare Provider Instructions for Use
-
Store refrigerated at 2°C to 8°C
(36°F to 46°F) - Keep in carton to protect from light
- Do not freeze. Do not shake
