FULL PRESCRIBING INFORMATION
WARNING: PERMANENT VISION LOSS
- SABRIL can cause permanent bilateral concentric visual field constriction, including tunnel vision that can result in disability. In some cases, SABRIL also can damage the central retina and may decrease visual acuity [see Warnings and Precautions (5.1)].
- The onset of vision loss from SABRIL is unpredictable, and can occur within weeks of starting treatment or sooner, or at any time after starting treatment, even after months or years.
- Symptoms of vision loss from SABRIL are unlikely to be recognized by patients or caregivers before vision loss is severe. Vision loss of milder severity, while often unrecognized by the patient or caregiver, can still adversely affect function.
- The risk of vision loss increases with increasing dose and cumulative exposure, but there is no dose or exposure known to be free of risk of vision loss.
- Vision assessment is recommended at baseline (no later than 4 weeks after starting SABRIL), at least every 3 months during therapy, and about 3 to 6 months after the discontinuation of therapy.
- Once detected, vision loss due to SABRIL is not reversible. It is expected that, even with frequent monitoring, some patients will develop severe vision loss.
- Consider drug discontinuation, balancing benefit and risk, if visual loss is documented.
- Risk of new or worsening vision loss continues as long as SABRIL is used. It is possible that vision loss can worsen despite discontinuation of SABRIL.
- Because of the risk of visual loss, SABRIL should be withdrawn from patients with refractory complex partial seizures who fail to show substantial clinical benefit within 3 months of initiation and within 2-4 weeks of initiation for patients with infantile spasms, or sooner if treatment failure becomes obvious. Patient response to and continued need for SABRIL should be periodically reassessed.
- SABRIL should not be used in patients with, or at high risk of, other types of irreversible vision loss unless the benefits of treatment clearly outweigh the risks.
- SABRIL should not be used with other drugs associated with serious adverse ophthalmic effects such as retinopathy or glaucoma unless the benefits clearly outweigh the risks.
- Use the lowest dosage and shortest exposure to SABRIL consistent with clinical objectives [see Dosage and Administration (2.1)].
Because of the risk of permanent vision loss, SABRIL is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the Vigabatrin REMS Program [see Warnings and Precautions (5.2)]. Further information is available at www.vigabatrinREMS.com or 1-866-244-8175.
1 INDICATIONS AND USAGE
1.1 Refractory Complex Partial Seizures (CPS)
SABRIL is indicated as adjunctive therapy for adults and pediatric patients 2 years of age and older with refractory complex partial seizures who have inadequately responded to several alternative treatments and for whom the potential benefits outweigh the risk of vision loss [see Warnings and Precautions (5.1)]. SABRIL is not indicated as a first line agent for complex partial seizures.
1.2 Infantile Spasms (IS)
SABRIL is indicated as monotherapy for pediatric patients with infantile spasms 1 month to 2 years of age for whom the potential benefits outweigh the potential risk of vision loss [see Warnings and Precautions (5.1)].
2 DOSAGE AND ADMINISTRATION
2.1 Important Dosing and Administration Instructions
Dosing
Use the lowest dosage and shortest exposure to SABRIL consistent with clinical objectives [see Warnings and Precautions (5.1)].
The SABRIL dosing regimen depends on the indication, age group, weight, and dosage form (tablets or for oral solution) [see Dosage and Administration (2.2, 2.3)]. Patients with impaired renal function require dose adjustment [see Dosage and Administration (2.4)].
Monitoring of SABRIL plasma concentrations to optimize therapy is not helpful.
Administration
SABRIL is given orally with or without food.
SABRIL for oral solution should be mixed with water prior to administration [see Dosage and Administration (2.5)]. A calibrated measuring device is recommended to measure and deliver the prescribed dose accurately. A household teaspoon or tablespoon is not an adequate measuring device.
If a decision is made to discontinue SABRIL, the dose should be gradually reduced [see Dosage and Administration (2.2, 2.3) and Warnings and Precautions (5.6)].
2.2 Refractory Complex Partial Seizures
Adults (Patients 17 Years of Age and Older)
Treatment should be initiated at 1000 mg/day (500 mg twice daily). Total daily dose may be increased in 500 mg increments at weekly intervals depending on response. The recommended dose of SABRIL in adults is 3000 mg/day (1500 mg twice daily). A 6000 mg/day dose has not been shown to confer additional benefit compared to the 3000 mg/day dose and is associated with an increased incidence of adverse events.
In controlled clinical studies in adults with complex partial seizures, SABRIL was tapered by decreasing the daily dose 1000 mg/day on a weekly basis until discontinued [see Warnings and Precautions (5.6)].
Pediatric (Patients 2 to 16 Years of Age)
The recommended dosage is based on body weight and administered as two divided doses, as shown in Table 1. The dosage may be increased in weekly intervals to the total daily maintenance dosage, depending on response.
Pediatric patients weighing more than 60 kg should be dosed according to adult recommendations.
|
Table 1. CPS Dosing Recommendations for Pediatric Patients Weighing 10 kg up to 60 kg†† |
||||
|
Body Weight [kg] |
Total Daily* Starting Dose [mg/day] |
Total Daily* Maintenance Dose† [mg/day] | ||
|
10 kg to 15 kg |
350 mg |
1050 mg | ||
|
Greater than 15 kg to 20 kg |
450 mg |
1300 mg | ||
|
Greater than 20 kg to 25 kg |
500 mg |
1500 mg | ||
|
Greater than 25 kg to 60 kg |
500 mg |
2000 mg | ||
|
* Administered in two divided doses † Maintenance dose is based on 3000 mg/day adult-equivalent dose †† Patients weighing more than 60 kg should be dosed according to adult recommendations | ||||
In patients with refractory complex partial seizures, SABRIL should be withdrawn if a substantial clinical benefit is not observed within 3 months of initiating treatment. If, in the clinical judgment of the prescriber, evidence of treatment failure becomes obvious earlier than 3 months, treatment should be discontinued at that time [see Warnings and Precautions (5.1)].
In a controlled study in pediatric patients with complex partial seizures, SABRIL was tapered by decreasing the daily dose by one third every week for three weeks [see Warnings and Precautions (5.6)].
2.3 Infantile Spasms
The initial daily dosing is 50 mg/kg/day given in two divided doses (25 mg/kg twice daily); subsequent dosing can be titrated by 25 mg/kg/day to 50 mg/kg/day increments every 3 days, up to a maximum of 150 mg/kg/day given in 2 divided doses (75 mg/kg twice daily) [see Use in Specific Populations (8.4)].
Table 2 provides the volume of the 50 mg/mL dosing solution that should be administered as individual doses in infants of various weights.
| Table 2. Infant Dosing Table | ||||
| Weight [kg] | Starting Dose 50 mg/kg/day | Maximum Dose 150 mg/kg/day |
||
| 3 | 1.5 mL twice daily | 4.5 mL twice daily | ||
| 4 | 2 mL twice daily | 6 mL twice daily | ||
| 5 | 2.5 mL twice daily | 7.5 mL twice daily | ||
| 6 | 3 mL twice daily | 9 mL twice daily | ||
| 7 | 3.5 mL twice daily | 10.5 mL twice daily | ||
| 8 | 4 mL twice daily | 12 mL twice daily | ||
| 9 | 4.5 mL twice daily | 13.5 mL twice daily | ||
| 10 | 5 mL twice daily | 15 mL twice daily | ||
| 11 | 5.5 mL twice daily | 16.5 mL twice daily | ||
| 12 | 6 mL twice daily | 18 mL twice daily | ||
| 13 | 6.5 mL twice daily | 19.5 mL twice daily | ||
| 14 | 7 mL twice daily | 21 mL twice daily | ||
| 15 | 7.5 mL twice daily | 22.5 mL twice daily | ||
| 16 | 8 mL twice daily | 24 mL twice daily | ||
In patients with infantile spasms, SABRIL should be withdrawn if a substantial clinical benefit is not observed within 2 to 4 weeks. If, in the clinical judgment of the prescriber, evidence of treatment failure becomes obvious earlier than 2 to 4 weeks, treatment should be discontinued at that time [see Warnings and Precautions (5.1)].
In a controlled clinical study in patients with infantile spasms, SABRIL was tapered by decreasing the daily dose at a rate of 25 mg/kg to 50 mg/kg every 3 to 4 days [see Warnings and Precautions (5.6)].
2.4 Patients with Renal Impairment
SABRIL is primarily eliminated through the kidney.
Infants
Information about how to adjust the dose in infants with renal impairment is unavailable.
Adult and pediatric patients 2 years and older
- Mild renal impairment (CLcr >50 to 80 mL/min): dose should be decreased by 25%
- Moderate renal impairment (CLcr >30 to 50 mL/min): dose should be decreased by 50%
- Severe renal impairment (CLcr >10 to 30 mL/min): dose should be decreased by 75%
CLcr in mL/min may be estimated from serum creatinine (mg/dL) using the following formulas:
- Patients 2 to <12 years old: CLcr (mL/min/1.73 m2) = (K × Ht) / Scr
height (Ht) in cm; serum creatinine (Scr) in mg/dL
K (proportionality constant): Female Child (<12 years): K=0.55;
Male Child (<12 years): K=0.70
- Adult and pediatric patients 12 years or older: CLcr (mL/min) = [140-age (years)] × weight (kg) / [72 × serum creatinine (mg/dL)] (× 0.85 for female patients)
The effect of dialysis on SABRIL clearance has not been adequately studied [see Clinical Pharmacology (12.3) and Use in Specific Populations (8.6)].
2.5 Preparation and Administration Instructions for SABRIL for oral solution
If using SABRIL for oral solution, physicians should review and discuss the Medication Guide and instructions for mixing and giving SABRIL with the patient or caregiver(s). Physicians should confirm that patients or caregiver(s) understand how to mix SABRIL powder with water and administer the correct daily dose.
Empty the entire contents of each 500 mg packet into a clean cup, and dissolve in 10 mL of cold or room temperature water per packet. Administer the resulting solution using the 3 mL or 10 mL oral syringe provided by the pharmacy [see How Supplied/Storage and Handling (16.1)]. The concentration of the final solution is 50 mg/mL.
Table 3 below describes how many packets and how many milliliters (mL) of water will be needed to prepare each individual dose. The concentration after reconstitution is 50 mg/mL.
| Table 3. Number of SABRIL Packets and mL of Water Needed for Each Individual Dose | ||
|
Individual Dose [mg] [Given Twice Daily] |
Total Number of SABRIL Packets |
Total mL of Water Required for Dissolving |
| 0 to 500 | 1 Packet | 10 mL |
| 501 to 1000 | 2 Packets | 20 mL |
| 1001 to 1500 | 3 Packets | 30 mL |
Discard the resulting solution if it is not clear (or free of particles) and colorless. Each individual dose should be prepared and used immediately. Discard any unused portion of the solution after administering the correct dose.
3 DOSAGE FORMS AND STRENGTHS
Tablet: 500 mg: white, oval, film-coated, biconvex, scored on one side, and debossed with OV 111 on the other.
For oral solution: 500 mg packet containing a white to off-white granular powder.
5 WARNINGS AND PRECAUTIONS
5.1 Permanent Vision Loss
SABRIL can cause permanent vision loss. Because of this risk and because, when it is effective, SABRIL provides an observable symptomatic benefit; patient response and continued need for treatment should be periodically assessed.
Based upon adult studies, 30 percent or more of patients can be affected with bilateral concentric visual field constriction ranging in severity from mild to severe. Severe cases may be characterized by tunnel vision to within 10 degrees of visual fixation, which can result in disability. In some cases, SABRIL also can damage the central retina and may decrease visual acuity. Symptoms of vision loss from SABRIL are unlikely to be recognized by patients or caregivers before vision loss is severe. Vision loss of milder severity, while often unrecognized by the patient or caregiver, can still adversely affect function.
Because assessing vision may be difficult in infants and children, the frequency and extent of vision loss is poorly characterized in these patients. For this reason, the understanding of the risk is primarily based on the adult experience. The possibility that vision loss from SABRIL may be more common, more severe, or have more severe functional consequences in infants and children than in adults cannot be excluded.
The onset of vision loss from SABRIL is unpredictable and can occur within weeks of starting treatment or sooner, or at any time after starting treatment, even after months or years.
The risk of vision loss increases with increasing dose and cumulative exposure, but there is no dose or exposure known to be free of risk of vision loss.
In patients with refractory complex partial seizures, SABRIL should be withdrawn if a substantial clinical benefit is not observed within 3 months of initiating treatment. If, in the clinical judgment of the prescriber, evidence of treatment failure becomes obvious earlier than 3 months, treatment should be discontinued at that time [see Dosage and Administration (2.2) and Warnings and Precautions (5.6)].
In patients with infantile spasms, SABRIL should be withdrawn if a substantial clinical benefit is not observed within 2 to 4 weeks. If, in the clinical judgment of the prescriber, evidence of treatment failure becomes obvious earlier than 2 to 4 weeks, treatment should be discontinued at that time [see Dosage and Administration (2.3) and Warnings and Precautions (5.6)].
SABRIL should not be used in patients with, or at high risk of, other types of irreversible vision loss unless the benefits of treatment clearly outweigh the risks. The interaction of other types of irreversible vision damage with vision damage from SABRIL has not been well-characterized, but is likely adverse.
SABRIL should not be used with other drugs associated with serious adverse ophthalmic effects such as retinopathy or glaucoma unless the benefits clearly outweigh the risks.
Monitoring of Vision
Monitoring of vision by an ophthalmic professional with expertise in visual field interpretation and the ability to perform dilated indirect ophthalmoscopy of the retina is recommended [see Warnings and Precautions (5.2)]. Because vision testing in infants is difficult, vision loss may not be detected until it is severe. For patients receiving SABRIL, vision assessment is recommended at baseline (no later than 4 weeks after starting SABRIL), at least every 3 months while on therapy, and about 3-6 months after the discontinuation of therapy. The diagnostic approach should be individualized for the patient and clinical situation.
In adults and cooperative pediatric patients, perimetry is recommended, preferably by automated threshold visual field testing. Additional testing may also include electrophysiology (e.g., electroretinography [ERG]), retinal imaging (e.g., optical coherence tomography [OCT]), and/or other methods appropriate for the patient. In patients who cannot be tested, treatment may continue according to clinical judgment, with appropriate patient counseling. Because of variability, results from ophthalmic monitoring must be interpreted with caution, and repeat assessment is recommended if results are abnormal or uninterpretable. Repeat assessment in the first few weeks of treatment is recommended to establish if, and to what degree, reproducible results can be obtained, and to guide selection of appropriate ongoing monitoring for the patient.
The onset and progression of vision loss from SABRIL is unpredictable, and it may occur or worsen precipitously between assessments. Once detected, vision loss due to SABRIL is not reversible. It is expected that even with frequent monitoring, some SABRIL patients will develop severe vision loss. Consider drug discontinuation, balancing benefit and risk, if vision loss is documented. It is possible that vision loss can worsen despite discontinuation of SABRIL.
5.2 Vigabatrin REMS Program
SABRIL is available only through a restricted distribution program called the Vigabatrin REMS Program, because of the risk of permanent vision loss.
Notable requirements of the Vigabatrin REMS Program include the following:
-
Prescribers must be certified by enrolling in the program, agreeing to counsel patients on the risk of vision loss and the need for periodic monitoring of vision, and reporting any event suggestive of vision loss to Lundbeck.
-
Patients must enroll in the program.
-
Pharmacies must be certified and must only dispense to patients authorized to receive SABRIL.
Further information is available at www.vigabatrinREMS.com, or call 1-866-244-8175.
5.3 Magnetic Resonance Imaging (MRI) Abnormalities in Infants
Abnormal MRI signal changes characterized by increased T2 signal and restricted diffusion in a symmetric pattern involving the thalamus, basal ganglia, brain stem, and cerebellum have been observed in some infants treated with vigabatrin.
In a retrospective epidemiologic study in infants with infantile spasms (N=205), the prevalence of MRI changes was 22% in vigabatrin-treated patients versus 4% in patients treated with other therapies. In this study, in post marketing experience, and in published literature reports, these changes generally resolved with discontinuation of treatment. In a few patients, the lesion resolved despite continued use. It has been reported that some infants exhibited coincident motor abnormalities, but no causal relationship has been established and the potential for long-term clinical sequelae has not been adequately studied.
Neurotoxicity (brain histopathology and neurobehavioral abnormalities) was observed in rats exposed to vigabatrin during late gestation and the neonatal and juvenile periods of development, and brain histopathological changes were observed in dogs exposed to vigabatrin during the juvenile period of development. The relationship between these findings and the abnormal MRI findings in infants treated with vigabatrin for infantile spasms is unknown [see Warnings and Precautions (5.4) and Use in Specific Populations (8.1)].
The specific pattern of signal changes observed in patients 6 years and younger was not observed in older pediatric and adult patients treated with vigabatrin. In a blinded review of MRI images obtained in prospective clinical trials in patients with refractory complex partial seizures (CPS) 3 years and older (N=656), no difference was observed in anatomic distribution or prevalence of MRI signal changes between vigabatrin treated and placebo treated patients. In the postmarketing setting, MRI changes have also been reported in patients 6 years of age and younger being treated for refractory CPS.
For adults treated with SABRIL, routine MRI surveillance is unnecessary as there is no evidence that vigabatrin causes MRI changes in this population.
5.4 Neurotoxicity
Intramyelinic Edema (IME) has been reported in postmortem examination of infants being treated for infantile spasms with vigabatrin.
Abnormal MRI signal changes characterized by increased T2 signal and restricted diffusion in a symmetric pattern involving the thalamus, basal ganglia, brain stem, and cerebellum have also been observed in some infants treated for IS with vigabatrin. Studies of the effects of vigabatrin on MRI and evoked potentials (EP) in adult epilepsy patients have demonstrated no clear-cut abnormalities [see Warnings and Precautions (5.3)].
Vacuolation, characterized by fluid accumulation and separation of the outer layers of myelin, has been observed in brain white matter tracts in adult and juvenile rats and adult mice, dogs, and possibly monkeys following administration of vigabatrin. This lesion, referred to as intramyelinic edema (IME), was seen in animals at doses within the human therapeutic range. A no-effect dose was not established in rodents or dogs. In the rat and dog, vacuolation was reversible following discontinuation of vigabatrin treatment, but, in the rat, pathologic changes consisting of swollen or degenerating axons, mineralization, and gliosis were seen in brain areas in which vacuolation had been previously observed. Vacuolation in adult animals was correlated with alterations in MRI and changes in visual and somatosensory EP.
Administration of vigabatrin to rats during the neonatal and juvenile periods of development produced vacuolar changes in the brain gray matter (including the thalamus, midbrain, deep cerebellar nuclei, substantia nigra, hippocampus, and forebrain) which are considered distinct from the IME observed in vigabatrin treated adult animals. Decreased myelination and evidence of oligodendrocyte injury were additional findings in the brains of vigabatrin-treated rats. An increase in apoptosis was seen in some brain regions following vigabatrin exposure during the early postnatal period. Long-term neurobehavioral abnormalities (convulsions, neuromotor impairment, learning deficits) were also observed following vigabatrin treatment of young rats. Administration of vigabatrin to juvenile dogs produced vacuolar changes in the brain gray matter (including the septal nuclei, hippocampus, hypothalamus, thalamus, cerebellum, and globus pallidus). Neurobehavioral effects of vigabatrin were not assessed in the juvenile dog. These effects in young animals occurred at doses lower than those producing neurotoxicity in adult animals and were associated with plasma vigabatrin levels substantially lower than those achieved clinically in infants and children [see Use in Specific Populations (8.1, 8.4)].
In a published study, vigabatrin (200, 400 mg/kg/day) induced apoptotic neurodegeneration in the brain of young rats when administered by intraperitoneal injection on postnatal days 5-7.
Administration of vigabatrin to female rats during pregnancy and lactation at doses below those used clinically resulted in hippocampal vacuolation and convulsions in the mature offspring.
5.5 Suicidal Behavior and Ideation
Antiepileptic drugs (AEDs), including SABRIL, increase the risk of suicidal thoughts or behavior in patients taking these drugs for any indication. Patients treated with any AED for any indication should be monitored for the emergence or worsening of depression, suicidal thoughts or behavior, and/or any unusual changes in mood or behavior.
Pooled analyses of 199 placebo-controlled clinical trials (mono- and adjunctive therapy) of 11 different AEDs showed that patients randomized to one of the AEDs had approximately twice the risk (adjusted Relative Risk 1.8, 95% CI: 1.2, 2.7) of suicidal thinking or behavior compared to patients randomized to placebo. In these trials, which had a median treatment duration of 12 weeks, the estimated incidence rate of suicidal behavior or ideation among 27,863 AED treated patients was 0.43%, compared to 0.24% among 16,029 placebo-treated patients, representing an increase of approximately one case of suicidal thinking or behavior for every 530 patients treated. There were four suicides in drug treated patients in the trials and none in placebo treated patients, but the number is too small to allow any conclusion about drug effect on suicide.
The increased risk of suicidal thoughts or behavior with AEDs was observed as early as one week after starting drug treatment with AEDs and persisted for the duration of treatment assessed. Because most trials included in the analysis did not extend beyond 24 weeks, the risk of suicidal thoughts or behavior beyond 24 weeks could not be assessed.
The risk of suicidal thoughts or behavior was generally consistent among drugs in the data analyzed. The finding of increased risk with AEDs of varying mechanisms of action and across a range of indications suggests that the risk applies to all AEDs used for any indication. The risk did not vary substantially by age (5-100 years) in the clinical trials analyzed. Table 4 shows absolute and relative risk by indication for all evaluated AEDs.
| Indication | Placebo Patients with Events per 1000 Patients | Drug Patients with Events per 1000 Patients |
Relative Risk: Incidence of Drug Events in Drug Patients/Incidence in Placebo Patients |
Risk Difference: |
| Epilepsy | 1.0 | 3.4 | 3.5 | 2.4 |
| Psychiatric | 5.7 | 8.5 | 1.5 | 2.9 |
| Other | 1.0 | 1.8 | 1.9 | 0.9 |
| Total | 2.4 | 4.3 | 1.8 | 1.9 |
The relative risk for suicidal thoughts or behavior was higher in clinical trials for epilepsy than in clinical trials for psychiatric or other conditions, but the absolute risk differences were similar for the epilepsy and psychiatric indications.
Anyone considering prescribing SABRIL or any other AED must balance the risk of suicidal thoughts or behavior with the risk of untreated illness. Epilepsy and many other illnesses for which AEDs are prescribed are themselves associated with morbidity and mortality and an increased risk of suicidal thoughts and behavior. Should suicidal thoughts and behavior emerge during treatment, the prescriber needs to consider whether the emergence of these symptoms in any given patient may be related to the illness being treated.
Patients, their caregivers, and families should be informed that AEDs increase the risk of suicidal thoughts and behavior and should be advised of the need to be alert for the emergence or worsening of the signs and symptoms of depression, any unusual changes in mood or behavior, or the emergence of suicidal thoughts, behavior, or thoughts about self-harm. Behaviors of concern should be reported immediately to healthcare providers.
5.6 Withdrawal of Antiepileptic Drugs (AEDs)
As with all AEDs, SABRIL should be withdrawn gradually. However, if withdrawal is needed because of a serious adverse event, rapid discontinuation can be considered. Patients and caregivers should be told not to suddenly discontinue SABRIL therapy.
In controlled clinical studies in adults with complex partial seizures, SABRIL was tapered by decreasing the daily dose 1000 mg/day on a weekly basis until discontinued.
In a controlled study in pediatric patients with complex partial seizures, SABRIL was tapered by decreasing the daily dose by one third every week for three weeks.
In a controlled clinical study in patients with infantile spasms, SABRIL was tapered by decreasing the daily dose at a rate of 25-50 mg/kg every 3-4 days.
5.7 Anemia
In North American controlled trials in adults, 6% of patients (16/280) receiving SABRIL and 2% of patients (3/188) receiving placebo had adverse events of anemia and/or met criteria for potentially clinically important hematology changes involving hemoglobin, hematocrit, and/or RBC indices. Across U.S. controlled trials, there were mean decreases in hemoglobin of about 3% and 0% in SABRIL and placebo-treated patients, respectively, and a mean decrease in hematocrit of about 1% in SABRIL-treated patients compared to a mean gain of about 1% in patients treated with placebo.
In controlled and open-label epilepsy trials in adults and pediatric patients, 3 SABRIL patients (0.06%, 3/4855) discontinued for anemia and 2 SABRIL patients experienced unexplained declines in hemoglobin to below 8 g/dL and/or hematocrit below 24%.
5.8 Somnolence and Fatigue
SABRIL causes somnolence and fatigue. Patients should be advised not to drive a car or operate other complex machinery until they are familiar with the effects of SABRIL on their ability to perform such activities.
Pooled data from two SABRIL controlled trials in adults demonstrated that 24% (54/222) of SABRIL patients experienced somnolence compared to 10% (14/135) of placebo patients. In those same studies, 28% of SABRIL patients experienced fatigue compared to 15% (20/135) of placebo patients. Almost 1% of SABRIL patients discontinued from clinical trials for somnolence and almost 1% discontinued for fatigue.
Pooled data from three SABRIL controlled trials in pediatric patients demonstrated that 6% (10/165) of SABRIL patients experienced somnolence compared to 5% (5/104) of placebo patients. In those same studies, 10% (17/165) of SABRIL patients experienced fatigue compared to 7% (7/104) of placebo patients. No SABRIL patients discontinued from clinical trials due to somnolence or fatigue.
5.9 Peripheral Neuropathy
SABRIL causes symptoms of peripheral neuropathy in adults. Pediatric clinical trials were not designed to assess symptoms of peripheral neuropathy, but observed incidence of symptoms based on pooled data from controlled pediatric studies appeared similar for pediatric patients on vigabatrin and placebo. In a pool of North American controlled and uncontrolled epilepsy studies, 4.2% (19/457) of SABRIL patients developed signs and/or symptoms of peripheral neuropathy. In the subset of North American placebo-controlled epilepsy trials, 1.4% (4/280) of SABRIL treated patients and no (0/188) placebo patients developed signs and/or symptoms of peripheral neuropathy. Initial manifestations of peripheral neuropathy in these trials included, in some combination, symptoms of numbness or tingling in the toes or feet, signs of reduced distal lower limb vibration or position sensation, or progressive loss of reflexes, starting at the ankles. Clinical studies in the development program were not designed to investigate peripheral neuropathy systematically and did not include nerve conduction studies, quantitative sensory testing, or skin or nerve biopsy. There is insufficient evidence to determine if development of these signs and symptoms was related to duration of SABRIL treatment, cumulative dose, or if the findings of peripheral neuropathy were completely reversible upon discontinuation of SABRIL.
5.10 Weight Gain
SABRIL causes weight gain in adult and pediatric patients.
Data pooled from randomized controlled trials in adults found that 17% (77/443) of SABRIL patients versus 8% (22/275) of placebo patients gained ≥7% of baseline body weight. In these same trials, the mean weight change among SABRIL patients was 3.5 kg compared to 1.6 kg for placebo patients.
Data pooled from randomized controlled trials in pediatric patients with refractory complex partial seizures found that 47% (77/163) of SABRIL patients versus 19% (19/102) of placebo patients gained ≥7% of baseline body weight.
In all epilepsy trials, 0.6% (31/4855) of SABRIL patients discontinued for weight gain. The long term effects of SABRIL related weight gain are not known. Weight gain was not related to the occurrence of edema.
5.11 Edema
SABRIL causes edema in adults. Pediatric clinical trials were not designed to assess edema, but observed incidence of edema-based pooled data from controlled pediatric studies appeared similar for pediatric patients on vigabatrin and placebo.
Pooled data from controlled trials demonstrated increased risk among SABRIL patients compared to placebo patients for peripheral edema (SABRIL 2%, placebo 1%), and edema (SABRIL 1%, placebo 0%). In these studies, one SABRIL and no placebo patients discontinued for an edema related AE. In adults, there was no apparent association between edema and cardiovascular adverse events such as hypertension or congestive heart failure. Edema was not associated with laboratory changes suggestive of deterioration in renal or hepatic function.
6 ADVERSE REACTIONS
The following serious and otherwise important adverse reactions are described elsewhere in labeling:
- Permanent Vision Loss [see BOXED WARNING and Warnings and Precautions (5.1)]
- Magnetic Resonance Imaging (MRI) Abnormalities in Infants [see Warnings and Precautions (5.3)]
- Neurotoxicity [see Warnings and Precautions (5.4)]
- Suicidal Behavior and Ideation [see Warnings and Precautions (5.5)]
- Withdrawal of Antiepileptic Drugs (AEDs) [see Warnings and Precautions (5.6)]
- Anemia [see Warnings and Precautions (5.7)]
- Somnolence and Fatigue [see Warnings and Precautions (5.8)]
- Peripheral Neuropathy [see Warnings and Precautions (5.9)]
- Weight Gain [see Warnings and Precautions (5.10)]
- Edema [see Warnings and Precautions (5.11)]
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In U.S. and primary non-U.S. clinical studies of 4,079 SABRIL-treated patients, the most common (≥5%) adverse reactions associated with the use of SABRIL in combination with other AEDs were headache, somnolence, fatigue, dizziness, convulsion, nasopharyngitis, weight gain, upper respiratory tract infection, visual field defect, depression, tremor, nystagmus, nausea, diarrhea, memory impairment, insomnia, irritability, abnormal coordination, blurred vision, diplopia, vomiting, influenza, pyrexia, and rash.
The adverse reactions most commonly associated with SABRIL treatment discontinuation in ≥1% of patients were convulsion and depression.
In patients with infantile spasms, the adverse reactions most commonly associated with SABRIL treatment discontinuation in ≥1% of patients were infections, status epilepticus, developmental coordination disorder, dystonia, hypotonia, hypertonia, weight gain, and insomnia.
Refractory Complex Partial Seizures
Adults
Table 5 lists the adverse reactions that occurred in ≥2% and more than one patient per SABRIL-treated group and that occurred more frequently than in placebo patients from 2 U.S. adjunctive clinical studies of refractory CPS in adults.
| Table 5. Adverse Reactions in Pooled, Adjunctive Trials in Adults with Refractory Complex Partial Seizures | |||
| SABRIL dosage (mg/day) | |||
| Body System
Adverse Reaction | 3000 [N=134] % | 6000 [N=43] % | Placebo [N=135] % |
| Ear Disorders | |||
| Tinnitus | 2 | 0 | 1 |
| Vertigo | 2 | 5 | 1 |
| Eye Disorders | |||
| Blurred vision | 13 | 16 | 5 |
| Diplopia | 7 | 16 | 3 |
| Asthenopia | 2 | 2 | 0 |
| Eye pain | 0 | 5 | 0 |
| Gastrointestinal Disorders | |||
| Diarrhea | 10 | 16 | 7 |
| Nausea | 10 | 2 | 8 |
| Vomiting | 7 | 9 | 6 |
| Constipation | 8 | 5 | 3 |
| Upper abdominal pain | 5 | 5 | 1 |
| Dyspepsia | 4 | 5 | 3 |
| Stomach discomfort | 4 | 2 | 1 |
| Abdominal pain | 3 | 2 | 1 |
| Toothache | 2 | 5 | 2 |
| Abdominal distension | 2 | 0 | 1 |
| General Disorders | |||
| Fatigue | 23 | 40 | 16 |
| Gait disturbance | 6 | 12 | 7 |
| Asthenia | 5 | 7 | 1 |
| Edema peripheral | 5 | 7 | 1 |
| Fever | 4 | 7 | 3 |
| Chest pain | 1 | 5 | 1 |
| Thirst | 2 | 0 | 0 |
| Malaise | 0 | 5 | 0 |
| Infections | |||
| Nasopharyngitis | 14 | 9 | 10 |
| Upper respiratory tract infection | 7 | 9 | 6 |
| Influenza | 5 | 7 | 4 |
| Urinary tract infection | 4 | 5 | 0 |
| Bronchitis | 0 | 5 | 1 |
| Injury | |||
| Contusion | 3 | 5 | 2 |
| Joint sprain | 1 | 2 | 1 |
| Muscle strain | 1 | 2 | 1 |
| Wound secretion | 0 | 2 | 0 |
| Metabolism and Nutrition Disorders | |||
| Increased appetite | 1 | 5 | 1 |
| Weight gain | 6 | 14 | 3 |
| Musculoskeletal Disorders | |||
| Arthralgia | 10 | 5 | 3 |
| Back pain | 4 | 7 | 2 |
| Pain in extremity | 6 | 2 | 4 |
| Myalgia | 3 | 5 | 1 |
| Muscle twitching | 1 | 9 | 1 |
| Muscle spasms | 3 | 0 | 1 |
| Nervous System Disorders | |||
| Headache | 33 | 26 | 31 |
| Somnolence | 22 | 26 | 13 |
| Dizziness | 24 | 26 | 17 |
| Nystagmus | 13 | 19 | 9 |
| Tremor | 15 | 16 | 8 |
| Memory impairment | 7 | 16 | 3 |
| Abnormal coordination | 7 | 16 | 2 |
| Disturbance in attention | 9 | 0 | 1 |
| Sensory disturbance | 4 | 7 | 2 |
| Hyporeflexia | 4 | 5 | 1 |
| Paraesthesia | 7 | 2 | 1 |
| Lethargy | 4 | 7 | 2 |
| Hyperreflexia | 4 | 2 | 3 |
| Hypoaesthesia | 4 | 5 | 1 |
| Sedation | 4 | 0 | 0 |
| Status epilepticus | 2 | 5 | 0 |
| Dysarthria | 2 | 2 | 1 |
| Postictal state | 2 | 0 | 1 |
| Sensory loss | 0 | 5 | 0 |
| Psychiatric Disorders | |||
| Irritability | 7 | 23 | 7 |
| Depression | 6 | 14 | 3 |
| Confusional state | 4 | 14 | 1 |
| Anxiety | 4 | 0 | 3 |
| Depressed mood | 5 | 0 | 1 |
| Abnormal thinking | 3 | 7 | 0 |
| Abnormal behavior | 3 | 5 | 1 |
| Expressive language disorder | 1 | 7 | 1 |
| Nervousness | 2 | 5 | 2 |
| Abnormal dreams | 1 | 5 | 1 |
| Reproductive System | |||
| Dysmenorrhea | 9 | 5 | 3 |
| Erectile dysfunction | 0 | 5 | 0 |
| Respiratory and Thoracic Disorders | |||
| Pharyngolaryngeal pain | 7 | 14 | 5 |
| Cough | 2 | 14 | 7 |
| Pulmonary congestion | 0 | 5 | 1 |
| Sinus headache | 6 | 2 | 1 |
| Skin and Subcutaneous Tissue Disorders | |||
| Rash | 4 | 5 | 4 |
Pediatrics 2 to 16 years of age
Table 6 lists adverse reactions from controlled clinical studies of pediatric patients receiving SABRIL or placebo as adjunctive therapy for refractory complex partial seizures. Adverse reactions that are listed occurred in at least 2% of SABRIL-treated patients and more frequently than placebo. The median SABRIL dose was 49.4 mg/kg (range of 8.0 – 105.9 mg/kg).
|
Table 6. Adverse Reactions in Pooled, Adjunctive Trials in Pediatric Patients 3 to 16 Years of Age with Refractory Complex Partial Seizures |
||
|
Body System Adverse Reaction |
All SABRIL |
Placebo |
|
Eye Disorders |
|
|
|
Diplopia |
3 |
2 |
|
Blurred vision |
2 |
0 |
|
Gastrointestinal Disorders |
|
|
|
Upper abdominal pain |
4 |
3 |
|
Constipation |
2 |
1 |
|
General Disorders |
|
|
|
Fatigue |
10 |
7 |
|
Infections and Infestations |
|
|
|
Upper respiratory tract infection |
15 |
11 |
|
Influenza |
7 |
3 |
|
Otitis media |
6 |
4 |
|
Streptococcal pharyngitis |
4 |
3 |
|
Viral gastroenteritis |
2 |
0 |
|
Investigations |
|
|
|
Weight gain |
15 |
2 |
|
Nervous System Disorders |
|
|
|
Somnolence |
6 |
5 |
|
Nystagmus |
4 |
3 |
|
Tremor |
4 |
2 |
|
Status epilepticus |
2 |
1 |
|
Psychiatric Disorders |
|
|
|
Abnormal behavior |
7 |
6 |
|
Aggression |
6 |
2 |
|
Disorientation |
3 |
0 |
Safety of SABRIL for the treatment of refractory CPS in patients 2 years of age is expected to be similar to pediatric patients 3 to 16 years of age.
Infantile Spasms
In a randomized, placebo-controlled IS study with a 5 day double-blind treatment phase (n=40), the adverse reactions that occurred in >5% of patients receiving SABRIL and that occurred more frequently than in placebo patients were somnolence (SABRIL 45%, placebo 30%), bronchitis (SABRIL 30%, placebo 15%), ear infection (SABRIL 10%, placebo 5%), and acute otitis media (SABRIL 10%, placebo 0%).
In a dose response study of low-dose (18-36 mg/kg/day) versus high-dose (100-148 mg/kg/day) SABRIL, no clear correlation between dose and incidence of adverse reactions was observed. The adverse reactions (≥5% in either dose group) are summarized in Table 7.
| Table 7. Adverse Reactions in a Placebo-Controlled Trial in Patients with Infantile Spasms | ||
|---|---|---|
| SABRIL Low Dose | SABRIL High Dose |
|
| Body System Adverse Reaction | [N=114] % | [N=108] % |
| Eye Disorders (other than field or acuity changes) | ||
| Strabismus | 5 | 5 |
| Conjunctivitis | 5 | 2 |
| Gastrointestinal Disorders | ||
| Vomiting | 14 | 20 |
| Constipation | 14 | 12 |
| Diarrhea | 13 | 12 |
| General Disorders | ||
| Fever | 29 | 19 |
| Infections | ||
| Upper respiratory tract infection | 51 | 46 |
| Otitis media | 44 | 30 |
| Viral infection | 20 | 19 |
| Pneumonia | 13 | 11 |
| Candidiasis | 8 | 3 |
| Ear infection | 7 | 14 |
| Gastroenteritis viral | 6 | 5 |
| Sinusitis | 5 | 9 |
| Urinary tract infection | 5 | 6 |
| Influenza | 5 | 3 |
| Croup infectious | 5 | 1 |
| Metabolism & Nutrition Disorders | ||
| Decreased appetite | 9 | 7 |
| Nervous System Disorders | ||
| Sedation | 19 | 17 |
| Somnolence | 17 | 19 |
| Status epilepticus | 6 | 4 |
| Lethargy | 5 | 7 |
| Convulsion | 4 | 7 |
| Hypotonia | 4 | 6 |
| Psychiatric Disorders | ||
| Irritability | 16 | 23 |
| Insomnia | 10 | 12 |
| Respiratory Disorders | ||
| Nasal congestion | 13 | 4 |
| Cough | 3 | 8 |
| Skin and Subcutaneous Tissue Disorders | ||
| Rash | 8 | 11 |
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of SABRIL. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure. Adverse reactions are categorized by system organ class.
Birth Defects: Congenital cardiac defects, congenital external ear anomaly, congenital hemangioma, congenital hydronephrosis, congenital male genital malformation, congenital oral malformation, congenital vesicoureteric reflux, dentofacial anomaly, dysmorphism, fetal anticonvulsant syndrome, hamartomas, hip dysplasia, limb malformation, limb reduction defect, low set ears, renal aplasia, retinitis pigmentosa, supernumerary nipple, talipes
Ear Disorders: Deafness
Endocrine Disorders: Delayed puberty
Gastrointestinal Disorders: Gastrointestinal hemorrhage, esophagitis
General Disorders: Developmental delay, facial edema, malignant hyperthermia, multi-organ failure
Hepatobiliary Disorders: Cholestasis
Nervous System Disorders: Dystonia, encephalopathy, hypertonia, hypotonia, muscle spasticity, myoclonus, optic neuritis, dyskinesia
Psychiatric Disorders: Acute psychosis, apathy, delirium, hypomania, neonatal agitation, psychotic disorder
Respiratory Disorders: Laryngeal edema, pulmonary embolism, respiratory failure, stridor
Skin and Subcutaneous Tissue Disorders: Angioedema, maculo-papular rash, pruritus, Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN), alopecia
7 DRUG INTERACTIONS
7.1 Antiepileptic Drugs
Phenytoin
Although phenytoin dose adjustments are not routinely required, dose adjustment of phenytoin should be considered if clinically indicated, since SABRIL may cause a moderate reduction in total phenytoin plasma levels [see Clinical Pharmacology (12.3)].
Clonazepam
SABRIL may moderately increase the Cmax of clonazepam resulting in an increase of clonazepam-associated adverse reactions [see Clinical Pharmacology (12.3)].
Other AEDs
There are no clinically significant pharmacokinetic interactions between SABRIL and either phenobarbital or sodium valproate. Based on population pharmacokinetics, carbamazepine, clorazepate, primidone, and sodium valproate appear to have no effect on plasma concentrations of vigabatrin [see Clinical Pharmacology (12.3)].
7.2 Oral Contraceptives
SABRIL is unlikely to affect the efficacy of steroid oral contraceptives [see Clinical Pharmacology (12.3)].
7.3 Drug-Laboratory Test Interactions
SABRIL decreases alanine transaminase (ALT) and aspartate transaminase (AST) plasma activity in up to 90% of patients. In some patients, these enzymes become undetectable. The suppression of ALT and AST activity by SABRIL may preclude the use of these markers, especially ALT, to detect early hepatic injury.
SABRIL may increase the amount of amino acids in the urine, possibly leading to a false positive test for certain rare genetic metabolic diseases (e.g., alpha aminoadipic aciduria).
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to AEDs, including SABRIL, during pregnancy. Encourage women who are taking SABRIL during pregnancy to enroll in the North American Antiepileptic Drug (NAAED) Pregnancy Registry. This can be done by calling the toll-free number 1-888-233-2334 or visiting the website, http://www.aedpregnancyregistry.org/. This must be done by the patient herself.
Risk Summary
There are no adequate data on the developmental risk associated with the use of SABRIL in pregnant women. Limited available data from case reports and cohort studies pertaining to SABRIL use in pregnant women have not established a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. However, based on animal data, SABRIL use in pregnant women may result in fetal harm.
When administered to pregnant animals, vigabatrin produced developmental toxicity, including an increase in fetal malformations and offspring neurobehavioral and neurohistopathological effects, at clinically relevant doses. In addition, developmental neurotoxicity was observed in rats treated with vigabatrin during a period of postnatal development corresponding to the third trimester of human pregnancy (see Data).
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively. The background risk of major birth defects and miscarriage for the indicated population is unknown.
Data
Animal Data
Administration of vigabatrin (oral doses of 50 to 200 mg/kg/day) to pregnant rabbits throughout the period of organogenesis was associated with an increased incidence of malformations (cleft palate) and embryofetal death; these findings were observed in two separate studies. The no-effect dose for adverse effects on embryofetal development in rabbits (100 mg/kg/day) is approximately 1/2 the maximum recommended human dose (MRHD) of 3 g/day on a body surface area (mg/m2) basis. In rats, oral administration of vigabatrin (50, 100, or 150 mg/kg/day) throughout organogenesis resulted in decreased fetal body weights and increased incidences of fetal anatomic variations. The no-effect dose for adverse effects on embryo-fetal development in rats (50 mg/kg/day) is approximately 1/5 the MRHD on a mg/m2 basis. Oral administration of vigabatrin (50, 100, 150 mg/kg/day) to rats from the latter part of pregnancy through weaning produced long-term neurohistopathological (hippocampal vacuolation) and neurobehavioral (convulsions) abnormalities in the offspring. A no-effect dose for developmental neurotoxicity in rats was not established; the low-effect dose (50 mg/kg/day) is approximately 1/5 the MRHD on a mg/m2 basis.
In a published study, vigabatrin (300 or 450 mg/kg) was administered by intraperitoneal injection to a mutant mouse strain on a single day during organogenesis (day 7, 8, 9, 10, 11, or 12). An increase in fetal malformations (including cleft palate) was observed at both doses.
Oral administration of vigabatrin (5, 15, or 50 mg/kg/day) to young rats during the neonatal and juvenile periods of development (postnatal days 4-65) produced neurobehavioral (convulsions, neuromotor impairment, learning deficits) and neurohistopathological (brain vacuolation, decreased myelination, and retinal dysplasia) abnormalities in treated animals. The early postnatal period in rats is generally thought to correspond to late pregnancy in humans in terms of brain development. The no-effect dose for developmental neurotoxicity in juvenile rats (5 mg/kg/day) was associated with plasma vigabatrin exposures (AUC) less than 1/30 of those measured in pediatric patients receiving an oral dose of 50 mg/kg.
8.2 Lactation
Risk Summary
Vigabatrin is excreted in human milk. The effects of SABRIL on the breastfed infant and on milk production are unknown. Because of the potential for serious adverse reactions from vigabatrin in nursing infants, breastfeeding is not recommended. If exposing a breastfed infant to SABRIL, observe for any potential adverse effects [see Warnings and Precautions (5.1, 5.3, 5.4, 5.8)].
8.4 Pediatric Use
The safety and effectiveness of SABRIL as adjunctive treatment of refractory complex partial seizures in pediatric patients 2 to 16 years of age have been established and is supported by three double-blind, placebo-controlled studies in patients 3 to 16 years of age, adequate and well-controlled studies in adult patients, pharmacokinetic data from patients 2 years of age and older, and additional safety information in patients 2 years of age [see Clinical Pharmacology (12.3) and Clinical Studies (14.1)]. The dosing recommendation in this population varies according to age group and is weight-based [see Dosage and Administration (2.2)]. Adverse reactions in this pediatric population are similar to those observed in the adult population [see Adverse Reactions (6.1)]. The safety and effectiveness of SABRIL as monotherapy for pediatric patients with infantile spasms (1 month to 2 years of age) have been established [see Dosage and Administration (2.3) and Clinical Studies (14.2)].
Safety and effectiveness as adjunctive treatment of refractory complex partial seizures in pediatric patients below the age of 2 and as monotherapy for the treatment of infantile spasms in pediatric patients below the age of 1 month have not been established.
Duration of therapy for infantile spasms was evaluated in a post hoc analysis of a Canadian Pediatric Epilepsy Network (CPEN) study of developmental outcomes in infantile spasms patients. This analysis suggests that a total duration of 6 months of vigabatrin therapy is adequate for the treatment of infantile spasms. However, prescribers must use their clinical judgment as to the most appropriate duration of use [see Clinical Studies (14.2)].
Abnormal MRI signal changes and Intramyelinic Edema (IME) in infants and young children being treated with SABRIL have been observed [see Warnings and Precautions (5.3, 5.4)].
Juvenile Animal Toxicity Data
Oral administration of vigabatrin (5, 15, or 50 mg/kg/day) to young rats during the neonatal and juvenile periods of development (postnatal days 4-65) produced neurobehavioral (convulsions, neuromotor impairment, learning deficits) and neurohistopathological (brain gray matter vacuolation, decreased myelination, and retinal dysplasia) abnormalities. The no-effect dose for developmental neurotoxicity in juvenile rats (the lowest dose tested) was associated with plasma vigabatrin exposures (AUC) substantially less than those measured in pediatric patients at recommended doses. In dogs, oral administration of vigabatrin (30 or 100 mg/kg/day) during selected periods of juvenile development (postnatal days 22-112) produced neurohistopathological abnormalities (brain gray matter vacuolation). Neurobehavioral effects of vigabatrin were not assessed in the juvenile dog. A no-effect dose for neurohistopathology was not established in juvenile dogs; the lowest effect dose (30 mg/kg/day) was associated with plasma vigabatrin exposures lower than those measured in pediatric patients at recommended doses [see Warnings and Precautions (5.4)].
8.5 Geriatric Use
Clinical studies of vigabatrin did not include sufficient numbers of patients aged 65 and over to determine whether they responded differently from younger patients.
Vigabatrin is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function.
Oral administration of a single dose of 1.5 g of vigabatrin to elderly (≥65 years) patients with reduced creatinine clearance (<50 mL/min) was associated with moderate to severe sedation and confusion in 4 of 5 patients, lasting up to 5 days. The renal clearance of vigabatrin was 36% lower in healthy elderly subjects (≥65 years) than in young healthy males. Adjustment of dose or frequency of administration should be considered. Such patients may respond to a lower maintenance dose [see Dosage and Administration (2.4) and Clinical Pharmacology (12.3)].
Other reported clinical experience has not identified differences in responses between the elderly and younger patients.
8.6 Renal Impairment
Dose adjustment, including initiating treatment with a lower dose, is necessary in pediatric patients 2 years of age and older and adults with mild (creatinine clearance >50 to 80 mL/min), moderate (creatinine clearance >30 to 50 mL/min) and severe (creatinine clearance >10 to 30 mL/min) renal impairment [see Dosage and Administration (2.4) and Clinical Pharmacology (12.3)].
9 DRUG ABUSE AND DEPENDENCE
9.2 Abuse
Vigabatrin did not produce adverse events or overt behaviors associated with abuse when administered to humans or animals. It is not possible to predict the extent to which a CNS active drug will be misused, diverted, and/or abused once marketed. Consequently, physicians should carefully evaluate patients for history of drug abuse and follow such patients closely, observing them for signs of misuse or abuse of vigabatrin (e.g., incrementation of dose, drug-seeking behavior).
9.3 Dependence
Following chronic administration of vigabatrin to animals, there were no apparent withdrawal signs upon drug discontinuation. However, as with all AEDs, vigabatrin should be withdrawn gradually to minimize increased seizure frequency [see Warnings and Precautions (5.6)].
10 OVERDOSAGE
10.1 Signs, Symptoms, and Laboratory Findings of Overdosage
Confirmed and/or suspected vigabatrin overdoses have been reported during clinical trials and in post marketing surveillance. No vigabatrin overdoses resulted in death. When reported, the vigabatrin dose ingested ranged from 3 g to 90 g, but most were between 7.5 g and 30 g. Nearly half the cases involved multiple drug ingestions including carbamazepine, barbiturates, benzodiazepines, lamotrigine, valproic acid, acetaminophen, and/or chlorpheniramine.
Coma, unconsciousness, and/or drowsiness were described in the majority of cases of vigabatrin overdose. Other less commonly reported symptoms included vertigo, psychosis, apnea or respiratory depression, bradycardia, agitation, irritability, confusion, headache, hypotension, abnormal behavior, increased seizure activity, status epilepticus, and speech disorder. These symptoms resolved with supportive care.
10.2 Management of Overdosage
There is no specific antidote for SABRIL overdose. Standard measures to remove unabsorbed drug should be used, including elimination by emesis or gastric lavage. Supportive measures should be employed, including monitoring of vital signs and observation of the clinical status of the patient.
In an in vitro study, activated charcoal did not significantly adsorb vigabatrin.
The effectiveness of hemodialysis in the treatment of SABRIL overdose is unknown. In isolated case reports in renal failure patients receiving therapeutic doses of vigabatrin, hemodialysis reduced vigabatrin plasma concentrations by 40% to 60%.
11 DESCRIPTION
SABRIL (vigabatrin) is an oral antiepileptic drug and is available as white film-coated 500 mg tablets and as a white to off-white granular powder for oral solution in packets of 500 mg.
The chemical name of vigabatrin, a racemate consisting of two enantiomers, is (±) 4-amino-5-hexenoic acid. The molecular formula is C6H11NO2 and the molecular weight is 129.16. It has the following structural formula:
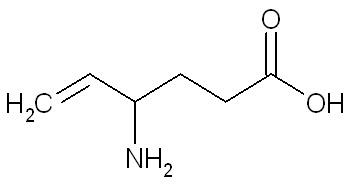
Vigabatrin is a white to off-white powder which is freely soluble in water, slightly soluble in methyl alcohol, very slightly soluble in ethyl alcohol and chloroform, and insoluble in toluene and hexane. The pH of a 1% aqueous solution is about 6.9. The n-octanol/water partition coefficient of vigabatrin is about 0.011 (log P=-1.96) at physiologic pH. Vigabatrin melts with decomposition in a 3-degree range within the temperature interval of 171ºC to 176ºC. The dissociation constants (pKa) of vigabatrin are 4 and 9.7 at room temperature (25ºC).
Each SABRIL tablet contains 500 mg of vigabatrin. The inactive ingredients are hydroxypropyl methylcellulose, magnesium stearate, microcrystalline cellulose, polyethylene glycols, povidone, sodium starch glycolate, and titanium dioxide.
SABRIL for oral solution is available as a white to off-white granular powder. Each packet contains 500 mg of vigabatrin. The inactive ingredient is povidone.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The precise mechanism of vigabatrin’s anti-seizure effect is unknown, but it is believed to be the result of its action as an irreversible inhibitor of γ-aminobutyric acid transaminase (GABA-T), the enzyme responsible for the metabolism of the inhibitory neurotransmitter GABA. This action results in increased levels of GABA in the central nervous system.
No direct correlation between plasma concentration and efficacy has been established. The duration of drug effect is presumed to be dependent on the rate of enzyme re-synthesis rather than on the rate of elimination of the drug from the systemic circulation.
12.2 Pharmacodynamics
Effects on Electrocardiogram
There is no indication of a QT/QTc prolonging effect of SABRIL in single doses up to 6.0 g. In a randomized, placebo-controlled, crossover study, 58 healthy subjects were administered a single oral dose of SABRIL (3 g and 6 g) and placebo. Peak concentrations for 6.0 g SABRIL were approximately 2-fold higher than the peak concentrations following the 3.0 g single oral dose.
12.3 Pharmacokinetics
Vigabatrin displayed linear pharmacokinetics after administration of single doses ranging from 0.5 g to 4 g, and after administration of repeated doses of 0.5 g and 2.0 g twice daily. Bioequivalence has been established between the oral solution and tablet formulations. The following PK information (Tmax, half-life, and clearance) of vigabatrin was obtained from stand-alone PK studies and population PK analyses.
Absorption
Following oral administration, vigabatrin is essentially completely absorbed. The time to maximum concentration (Tmax) is approximately 1 hour for children and adolescents (3 years to 16 years of age) and adults, and approximately 2.5 hours for infants (5 months to 2 years of age). There was little accumulation with multiple dosing in adult and pediatric patients. A food effect study involving administration of vigabatrin to healthy volunteers under fasting and fed conditions indicated that the Cmax was decreased by 33%, Tmax was increased to 2 hours, and AUC was unchanged under fed conditions.
Distribution
Vigabatrin does not bind to plasma proteins. Vigabatrin is widely distributed throughout the body; mean steady-state volume of distribution is 1.1 L/kg (CV = 20%).
Metabolism and Elimination
Vigabatrin is not significantly metabolized; it is eliminated primarily through renal excretion. The terminal half-life of vigabatrin is about 5.7 hours for infants (5 months to 2 years of age), 6.8 hours for children (3 to 9 years of age), 9.5 hours for children and adolescents (10 to 16 years of age), and 10.5 hours for adults. Following administration of [14]C-vigabatrin to healthy male volunteers, about 95% of total radioactivity was recovered in the urine over 72 hours with the parent drug representing about 80% of this. Vigabatrin induces CYP2C9, but does not induce other hepatic cytochrome P450 enzyme systems.
Specific Populations
Geriatric
The renal clearance of vigabatrin in healthy elderly patients (≥65 years of age) was 36% less than those in healthy younger patients. This finding is confirmed by an analysis of data from a controlled clinical trial [see Use in Specific Populations (8.5)].
Pediatric
The clearance of vigabatrin is 2.4 L/hr for infants (5 months to 2 years of age), 5.1 L/hr for children (3 to 9 years of age), 5.8 L/hr for children and adolescents (10 to 16 years of age) and 7 L/hr for adults.
Gender
No gender differences were observed for the pharmacokinetic parameters of vigabatrin in patients.
Race
No specific study was conducted to investigate the effects of race on vigabatrin pharmacokinetics. A cross study comparison between 23 Caucasian and 7 Japanese patients who received 1, 2, and 4 g of vigabatrin indicated that the AUC, Cmax, and half-life were similar for the two populations. However, the mean renal clearance of Caucasians (5.2 L/hr) was about 25% higher than the Japanese (4.0 L/hr). Inter-subject variability in renal clearance was 20% in Caucasians and was 30% in Japanese.
Renal Impairment
Mean AUC increased by 30% and the terminal half-life increased by 55% (8.1 hr vs 12.5 hr) in adult patients with mild renal impairment (CLcr from >50 to 80 mL/min) in comparison to normal subjects.
Mean AUC increased by two-fold and the terminal half-life increased by two-fold in adult patients with moderate renal impairment (CLcr from >30 to 50 mL/min) in comparison to normal subjects.
Mean AUC increased by 4.5-fold and the terminal half-life increased by 3.5-fold in adult patients with severe renal impairment (CLcr from >10 to 30 mL/min) in comparison to normal subjects.
Adult patients with renal impairment
Dosage adjustment, including starting at a lower dose, is recommended for adult patients with any degree of renal impairment [see Use in Specific Populations (8.6) and Dosage and Administration (2.4)].
Infants with renal impairment
Information about how to adjust the dose in infants with renal impairment is unavailable.
Pediatric patients 2 years and older with renal impairment
Although information is unavailable on the effects of renal impairment on vigabatrin clearance in pediatric patients 2 years and older, dosing can be calculated based upon adult data and an established formula [see Use in Specific Populations (8.6) and Dosage and Administration (2.4)].
Hepatic Impairment
Vigabatrin is not significantly metabolized. The pharmacokinetics of vigabatrin in patients with impaired liver function has not been studied.
Drug Interactions
Phenytoin
A 16% to 20% average reduction in total phenytoin plasma levels was reported in adult controlled clinical studies. In vitro drug metabolism studies indicate that decreased phenytoin concentrations upon addition of vigabatrin therapy are likely to be the result of induction of cytochrome P450 2C enzymes in some patients. Although phenytoin dose adjustments are not routinely required, dose adjustment of phenytoin should be considered if clinically indicated [see Drug Interactions (7.1)].
Clonazepam
In a study of 12 healthy adult volunteers, clonazepam (0.5 mg) co-administration had no effect on SABRIL (1.5 g twice daily) concentrations. SABRIL increases the mean Cmax of clonazepam by 30% and decreases the mean Tmax by 45% [see Drug Interactions (7.1)].
Other AEDs
When co-administered with vigabatrin, phenobarbital concentration (from phenobarbital or primidone) was reduced by an average of 8% to 16%, and sodium valproate plasma concentrations were reduced by an average of 8%. These reductions did not appear to be clinically relevant. Based on population pharmacokinetics, carbamazepine, clorazepate, primidone, and sodium valproate appear to have no effect on plasma concentrations of vigabatrin [see Drug Interactions (7.1)].
Alcohol
Co-administration of ethanol (0.6 g/kg) with vigabatrin (1.5 g twice daily) indicated that neither drug influences the pharmacokinetics of the other.
Oral Contraceptives
In a double-blind, placebo-controlled study using a combination oral contraceptive containing 30 mcg ethinyl estradiol and 150 mcg levonorgestrel, vigabatrin (3 g/day) did not interfere significantly with the cytochrome P450 isoenzyme (CYP3A)-mediated metabolism of the contraceptive tested. Based on this study, vigabatrin is unlikely to affect the efficacy of steroid oral contraceptives. Additionally, no significant difference in pharmacokinetic parameters (elimination half-life, AUC, Cmax, apparent oral clearance, time to peak, and apparent volume of distribution) of vigabatrin were found after treatment with ethinyl estradiol and levonorgestrel [see Drug Interactions (7.2)].
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Vigabatrin showed no carcinogenic potential in mouse or rat when given in the diet at doses up to 150 mg/kg/day for 18 months (mouse) or at doses up to 150 mg/kg/day for 2 years (rat). These doses are less than the maximum recommended human dose (MRHD) for infantile spasms (150 mg/kg/day) and for refractory complex partial seizures (3 g/day) on a mg/m2 basis.
Vigabatrin was negative in in vitro (Ames, CHO/HGPRT mammalian cell forward gene mutation, chromosomal aberration in rat lymphocytes) and in in vivo (mouse bone marrow micronucleus) assays.
No adverse effects on male or female fertility were observed in rats at oral doses up to 150 mg/kg/day (approximately 1/2 the MRHD of 3 g/day on a mg/m2 basis for refractory complex partial seizures).
14 CLINICAL STUDIES
14.1 Complex Partial Seizures
Adults
The effectiveness of SABRIL as adjunctive therapy in adult patients was established in two U.S. multicenter, double-blind, placebo-controlled, parallel-group clinical studies. A total of 357 adults (age 18 to 60 years) with complex partial seizures, with or without secondary generalization were enrolled (Studies 1 and 2). Patients were required to be on an adequate and stable dose of an anticonvulsant, and have a history of failure on an adequate regimen of carbamazepine or phenytoin. Patients had a history of about 8 seizures per month (median) for about 20 years (median) prior to entrance into the study. These studies were not capable by design of demonstrating direct superiority of SABRIL over any other anticonvulsant added to a regimen to which the patient had not adequately responded. Further, in these studies patients had previously been treated with a limited range of anticonvulsants.
The primary measure of efficacy was the patient’s reduction in mean monthly frequency of complex partial seizures plus partial seizures secondarily generalized at end of study compared to baseline.
Study 1
Study 1 (N=174) was a randomized, double-blind, placebo-controlled, dose-response study consisting of an 8-week baseline period followed by an 18-week treatment period. Patients were randomized to receive placebo or 1, 3, or 6 g/day vigabatrin administered twice daily. During the first 6 weeks following randomization, the dose was titrated upward beginning with 1 g/day and increasing by 0.5 g/day on days 1 and 5 of each subsequent week in the 3 g/day and 6 g/day groups, until the assigned dose was reached.
Results for the primary measure of effectiveness, reduction in monthly frequency of complex partial seizures, are shown in Table 8. The 3 g/day and 6 g/day dose groups were statistically significantly superior to placebo, but the 6 g/day dose was not superior to the 3 g/day dose.
| Table 8. Median Monthly Frequency of Complex Partial Seizures+ | |||
| N | Baseline | Endstudy | |
| Placebo | 45 | 9.0 | 8.8 |
| 1 g/day SABRIL | 45 | 8.5 | 7.7 |
| 3 g/day SABRIL | 41 | 8.5 | 3.7* |
| 6 g/day SABRIL | 43 | 8.5 | 4.5* |
| *p<0.05 compared to placebo +Including one patient with simple partial seizures with secondary generalization only |
|||
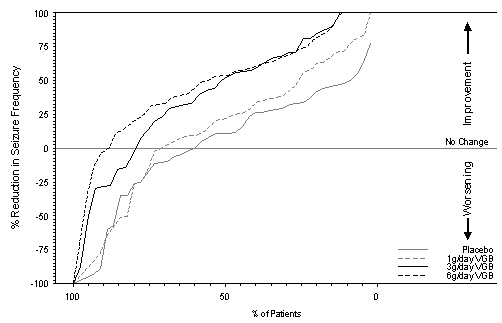
Figure 1 presents the percentage of patients (X-axis) with a percent reduction in seizure frequency (responder rate) from baseline to the maintenance phase at least as great as that represented on the Y-axis. A positive value on the Y-axis indicates an improvement from baseline (i.e., a decrease in complex partial seizure frequency), while a negative value indicates a worsening from baseline (i.e., an increase in complex partial seizure frequency). Thus, in a display of this type, a curve for an effective treatment is shifted to the left of the curve for placebo. The proportion of patients achieving any particular level of reduction in complex partial seizure frequency was consistently higher for the SABRIL 3 and 6 g/day groups compared to the placebo group. For example, 51% of patients randomized to SABRIL 3 g/day and 53% of patients randomized to SABRIL 6 g/day experienced a 50% or greater reduction in seizure frequency, compared to 9% of patients randomized to placebo. Patients with an increase in seizure frequency >100% are represented on the Y-axis as equal to or greater than -100%.
Figure 1. Percent Reduction from Baseline in Seizure Frequency
Study 2
Study 2 (N=183 randomized, 182 evaluated for efficacy) was a randomized, double-blind, placebo-controlled, parallel study consisting of an 8-week baseline period and a 16-week treatment period. During the first 4 weeks following randomization, the dose of vigabatrin was titrated upward beginning with 1 g/day and increased by 0.5 g/day on a weekly basis to the maintenance dose of 3 g/day.
Results for the primary measure of effectiveness, reduction in monthly complex partial seizure frequency, are shown in Table 9. Vigabatrin 3 g/day was statistically significantly superior to placebo in reducing seizure frequency.
| Table 9. Median Monthly Frequency of Complex Partial Seizures | |||
| N | Baseline | Endstudy | |
| Placebo | 90 | 9.0 | 7.5 |
| 3 g/day SABRIL | 92 | 8.3 | 5.5* |
| *p<0.05 compared to placebo | |||
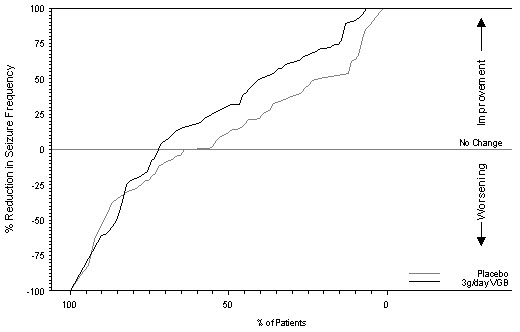
Figure 2 presents the percentage of patients (X-axis) with a percent reduction in seizure frequency (responder rate) from baseline to the maintenance phase at least as great as that represented on the Y-axis. A positive value on the Y-axis indicates an improvement from baseline (i.e., a decrease in complex partial seizure frequency), while a negative value indicates a worsening from baseline (i.e., an increase in complex partial seizure frequency). Thus, in a display of this type, a curve for an effective treatment is shifted to the left of the curve for placebo. The proportion of patients achieving any particular level of reduction in seizure frequency was consistently higher for the SABRIL 3 g/day group compared to the placebo group. For example, 39% of patients randomized to SABRIL (3 g/day) experienced a 50% or greater reduction in complex partial seizure frequency, compared to 21% of patients randomized to placebo. Patients with an increase in seizure frequency >100% are represented on the Y-axis as equal to or greater than -100%.
Figure 2. Percent Reduction from Baseline in Seizure Frequency
For both studies, there was no difference in the effectiveness of vigabatrin between male and female patients. Analyses of age and race were not possible as nearly all patients were between the ages of 18 to 65 and Caucasian.
Pediatric patients 3 to 16 years of age
SABRIL was studied in three double-blind, placebo-controlled, parallel-group studies in 269 patients who received SABRIL and 104 patients who received placebo. No individual study was considered adequately powered to determine efficacy in pediatric patients age 3 years and above. The data from all three pediatric studies were pooled and used in a pharmacometric bridging analysis using weight-normalized doses to establish efficacy and determine appropriate dosing. All three studies were randomized, double-blind, placebo-controlled, parallel-group, adjunctive-treatment studies in patients aged 3-16 years with uncontrolled complex partial seizures with or without secondary generalization. The study period included a 6 to 10 week baseline phase and a 14 to 17 week treatment phase (composed of a titration and maintenance period).
The pharmacometric bridging approach consisted of defining a weight-normalized dose-response, and showing that a similar dose-response relationship exists between pediatric patients and adults patients when SABRIL was given as adjunctive therapy for complex partial seizures. Dosing recommendations in pediatric patients 2 to 16 years of age were derived from simulations utilizing these pharmacometric dose-response analyses [see Dosage and Administration (2.2)].
14.2 Infantile Spasms
The effectiveness of SABRIL as monotherapy was established for infantile spasms in two multicenter controlled studies. Both studies were similar in terms of disease characteristics and prior treatments of patients and all enrolled infants had a confirmed diagnosis of infantile spasms.
Study 1
Study 1 (N=221) was a multicenter, randomized, low-dose high-dose, parallel-group, partially-blind (caregivers knew the actual dose but not whether their child was classified as low or high dose; EEG reader was blinded but investigators were not blinded) study to evaluate the safety and efficacy of vigabatrin in patients <2 years of age with new-onset infantile spasms. Patients with both symptomatic and cryptogenic etiologies were studied. The study was comprised of two phases. The first phase was a 14 to 21 day partially-blind phase in which patients were randomized to receive either low-dose (18-36 mg/kg/day) or high-dose (100-148 mg/kg/day) vigabatrin. Study drug was titrated over 7 days, followed by a constant dose for 7 days. If the patient became spasm-free on or before day 14, another 7 days of constant dose was administered. The primary efficacy endpoint of this study was the proportion of patients who were spasm-free for 7 consecutive days beginning within the first 14 days of vigabatrin therapy. Patients considered spasm-free were defined as those patients who remained free of spasms (evaluated according to caregiver response to direct questioning regarding spasm frequency) and who had no indication of spasms or hypsarrhythmia during 8 hours of CCTV EEG recording (including at least one sleep-wake-sleep cycle) performed within 3 days of the seventh day of spasm freedom and interpreted by a blinded EEG reader. Seventeen patients in the high-dose group achieved spasm freedom compared with 8 patients in the low dose group. This difference was statistically significant (p=0.0375). Primary efficacy results are shown in Table 10.
| Table 10. Spasm Freedom by Primary Criteria (Study 1) | |||
| SABRIL Treatment Group | |||
| 18-36 mg/kg/day [N=114] n (%) | 100-148 mg/kg/day [N=107] n (%) |
||
|
Patients who Achieved Spasm Freedom |
8 (7.0) |
17 (15.9) |
|
| p=0.0375 |
|||
Study 2
Study 2 (N=40) was a multicenter, randomized, double-blind, placebo-controlled, parallel-group study consisting of a pre-treatment (baseline) period of 2-3 days, followed by a 5-day double-blind treatment phase during which patients were treated with vigabatrin (initial dose of 50 mg/kg/day with titration allowed to 150 mg/kg/day) or placebo. The primary efficacy endpoint in this study was the average percent change in daily spasm frequency, assessed during a pre-defined and consistent 2-hour window of evaluation, comparing baseline to the final 2 days of the 5-day double-blind treatment phase. No statistically significant differences were observed in the average frequency of spasms using the 2-hour evaluation window. However, a post-hoc alternative efficacy analysis, using a 24-hour clinical evaluation window found a statistically significant difference in the overall percentage of reductions in spasms between the vigabatrin group (68.9%) and the placebo group (17.0%) (p=0.030).
Duration of therapy for infantile spasms was evaluated in a post hoc analysis of a Canadian Pediatric Epilepsy Network (CPEN) study of developmental outcomes in infantile spasms patients. The 38/68 infants in the study who had responded to vigabatrin therapy (complete cessation of spasms and hypsarrhythmia) continued vigabatrin therapy for a total duration of 6 months therapy. The 38 infants who responded were then followed for an additional 18 months after discontinuation of vigabatrin to determine their clinical outcome. A post hoc analysis indicated no observed recurrence of infantile spasms in any of these 38 infants.
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
SABRIL 500 mg tablets are white, film-coated, oval, biconvex, scored on one side, and debossed with OV 111 on the other. They are supplied as bottles of 100 (NDC 67386-111-01).
SABRIL for oral solution is available as 500 mg packets containing a white to off-white granular powder. They are supplied in packages of 50 (NDC 67386-211-65).
The oral syringes are provided separately by the pharmacy.
17 PATIENT COUNSELING INFORMATION
Advise patients and caregivers to read the FDA-approved patient labeling (Medication Guide and Instructions for Use).
Administration Instructions for SABRIL for oral solution
Physicians should confirm that caregivers(s) understand how to mix SABRIL for oral solution and to administer the correct dose to their infants and pediatric patients [see Dosage and Administration (2.5)].
Permanent Vision Loss
Inform patients and caregivers of the risk of permanent vision loss, particularly loss of peripheral vision, from SABRIL, and the need for monitoring vision [see Warnings and Precautions (5.1)].
Monitoring of vision, including assessment of visual fields and visual acuity, is recommended at baseline (no later than 4 weeks after starting SABRIL), at least every 3 months while on therapy, and about 3-6 months after discontinuation of therapy. In patients for whom vision testing is not possible, treatment may continue without recommended testing according to clinical judgment with appropriate patient or caregiver counseling. Patients or caregivers should be informed that if baseline or subsequent vision is not normal, SABRIL should only be used if the benefits of SABRIL treatment clearly outweigh the risks of additional vision loss.
Advise patients and caregivers that vision testing may be insensitive and may not detect vision loss before it is severe. Also advise patients and caregivers that if vision loss is documented, such loss is irreversible. Ensure that both of these points are understood by patients and caregivers.
Patients and caregivers should be informed that if changes in vision are suspected, they should notify their physician immediately.
Vigabatrin REMS Program
SABRIL is available only through a restricted program called the Vigabatrin REMS Program [see Warnings and Precautions (5.2)]. Inform patients/caregivers of the following:
- Patients/caregivers must be enrolled in the program.
- SABRIL is only available through pharmacies that are enrolled in the Vigabatrin REMS Program.
MRI Abnormalities in Infants
Inform caregiver(s) of the possibility that infants may develop an abnormal MRI signal of unknown clinical significance [see Warnings and Precautions (5.3)].
Suicidal Thinking and Behavior
Counsel patients, their caregiver(s), and families that AEDs, including SABRIL, may increase the risk of suicidal thoughts and behavior. Also advise patients and caregivers of the need to be alert for the emergence or worsening of symptoms of depression, any unusual changes in mood or behavior, or the emergence of suicidal thoughts, behavior, or thoughts of self-harm. Behaviors of concern should be reported immediately to healthcare providers [see Warnings and Precautions (5.5)].
Pregnancy
Advise pregnant women and women of child-bearing potential that the use of SABRIL during pregnancy can cause fetal harm which may occur early in pregnancy before many women know they are pregnant. Instruct patients to notify their physician if they become pregnant or intend to become pregnant during therapy. Advise patients that there is a pregnancy exposure registry that collects information about the safety of antiepileptic drugs during pregnancy [see Use in Specific Populations (8.1)].
Nursing
Counsel patients that SABRIL is excreted in breast milk. Because of the potential for serious adverse reactions in nursing infants from SABRIL, breastfeeding is not recommended. If a decision is made to breastfeed, nursing mothers should be counseled to observe their infants for signs of vision loss, sedation and poor sucking [see Use in Specific Populations (8.2)].
Withdrawal of SABRIL Therapy
Instruct patients and caregivers not to suddenly discontinue SABRIL therapy without consulting with their healthcare provider. As with all AEDs, withdrawal should be gradual [see Warnings and Precautions (5.6)].
Manufactured by: Patheon
Cincinnati, OH 45237, U.S.A.
For: Lundbeck
Deerfield, IL 60015, U.S.A.
SABRIL is a registered trademark of Lundbeck
MEDICATION GUIDE
SABRIL®(SAY-bril)
(vigabatrin)
Tablets
SABRIL®(SAY-bril)
(vigabatrin)
for oral solution
What is the most important information I should know about SABRIL?
SABRIL can cause serious side effects, including:
- Permanent vision loss
- Magnetic resonance imaging (MRI) changes in babies with infantile spasms (IS)
- Risk of suicidal thoughts or actions
1. Permanent vision loss:
SABRIL can damage the vision of anyone who takes it. Some people can have severe loss particularly to their ability to see to the side when they look straight ahead (peripheral vision). With severe vision loss, you may only be able to see things straight in front of you (sometimes called “tunnel vision”). You may also have blurry vision. If this happens, it will not get better.
- Vision loss and use of SABRIL in adults and children 2 years and older: Because of the risk of vision loss, SABRIL is used to treat complex partial seizures (CPS) only in people who do not respond well enough to several other medicines.
Tell your healthcare provider right away if you (or your child):
- might not be seeing as well as before starting SABRIL.
- start to trip, bump into things, or are more clumsy than usual.
- are surprised by people or things coming in front of you that seem to come out of nowhere.
- These changes can mean that you (or your child) have damage to your vision.
- It is recommended that your healthcare provider test your (or your child’s) vision (including peripheral vision) and visual acuity (ability to read an eye chart) before you (or your child) start SABRIL or within 4 weeks after starting SABRIL, and at least every 3 months after that until SABRIL is stopped. It is also recommended that you (or your child) have a vision test about 3 to 6 months after SABRIL is stopped. Your vision loss may get worse after you stop taking Sabril.
- Some people are not able to complete testing of vision. Your healthcare provider will determine if you (or your child) can be tested. If you (or your child) cannot complete vision testing, your healthcare provider may continue prescribing SABRIL, but your healthcare provider will not be able to watch for any vision loss you (or your child) may get.
- Even if your vision (or your child’s vision) seems fine, it is important that you (or your child) get these regular vision tests because vision damage can happen before you (or your child) notice any changes.
- These vision tests cannot prevent the vision damage that can happen with SABRIL, but they do allow the healthcare provider to decide if you (or your child) should stop SABRIL if your vision has gotten worse.
- Vision testing may not detect vision loss before it is severe.
- If you do not have these vision tests regularly, your healthcare provider may stop prescribing SABRIL.
- If you drive and your vision is damaged by SABRIL, driving might be more dangerous, or you may not be able to drive safely at all. Talk about this with your healthcare provider.
- Vision loss in babies: Because of the risk of vision loss, SABRIL is used in babies 1 month to 2 years of age with infantile spasms (IS) only when you and your healthcare provider decide that the possible benefits of SABRIL are more important than the risks.
- Parents or caregivers are not likely to recognize the symptoms of vision loss in babies until it is severe. Healthcare providers may not find vision loss in babies until it is severe.
- It is difficult to test vision in babies, but, to the extent possible, all babies should have their vision tested before starting SABRIL or within 4 weeks after starting SABRIL, and every 3 months after that until SABRIL is stopped. Your baby should also have a vision test about 3 to 6 months after SABRIL is stopped.
- Your baby may not be able to be tested. Your healthcare provider will determine if your baby can be tested. If your baby cannot be tested, your healthcare provider may continue prescribing SABRIL, but your healthcare provider will not be able to watch for any vision loss.
Tell your healthcare provider right away if you think that your baby is:
- not seeing as well as before taking SABRIL.
- acting differently than normal.
- Even if your baby’s vision seems fine, it is important to get regular vision tests because damage can happen before your baby acts differently. Even these regular vision exams may not show the damage to your baby’s vision before it is severe and permanent.
All people who take SABRIL:
|
- Because SABRIL might cause permanent vision loss, it is available to healthcare providers and patients only under a special program called the Vigabatrin Risk Evaluation and Mitigation Strategy (REMS) Program. SABRIL can only be prescribed to people who are enrolled in this program. As part of the Vigabatrin REMS Program, it is recommended that your healthcare provider test your (or your child’s) vision from time to time (periodically) while you (or your child) are being treated with SABRIL, and even after you (or your child) stop treatment. Your healthcare provider will explain the details of the Vigabatrin REMS Program to you. For more information, go to www.vigabatrinREMS.com or call 1-866-244-8175.
2. Magnetic resonance imaging (MRI) changes in babies with infantile spasms:
Brain pictures taken by magnetic resonance imaging (MRI) show changes in some babies after they are given SABRIL. It is not known if these changes are harmful.
3. Risk of suicidal thoughts or actions:
Like other antiepileptic drugs, SABRIL may cause suicidal thoughts or actions in a very small number of people, about 1 in 500 people taking it. Call a healthcare provider right away if you or your child have any of these symptoms, especially if they are new, worse, or worry you:
- thoughts about suicide or dying
- attempts to commit suicide
- new or worse depression
- new or worse anxiety
- feeling agitated or restless
- panic attacks
- trouble sleeping (insomnia)
- new or worse irritability
- acting aggressive, being angry, or violent
- acting on dangerous impulses
- an extreme increase in activity and talking (mania)
- other unusual changes in behavior or mood
Suicidal thoughts or actions can be caused by things other than medicines. If you or your child have suicidal thoughts or actions, your healthcare provider may check for other causes.
How can I watch for early symptoms of suicidal thoughts and actions?
- Pay attention to any changes, especially sudden changes, in mood, behaviors, thoughts, or feelings.
- Keep all follow-up visits with your healthcare provider as scheduled.
- Call your healthcare provider between visits as needed, especially if you are worried about symptoms.
- Do not stop SABRIL without first talking to a healthcare provider.
- Stopping SABRIL suddenly can cause serious problems. Stopping a seizure medicine suddenly can cause seizures that will not stop (status epilepticus) in people who are being treated for seizures.
What is SABRIL?
- SABRIL is a prescription medicine used along with other treatments to treat adults and children 2 years and older with complex partial seizures (CPS) if:
○ the CPS do not respond well enough to several other treatments, and
○ you and your healthcare provider decide the possible benefit of taking SABRIL is more important than the risk of vision loss.
SABRIL should not be the first medicine used to treat CPS.
- SABRIL is also used to treat babies 1 month to 2 years of age who have infantile spasms (IS) if you and your healthcare provider decide the possible benefits of taking SABRIL are more important than the possible risk of vision loss.
What should I tell my healthcare provider before starting SABRIL?
If you or your child has CPS, before taking SABRIL tell your healthcare provider about all of your medical conditions, including if you or your child:
- have or had an allergic reaction to SABRIL, such as hives, itching, or trouble breathing.
- have or had any vision problems.
- have or had any kidney problems.
- have or had low red blood cell counts (anemia).
- have or had any nervous or mental illnesses such as depression, mood problems, thoughts of suicide, or attempts at suicide.
- are breastfeeding or planning to breastfeed. SABRIL can pass into breast milk and may harm your baby. Talk to your healthcare provider about the best way to feed your baby if you take SABRIL.
- are pregnant or plan to become pregnant. SABRIL can cause harm to your unborn baby. You and your healthcare provider will have to decide if you should take SABRIL while you are pregnant.
Pregnancy Registry:
If you become pregnant while taking SABRIL, talk to your healthcare provider about registering with the North American Antiepileptic Drug Pregnancy Registry. You can enroll in this registry by calling 1-888-233-2334. Information on the registry can also be found at the website http://www.aedpregnancyregistry.org/. The purpose of this registry is to collect information about the safety of antiepileptic medicine during pregnancy.
If you are a parent or caregiver whose baby has IS, before giving SABRIL to your baby, tell your healthcare provider about all of your baby’s medical conditions, including if your baby has or ever had:
- an allergic reaction to SABRIL, such as hives, itching, or trouble breathing.
- any vision problems.
- any kidney problems.
Tell your healthcare provider about all the medicines you or your child take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. SABRIL and other medicines may affect each other causing side effects.
How should I take SABRIL?
- SABRIL comes as tablets or as packets containing powder for mixing with water to make an oral solution.
- You or your child will receive SABRIL from a specialty pharmacy.
- Take SABRIL exactly as your healthcare provider tells you to. SABRIL is usually taken 2 times each day.
- SABRIL may be taken with or without food.
- Before starting to take SABRIL, talk to your healthcare provider about what you or your child should do if a SABRIL dose is missed.
- If you or your child are taking SABRIL for CPS and the seizures do not improve enough within 3 months, your healthcare provider will stop prescribing SABRIL.
- If your child is taking SABRIL for IS and the seizures do not improve within 2 to 4 weeks, your healthcare provider will stop prescribing SABRIL.
- Do not stop taking SABRIL suddenly. This can cause serious problems. Stopping SABRIL or any seizure medicine suddenly can cause seizures that will not stop (status epilepticus) in people who are being treated for seizures. You should follow your healthcare provider’s instructions on how to stop taking SABRIL.
- Tell your healthcare provider right away about any increase in seizures when SABRIL treatment is being stopped. Before your child starts taking SABRIL, speak to your child’s healthcare provider about what to do if your baby misses a dose, vomits, spits up, or only takes part of the dose of SABRIL.
- Do not stop taking SABRIL without talking to your healthcare provider. If SABRIL improves your (or your child’s) seizures, you and your healthcare provider should talk about whether the benefit of taking SABRIL is more important than the risk of vision loss, and decide if you (or your child) will continue to take SABRIL.
- If you are giving SABRIL for oral solution to your child, it can be given at the same time as their meal. SABRIL powder should be mixed with water only.
- See “Instructions for Use” for detailed information about how to mix and give SABRIL for oral solution to your child the right way.
What should I avoid while taking SABRIL?
SABRIL causes sleepiness and tiredness. Adults taking SABRIL should not drive, operate machinery, or perform any hazardous task, unless you and your healthcare provider have decided that you can do these things safely.
What are the possible side effects of SABRIL?
SABRIL can cause serious side effects, including:
- See “What is the most important information I should know about SABRIL?”
- sleepiness and tiredness. See “What should I avoid while taking SABRIL?”
- SABRIL may cause your baby to be sleepy. Sleepy babies may have a harder time suckling and feeding, or may be irritable.
- weight gain that happens without swelling.
The following serious side effects happen in adults. It is not known if these side effects also happen in babies who take SABRIL.
- low red blood cell counts (anemia).
- nerve problems. Symptoms of a nerve problem can include numbness and tingling in your toes or feet. It is not known if nerve problems will go away after you stop taking SABRIL.
- swelling.
If you or your child has CPS, SABRIL may make certain types of seizures worse. Tell your healthcare provider right away if your (or your child's) seizures get worse.
The most common side effect of SABRIL in adults include blurred vision, sleepiness, dizziness, problems walking or feeling uncoordinated, shaking (tremor), and tiredness.
The most common side effect of SABRIL in children 3 to 16 years of age is weight gain. Also expect side effects like those seen in adults.
If you are giving SABRIL to your baby for IS:
SABRIL may make certain types of seizures worse. You should tell your baby’s healthcare provider right away if your baby’s seizures get worse. Tell your baby’s healthcare provider if you see any changes in your baby’s behavior.
The most common side effects of SABRIL in babies include:
- sleepiness - SABRIL may cause your baby to be sleepy. Sleepy babies may have a harder time suckling and feeding or may be irritable.
- swelling in the bronchial tubes (bronchitis)
- ear infection
- irritability
Tell your healthcare provider if you or your child have any side effect that bothers you or that does not go away. These are not all the possible side effects of SABRIL.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store SABRIL?
- Store SABRIL tablets and SABRIL packets at room temperature between 68ºF to 77ºF (20ºC to 25ºC).
Keep SABRIL and all medicines out of the reach of children.
General information about the safe and effective use of SABRIL.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. You can ask your pharmacist or healthcare provider for information about SABRIL that is written for health professionals. Do not use SABRIL for a condition for which it was not prescribed. Do not give SABRIL to other people, even if they have the same symptoms that you have. It may harm them.
What are the ingredients in SABRIL?
Active Ingredient: vigabatrin
Inactive Ingredients:
- Tablets: hydroxypropyl methylcellulose, magnesium stearate, microcrystalline cellulose, polyethylene glycols, povidone, sodium starch glycolate, and titanium dioxide
- For oral solution: povidone
Marketed by: Lundbeck, Deerfield, IL 60015, U.S.A.
SABRIL is a registered trademark of Lundbeck
For more information, go to www.SABRIL.net or call 1-866-402-8520.
This Medication Guide has been approved by the U.S. Food and Drug Administration. Revised: 10/2021
INSTRUCTIONS FOR USE
SABRIL®(SAY-bril)
(vigabatrin)
for oral solution
Read this Instructions for Use before your child starts taking SABRIL and each time you get a refill. There may be new information. This information does not take the place of talking with your healthcare provider about your child’s medical condition or treatment. Talk to your healthcare provider if you have any questions about the right dose of medicine to give your child or how to mix it.
Important Note:
- SABRIL for oral solution comes in a packet
- Sabril packet contains 500 mg of powder
- SABRIL must be mixed with water only. The water may be cold or at room temperature.
- Your healthcare provider will tell you:
- how many packets of SABRIL you will need for each dose
- how many milliliters (mL) of water to use to mix one dose of SABRIL
- how many milliliters (mL) of the powder and water mixture you will need for each dose of medicine
- SABRIL should be given right away after it is mixed
- Use the oral syringes, provided by the pharmacy, to measure and give the correct dose. Do not use a household teaspoon or tablespoon.
Supplies you will need to mix 1 dose of SABRIL:
- The number of packets of SABRIL needed for each dose
- 2 clean cups: 1 for mixing and 1 for water. The cup used for mixing SABRIL should be clear so you can see if the powder is dissolved
- Water to mix with the SABRIL powder
- One small 3 mL oral syringe and one large 10 mL oral syringe which are provided by the pharmacy
- Small spoon or other clean utensil to stir the mixture
- Scissors
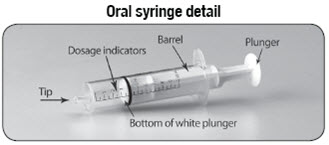
Step 1: Start with 1 of the empty cups and the total number of packets you will need for 1 dose.
Step 2: Before you open the packet, tap it to settle all the powder to the bottom of the packet.
Step 3: Use a pair of scissors to cut open the SABRIL packet along the dotted line.
Step 4: Empty the entire contents of the SABRIL packet into 1 of the clean empty cups (see Figure A).

- Repeat steps 2 to 4 above to open all of the packets needed for 1 dose of SABRIL.
Step 5: Take the second cup and fill it half way with water (see Figure B).
Do not mix SABRIL with anything other than water.

- You will use the larger oral syringe (10 mL) to draw up the water needed to mix with the powder from the packets. You will need 10 mL of water for each packet of SABRIL.
For example:
- If you are using 1 packet of SABRIL, you will need to use 10 mL of water (fill the 10 mL oral syringe 1 time)
- If you are using 2 packets of SABRIL, you will need to use 20 mL of water (fill the 10 mL oral syringe 2 times)
- If you are using 3 packets of SABRIL, you will need to use 30 mL of water (fill the 10 mL oral syringe 3 times)
Step 6: Use the 10 mL oral syringe to draw up 10 mL of water. To do this, put the tip of the oral syringe all the way into the water in your cup. Then pull the plunger up towards you until the edge of the plunger is at the 10 mL line on the barrel of the oral syringe (see Figure C).
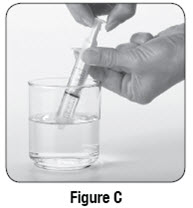
- If you see bubbles of air in the oral syringe after drawing up the water, turn the oral syringe so the tip is pointing up (see Figure D). The air will move to the top of the oral syringe. Pull the plunger back towards you and then push it back gently into the oral syringe to get rid of the bubbles. Tiny bubbles are normal.
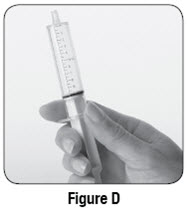
Step 7: Check the oral syringe to make sure it is filled with water up to the 10 mL line (see Figure E).
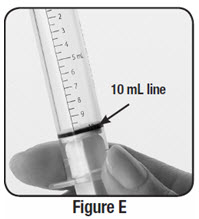
Step 8: Get the second cup that contains the SABRIL needed for your dose.
Step 9: Hold the 10mL oral syringe that is filled with water with the tip pointing down over the SABRIL.
Step 10: Slowly push the oral syringe plunger all the way down to empty the water from the oral syringe straight into the cup containing the SABRIL (see Figure F).

Repeat steps 6 through 10 until all of the water that is needed to mix 1 dose of SABRIL has been added to the cup containing the powder.
Step 11: Stir the mixture with the small spoon or other clean utensil until the solution is clear (see Figure G). This means that all of the powder is dissolved and ready for use.

- To give a dose of SABRIL to your child, you should use the oral syringe to draw up the total number of mLs of the mixture that your healthcare provider tells you to.
- If you are giving 3 mL or less of the mixture, use the smaller 3 mL oral syringe.
- If you are giving more than 3 mL of the mixture, use the larger 10 mL oral syringe (this is the oral syringe that you just used to add the water).
Step 12: Put the tip of the oral syringe all the way into the mixture. Pull the plunger up towards you to draw up the mixture. Stop when the edge of the plunger lines up with markings on the barrel of the oral syringe that matches the number of mLs of mixture your healthcare provider told you to give (see Figure H).

- If you see bubbles of air in the oral syringe after drawing up the mixture, turn the oral syringe so the tip is pointing up (see Figure I). The air will move to the top of the oral syringe. Pull the plunger back towards you and then gently push it back in the oral syringe in order to get rid of the bubbles. Tiny bubbles are normal.
Step 13: Place the tip of the oral syringe into your child’s mouth and point the oral syringe towards either cheek (see Figure J). Push on the plunger slowly, a small amount at a time, until all of the mixture in the oral syringe is given.
- If the dose you are giving your child is more than 10 mLs, repeat steps 12 and 13 until you give the total dose of mixture prescribed by your healthcare provider.
Step 14: Throw away any mixture that is left over. Do not save or reuse any leftover mixture.
Step 15: Wash the oral syringes and mixing cups in warm water. To clean the oral syringes, remove the plunger by gently pulling it straight out of the barrel. The barrel and plunger can be hand washed with soap and water, rinsed, and allowed to dry.
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Marketed by: Lundbeck, Deerfield, IL 60015, U.S.A.
SABRIL is a registered trademark of Lundbeck
Revised: 10/2021