FULL PRESCRIBING INFORMATION
WARNING: SERIOUS ADVERSE REACTIONS INCLUDING TENDINITIS, TENDON RUPTURE, PERIPHERAL NEUROPATHY, CENTRAL NERVOUS SYSTEM EFFECTS AND EXACERBATION OF MYASTHENIA GRAVIS
-
Fluoroquinolones, including ciprofloxacin, have been associated with disabling and potentially irreversible serious adverse reactions that have occurred together
[see
Warnings and Precautions (5.1)]
including:
- Tendinitis and tendon rupture [see Warnings and Precautions (5.2)]
- Peripheral neuropathy [see Warnings and Precautions (5.3)]
- Central nervous system effects [see Warnings and Precautions (5.4)]
- Discontinue ciprofloxacin immediately and avoid the use of fluoroquinolones, including ciprofloxacin tablets, in patients who experience any of these serious adverse reactions [see Warnings and Precautions (5.1)]. Fluoroquinolones, including ciprofloxacin, may exacerbate muscle weakness in patients with myasthenia gravis. Avoid ciprofloxacin in patients with known history of myasthenia gravis [see Warnings and Precautions (5.5)] .
-
Because fluoroquinolones, including ciprofloxacin, have been associated with serious adverse reactions
[see
Warnings and Precautions (5.1-5.16)]
, reserve ciprofloxacin tablets for use in patients who have no alternative treatment options for the following indications:
- Acute exacerbation of chronic bronchitis [see Indications and Usage (1.10)]
- Acute uncomplicated cystitis [see Indications and Usage (1.11)]
- Acute sinusitis [see Indications and Usage (1.12)]
1 INDICATIONS AND USAGE
1.1 Skin and Skin Structure Infections
Ciprofloxacin is indicated in adult patients for treatment of skin and skin structure infections caused by Escherichia coli, Klebsiella pneumoniae, Enterobacter cloacae, Proteus mirabilis, Proteus vulgaris, Providencia stuartii, Morganella morganii, Citrobacter freundii, Pseudomonas aeruginosa, methicillin-susceptible Staphylococcus aureus, methicillin-susceptible Staphylococcus epidermidis, or Streptococcus pyogenes.
1.2 Bone and Joint Infections
Ciprofloxacin is indicated in adult patients for treatment of bone and joint infections caused by Enterobacter cloacae, Serratia marcescens, or Pseudomonas aeruginosa.
1.3 Complicated Intra-Abdominal Infections
Ciprofloxacin is indicated in adult patients for treatment of complicated intra-abdominal infections (used in combination with metronidazole) caused by Escherichia coli, Pseudomonas aeruginosa, Proteus mirabilis, Klebsiella pneumoniae, or Bacteroides fragilis.
1.4 Infectious Diarrhea
Ciprofloxacin is indicated in adult patients for treatment of infectious diarrhea caused by
Escherichia coli (enterotoxigenic isolates),
Campylobacter jejuni,
Shigella boydii
†,
Shigella dysenteriae,
Shigella flexneri or
Shigella sonnei
† when antibacterial therapy is indicated.
†Although treatment of infections due to this organism in this organ system demonstrated a clinically significant outcome, efficacy was studied in fewer than 10 patients.
1.5 Typhoid Fever (Enteric Fever)
Ciprofloxacin is indicated in adult patients for treatment of typhoid fever (enteric fever) caused by Salmonella typhi. The efficacy of ciprofloxacin in the eradication of the chronic typhoid carrier state has not been demonstrated.
1.6 Uncomplicated Cervical and Urethral Gonorrhea
Ciprofloxacin is indicated in adult patients for treatment of uncomplicated cervical and urethral gonorrhea due to Neisseria gonorrhoeae [see Warnings and Precautions (5.17)].
1.7 Inhalational Anthrax (Post-Exposure)
Ciprofloxacin is indicated in adults and pediatric patients from birth to 17 years of age for inhalational anthrax (post-exposure) to reduce the incidence or progression of disease following exposure to aerosolized Bacillus anthracis.
Ciprofloxacin serum concentrations achieved in humans served as a surrogate endpoint reasonably likely to predict clinical benefit and provided the initial basis for approval of this indication. 1Supportive clinical information for ciprofloxacin for anthrax post-exposure prophylaxis was obtained during the anthrax bioterror attacks of October 2001 [see Clinical Studies (14.2)].
1.8 Plague
Ciprofloxacin is indicated for treatment of plague, including pneumonic and septicemic plague, due to Yersinia pestis (Y. pestis) and prophylaxis for plague in adults and pediatric patients from birth to 17 years of age. Efficacy studies of ciprofloxacin could not be conducted in humans with plague for feasibility reasons. Therefore this indication is based on an efficacy study conducted in animals only [see Clinical Studies (14.3)] .
1.9 Chronic Bacterial Prostatitis
Ciprofloxacin is indicated in adult patients for treatment of chronic bacterial prostatitis caused by Escherichia coli or Proteus mirabilis.
1.10 Lower Respiratory Tract Infections
Ciprofloxacin is indicated in adult patients for treatment of lower respiratory tract infections caused by
Escherichia coli,
Klebsiella pneumoniae,
Enterobacter cloacae,
Proteus mirabilis,
Pseudomonas aeruginosa,
Haemophilus influenzae,
Haemophilus parainfluenzae, or
Streptococcus pneumoniae.
Ciprofloxacin is not a drug of first choice in the treatment of presumed or confirmed pneumonia secondary to
Streptococcus pneumoniae.
Ciprofloxacin is indicated for the treatment of acute exacerbations of chronic bronchitis (AECB) caused by
Moraxella catarrhalis.
Because fluoroquinolones, including ciprofloxacin, have been associated with serious adverse reactions
[see
Warnings and Precautions (5.1–5.16)]
and for some patients AECB is self-limiting, reserve ciprofloxacin for treatment of AECB in patients who have no alternative treatment options.
1.11 Urinary Tract Infections
Urinary Tract Infections in Adults
Ciprofloxacin is indicated in adult patients for treatment of urinary tract infections caused by
Escherichia coli,
Klebsiella pneumoniae,
Enterobacter cloacae,
Serratia marcescens,
Proteus mirabilis,
Providencia rettgeri,
Morganella morganii,
Citrobacter koseri,
Citrobacter freundii,
Pseudomonas aeruginosa, methicillin-susceptible
Staphylococcus epidermidis,
Staphylococcus saprophyticus, or
Enterococcus faecalis.
Acute Uncomplicated Cystitis
Ciprofloxacin is indicated in adult female patients for treatment of acute uncomplicated cystitis caused by
Escherichia coli or
Staphylococcus saprophyticus.
Because fluoroquinolones, including ciprofloxacin, have been associated with serious adverse reactions
[see
Warnings and Precautions (5.1-5.16)]
and for some patients acute uncomplicated cystitis is self-limiting, reserve ciprofloxacin tablets for treatment of acute uncomplicated cystitis in patients who have no alternative treatment options.
Complicated Urinary Tract Infection and Pyelonephritis in Pediatric Patients
Ciprofloxacin is indicated in pediatric patients aged one to 17 years of age for treatment of complicated urinary tract infections (cUTI) and pyelonephritis due to
Escherichia coli [see
Use in Specific Populations (8.4)].
Although effective in clinical trials, ciprofloxacin is not a drug of first choice in the pediatric population due to an increased incidence of adverse reactions compared to controls, including reactions related to joints and/or surrounding tissues. Ciprofloxacin, like other fluoroquinolones, is associated with arthropathy and histopathological changes in weight-bearing joints of juvenile animals
[see
Warnings and Precautions (5.13),
Adverse Reactions (6.1),
Use in Specific Populations (8.4) and
Nonclinical Toxicology (13.2)].
1.12 Acute Sinusitis
Ciprofloxacin is indicated in adult patients for treatment of acute sinusitis caused by
Haemophilus influenzae,
Streptococcus pneumoniae, or
Moraxella catarrhalis.
Because fluoroquinolones, including ciprofloxacin, have been associated with serious adverse reactions
[see
Warnings and Precautions (5.1-5.16)]
and for some patients acute sinusitis is self-limiting, reserve ciprofloxacin for treatment of acute sinusitis in patients who have no alternative treatment options.
1.13 Usage
To reduce the development of drug-resistant bacteria and maintain the effectiveness of ciprofloxacin and other antibacterial drugs, ciprofloxacin should be used only to treat or prevent infections that are proven or strongly suspected to be caused by susceptible bacteria. When culture and susceptibility information are available, they should be considered in selecting or modifying antibacterial therapy. In the absence of such data, local epidemiology and susceptibility patterns may contribute to the empiric selection of therapy.
If anaerobic organisms are suspected of contributing to the infection, appropriate therapy should be administered. Appropriate culture and susceptibility tests should be performed before treatment in order to isolate and identify organisms causing infection and to determine their susceptibility to ciprofloxacin. Therapy with ciprofloxacin may be initiated before results of these tests are known; once results become available appropriate therapy should be continued.
As with other drugs, some isolates of
Pseudomonas aeruginosa may develop resistance fairly rapidly during treatment with ciprofloxacin. Culture and susceptibility testing performed periodically during therapy will provide information not only on the therapeutic effect of the antimicrobial agent but also on the possible emergence of bacterial resistance.
2 DOSAGE AND ADMINISTRATION
Ciprofloxacin tablets should be administered orally as described in the appropriate Dosage Guidelines tables.
2.1 Dosage in Adults
The determination of dosage and duration for any particular patient must take into consideration the severity and nature of the infection, the susceptibility of the causative microorganism, the integrity of the patient's host-defense mechanisms, and the status of renal and hepatic function. Ciprofloxacin tablets may be administered to adult patients when clinically indicated at the discretion of the physician.
|
Table 1: Adult Dosage Guidelines |
|||
| Infection
| Dose
| Frequency
| Usual Durations1
|
| Skin and Skin Structure
| 500-750 mg
| every 12 hours
| 7 to 14 days
|
| Bone and Joint
| 500-750 mg
| every 12 hours
| 4 to 8 weeks
|
| Complicated Intra-Abdominal
2
| 500 mg
| every 12 hours
| 7 to 14 days
|
| Infectious Diarrhea
| 500 mg
| every 12 hours
| 5 to 7 days
|
| Typhoid Fever
| 500 mg
| every 12 hours
| 10 days
|
| Uncomplicated Urethral and Cervical Gonococcal Infections
| 250 mg
| single dose
| single dose
� |
| Inhalational anthrax (post-exposure)
3
| 500 mg
| every 12 hours
| 60 days
|
| Plague
3
| 500-750 mg
| every 12 hours
| 14 days
|
| Chronic Bacterial Prostatitis
| 500 mg
| every 12 hours
| 28 days
|
| Lower Respiratory Tract Infections
| 500-750 mg
| every 12 hours
| 7 to 14 days
|
| Urinary Tract Infections
| 250-500 mg
| every 12 hours
| 7 to 14 days
|
| Acute Uncomplicated Cystitis
| 250 mg
| every 12 hours
| 3 days
|
| Acute Sinusitis
| 500 mg
| every 12 hours
| 10 days
|
1. Generally ciprofloxacin should be continued for at least 2 days after the signs and symptoms of infection have disappeared, except for inhalational anthrax (post-exposure).
2. Used in conjunction with metronidazole.
3. Begin drug administration as soon as possible after suspected or confirmed exposure.
Conversion of IV to Oral Dosing in Adults
Patients whose therapy is started with ciprofloxacin IV may be switched to ciprofloxacin tablets or oral suspension when clinically indicated at the discretion of the physician (Table 2)
[see
Clinical Pharmacology (12.3)]
.
| Table 2: Equivalent AUC Dosing Regimens
|
|
| Ciprofloxacin Oral Dosage
| Equivalent Ciprofloxacin IV Dosage
|
| 250 mg Tablet every 12 hours
| 200 mg intravenous every 12 hours
|
| 500 mg Tablet every 12 hours
| 400 mg intravenous every 12 hours
|
| 750 mg Tablet every 12 hours
| 400 mg intravenous every 8 hours
|
2.2 Dosage in Pediatric Patients
Dosing and initial route of therapy (that is, IV or oral) for cUTI or pyelonephritis should be determined by the severity of the infection. Ciprofloxacin should be administered as described in Table 3.
| 1. The total duration of therapy for cUTI and pyelonephritis in the clinical trial was determined by the physician. The mean duration of treatment was 11 days (range 10 to 21 days).
2. Begin drug administration as soon as possible after suspected or confirmed exposure. 3. Begin drug administration as soon as possible after suspected or confirmed exposure to Y. pestis. |
|||||||||||||||||||
| Table 3: Pediatric Dosage Guidelines
|
|||||||||||||||||||
|
Infection |
Dose |
Frequency |
Total Duration |
||||||||||||||||
|
Complicated Urinary Tract or Pyelonephritis (patients from 1 to 17 years of age) |
10 mg/kg to 20 mg/kg (maximum 750 mg per dose; not to be exceeded even in patients weighing more than 51 kg) |
Every 12 hours |
10-21 days 1 |
||||||||||||||||
|
Inhalational Anthrax (Post-Exposure) 2 |
15 mg/kg (maximum 500 mg per dose) |
Every 12 hours |
60 days |
||||||||||||||||
|
Plague 2,3 |
15 mg/kg (maximum 500 mg per dose) |
Every 8 to 12 hours |
14 days |
||||||||||||||||
2.3 Dosage Modifications in Patients with Renal Impairment
Ciprofloxacin is eliminated primarily by renal excretion; however, the drug is also metabolized and partially cleared through the biliary system of the liver and through the intestine. These alternative pathways of drug elimination appear to compensate for the reduced renal excretion in patients with renal impairment. Nonetheless, some modification of dosage is recommended, particularly for patients with severe renal dysfunction. Dosage guidelines for use in patients with renal impairment are shown in Table 4.
|
Table 4: Recommended Starting and Maintenance Doses for Adult Patients with Impaired Renal Function |
|
|
Creatinine Clearance (mL/min) |
Dose |
|
>50 |
See Usual Dosage |
|
30 -50 |
250 – 500 mg every 12 hours |
|
5 - 29 |
250 – 500 mg every 18 hours |
|
Patients on hemodialysis or Peritoneal dialysis |
250 – 500 mg every 24 hours (after dialysis) |
When only the serum creatinine concentration is known, the following formulas may be used to estimate creatinine clearance:
Men - Creatinine clearance (mL/min) =
Weight (kg) x (140-age)
72 x serum creatinine (mg/dL)
Women - 0.85 x the value calculated for men.
The serum creatinine should represent a steady state of renal function.
In patients with severe infections and severe renal impairment, a unit dose of 750 mg may be administered at the intervals noted above. Patients should be carefully monitored.
Pediatric patients with moderate to severe renal insufficiency were excluded from the clinical trial of cUTI and pyelonephritis. No information is available on dosing adjustments necessary for pediatric patients with moderate to severe renal insufficiency (that is, creatinine clearance of < 50 mL/min/1.73m 2).
2.4 Important Administration Instructions
With Multivalent Cations
Administer ciprofloxacin tablets at least 2 hours before or 6 hours after magnesium/aluminum antacids; polymeric phosphate binders (for example, sevelamer, lanthanum carbonate) or sucralfate; Videx ® (didanosine) chewable/buffered tablets or pediatric powder for oral solution; other highly buffered drugs; or other products containing calcium, iron or zinc.
With Dairy Products
Concomitant administration of ciprofloxacin tablets with dairy products (like milk or yogurt) or calcium-fortified juices alone should be avoided since decreased absorption is possible; however, ciprofloxacin tablets may be taken with a meal that contains these products.
Hydration of Patients Receiving Ciprofloxacin Tablets
Assure adequate hydration of patients receiving ciprofloxacin tablets to prevent the formation of highly concentrated urine. Crystalluria has been reported with quinolones.
Instruct the patient of the appropriate ciprofloxacin tablet administration [see Patient Counseling Information (17)].
Missed Doses
If a dose is missed, it should be taken anytime but not later than 6 hours prior to the next scheduled dose. If less than 6 hours remain before the next dose, the missed dose should not be taken and treatment should be continued as prescribed with the next scheduled dose. Double doses should not be taken to compensate for a missed dose.
3 DOSAGE FORMS AND STRENGTHS
3.1 Tablets
- For 250 mg: White colored, biconvex, circular, film coated tablets, debossed 'P' on one side and '250' on the other side.
- For 500 mg: White colored, biconvex, capsule shaped, film coated tablets, debossed 'P' on one side and '500' on the other side.
- For 750 mg: White colored, biconvex, capsule shaped, film coated tablets, debossed 'P' on one side and '750' on the other side.
4 CONTRAINDICATIONS
4.1 Hypersensitivity
Ciprofloxacin tablet is contraindicated in persons with a history of hypersensitivity to ciprofloxacin, any member of the quinolone class of antibacterials, or any of the product components [see Warnings and Precautions (5.7)].
4.2 Tizanidine
Concomitant administration with tizanidine is contraindicated [see Drug Interactions (7)].
5 WARNINGS AND PRECAUTIONS
5.1 Disabling and Potentially Irreversible Serious Adverse Reactions Including Tendinitis and Tendon Rupture, Peripheral Neuropathy, and Central Nervous System Effects
Fluoroquinolones, including ciprofloxacin, have been associated with disabling and potentially irreversible serious adverse reactions from different body systems that can occur together in the same patient. Commonly seen adverse reactions include tendinitis, tendon rupture, arthralgia, myalgia, peripheral neuropathy, and central nervous system effects (hallucinations, anxiety, depression, insomnia, severe headaches, and confusion). These reactions can occur within hours to weeks after starting ciprofloxacin tablets. Patients of any age or without pre-existing risk factors have experienced these adverse reactions
[see Warnings and Precautions (
5.2,
5.3,
5.4)].
Discontinue ciprofloxacin tablets immediately at the first signs or symptoms of any serious adverse reaction. In addition, avoid the use of fluoroquinolones, including ciprofloxacin, in patients who have experienced any of these serious adverse reactions associated with fluoroquinolones.
5.2 Tendinitis and Tendon Rupture
Fluoroquinolones, including ciprofloxacin, have been associated with an increased risk of tendinitis and tendon rupture in all ages
[see
Warnings and Precautions (5.1) and
Adverse Reactions (6.2)]
. This adverse reaction most frequently involves the Achilles tendon, and has also been reported with the rotator cuff (the shoulder), the hand, the biceps, the thumb, and other tendons. Tendinitis or tendon rupture can occur, within hours or days of starting ciprofloxacin tablets, or as long as several months after completion of fluoroquinolone therapy. Tendinitis and tendon rupture can occur bilaterally.
The risk of developing fluoroquinolone-associated tendinitis and tendon rupture is increased in patients over 60 years of age, in patients taking corticosteroid drugs, and in patients with kidney, heart or lung transplants. Other factors that may independently increase the risk of tendon rupture include strenuous physical activity, renal failure, and previous tendon disorders such as rheumatoid arthritis. Tendinitis and tendon rupture have also occurred in patients taking fluoroquinolones who do not have the above risk factors. Discontinue ciprofloxacin tablets immediately if the patient experiences pain, swelling, inflammation or rupture of a tendon. Avoid fluoroquinolones, including ciprofloxacin, in patients who have a history of tendon disorders or have experienced tendinitis or tendon rupture
[see
Adverse Reactions (6.2)].
5.3 Peripheral Neuropathy
Fluoroquinolones, including ciprofloxacin, have been associated with an increased risk of peripheral neuropathy. Cases of sensory or sensorimotor axonal polyneuropathy affecting small and/or large axons resulting in paresthesias, hypoesthesias, dysesthesias and weakness have been reported in patients receiving fluoroquinolones, including ciprofloxacin. Symptoms may occur soon after initiation of ciprofloxacin tablets and may be irreversible in some patients
[see Warnings and Precautions (
5.1) and Adverse Reactions (
6.1,
6.2)]
.
Discontinue ciprofloxacin tablets immediately if the patient experiences symptoms of peripheral neuropathy including pain, burning, tingling, numbness, and/or weakness, or other alterations in sensations including light touch, pain, temperature, position sense and vibratory sensation, and/or motor strength in order to minimize the development of an irreversible condition. Avoid fluoroquinolones, including ciprofloxacin, in patients who have previously experienced peripheral neuropathy
[see Adverse Reactions (
6.1,
6.2)].
5.4 Central Nervous System Effects
Psychiatric Adverse Reactions
Fluoroquinolones, including ciprofloxacin, have been associated with an increased risk of psychiatric adverse reactions, including: toxic psychosis, psychotic reactions progressing to suicidal ideations/thoughts, hallucinations, or paranoia; depression, or self-injurious behavior such as attempted or completed suicide; anxiety, agitation, or nervousness; confusion, delirium, disorientation, or disturbances in attention; insomnia or nightmares; memory impairment. These reactions may occur following the first dose. Advise patients receiving ciprofloxacin to inform their healthcare provider immediately if these reactions occur, discontinue the drug, and institute appropriate care.
Central Nervous System Adverse Reactions
Fluoroquinolones, including ciprofloxacin, have been associated with an increased risk of seizures (convulsions), increased intracranial pressure (pseudotumor cerebri), dizziness, and tremors. Ciprofloxacin, like other fluoroquinolones, is known to trigger seizures or lower the seizure threshold. Cases of status epilepticus have been reported. As with all fluoroquinolones, use ciprofloxacin with caution in epileptic patients and patients with known or suspected CNS disorders that may predispose to seizures or lower the seizure threshold (for example, severe cerebral arteriosclerosis, previous history of convulsion, reduced cerebral blood flow, altered brain structure, or stroke), or in the presence of other risk factors that may predispose to seizures or lower the seizure threshold (for example, certain drug therapy, renal dysfunction). If seizures occur, discontinue ciprofloxacin and institute appropriate care
[see
Adverse Reactions (6.1) and
Drug Interactions (7)].
5.5 Exacerbation of Myasthenia Gravis
Fluoroquinolones, including ciprofloxacin, have neuromuscular blocking activity and may exacerbate muscle weakness in patients with myasthenia gravis. Postmarketing serious adverse reactions, including deaths and requirement for ventilatory support, have been associated with fluoroquinolone use in patients with myasthenia gravis. Avoid ciprofloxacin in patients with known history of myasthenia gravis [see Adverse Reactions (6.2)] .
5.6 Other Serious and Sometimes Fatal Adverse Reactions
Other serious and sometimes fatal adverse reactions, some due to hypersensitivity, and some due to uncertain etiology, have been reported in patients receiving therapy with quinolones, including ciprofloxacin. These events may be severe and generally occur following the administration of multiple doses. Clinical manifestations may include one or more of the following:
- Fever, rash, or severe dermatologic reactions (for example, toxic epidermal necrolysis, Stevens-Johnson syndrome);
- Vasculitis; arthralgia; myalgia; serum sickness;
- Allergic pneumonitis;
- Interstitial nephritis; acute renal insufficiency or failure;
- Hepatitis; jaundice; acute hepatic necrosis or failure;
- Anemia, including hemolytic and aplastic; thrombocytopenia, including thrombotic thrombocytopenic purpura; leukopenia; agranulocytosis; pancytopenia; and/or other hematologic abnormalities.
Discontinue ciprofloxacin tablets immediately at the first appearance of a skin rash, jaundice, or any other sign of hypersensitivity and supportive measures instituted [see Adverse Reactions (6.1, 6.2)] .
5.7 Hypersensitivity Reactions
Serious and occasionally fatal hypersensitivity (anaphylactic) reactions, some following the first dose, have been reported in patients receiving fluoroquinolone therapy, including ciprofloxacin. Some reactions were accompanied by cardiovascular collapse, loss of consciousness, tingling, pharyngeal or facial edema, dyspnea, urticaria, and itching. Only a few patients had a history of hypersensitivity reactions. Serious anaphylactic reactions require immediate emergency treatment with epinephrine and other resuscitation measures, including oxygen, intravenous fluids, intravenous antihistamines, corticosteroids, pressor amines, and airway management, including intubation, as indicated [see Adverse Reactions (6.1)].
5.8 Hepatotoxicity
Cases of severe hepatotoxicity, including hepatic necrosis, life-threatening hepatic failure, and fatal events, have been reported with ciprofloxacin. Acute liver injury is rapid in onset (range 1 -39 days), and is often associated with hypersensitivity. The pattern of injury can be hepatocellular, cholestatic, or mixed. Most patients with fatal outcomes were older than 55 years old. In the event of any signs and symptoms of hepatitis (such as anorexia, jaundice, dark urine, pruritus, or tender abdomen), discontinue treatment immediately.
There can be a temporary increase in transaminases, alkaline phosphatase, or cholestatic jaundice, especially in patients with previous liver damage, who are treated with ciprofloxacin [see Adverse Reactions (6.2, 6.3)].
5.9 Risk of Aortic Aneurysm and Dissection
Epidemiologic studies report an increased rate of aortic aneurysm and dissection within two months following use of fluoroquinolones, particularly in elderly patients. The cause for the increased risk has not been identified. In patients with a known aortic aneurysm or patients who are at greater risk for aortic aneurysms, reserve ciprofloxacin tablets for use only when there are no alternative antibacterial treatments available.
5.10 Serious Adverse Reactions with Concomitant Theophylline
Serious and fatal reactions have been reported in patients receiving concurrent administration of ciprofloxacin and theophylline. These reactions have included cardiac arrest, seizure, status epilepticus, and respiratory failure. Instances of nausea, vomiting, tremor, irritability, or palpitation have also occurred.
Although similar serious adverse reactions have been reported in patients receiving theophylline alone, the possibility that these reactions may be potentiated by ciprofloxacin cannot be eliminated. If concomitant use cannot be avoided, monitor serum levels of theophylline and adjust dosage as appropriate [see Drug Interactions (7)].
5.11 Clostridioides difficile -Associated Diarrhea
Clostridioides difficile (C. difficile)-associated diarrhea (CDAD) has been reported with use of nearly all antibacterial agents, including ciprofloxacin, and may range in severity from mild diarrhea to fatal colitis. Treatment with antibacterial agents alters the normal flora of the colon leading to overgrowth of C. difficile.
C. difficile produces toxins A and B which contribute to the development of CDAD. Hypertoxin producing isolates of C. difficile cause increased morbidity and mortality, as these infections can be refractory to antimicrobial therapy and may require colectomy. CDAD must be considered in all patients who present with diarrhea following antibacterial use. Careful medical history is necessary since CDAD has been reported to occur over two months after the administration of antibacterial agents.
If CDAD is suspected or confirmed, ongoing antibacterial use not directed against C. difficile may need to be discontinued. Appropriate fluid and electrolyte management, protein supplementation, antibacterial treatment of C. difficile, and institute surgical evaluation as clinically indicated [see Adverse Reactions (6.1)].
5.12 Prolongation of the QT Interval
Some fluoroquinolones, including ciprofloxacin, have been associated with prolongation of the QT interval on the electrocardiogram and cases of arrhythmia. Cases of torsade de pointes have been reported during postmarketing surveillance in patients receiving fluoroquinolones, including ciprofloxacin.
Avoid ciprofloxacin in patients with known prolongation of the QT interval, risk factors for QT prolongation or torsade de pointes (for example, congenital long QT syndrome, uncorrected electrolyte imbalance, such as hypokalemia or hypomagnesemia and cardiac disease, such as heart failure, myocardial infarction, or bradycardia), and patients receiving Class IA antiarrhythmic agents (quinidine, procainamide), or Class III antiarrhythmic agents (amiodarone, sotalol), tricyclic antidepressants, macrolides, and antipsychotics. Elderly patients may also be more susceptible to drug-associated effects on the QT interval
[see
Adverse Reactions (6.2),
Use in Specific Populations (8.5)].
5.13 Musculoskeletal Disorders in Pediatric Patients and Arthropathic Effects in Animals
Ciprofloxacin is indicated in pediatric patients (less than 18 years of age) only for cUTI, prevention of inhalational anthrax (post exposure), and plague [see Indications and Usage ( 1.7, 1.8, 1.11)] . An increased incidence of adverse reactions compared to controls, including reactions related to joints and/or surrounding tissues, has been observed [see Adverse Reactions (6.1)] .
In pre-clinical studies, oral administration of ciprofloxacin caused lameness in immature dogs. Histopathological examination of the weight-bearing joints of these dogs revealed permanent lesions of the cartilage. Related quinolone-class drugs also produce erosions of cartilage of weight-bearing joints and other signs of arthropathy in immature animals of various species [see Use in Specific Populations (8.4) and Nonclinical Toxicology (13.2)].
5.14 Photosensitivity/Phototoxicity
Moderate to severe photosensitivity/phototoxicity reactions, the latter of which may manifest as exaggerated sunburn reactions (for example, burning, erythema, exudation, vesicles, blistering, edema) involving areas exposed to light (typically the face, "V" area of the neck, extensor surfaces of the forearms, dorsa of the hands), can be associated with the use of quinolones including ciprofloxacin after sun or UV light exposure. Therefore, avoid excessive exposure to these sources of light. Discontinue ciprofloxacin if phototoxicity occurs [see Adverse Reactions (6.1)].
5.15 Development of Drug Resistant Bacteria
Prescribing ciprofloxacin tablets in the absence of a proven or strongly suspected bacterial infection or a prophylactic indication is unlikely to provide benefit to the patient and increases the risk of the development of drug-resistant bacteria.
5.16 Potential Risks With Concomitant Use of Drugs Metabolized by Cytochrome P450 1A2 Enzymes
Ciprofloxacin is an inhibitor of the hepatic CYP1A2 enzyme pathway. Co-administration of ciprofloxacin and other drugs primarily metabolized by CYP1A2 (for example, theophylline, methylxanthines, caffeine, tizanidine, ropinirole, clozapine, olanzapine and zolpidem), results in increased plasma concentrations of the co-administered drug and could lead to clinically significant pharmacodynamic adverse reactions of the co-administered drug [see Drug Interactions (7) and Clinical Pharmacology (12.3)].
5.17 Interference with Timely Diagnosis of Syphilis
Ciprofloxacin has not been shown to be effective in the treatment of syphilis. Antimicrobial agents used in high dose for short periods of time to treat gonorrhea may mask or delay the symptoms of incubating syphilis. Perform a serologic test for syphilis in all patients with gonorrhea at the time of diagnosis. Perform follow-up serologic test for syphilis three months after ciprofloxacin treatment.
5.18 Crystalluria
Crystals of ciprofloxacin have been observed rarely in the urine of human subjects but more frequently in the urine of laboratory animals, which is usually alkaline [see Nonclinical Toxicology (13.2)]. Crystalluria related to ciprofloxacin has been reported only rarely in humans because human urine is usually acidic. Avoid alkalinity of the urine in patients receiving ciprofloxacin. Hydrate patients well to prevent the formation of highly concentrated urine [see Dosage and Administration (2.4)] .
5.19 Blood Glucose Disturbances
Fluoroquinolones, including ciprofloxacin, have been associated with disturbances of blood glucose, including symptomatic hyperglycemia and hypoglycemia, usually in diabetic patients receiving concomitant treatment with an oral hypoglycemic agent (for example, glyburide) or with insulin. In these patients, careful monitoring of blood glucose is recommended. Severe cases of hypoglycemia resulting in coma or death have been reported. If a hypoglycemic reaction occurs in a patient being treated with ciprofloxacin, discontinue ciprofloxacin and initiate appropriate therapy immediately [ see Adverse Reactions (6.1), Drug Interactions (7)].
6 ADVERSE REACTIONS
The following serious and otherwise important adverse drug reactions are discussed in greater detail in other sections of labeling:
- Disabling and Potentially Irreversible Serious Adverse Reactions [see Warnings and Precautions (5.1)]
- Tendinitis and Tendon Rupture [see Warnings and Precautions (5.2)]
- Peripheral Neuropathy [see Warnings and Precautions (5.3)]
- Central Nervous System Effects [see Warnings and Precautions (5.4)]
- Exacerbation of Myasthenia Gravis [see Warnings and Precautions (5.5)]
- Other Serious and Sometimes Fatal Adverse Reactions [see Warnings and Precautions (5.6)]
- Hypersensitivity Reactions [see Warnings and Precautions (5.7)]
- Hepatotoxicity [see Warnings and Precautions (5.8)]
- Risk of Aortic Aneurysm and Dissection [see Warnings and Precautions (5.9)]
- Serious Adverse Reactions with Concomitant Theophylline [see Warnings and Precautions (5.10)]
- Clostridioides difficile-Associated Diarrhea [see Warnings and Precautions (5.11)]
- Prolongation of the QT Interval [see Warnings and Precautions (5.12)]
- Musculoskeletal Disorders in Pediatric Patients [see Warnings and Precautions (5.13)]
- Photosensitivity/Phototoxicity [see Warnings and Precautions (5.14)]
- Development of Drug Resistant Bacteria [see Warnings and Precautions (5.15)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adult Patients
During clinical investigations with oral and parenteral ciprofloxacin, 49,038 patients received courses of the drug.
The most frequently reported adverse reactions, from clinical trials of all formulations, all dosages, all drug-therapy durations, and for all indications of ciprofloxacin therapy were nausea (2.5%), diarrhea (1.6%), liver function tests abnormal (1.3%), vomiting (1%), and rash (1%).
Table 8: Medically Important Adverse Reactions That Occurred In less than 1% of Ciprofloxacin Patients
|
System Organ Class |
Adverse Reactions |
|---|---|
|
Body as a Whole |
Headache Abdominal Pain/Discomfort Pain |
|
Cardiovascular |
Syncope Angina Pectoris Myocardial Infarction Cardiopulmonary Arrest Tachycardia Hypotension |
|
Central Nervous System |
Restlessness Dizziness Insomnia Nightmares Hallucinations Paranoia Psychosis (toxic) Manic Reaction Irritability Tremor Ataxia Seizures (including Status Epilepticus) Malaise Anorexia Phobia Depersonalization Depression (potentially culminating in self-injurious behavior (such as suicidal ideations/ thoughts and attempted or completed suicide) Paresthesia Abnormal Gait Migraine |
|
Gastrointestinal |
Intestinal Perforation Gastrointestinal Bleeding Cholestatic Jaundice Hepatitis Pancreatitis |
|
Hemic/Lymphatic |
Petechia |
|
Metabolic/Nutritional |
Hyperglycemia Hypoglycemia |
|
Musculoskeletal |
Arthralgia Joint Stiffness Muscle Weakness |
|
Renal/Urogenital |
Interstitial Nephritis Renal Failure |
|
Respiratory |
Dyspnea Laryngeal Edema Hemoptysis Bronchospasm |
|
Skin/Hypersensitivity |
Anaphylactic Reactions including life-threatening anaphylactic shock Erythema Multiforme/Stevens-Johnson Syndrome Exfoliative Dermatitis Toxic Epidermal Necrolysis Pruritus Urticaria Photosensitivity/Phototoxicity reaction Flushing Fever Angioedema Erythema Nodosum Sweating |
|
Special Senses |
Blurred Vision Disturbed Vision (chromatopsia and photopsia) Decreased Visual Acuity Diplopia Tinnitus Hearing Loss Bad Taste |
In randomized, double-blind controlled clinical trials comparing ciprofloxacin tablets [500 mg two times daily (BID)] to cefuroxime axetil (250 mg–500 mg BID) and to clarithromycin (500 mg BID) in patients with respiratory tract infections, ciprofloxacin demonstrated a CNS adverse reaction profile comparable to the control drugs.
Pediatric Patients
Short (6 weeks) and long term (1 year) musculoskeletal and neurological safety of oral/ intravenous ciprofloxacin, was compared to a cephalosporin for treatment of cUTI or pyelonephritis in pediatric patients 1 to 17 years of age (mean age of 6 ± 4 years) in an international multicenter trial. The duration of therapy was 10 to 21 days (mean duration of treatment was 11 days with a range of 1 to 88 days). A total of 335 ciprofloxacin- and 349 comparator-treated patients were enrolled.
An Independent Pediatric Safety Committee (IPSC) reviewed all cases of musculoskeletal adverse reactions including abnormal gait or abnormal joint exam (baseline or treatment-emergent). Within 6 weeks of treatment initiation, the rates of musculoskeletal adverse reactions were 9.3% (31/335) in the ciprofloxacin-treated group versus 6% (21/349) in comparator-treated patients. All musculoskeletal adverse reactions occurring by 6 weeks resolved (clinical resolution of signs and symptoms), usually within 30 days of end of treatment. Radiological evaluations were not routinely used to confirm resolution of the adverse reactions. Ciprofloxacin-treated patients were more likely to report more than one adverse reaction and on more than one occasion compared to control patients. The rate of musculoskeletal adverse reactions was consistently higher in the ciprofloxacin group compared to the control group across all age subgroups. At the end of 1 year, the rate of these adverse reactions reported at any time during that period was 13.7% (46/335) in the ciprofloxacin-treated group versus 9.5% (33/349) in the comparator-treated patients (Table 9).
Table 9: Musculoskeletal Adverse Reactions 1 as Assessed by the IPSC
|
| Ciprofloxacin Tablets
| Comparator
|
| All Patients (within 6 weeks)
| 31/335 (9.3%)
| 21/349 (6%)
|
| 95% Confidence Interval
2
| (-0.8%, +7.2%)
|
|
| Age Group
|
||
| 12 months < 24 months
| 1/36 (2.8%)
| 0/41
|
| 2 years < 6 years
| 5/124 (4%)
| 3/118 (2.5%)
|
| 6 years < 12 years
| 18/143 (12.6%)
| 12/153 (7.8%)
|
| 12 years to 17 years
| 7/32 (21.9%)
| 6/37 (16.2%)
|
|
All Patients (within 1 year) |
46/335 (13.7%) |
33/349 (9.5%) |
| 95% Confidence Interval
1
| (-0.6%, + 9.1%)
|
|
- Included: arthralgia, abnormal gait, abnormal joint exam, joint sprains, leg pain, back pain, arthrosis, bone pain, pain, myalgia, arm pain, and decreased range of motion in a joint (knee, elbow, ankle, hip, wrist, and shoulder)
- The study was designed to demonstrate that the arthropathy rate for the ciprofloxacin group did not exceed that of the control group by more than + 6%. At both the 6 week and 1 year evaluations, the 95% confidence interval indicated that it could not be concluded that the ciprofloxacin group had findings comparable to the control group.
The incidence rates of neurological adverse reactions within 6 weeks of treatment initiation were 3% (9/335) in the ciprofloxacin group versus 2% (7/349) in the comparator group and included dizziness, nervousness, insomnia, and somnolence.
In this trial, the overall incidence rates of adverse reactions within 6 weeks of treatment initiation were 41% (138/335) in the ciprofloxacin group versus 31% (109/349) in the comparator group. The most frequent adverse reactions were gastrointestinal: 15% (50/335) of ciprofloxacin patients compared to 9% (31/349) of comparator patients. Serious adverse reactions were seen in 7.5% (25/335) of ciprofloxacin-treated patients compared to 5.7% (20/349) of control patients. Discontinuation of drug due to an adverse reaction was observed in 3% (10/335) of ciprofloxacin-treated patients versus 1.4% (5/349) of comparator patients. Other adverse reactions that occurred in at least 1% of ciprofloxacin patients were diarrhea 4.8%, vomiting 4.8%, abdominal pain 3.3%, dyspepsia 2.7%, nausea 2.7%, fever 2.1%, asthma 1.8% and rash 1.8%.
Short-term safety data for ciprofloxacin was also collected in a randomized, double-blind clinical trial for the treatment of acute pulmonary exacerbations in cystic fibrosis patients (ages 5–17 years). Sixty-seven patients received ciprofloxacin IV 10 mg/kg/dose every 8 hours for one week followed by ciprofloxacin tablets 20 mg/kg/dose every 12 hours to complete 10–21 days treatment and 62 patients received the combination of ceftazidime intravenous 50 mg/kg/dose every 8 hours and tobramycin intravenous 3 mg/kg/dose every 8 hours for a total of 10–21 days. Periodic musculoskeletal assessments were conducted by treatment-blinded examiners. Patients were followed for an average of 23 days after completing treatment (range 0–93 days). Musculoskeletal adverse reactions were reported in 22% of the patients in the ciprofloxacin group and 21% in the comparison group. Decreased range of motion was reported in 12% of the subjects in the ciprofloxacin group and 16% in the comparison group. Arthralgia was reported in 10% of the patients in the ciprofloxacin group and 11% in the comparison group. Other adverse reactions were similar in nature and frequency between treatment arms. The efficacy of ciprofloxacin tablets for the treatment of acute pulmonary exacerbations in pediatric cystic fibrosis patients has not been established.
In addition to the adverse reactions reported in pediatric patients in clinical trials, it should be expected that adverse reactions reported in adults during clinical trials or postmarketing experience may also occur in pediatric patients.
6.2 Postmarketing Experience
The following adverse reactions have been reported from worldwide marketing experience with fluoroquinolones, including ciprofloxacin tablets. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure (Table 10).
| Table 10: Postmarketing Reports of Adverse Drug Reactions
|
|
| System Organ Class
| Adverse Reactions
|
| Cardiovascular
| QT prolongation
Torsade de Pointes Vasculitis and ventricular arrhythmia |
| Central Nervous System
| Hypertonia
Myasthenia Exacerbation of myasthenia gravis Peripheral neuropathy Polyneuropathy Twitching |
| Eye Disorders
| Nystagmus
|
| Gastrointestinal
| Pseudomembranous colitis
|
| Hemic/Lymphatic
| Pancytopenia (life threatening or fatal outcome)
Methemoglobinemia |
| Hepatobiliary
| Hepatic failure (including fatal cases)
|
| Infections and Infestations
| Candidiasis (oral, gastrointestinal, vaginal)
|
| Investigations
| Prothrombin time prolongation or decrease
Cholesterol elevation (serum) Potassium elevation (serum) |
| Musculoskeletal
| Myalgia
Myoclonus Tendinitis Tendon rupture |
| Psychiatric Disorders
| Agitation
Confusion Delirium |
| Skin/Hypersensitivity
| Acute generalize exanthematous pustulosis (AGEP)
Fixed eruption Serum sickness-like reaction |
| Special Senses
| Anosmia
Hyperesthesia Hypesthesia Taste loss |
6.3 Adverse Laboratory Changes
Changes in laboratory parameters while on ciprofloxacin are listed below:
Hepatic–Elevations of ALT (SGPT), AST (SGOT), alkaline phosphatase, LDH, serum bilirubin. Hematologic–Eosinophilia, leukopenia, decreased blood platelets, elevated blood platelets, pancytopenia. Renal–Elevations of serum creatinine, BUN, crystalluria, cylindruria, and hematuria have been reported.
Other changes occurring were: elevation of serum gammaglutamyl transferase, elevation of serum amylase, reduction in blood glucose, elevated uric acid, decrease in hemoglobin, anemia, bleeding diathesis, increase in blood monocytes, and leukocytosis.
7 DRUG INTERACTIONS
Ciprofloxacin is an inhibitor of human cytochrome P450 1A2 (CYP1A2) mediated metabolism. Co-administration of ciprofloxacin with other drugs primarily metabolized by CYP1A2 results in increased plasma concentrations of these drugs and could lead to clinically significant adverse events of the co-administered drug.
| Table 11: Drugs That are Affected by and Affecting Ciprofloxacin
|
||
|
Drugs That are Affected by Ciprofloxacin |
||
| Drug(s)
| Recommendation
| Comments
|
| Tizanidine
| Contraindicated
| Concomitant administration of tizanidine and ciprofloxacin is contraindicated due to the potentiation of hypotensive and sedative effects of tizanidine
[see
Contraindications (4.2)]
.
|
| Theophylline
| Avoid Use
(Plasma Exposure Likely to be Increased and Prolonged) | Concurrent administration of ciprofloxacin with theophylline may result in increased risk of a patient developing central nervous system (CNS) or other adverse reactions. If concomitant use cannot be avoided, monitor serum levels of theophylline and adjust dosage as appropriate
[see
Warnings and Precautions (5.10)]
.
|
| Drugs Known to Prolong QT Interval
| Avoid Use
| Ciprofloxacin may further prolong the QT interval in patients receiving drugs known to prolong the QT interval (for example, class IA or III antiarrhythmics, tricyclic antidepressants, macrolides, antipsychotics)
[see
Warnings and Precautions (5.12) and
Use in Specific Populations (8.5)].
|
| Oral antidiabetic drugs
| Use with caution
Glucose-lowering effect potentiated | Hypoglycemia sometimes severe has been reported when ciprofloxacin and oral antidiabetic agents, mainly sulfonylureas (for example, glyburide, glimepiride), were co-administered, presumably by intensifying the action of the oral antidiabetic agent. Fatalities have been reported
. Monitor blood glucose when ciprofloxacin is co-administered with oral antidiabetic drugs
[see
Adverse Reactions (6.1)] .
|
| Phenytoin
| Use with caution
Altered serum levels of phenytoin (increased and decreased) | To avoid the loss of seizure control associated with decreased phenytoin levels and to prevent phenytoin overdose-related adverse reactions upon ciprofloxacin discontinuation in patients receiving both agents, monitor phenytoin therapy, including phenytoin serum concentration during and shortly after co-administration of ciprofloxacin with phenytoin.
|
| Cyclosporine
| Use with caution
(transient elevations in serum creatinine) | Monitor renal function (in particular serum creatinine) when ciprofloxacin is co-administered with cyclosporine.
|
| Anti-coagulant drugs
| Use with caution
(Increase in anticoagulant effect) | The risk may vary with the underlying infection, age and general status of the patient so that the contribution of ciprofloxacin to the increase in INR (international normalized ratio) is difficult to assess. Monitor prothrombin time and INR frequently during and shortly after co-administration of ciprofloxacin with an oral anti-coagulant (for example, warfarin).
|
| Methotrexate
| Use with caution
Inhibition of methotrexate renal tubular transport potentially leading to increased methotrexate plasma levels | Potential increase in the risk of methotrexate associated toxic reactions. Therefore, carefully monitor patients under methotrexate therapy when concomitant ciprofloxacin therapy is indicated.
|
| Ropinirole
| Use with caution
| Monitoring for ropinirole-related adverse reactions and appropriate dose adjustment of ropinirole is recommended during and shortly after co-administration with ciprofloxacin
[see
Warnings and Precautions (5.16)].
|
| Clozapine
| Use with caution
| Careful monitoring of clozapine associated adverse reactions and appropriate adjustment of clozapine dosage during and shortly after co-administration with ciprofloxacin are advised.
|
| NSAIDs
| Use with caution
| Non-steroidal anti-inflammatory drugs (but not acetyl salicylic acid) in combination of very high doses of quinolones have been shown to provoke convulsions in pre-clinical studies and in postmarketing.
|
| Sildenafil
| Use with caution
Two-fold increase in exposure | Monitor for sildenafil toxicity
[see
Clinical Pharmacology(12.3)].
|
| Duloxetine
| Avoid Use
Five-fold increase in duloxetine exposure | If unavoidable, monitor for duloxetine toxicity
|
| Caffeine/Xanthine Derivatives
| Use with caution
Reduced clearance resulting in elevated levels and prolongation of serum half-life | Ciprofloxacin inhibits the formation of paraxanthine after caffeine administration (or pentoxifylline containing products). Monitor for xanthine toxicity and adjust dose as necessary.
|
| Zolpidem
| Avoid Use
| Co-administration with ciprofloxacin may increase blood levels of zolpidem, concurrent use is not recommended.
|
|
Drug(s) Affecting Pharmacokinetics of Ciprofloxacin |
||
| Antacids, Sucralfate, Multivitamins and Other Products Containing Multivalent Cations (magnesium/aluminum antacids; polymeric phosphate binders (for example, sevelamer, lanthanum carbonate); sucralfate; Videx
® (didanosine) chewable/buffered tablets or pediatric powder; other highly buffered drugs; or products containing calcium, iron, or zinc and dairy products)
| Ciprofloxacin should be taken at least two hours before or six hours after Multivalent cation-containing products administration
[see
Dosage and Administration (2.4)]
| Decrease ciprofloxacin absorption, resulting in lower serum and urine levels
|
| Probenecid
| Use with caution
(interferes with renal tubular secretion of ciprofloxacin and increases ciprofloxacin serum levels) | Potentiation of ciprofloxacin toxicity may occur.
|
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Prolonged experience with ciprofloxacin in pregnant women over several decades, based on available published information from case reports, case control studies and observational studies on ciprofloxacin administered during pregnancy, have not identified any drug-associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes
(see Data). Oral administration of ciprofloxacin during organogenesis at doses up to 100 mg/kg to pregnant mice and rats, and up to 30 mg/kg to pregnant rabbits did not cause fetal malformations
(see Data). These doses were up to 0.3, 0.6, and 0.4 times the maximum recommended clinical oral dose in mice, rats, and rabbits, respectively, based on body surface area. The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risks of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Human Data
While available studies cannot definitively establish the absence of risk, published data from prospective observational studies over several decades have not established an association with ciprofloxacin use during pregnancy and major birth defects, miscarriage, or adverse maternal or fetal outcomes. Available studies have methodological limitations including small sample size and some of them are not specific for ciprofloxacin. A controlled prospective observational study followed 200 women exposed to fluoroquinolones (52.5% exposed to ciprofloxacin and 68% first trimester exposures) during gestation. In utero exposure to fluoroquinolones during embryogenesis was not associated with increased risk of major malformations. The reported rates of major congenital malformations were 2.2% for the fluoroquinolone group and 2.6% for the control group (background incidence of major malformations is 1–5%). Rates of spontaneous abortions, prematurity and low birth weight did not differ between the groups and there were no clinically significant musculoskeletal dysfunctions up to one year of age in the ciprofloxacin exposed children.
Another prospective follow-up study reported on 549 pregnancies with fluoroquinolone exposure (93% first trimester exposures). There were 70 ciprofloxacin exposures, all within the first trimester. The malformation rates among live-born babies exposed to ciprofloxacin and to fluoroquinolones overall were both within background incidence ranges. No specific patterns of congenital abnormalities were found. The study did not reveal any clear adverse reactions due to in utero exposure to ciprofloxacin.
No differences in the rates of prematurity, spontaneous abortions, or birth weight were seen in women exposed to ciprofloxacin during pregnancy. However, these small postmarketing epidemiology studies, of which most experience is from short term, first trimester exposure, are insufficient to evaluate the risk for less common defects or to permit reliable and definitive conclusions regarding the safety of ciprofloxacin in pregnant women and their developing fetuses.
Animal Data
Developmental toxicology studies have been performed with ciprofloxacin in rats, mice, and rabbits. In rats and mice, oral doses up to 100 mg/kg administered during organogenesis (Gestation Days, GD, 6-17) were not associated with adverse developmental outcomes, including embryofetal toxicity or malformations. In rats and mice, a 100 mg/kg dose is approximately 0.6 and 0.3 times the maximum daily human oral dose (1500 mg/day) based upon body surface area, respectively. In a series of rabbit developmental toxicology studies, does received oral or intravenous ciprofloxacin for one of the following 5 day periods: GD 6 to 10, GD 10 to 14, or GD 14 to 18, intended to cover the period of organogenesis. This was an attempt to mitigate the gastrointestinal intolerance observed in rabbits that receive antibacterials manifested by reduced maternal food consumption and weight loss, that can lead to embryofetal resorption or spontaneous abortion. An oral ciprofloxacin dose of 100 mg/kg (approximately 1.3 times the highest recommended clinical oral dose based on body surface area) caused excessive maternal toxicity confounding evaluation of the fetuses. A 30 mg/kg oral dose (approximately 0.4 times the highest recommended clinical oral dose) was associated with suppression of maternal and fetal body weight gain, but fetal malformations were not observed. Intravenous administration of doses up to 20 mg/kg (approximately 0.3 times the highest recommended clinical oral dose based upon body surface area) to pregnant rabbits was not maternally toxic and neither embryofetal toxicity nor fetal malformations were observed.
In peri-and post-natal studies, rats received ciprofloxacin doses up to 200 mg/kg/day (oral) or up to 30 mg/kg/day (subcutaneous) from GD 16 to 22 days postpartum. The 200 mg/kg dose is approximately 1.3-times the maximum recommended clinical oral dose based on body surface area. Neither maternal toxicity nor adverse effects on growth and development of the pups were observed, including no sign of arthropathy on the rear leg joints of the pups. Ciprofloxacin and other quinolones have been shown to cause arthropathy in immature animals of most species tested when administered directly [see Warnings and Precautions (5.13) and Nonclinical Toxicology 13.2].
8.2 Lactation
Risk Summary
Published literature reports that ciprofloxacin is present in human milk following intravenous and oral administration
. There is no information regarding effects of ciprofloxacin on milk production or the breastfed infant. Because of the potential risk of serious adverse reactions in breastfed infants, including arthropathy shown in juvenile animal studies
[see
Use in Specific Populations (8.4), (Clinical Considerations)],
for most indications a lactating woman may consider pumping and discarding breast milk during treatment with ciprofloxacin and an additional two days (five half-lives) after the last dose. Alternatively, advise a woman that breastfeeding is not recommended during treatment with ciprofloxacin and for an additional two days (five half-lives) after the last dose.
However, for inhalation anthrax (post exposure), during an incident resulting in exposure to anthrax, the risk-benefit assessment of continuing breastfeeding while the mother (and potentially the infant) is (are) on ciprofloxacin may be acceptable [see Dosage and Administration (2.2), Pediatric Use (8.4), and Clinical Studies (14.2)]. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for ciprofloxacin and any potential adverse effects on the breastfed child from ciprofloxacin or from the underlying maternal condition.
Clinical Considerations
Ciprofloxacin may cause intestinal flora alteration of the breastfeeding infant. Advise a woman to monitor the breastfed infant for loose or bloody stools and candidiasis (thrush, diaper rash).
8.4 Pediatric Use
Although effective in clinical trials, ciprofloxacin is not a drug of first choice in the pediatric population due to an increased incidence of adverse reactions compared to controls. Quinolones, including ciprofloxacin, cause arthropathy (arthralgia, arthritis), in juvenile animals [see Warnings and Precautions (5.13) and Nonclinical Toxicology (13.2)] .
Complicated Urinary Tract Infection and Pyelonephritis
Ciprofloxacin is indicated for the treatment of cUTI and pyelonephritis due to Escherichia coli in pediatric patients 1 to 17 years of age . Although effective in clinical trials, ciprofloxacin is not a drug of first choice in the pediatric population due to an increased incidence of adverse reactions compared to the controls, including events related to joints and/or surrounding tissues [see Adverse Reactions (6.1) and Clinical Studies (14.1)].
Inhalational Anthrax (Post-Exposure)
Ciprofloxacin is indicated in pediatric patients from birth to 17 years of age, for inhalational anthrax (post-exposure). The risk-benefit assessment indicates that administration of ciprofloxacin to pediatric patients is appropriate [see Dosage and Administration (2.2) and Clinical Studies (14.2)].
Plague
Ciprofloxacin is indicated in pediatric patients from birth to 17 years of age, for treatment of plague, including pneumonic and septicemic plague due to Yersinia pestis (Y. pestis) and prophylaxis for plague. Efficacy studies of ciprofloxacin could not be conducted in humans with pneumonic plague for feasibility reasons. Therefore, approval of this indication was based on an efficacy study conducted in animals. The risk-benefit assessment indicates that administration of ciprofloxacin to pediatric patients is appropriate [see Indications and Usage (1.8), Dosage and Administration (2.2) and Clinical Studies (14.3)].
8.5 Geriatric Use
Geriatric patients are at increased risk for developing severe tendon disorders including tendon rupture when being treated with a fluoroquinolone such as ciprofloxacin. This risk is further increased in patients receiving concomitant corticosteroid therapy. Tendinitis or tendon rupture can involve the Achilles, hand, shoulder, or other tendon sites and can occur during or after completion of therapy; cases occurring up to several months after fluoroquinolone treatment have been reported. Caution should be used when prescribing ciprofloxacin to elderly patients especially those on corticosteroids. Patients should be informed of this potential adverse reaction and advised to discontinue ciprofloxacin and contact their healthcare provider if any symptoms of tendinitis or tendon rupture occur
[see
Boxed Warning,
Warnings and Precautions (5.2), and
Adverse Reactions (6.2)].
Epidemiologic studies report an increased rate of aortic aneurysm and dissection within two months following use of fluoroquinolones, particularly in elderly patients
[see
Warnings and Precautions (5.9)].
In a retrospective analysis of 23 multiple-dose controlled clinical trials of ciprofloxacin encompassing over 3500 ciprofloxacin-treated patients, 25% of patients were greater than or equal to 65 years of age and 10% were greater than or equal to 75 years of age. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals on any drug therapy cannot be ruled out. Ciprofloxacin is known to be substantially excreted by the kidney, and the risk of adverse reactions may be greater in patients with impaired renal function. No alteration of dosage is necessary for patients greater than 65 years of age with normal renal function. However, since some older individuals experience reduced renal function by virtue of their advanced age, care should be taken in dose selection for elderly patients, and renal function monitoring may be useful in these patients
[see
Dosage and Administration (2.3) and
Clinical Pharmacology (12.3)].
In general, elderly patients may be more susceptible to drug-associated effects on the QT interval. Therefore, precaution should be taken when using ciprofloxacin with concomitant drugs that can result in prolongation of the QT interval (for example, class IA or class III antiarrhythmics) or in patients with risk factors for torsade de pointes (for example, known QT prolongation, uncorrected hypokalemia) [see Warnings and Precautions (5.12)].
8.6 Renal Impairment
Ciprofloxacin is eliminated primarily by renal excretion; however, the drug is also metabolized and partially cleared through the biliary system of the liver and through the intestine. These alternative pathways of drug elimination appear to compensate for the reduced renal excretion in patients with renal impairment. Nonetheless, some modification of dosage is recommended, particularly for patients with severe renal dysfunction [see Dosage and Administration (2.3) and Clinical Pharmacology (12.3)].
10 OVERDOSAGE
In the event of acute overdosage, reversible renal toxicity has been reported in some cases. Empty the stomach by inducing vomiting or by gastric lavage. Observe the patient carefully and give supportive treatment, including monitoring of renal function, urinary pH and acidify, if required, to prevent crystalluria and administration of magnesium, aluminum, or calcium containing antacids which can reduce the absorption of ciprofloxacin. Adequate hydration must be maintained. Only a small amount of ciprofloxacin (less than 10%) is removed from the body after hemodialysis or peritoneal dialysis.
11 DESCRIPTION
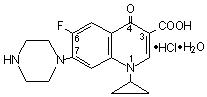
Ciprofloxacin (ciprofloxacin hydrochloride) tablets USP are synthetic antimicrobial agents for oral administration. Ciprofloxacin hydrochloride, USP, a fluoroquinolone, is the monohydrochloride monohydrate salt of 1-cyclopropyl-6-fluoro-1, 4-dihydro-4-oxo-7-(1-piperazinyl)-3-quinolinecarboxylic acid. It is a faintly yellowish to light yellow crystalline substance with a molecular weight of 385.8. Its empirical formula is C 17H 18FN 3O 3 •HCl •H 2O and its chemical structure is as follows:
Ciprofloxacin film-coated tablets USP are available in 250 mg, 500 mg and 750 mg (ciprofloxacin equivalent) strengths. Each ciprofloxacin film-coated tablet contains 250 mg (equivalent to 291 mg ciprofloxacin hydrochloride monohydrate), 500 mg of ciprofloxacin (equivalent to 582 mg ciprofloxacin hydrochloride monohydrate) or 750 mg of ciprofloxacin (equivalent to 873 mg ciprofloxacin hydrochloride monohydrate). Ciprofloxacin tablets USP are white film coated tablets. The inactive ingredients are colloidal silicon dioxide, crospovidone, hypromellose, magnesium stearate, microcrystalline cellulose, polyethylene glycol, pregelatinized starch and titanium dioxide.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Ciprofloxacin is a member of the fluoroquinolone class of antibacterial agents [see Microbiology (12.4)] .
12.3 Pharmacokinetics
Absorption
The absolute bioavailability of ciprofloxacin when given as an oral tablet is approximately 70% with no substantial loss by first pass metabolism. Ciprofloxacin maximum serum concentrations (C max) and area under the curve (AUC) are shown in the chart for the 250 mg to 1000 mg dose range (Table 12).
| Table 12: Ciprofloxacin C
max
and AUC Following Administration of Single Doses of Ciprofloxacin Tablets to Healthy Subjects
|
||
|
Dose (mg) |
C max (mcg/mL) |
AUC (mcg•hr/mL) |
|
250 |
1.2 |
4.8 |
|
500 |
2.4 |
11.6 |
|
750 |
4.3 |
20.2 |
|
1000 |
5.4 |
30.8 |
Maximum serum concentrations are attained 1 to 2 hours after oral dosing. Mean concentrations 12 hours after dosing with 250, 500, or 750 mg are 0.1, 0.2, and 0.4 mcg/mL, respectively. The serum elimination half-life in subjects with normal renal function is approximately 4 hours. Serum concentrations increase proportionately with doses up to 1000 mg.
A 500 mg oral dose given every 12 hours has been shown to produce an AUC equivalent to that produced by an intravenous infusion of 400 mg ciprofloxacin given over 60 minutes every 12 hours. A 750 mg oral dose given every 12 hours has been shown to produce an AUC at steady-state equivalent to that produced by an intravenous infusion of 400 mg given over 60 minutes every 8 hours. A 750 mg oral dose results in a C max similar to that observed with a 400 mg intravenous dose (Table 13). A 250 mg oral dose given every 12 hours produces an AUC equivalent to that produced by an infusion of 200 mg ciprofloxacin given every 12 hours.
| Table 13: Steady-state Pharmacokinetic Parameters Following Multiple Oral and Intravenous Doses (Adults)
|
||||
| Parameters
|
500 mg |
400 mg |
750 mg |
400 mg |
|
every 12 hours, orally |
every 12 hours, intravenously |
every 12 hours, orally |
every 8 hours, intravenously |
|
| AUC
0-24h,ss (μg•h/mL)
|
27.4* |
25.4* |
31.6* |
32.9** |
| C
max,ss (μg/mL)
|
2.97 |
4.56 |
3.59 |
4.07 |
*AUC
0–12h x 2
**AUC
0-8h x 3
Food
When ciprofloxacin tablet is given concomitantly with food, there is a delay in the absorption of the drug, resulting in peak concentrations that occur closer to 2 hours after dosing rather than 1 hour. The overall absorption of ciprofloxacin tablet, however, is not substantially affected. Avoid concomitant administration of ciprofloxacin with dairy products (like milk or yogurt) or calcium-fortified juices alone since decreased absorption is possible; however, ciprofloxacin may be taken with a meal that contains these products.
With oral administration, a 500 mg dose, given as 10 mL of the 5% ciprofloxacin suspension (containing 250 mg ciprofloxacin/5mL) is bioequivalent to the 500 mg tablet.
Distribution
The binding of ciprofloxacin to serum proteins is 20% to 40% which is not likely to be high enough to cause significant protein binding interactions with other drugs.
After oral administration, ciprofloxacin is widely distributed throughout the body. Tissue concentrations often exceed serum concentrations in both men and women, particularly in genital tissue including the prostate. Ciprofloxacin is present in active form in the saliva, nasal and bronchial secretions, mucosa of the sinuses, sputum, skin blister fluid, lymph, peritoneal fluid, bile, and prostatic secretions. Ciprofloxacin has also been detected in lung, skin, fat, muscle, cartilage, and bone. The drug diffuses into the cerebrospinal fluid (CSF); however, CSF concentrations are generally less than 10% of peak serum concentrations. Low levels of the drug have been detected in the aqueous and vitreous humors of the eye.
Metabolism
Four metabolites have been identified in human urine which together account for approximately 15% of an oral dose. The metabolites have antimicrobial activity, but are less active than unchanged ciprofloxacin. Ciprofloxacin is an inhibitor of human cytochrome P450 1A2 (CYP1A2) mediated metabolism. Co-administration of ciprofloxacin with other drugs primarily metabolized by CYP1A2 results in increased plasma concentrations of these drugs and could lead to clinically significant adverse events of the co-administered drug [see Contraindications (4.2), Warnings and Precautions ( 5.10 , 5.16), and Drug Interactions (7)].
Excretion
The serum elimination half-life in subjects with normal renal function is approximately 4 hours. Approximately 40 to 50% of an orally administered dose is excreted in the urine as unchanged drug. After a 250 mg oral dose, urine concentrations of ciprofloxacin usually exceed 200 mcg/mL during the first two hours and are approximately 30 mcg/mL at 8 to 12 hours after dosing. The urinary excretion of ciprofloxacin is virtually complete within 24 hours after dosing. The renal clearance of ciprofloxacin, which is approximately 300 mL/minute, exceeds the normal glomerular filtration rate of 120 mL/minute. Thus, active tubular secretion would seem to play a significant role in its elimination. Co-administration of probenecid with ciprofloxacin results in about a 50% reduction in the ciprofloxacin renal clearance and a 50% increase in its concentration in the systemic circulation.
Although bile concentrations of ciprofloxacin are several fold higher than serum concentrations after oral dosing, only a small amount of the dose administered is recovered from the bile as unchanged drug. An additional 1% to 2% of the dose is recovered from the bile in the form of metabolites. Approximately 20% to 35% of an oral dose is recovered from the feces within 5 days after dosing. This may arise from either biliary clearance or transintestinal elimination.
Specific Populations
Elderly
Pharmacokinetic studies of the oral (single dose) and intravenous (single and multiple dose) forms of ciprofloxacin indicate that plasma concentrations of ciprofloxacin are higher in elderly subjects (older than 65 years) as compared to young adults. Although the C max is increased 16% to 40%, the increase in mean AUC is approximately 30%, and can be at least partially attributed to decreased renal clearance in the elderly. Elimination half-life is only slightly (~20%) prolonged in the elderly. These differences are not considered clinically significant. [see Use in Specific Populations (8.5).]
Renal Impairment
In patients with reduced renal function, the half-life of ciprofloxacin is slightly prolonged. Dosage adjustments may be required [see Use in Specific Populations (8.6) and Dosage and Administration (2.3)].
Hepatic Impairment
In preliminary studies in patients with stable chronic liver cirrhosis, no significant changes in ciprofloxacin pharmacokinetics have been observed. The kinetics of ciprofloxacin in patients with acute hepatic insufficiency, have not been fully studied.
Drug-Drug Interactions
Antacids
Concurrent administration of antacids containing magnesium hydroxide or aluminum hydroxide may reduce the bioavailability of ciprofloxacin by as much as 90% [see Dosage and Administration (2.4) and Drug Interactions (7)].
Histamine H2-receptor antagonists
Histamine H 2-receptor antagonists appear to have no significant effect on the bioavailability of ciprofloxacin.
Metronidazole
The serum concentrations of ciprofloxacin and metronidazole were not altered when these two drugs were given concomitantly.
Tizanidine
In a pharmacokinetic study, systemic exposure of tizanidine (4 mg single dose) was significantly increased (C max 7-fold, AUC 10-fold) when the drug was given concomitantly with ciprofloxacin (500 mg twice a day for 3 days). Concomitant administration of tizanidine and ciprofloxacin is contraindicated due to the potentiation of hypotensive and sedative effects of tizanidine [see Contraindications (4.2)] .
Ropinirole
In a study conducted in 12 patients with Parkinson’s disease who were administered 6 mg ropinirole once daily with 500 mg ciprofloxacin twice-daily, the mean C max and mean AUC of ropinirole were increased by 60% and 84%, respectively. Monitoring for ropinirole-related adverse reactions and appropriate dose adjustment of ropinirole is recommended during and shortly after co-administration with ciprofloxacin [see Warnings and Precautions (5.10)].
Clozapine
Following concomitant administration of 250 mg ciprofloxacin with 304 mg clozapine for 7 days, serum concentrations of clozapine and N-desmethylclozapine were increased by 29% and 31%, respectively. Careful monitoring of clozapine associated adverse reactions and appropriate adjustment of clozapine dosage during and shortly after co-administration with ciprofloxacin are advised.
Sildenafil
Following concomitant administration of a single oral dose of 50 mg sildenafil with 500 mg ciprofloxacin to healthy subjects, the mean C max and mean AUC of sildenafil were both increased approximately two-fold. Use sildenafil with caution when co-administered with ciprofloxacin due to the expected two-fold increase in the exposure of sildenafil upon co-administration of ciprofloxacin.
Duloxetine
In clinical studies it was demonstrated that concomitant use of duloxetine with strong inhibitors of the CYP450 1A2 isozyme such as fluvoxamine, may result in a 5-fold increase in mean AUC and a 2.5-fold increase in mean C max of duloxetine.
Lidocaine
In a study conducted in 9 healthy volunteers, concomitant use of 1.5 mg/kg IV lidocaine with ciprofloxacin 500 mg twice daily resulted in an increase of lidocaine C max and AUC by 12% and 26%, respectively. Although lidocaine treatment was well tolerated at this elevated exposure, a possible interaction with ciprofloxacin and an increase in adverse reactions related to lidocaine may occur upon concomitant administration.
Metoclopramide
Metoclopramide significantly accelerates the absorption of oral ciprofloxacin resulting in a shorter time to reach maximum plasma concentrations. No significant effect was observed on the bioavailability of ciprofloxacin.
Omeprazole
When ciprofloxacin was administered as a single 1000 mg dose concomitantly with omeprazole (40 mg once daily for three days) to 18 healthy volunteers, the mean AUC and C max of ciprofloxacin were reduced by 20% and 23%, respectively. The clinical significance of this interaction has not been determined.
12.4 Microbiology
Mechanism of Action
The bactericidal action of ciprofloxacin results from inhibition of the enzymes topoisomerase II (DNA gyrase) and topoisomerase IV (both Type II topoisomerases), which are required for bacterial DNA replication, transcription, repair, and recombination.
Mechanism of Resistance
The mechanism of action of fluoroquinolones, including ciprofloxacin, is different from that of penicillins, cephalosporins, aminoglycosides, macrolides, and tetracyclines; therefore, microorganisms resistant to these classes of drugs may be susceptible to ciprofloxacin. Resistance to fluoroquinolones occurs primarily by either mutations in the DNA gyrases, decreased outer membrane permeability, or drug efflux. In vitro resistance to ciprofloxacin develops slowly by multiple step mutations. Resistance to ciprofloxacin due to spontaneous mutations occurs at a general frequency of between < 10 -9 to 1x10 -6 .
Cross Resistance
There is no known cross-resistance between ciprofloxacin and other classes of antimicrobials.
Ciprofloxacin has been shown to be active against most isolates of the following bacteria, both in vitro and in clinical infections [see Indications and Usage (1)].
Gram-positive bacteria
Bacillus anthracis
Enterococcus faecalis
Staphylococcus aureus (methicillin-susceptible isolates only)
Staphylococcus epidermidis (methicillin-susceptible isolates only)
Staphylococcus saprophyticus
Streptococcus pneumoniae
Streptococcus pyogenes
Gram-negative bacteria
Campylobacter jejuni
Citrobacter koseri
Citrobacter freundii
Enterobacter cloacae
Escherichia coli
Haemophilus influenzae
Haemophilus parainfluenzae
Klebsiella pneumoniae
Moraxella catarrhalis
Morganella morganii
Neisseria gonorrhoeae
Proteus mirabilis
Proteus vulgaris
Providencia rettgeri
Providencia stuartii
Pseudomonas aeruginosa
Salmonella typhi
Serratia marcescens
Shigella boydii
Shigella dysenteriae
Shigella flexneri
Shigella sonnei
Yersinia pestis
The following in vitro data are available, but their clinical significance is unknown. At least 90 percent of the following bacteria exhibit an in vitro minimum inhibitory concentration (MIC) less than or equal to the susceptible breakpoint for ciprofloxacin against isolates of similar genus or organism group. However, the efficacy of ciprofloxacin in treating clinical infections caused by these bacteria has not been established in adequate and well-controlled clinical trials.
Gram-positive bacteria
Staphylococcus haemolyticus (methicillin-susceptible isolates only)
Staphylococcus hominis (methicillin-susceptible isolates only)
Gram-negative bacteria
Acinetobacter lwoffi
Aeromonas hydrophila
Edwardsiella tarda
Enterobacter aerogenes
Klebsiella oxytoca
Legionella pneumophila
Pasteurella multocida
Salmonella enteritidis
Vibrio cholerae
Vibrio parahaemolyticus
Vibrio vulnificus
Yersinia enterocolitica
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Eight in vitro mutagenicity tests have been conducted with ciprofloxacin, and the test results are listed below:
-
Salmonella/Microsome Test (Negative)
-
E. coli DNA Repair Assay (Negative)
-
Mouse Lymphoma Cell Forward Mutation Assay (Positive)
-
Chinese Hamster V 79 Cell HGPRT Test (Negative)
-
Syrian Hamster Embryo Cell Transformation Assay (Negative)
-
Saccharomyces cerevisiae Point Mutation Assay (Negative)
-
Saccharomyces cerevisiae Mitotic Crossover and Gene Conversion Assay (Negative)
-
Rat Hepatocyte DNA Repair Assay (Positive)
Thus, 2 of the 8 tests were positive, but results of the following 3 in vivo test systems gave negative results:
• Rat Hepatocyte DNA Repair Assay
• Micronucleus Test (Mice)
• Dominant Lethal Test (Mice)
Long-term carcinogenicity studies in rats and mice resulted in no carcinogenic or tumorigenic effects due to ciprofloxacin at daily oral dose levels up to 250 mg/kg and 750 mg/kg to rats and mice, respectively (approximately 1.7- and 2.5- times the highest recommended therapeutic dose based upon body surface area, respectively).
Results from photo co-carcinogenicity testing indicate that ciprofloxacin does not reduce the time to appearance of UV-induced skin tumors as compared to vehicle control. Hairless (Skh-1) mice were exposed to UVA light for 3.5 hours five times every two weeks for up to 78 weeks while concurrently being administered ciprofloxacin. The time to development of the first skin tumors was 50 weeks in mice treated concomitantly with UVA and ciprofloxacin (mouse dose approximately equal to maximum recommended human dose based upon body surface area), as opposed to 34 weeks when animals were treated with both UVA and vehicle. The times to development of skin tumors ranged from 16 weeks to 32 weeks in mice treated concomitantly with UVA and other quinolones. 5
In this model, mice treated with ciprofloxacin alone did not develop skin or systemic tumors. There are no data from similar models using pigmented mice and/or fully haired mice. The clinical significance of these findings to humans is unknown.
Fertility studies performed in male and female rats at oral doses of ciprofloxacin up to 100 mg/kg (approximately 0.6-times the highest recommended therapeutic oral dose based upon body surface area) revealed no evidence of impairment. Male rats received oral ciprofloxacin for 10 weeks prior to mating and females were dosed for 3 weeks prior to mating through Gestation Day 7.
13.2 Animal Toxicology and/or Pharmacology
Ciprofloxacin and other quinolones have been shown to cause arthropathy in immature animals of most species tested [see Warnings and Precautions (5.13)] . Damage of weight bearing joints was observed in juvenile dogs and rats. In young beagles, 100 mg/kg ciprofloxacin, given daily for 4 weeks, caused degenerative articular changes of the knee joint. At 30 mg/kg, the effect on the joint was minimal. In a subsequent study in young beagle dogs, oral ciprofloxacin doses of 30 mg/kg and 90 mg/kg ciprofloxacin (approximately 1.3-times and 3.5-times the pediatric dose based upon comparative plasma AUCs) given daily for 2 weeks caused articular changes which were still observed by histopathology after a treatment-free period of 5 months. At 10 mg/kg (approximately 0.6-times the pediatric dose based upon comparative plasma AUCs), no effects on joints were observed. This dose was also not associated with arthrotoxicity after an additional treatment-free period of 5 months. In another study, removal of weight bearing from the joint reduced the lesions but did not totally prevent them.
Crystalluria, sometimes associated with secondary nephropathy, occurs in laboratory animals dosed with ciprofloxacin. This is primarily related to the reduced solubility of ciprofloxacin under alkaline conditions, which predominate in the urine of test animals; in man, crystalluria is rare since human urine is typically acidic. In rhesus monkeys, crystalluria without nephropathy was noted after single oral doses as low as 5 mg/kg. (approximately 0.07-times the highest recommended therapeutic dose based upon body surface area). After 6 months of intravenous dosing at 10 mg/kg/day, no nephropathological changes were noted; however, nephropathy was observed after dosing at 20 mg/kg/day for the same duration (approximately 0.2-times the highest recommended therapeutic dose based upon body surface area).
In dogs, ciprofloxacin at 3 mg/kg and 10 mg/kg by rapid intravenous injection (15 sec.) produces pronounced hypotensive effects. These effects are considered to be related to histamine release, since they are partially antagonized by pyrilamine, an antihistamine. In rhesus monkeys, rapid intravenous injection also produces hypotension but the effect in this species is inconsistent and less pronounced.
In mice, concomitant administration of nonsteroidal anti-inflammatory drugs such as phenylbutazone and indomethacin with quinolones has been reported to enhance the CNS stimulatory effect of quinolones.
Ocular toxicity seen with some related drugs has not been observed in ciprofloxacin-treated animals.
14 CLINICAL STUDIES
14.1 Complicated Urinary Tract Infection and Pyelonephritis-Efficacy in Pediatric Patients
Ciprofloxacin administered intravenously and/or orally was compared to a cephalosporin for treatment of cUTI and pyelonephritis in pediatric patients 1 to 17 years of age (mean age of 6 ± 4 years). The trial was conducted in the US, Canada, Argentina, Peru, Costa Rica, Mexico, South Africa, and Germany. The duration of therapy was 10 to 21 days (mean duration of treatment was 11 days with a range of 1 to 88 days). The primary objective of the study was to assess musculoskeletal and neurological safety.
Patients were evaluated for clinical success and bacteriological eradication of the baseline organism(s) with no new infection or superinfection at 5 to 9 days post-therapy (Test of Cure or TOC). The Per Protocol population had a causative organism(s) with protocol specified colony count(s) at baseline, no protocol violation, and no premature discontinuation or loss to follow-up (among other criteria).
The clinical success and bacteriologic eradication rates in the Per Protocol population were similar between ciprofloxacin and the comparator group as shown below.
| Table 15: Clinical Success and Bacteriologic Eradication at Test of Cure (5 to 9 Days Post-Therapy)
|
||
|
Ciprofloxacin |
Comparator |
|
| Randomized Patients
|
337 |
352 |
| Per Protocol Patients
|
211 |
231 |
| Clinical Response at 5 to 9 Days Post-Treatment
|
95.7% (202/211) |
92.6% (214/231) |
|
95% CI [-1.3%, 7.3%] |
||
| Bacteriologic Eradication by Patient at 5 to 9 Days Post-Treatment
1
|
84.4% (178/211) |
78.3% (181/231) |
|
95% CI [-1.3%, 13.1%] |
||
| Bacteriologic Eradication of the Baseline Pathogen at 5 to 9 Days Post-Treatment
| ||
| Escherichia coli
|
156/178 (88%) |
161/179 (90%) |
1Patients with baseline pathogen(s) eradicated and no new infections or superinfections/total number of patients. There were 5.5% (6/211) ciprofloxacin and 9.5% (22/231) comparator patients with superinfections or new infections.
14.2 Inhalational Anthrax in Adults and Pediatrics
The mean serum concentrations of ciprofloxacin associated with a statistically significant improvement in survival in the rhesus monkey model of inhalational anthrax are reached or exceeded in adult and pediatric patients receiving oral and intravenous regimens. Ciprofloxacin pharmacokinetics have been evaluated in various human populations. The mean peak serum concentration achieved at steady-state in human adults receiving 500 mg orally every 12 hours is 2.97 mcg/mL, and 4.56 mcg/mL following 400 mg intravenously every 12 hours. The mean trough serum concentration at steady-state for both of these regimens is 0.2 mcg/mL. In a study of 10 pediatric patients between 6 and 16 years of age, the mean peak plasma concentration achieved is 8.3 mcg/mL and trough concentrations range from 0.09 mcg/mL to 0.26 mcg/mL, following two 30-minute intravenous infusions of 10 mg/kg administered 12 hours apart. After the second intravenous infusion patients switched to 15 mg/kg orally every 12 hours achieve a mean peak concentration of 3.6 mcg/mL after the initial oral dose. Long-term safety data, including effects on cartilage, following the administration of ciprofloxacin to pediatric patients are limited. Ciprofloxacin serum concentrations achieved in humans serve as a surrogate endpoint reasonably likely to predict clinical benefit and provide the basis for this indication. 1
A placebo-controlled animal study in rhesus monkeys exposed to an inhaled mean dose of 11 LD 50 (~5.5 x 10 5 spores (range 5 -30 LD 50) of B. anthracis was conducted. The minimal inhibitory concentration (MIC) of ciprofloxacin for the anthrax strain used in this study was 0.08 mcg/mL. In the animals studied, mean serum concentrations of ciprofloxacin achieved at expected T max (1 hour post-dose) following oral dosing to steady-state ranged from 0.98 mcg/mL to 1.69 mcg/mL. Mean steady-state trough concentrations at 12 hours post-dose ranged from 0.12 mcg/mL to 0.19 mcg/mL. 6 Mortality due to anthrax for animals that received a 30-day regimen of oral ciprofloxacin beginning 24 hours post-exposure was significantly lower (1/9), compared to the placebo group (9/10) [p= 0.001]. The one ciprofloxacin-treated animal that died of anthrax did so following the 30-day drug administration period. 7
More than 9300 persons were recommended to complete a minimum of 60 days of antibacterial prophylaxis against possible inhalational exposure to B. anthracis during 2001. Ciprofloxacin was recommended to most of those individuals for all or part of the prophylaxis regimen. Some persons were also given anthrax vaccine or were switched to alternative antibacterial drugs. No one who received ciprofloxacin or other therapies as prophylactic treatment subsequently developed inhalational anthrax. The number of persons who received ciprofloxacin as all or part of their post-exposure prophylaxis regimen is unknown.
14.3 Plague
A placebo-controlled animal study in African green monkeys exposed to an inhaled mean dose of 110 LD 50 (range 92 to 127 LD 50) of Yersinia pestis (CO92 strain) was conducted. The minimal inhibitory concentration (MIC) of ciprofloxacin for the Y. pestis strain used in this study was 0.015 mcg/mL. Mean peak serum concentrations of ciprofloxacin achieved at the end of a single 60 minute infusion were 3.49 ± mcg/mL 0.55 mcg/mL, 3.91 mcg/mL ± 0.58 mcg/mL and 4.03 mcg/mL ± 1.22 mcg/mL on Day 2, Day 6 and Day 10 of treatment in African green monkeys, respectively. All trough concentrations (Day 2, Day 6 and Day 10) were <0.5 mcg /mL. Animals were randomized to receive either a 10-day regimen of intravenous ciprofloxacin 15 mg/kg, or placebo beginning when animals were found to be febrile (a body temperature greater than 1.5 °C over baseline for two hours), or at 76 hours post-challenge, whichever occurred sooner. Mortality in the ciprofloxacin group was significantly lower (1/10) compared to the placebo group (2/2) [difference: -90.0%, 95% exact confidence interval: -99.8% to -5.8%]. The one ciprofloxacin-treated animal that died did not receive the proposed dose of ciprofloxacin due to a failure of the administration catheter. Circulating ciprofloxacin concentration was below 0.5 mcg/mL at all timepoints tested in this animal. It became culture negative on Day 2 of treatment, but had a resurgence of low grade bacteremia on Day 6 after treatment initiation. Terminal blood culture in this animal was negative. 8
15 REFERENCES
- 21 CFR 314.510 (Subpart H–Accelerated Approval of New Drugs for Life-Threatening Illnesses).
- Friedman J, Polifka J. Teratogenic effects of drugs: a resource for clinicians (TERIS). Baltimore, Maryland: Johns Hopkins University Press, 2000:149-195.
- Loebstein R, Addis A, Ho E, et al. Pregnancy outcome following gestational exposure to fluoroquinolones: a multicenter prospective controlled study. Antimicrob Agents Chemother. 1998; 42(6):1336-1339.
- Schaefer C, Amoura-Elefant E, Vial T, et al. Pregnancy outcome after prenatal quinolone exposure. Evaluation of a case registry of the European network of teratology information services (ENTIS). Eur J Obstet Gynecol Reprod Biol. 1996; 69:83-89.
- Report presented at the FDA’s Anti-Infective Drug and Dermatological Drug Product’s Advisory Committee meeting, March 31, 1993, Silver Spring, MD. Report available from FDA, CDER, Advisors and Consultants Staff, HFD-21, 1901 Chapman Avenue, Room 200, Rockville, MD 20852, USA.
- Kelly DJ, et al. Serum concentrations of penicillin, doxycycline, and ciprofloxacin during prolonged therapy in rhesus monkeys. J Infect Dis 1992; 166:1184-7.
- Friedlander AM, et al. Postexposure prophylaxis against experimental inhalational anthrax. J Infect Dis 1993; 167:1239-42.
- Anti-infective Drugs Advisory Committee Meeting, April 3, 2012 -The efficacy of Ciprofloxacin for treatment of Pneumonic Plague.
16 HOW SUPPLIED/STORAGE AND HANDLING
Ciprofloxacin tablets USP 500 mg are available as white colored, biconvex, capsule shaped, film coated tablets, debossed 'P' on one side and '500' on the other side containing ciprofloxacin hydrochloride, USP equivalent to 500 mg of ciprofloxacin.
NDC:
60760-815-20 bottle of 20
60760-815-10 bottle of 10
60760-815-30 bottle of 30
60760-815-14 bottle of 14 - inactive
60760-815-06 bottle of 6 - inactive
Store at 20° to 25°C (68° to 77° F); excursions permitted to 15° to 30°C (59° to 86° F) [see USP Controlled Room Temperature].
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide)
Serious Adverse Reactions
Advise patients to stop taking ciprofloxacin tablets if they experience an adverse reaction and to call their healthcare provider for advice on completing the full course of treatment with another antibacterial drug.
Inform patients of the following serious adverse reactions that have been associated with ciprofloxacin tablets or other fluoroquinolone use:
- Disabling and potentially irreversible serious adverse reactions that may occur together: Inform patients that disabling and potentially irreversible serious adverse reactions, including tendinitis and tendon rupture, peripheral neuropathies, and central nervous system effects, have been associated with use of ciprofloxacin tablets and may occur together in the same patient. Inform patients to stop taking ciprofloxacin tablets immediately if they experience an adverse reaction and to call their healthcare provider.
- Tendinitis and tendon rupture: Instruct patients to contact their healthcare provider if they experience pain, swelling, or inflammation of a tendon, or weakness or inability to use one of their joints; rest and refrain from exercise; and discontinue ciprofloxacin tablets treatment. Symptoms may be irreversible. The risk of severe tendon disorder with fluoroquinolones is higher in older patients usually over 60 years of age, in patients taking corticosteroid drugs, and in patients with kidney, heart or lung transplants.
- Peripheral Neuropathies: Inform patients that peripheral neuropathies have been associated with ciprofloxacin use, symptoms may occur soon after initiation of therapy and may be irreversible. If symptoms of peripheral neuropathy including pain, burning, tingling, numbness and/or weakness develop, immediately discontinue ciprofloxacin tablets and tell them to contact their physician.
- Central nervous system effects (for example, convulsions, dizziness, lightheadedness, increased intracranial pressure): Inform patients that convulsions have been reported in patients receiving fluoroquinolones, including Ciprofloxacin. Instruct patients to notify their physician before taking this drug if they have a history of convulsions. Inform patients that they should know how they react to ciprofloxacin tablets before they operate an automobile or machinery or engage in other activities requiring mental alertness and coordination. Instruct patients to notify their physician if persistent headache with or without blurred vision occurs.
- Exacerbation of Myasthenia Gravis: Instruct patients to inform their physician of any history of myasthenia gravis. Instruct patients to notify their physician if they experience any symptoms of muscle weakness, including respiratory difficulties.
- Hypersensitivity Reactions: Inform patients that ciprofloxacin can cause hypersensitivity reactions, even following a single dose, and to discontinue the drug at the first sign of a skin rash, hives or other skin reactions, a rapid heartbeat, difficulty in swallowing or breathing, any swelling suggesting angioedema (for example, swelling of the lips, tongue, face, tightness of the throat, hoarseness), or other symptoms of an allergic reaction.
- Hepatotoxicity: Inform patients that severe hepatotoxicity (including acute hepatitis and fatal events) has been reported in patients taking ciprofloxacin tablets. Instruct patients to inform their physician if they experience any signs or symptoms of liver injury including: loss of appetite, nausea, vomiting, fever, weakness, tiredness, right upper quadrant tenderness, itching, yellowing of the skin and eyes, light colored bowel movements or dark colored urine.
- Aortic aneurysm and dissection: Inform patients to seek emergency medical care if they experience sudden chest, stomach, or back pain.
- Diarrhea: Diarrhea is a common problem caused by antibiotics which usually ends when the antibiotic is discontinued. Sometimes after starting treatment with antibiotics, patients can develop watery and bloody stools (with or without stomach cramps and fever) even as late as two or more months after having taken the last dose of the antibiotic. If this occurs, instruct patients to contact their physician as soon as possible.
- Prolongation of the QT Interval: Instruct patients to inform their physician of any personal or family history of QT prolongation or proarrhythmic conditions such as hypokalemia, bradycardia, or recent myocardial ischemia; if they are taking any Class IA (quinidine, procainamide), or Class III (amiodarone, sotalol) antiarrhythmic agents. Instruct patients to notify their physician if they have any symptoms of prolongation of the QT interval, including prolonged heart palpitations or a loss of consciousness.
- Musculoskeletal Disorders in Pediatric Patients: Instruct parents to inform their child's physician if the child has a history of joint-related problems before taking this drug. Inform parents of pediatric patients to notify their child’s physician of any joint-related problems that occur during or following ciprofloxacin therapy [see Warnings and Precautions (5.13) and Use in Specific Populations (8.4)].
- Tizanidine: Instruct patients not to use ciprofloxacin if they are already taking tizanidine. Ciprofloxacin increases the effects of tizanidine (Zanaflex ®).
- Theophylline: Inform patients that ciprofloxacin may increase the effects of theophylline. Life-threatening CNS effects and arrhythmias can occur. Advise the patients to immediately seek medical help if they experience seizures, palpitations, or difficulty breathing.
- Caffeine: Inform patients that ciprofloxacin may increase the effects of caffeine. There is a possibility of caffeine accumulation when products containing caffeine are consumed while taking quinolones.
- Photosensitivity/Phototoxicity: Inform patients that photosensitivity/phototoxicity has been reported in patients receiving fluoroquinolones. Inform patients to minimize or avoid exposure to natural or artificial sunlight (tanning beds or UVA/B treatment) while taking quinolones. If patients need to be outdoors while using quinolones, instruct them to wear loose-fitting clothes that protect skin from sun exposure and discuss other sun protection measures with their physician. If a sunburn-like reaction or skin eruption occurs, instruct patients to contact their physician.
- Blood Glucose Disturbances: Inform the patients that if they are diabetic and are being treated with insulin or an oral hypoglycemic agent and a hypoglycemic reaction occurs, they should discontinue ciprofloxacin tablets and consult a physician.
- Lactation: For indications other than inhalational anthrax (post exposure), advise a woman that breastfeeding is not recommended during treatment with ciprofloxacin tablets and for an additional 2 days after the last dose. Alternatively, a woman may pump and discard during treatment and for additional 2 days after the last dose [ see Use in Specific Populations (8.2)].
Antibacterial Resistance
Inform patients that antibacterial drugs including ciprofloxacin tablets should only be used to treat bacterial infections. They do not treat viral infections (for example, the common cold). When ciprofloxacin tablets are prescribed to treat a bacterial infection, patients should be told that although it is common to feel better early in the course of therapy, the medication should be taken exactly as directed. Skipping doses or not completing the full course of therapy may (1) decrease the effectiveness of the immediate treatment and (2) increase the likelihood that bacteria will develop resistance and will not be treatable by ciprofloxacin tablets or other antibacterial drugs in the future.
Administration Instructions
Inform patients that ciprofloxacin tablets may be taken with or without food.
Inform patients to drink fluids liberally while taking ciprofloxacin tablets to avoid formation of highly concentrated urine and crystal formation in the urine.
Inform patients that antacids containing magnesium, or aluminum, as well as sucralfate, metal cations such as iron, and multivitamin preparations with zinc or didanosine should be taken at least two hours before or six hours after ciprofloxacin tablets administration. Ciprofloxacin tablets should not be taken with dairy products (like milk or yogurt) or calcium-fortified juices alone since absorption of ciprofloxacin may be significantly reduced; however, ciprofloxacin tablets may be taken with a meal that contains these products.
Advise patients that if a dose is missed, it should be taken anytime but not later than 6 hours prior to the next scheduled dose. If less than 6 hours remain before the next dose, the missed dose should not be taken and treatment should be continued as prescribed with the next scheduled dose. Double doses should not be taken to compensate for a missed dose.
Drug Interactions Oral Antidiabetic Agents
Inform patients that hypoglycemia has been reported when ciprofloxacin and oral antidiabetic agents were co-administered; if low blood sugar occurs with ciprofloxacin, instruct them to consult their physician and that their antibacterial medicine may need to be changed.
Anthrax and Plague Studies
Inform patients given ciprofloxacin for these conditions that efficacy studies could not be conducted in humans for feasibility reasons. Therefore, approval for these conditions was based on efficacy studies conducted in animals.
Pharmacist: Dispense with Medication Guide available at: www.risingpharma.com/Medguides/ciprofloxacin-tablets.pdf
Manufactured by:
Unique Pharmaceutical Laboratories
(A Div. of J. B. Chemicals & Pharmaceuticals Ltd.),
Mumbai 400 030, India.
Rx only
Distributed by:

Rising Pharma Holdings, Inc.
East Brunswick, NJ 08816
132754
Revised: Jan. 2022
Dispense with Medication Guide available at: www.risingpharma.com/Medguides/ciprofloxacin-tablets.pdf
MEDICATION GUIDE
Ciprofloxacin (sip"row flox"a sin) Tablets USP for oral use
Read this Medication Guide before you start taking ciprofloxacin tablets and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or your treatment.
What is the most important information I should know about ciprofloxacin tablets?
Ciprofloxacin tablets, a fluoroquinolone antibacterial medicine, can cause serious side effects. Some of these serious side effects can happen at the same time and could result in death.
If you get any of the following serious side effects while you take ciprofloxacin tablets, you should stop taking ciprofloxacin tablets immediately and get medical help right away.
1. Tendon rupture or swelling of the tendon (tendinitis).
-
Tendon problems can happen in people of all ages who take ciprofloxacin tablets. Tendons are tough cords of tissue that connect muscles to bones.
Symptoms of tendon problems may include:
- pain
- swelling
- tears and swelling of the tendons including the back of the ankle (Achilles), shoulder, hand, thumb, or other tendon sites.
-
The risk of getting tendon problems while you take ciprofloxacin tablets is higher if you:
- are over 60 years of age
- are taking steroids (corticosteroids)
- have had a kidney, heart or lung transplant.
- Tendon problems can happen in people who do not have the above risk factors when they take ciprofloxacin tablets.
-
Other reasons that can increase your risk of tendon problems can include:
- physical activity or exercise
- kidney failure
- tendon problems in the past, such as in people with rheumatoid arthritis (RA).
- Stop taking ciprofloxacin tablets immediately and get medical help right away at the first sign of tendon pain, swelling or inflammation. The most common area of pain and swelling is the Achilles tendon at the back of your ankle. This can also happen with other tendons.
- Tendon rupture can happen while you are taking or after you have finished taking ciprofloxacin tablets. Tendon ruptures can happen within hours or days of taking ciprofloxacin tablets and have happened up to several months after people have finished taking their fluoroquinolone.
-
Stop taking ciprofloxacin tablets immediately and get medical help right away if you get any of the following signs or symptoms of a tendon rupture:
- hear or feel a snap or pop in a tendon area
- bruising right after an injury in a tendon area
- unable to move the affected area or bear weight
The tendon problems may be permanent.
2. Changes in sensation and possible nerve damage (Peripheral Neuropathy).
Damage to the nerves in arms, hands, legs, or feet can happen in people who take fluoroquinolones, including ciprofloxacin tablets. Stop taking ciprofloxacin tablets immediately and talk to your healthcare provider right away if you get any of the following symptoms of peripheral neuropathy in your arms, hands, legs, or feet:
|
|
|
|
|
Ciprofloxacin tablets may need to be stopped to prevent permanent nerve damage.
3. Central Nervous System (CNS) effects.
Mental health problems and seizures have been reported in people who take fluoroquinolone antibacterial medicines, including ciprofloxacin tablets. Tell your healthcare provider if you have a history of seizures before you start taking ciprofloxacin tablets. CNS side effects may happen as soon as after taking the first dose of ciprofloxacin. Stop taking ciprofloxacin tablets immediately and talk to your healthcare provider right away if you get any of these side effects, or other changes in mood or behavior:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
These changes may be permanent.
4. Worsening of myasthenia gravis (a problem that causes muscle weakness). Fluoroquinolones like ciprofloxacin may cause worsening of myasthenia gravis symptoms, including muscle weakness and breathing problems. Tell your healthcare provider if you have a history of myasthenia gravis before you start taking ciprofloxacin tablets. Call your healthcare provider right away if you have any worsening muscle weakness or breathing problems.
What are ciprofloxacin tablets?
Ciprofloxacin tablets are a fluoroquinolone antibacterial medicine used in adults age 18 years and older to treat certain infections caused by certain germs called bacteria. These bacterial infections include:
| o urinary tract infection
| o bone and joint infection
| o cervical and urethral gonorrhea, uncomplicated
|
| o chronic prostate infection
| o nosocomial pneumonia
| o people with a low white blood cell count and a fever
|
| o lower respiratory tract infection
| o intra-abdominal infection, complicated
| o inhalational anthrax
|
| o sinus infection
| o infectious diarrhea
| o plague
|
| o skin infection
| o typhoid (enteric) fever
|
|
- Studies of ciprofloxacin tablets for use in the treatment of plague and anthrax were done in animals only, because plague and anthrax could not be studied in people.
- Ciprofloxacin should not be used in people with acute exacerbation of chronic bronchitis, acute uncomplicated cystitis, and sinus infections, if there are other treatment options available.
- Ciprofloxacin should not be used as the first choice of antibacterial medicine to treat lower respiratory tract infections caused by a certain type of bacterial called Streptococcus pneumoniae.
- Ciprofloxacin is also used in children younger than 18 years of age to treat complicated urinary tract and kidney infections or who may have breathed in anthrax germs, have plague or have been exposed to plague germs.
- Children younger than 18 years of age have a higher chance of getting bone, joint, or tendon (musculoskeletal) problems such as pain or swelling while taking ciprofloxacin. Ciprofloxacin should not be used as the first choice of antibacterial medicine in children under 18 years of age.
Who should not take ciprofloxacin tablets?
Do not take ciprofloxacin tablets if you:
- have ever had a severe allergic reaction to an antibacterial medicine known as a fluoroquinolone, or are allergic to ciprofloxacin hydrochloride or any of the ingredients in ciprofloxacin tablets. See the end of this Medication Guide for a complete list of ingredients in ciprofloxacin tablets.
- also take a medicine called tizanidine (Zanaflex ®).
Ask your healthcare provider if you are not sure.
What should I tell my healthcare provider before taking ciprofloxacin tablets?
Before you take ciprofloxacin tablets, tell your healthcare provider about all your medical conditions, including if you:
- have tendon problems. Ciprofloxacin tablets should not be used in people who have a history of tendon problems.
- have a disease that causes muscle weakness (myasthenia gravis). Ciprofloxacin tablets should not be used in people who have a known history of myasthenia gravis.
- have liver problems.
- have central nervous system problems (such as epilepsy).
- have nerve problems. Ciprofloxacin tablets should not be used in people who have a history of a nerve problem called peripheral neuropathy have or anyone in your family has an irregular heartbeat, or heart attack, especially a condition called “QT prolongation”.
- have low blood potassium (hypokalemia) or low magnesium (hypomagnesemia).
- have or have had seizures.
- have kidney problems. You may need a lower dose of ciprofloxacin tablets if your kidneys do not work well.
- have diabetes or problems with low blood sugar (hypoglycemia).
- have joint problems including rheumatoid arthritis (RA).
- have trouble swallowing pills.
- are pregnant or plan to become pregnant. It is not known if ciprofloxacin will harm your unborn baby.
- are breastfeeding or plan to breastfeed. Ciprofloxacin passes into your breast milk.
- You should not breastfeed during treatment with ciprofloxacin tablets and for 2 days after taking your last dose of ciprofloxacin tablets. You may pump your breast milk and throw it away during treatment with ciprofloxacin tablets and for 2 days after taking your last dose of ciprofloxacin tablets.
- If you are taking ciprofloxacin tablets for inhalation anthrax, you and your healthcare provider should decide whether you can continue breastfeeding while taking ciprofloxacin tablets.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
- Ciprofloxacin tablets and other medicines can affect each other causing side effects.
-
Especially tell your healthcare provider if you take:
- a steroid medicine.
- an anti-psychotic medicine.
- a tricyclic antidepressant.
- a water pill (diuretic).
- theophylline (such as Theo-24 ®, Elixophyllin ®, Theochron ®, Uniphyl ®, Theolair ®).
- a medicine to control your heart rate or rhythm (antiarrhythmics).
- an oral anti-diabetes medicine.
- phenytoin (Fosphenytoin Sodium ®, Cerebyx ®, Dilantin-125 ®, Dilantin ® , Extended Phenytoin Sodium ®, Prompt Phenytoin Sodium ®, Phenytek ®).
- cyclosporine (Gengraf ®, Neoral ®, Sandimmune ®, Sangcya ®).
- a blood thinner (such as warfarin, Coumadin ®, Jantoven ®).
- methotrexate (Trexall ®).
- ropinirole (Requip ®).
- clozapine (Clozaril ®, Fazaclo ® ODT ®).
- a Non-Steroidal Anti-Inflammatory Drug (NSAID). Many common medicines for pain relief are NSAIDs. Taking an NSAID while you take ciprofloxacin or other fluoroquinolones may increase your risk of central nervous system effects and seizures.
- sildenafil (Viagra ®, Revatio ®).
- duloxetine.
- products that contain caffeine.
- probenecid (Probalan ®, Col-probenecid ®).
- Certain medicines may keep ciprofloxacin tablets from working correctly. Take ciprofloxacin tablets either 2 hours before or 6 hours after taking these medicines, vitamins, or supplements:
- an antacid, multivitamin, or other medicine or supplements that has magnesium, calcium, aluminum, iron, or zinc.
- sucralfate (Carafate ®).
- didanosine (Videx ®, Videx EC ®).
Ask your healthcare provider for a list of these medicines if you are not sure.
Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine.
How should I take ciprofloxacin tablets?
- Take ciprofloxacin tablets exactly as your healthcare provider tells you to take it.
- Your healthcare provider will tell you how much ciprofloxacin tablets to take and when to take it.
- Ciprofloxacin tablets can be taken with or without food.
- Ciprofloxacin tablets should not be taken with dairy products (like milk or yogurt) or calcium-fortified juices alone but may be taken with a meal that contains these products.
- Drink plenty of fluids while taking ciprofloxacin tablets.
- Do not skip any doses of ciprofloxacin tablets, or stop taking it, even if you begin to feel better, until you finish your prescribed treatment unless:
- you have tendon problems. See “ What is the most important information I should know about ciprofloxacin tablets?”
- you have nerve problems. See “What is the most important information I should know about ciprofloxacin tablets?”
- you have central nervous system problems. See “What is the most important information I should know about ciprofloxacin tablets?”
- you have a serious allergic reaction. See “ What are the possible side effects of ciprofloxacin tablets?”
- your healthcare provider tells you to stop taking ciprofloxacin tablets.
Taking all of your ciprofloxacin doses will help make sure that all of the bacteria are killed. Taking all of your ciprofloxacin doses will help lower the chance that the bacteria will become resistant to ciprofloxacin. If you become resistant to ciprofloxacin, ciprofloxacin tablets and other antibacterial medicines may not work for you in the future.
- If you take too much ciprofloxacin tablets, call your healthcare provider or get medical help right away.
- Ciprofloxacin is supplied as ciprofloxacin tablets. Read and follow the instructions below for ciprofloxacin tablets your healthcare provider has prescribed for you.
Ciprofloxacin Tablets:
- Ciprofloxacin tablets comes as 250 mg, 500 mg and 750 mg tablets that should be taken whole. Your healthcare provider will tell you how much ciprofloxacin tablets to take.
- Take ciprofloxacin tablets in the morning and evening at about the same time each day. Do not crush or chew the tablet. Tell your healthcare provider if you cannot swallow the tablet.
- If you miss a dose of ciprofloxacin tablets and it is:
- 6 hours or more until your next scheduled dose, take your missed dose right away. Then take the next dose at your regular time.
- less than 6 hours until your next scheduled dose, do not take the missed dose. Take the next dose at your regular time.
- Do not take 2 doses of ciprofloxacin tablets at 1 time to make up for a missed dose. If you are not sure about when to take ciprofloxacin tablets after a missed dose, ask your healthcare provider or pharmacist.
What should I avoid while taking ciprofloxacin tablets?
- Ciprofloxacin tablets can make you feel dizzy and lightheaded. Do not drive, operate machinery, or do other activities that require mental alertness or coordination until you know how ciprofloxacin tablets affects you.
- Avoid sunlamps, tanning beds, and try to limit your time in the sun. Ciprofloxacin tablets can make your skin sensitive to the sun (photosensitivity) and the light from sunlamps and tanning beds. You could get a severe sunburn, blisters or swelling of your skin. If you get any of these symptoms while you take ciprofloxacin tablets, call your healthcare provider right away. You should use a sunscreen and wear a hat and clothes that cover your skin if you have to be in sunlight.
What are the possible side effects of ciprofloxacin tablets?
Ciprofloxacin tablets may cause serious side effects, including:
- See "What is the most important information I should know about ciprofloxacin tablets?"
- Serious allergic reactions. Serious allergic reactions, including death, can happen in people taking fluoroquinolones, including ciprofloxacin tablets, even after only 1 dose. Stop taking ciprofloxacin tablets and get emergency medical help right away if you get any of the following symptoms of a severe allergic reaction:
| o hives
| o rapid heartbeat
|
| o trouble breathing or swallowing
| o faint
|
| o swelling of the lips, tongue, face
| o skin rash
|
| o throat tightness, hoarseness
|
|
Skin rash may happen in people taking ciprofloxacin tablets even after only 1 dose. Stop taking ciprofloxacin tablets at the first sign of a skin rash and call your healthcare provider. Skin rash may be a sign of a more serious reaction to ciprofloxacin.
- Liver damage (hepatotoxicity). Hepatotoxicity can happen in people who take ciprofloxacin tablets. Call your healthcare provider right away if you have unexplained symptoms such as:
| o nausea or vomiting
| o unusual tiredness
|
| o stomach pain
| o loss of appetite
|
| o fever
| o light colored bowel movements
|
| o weakness
| o dark colored urine
|
| o abdominal pain or tenderness
| o yellowing of the skin and whites of your eyes
|
| o itching
|
|
Stop taking ciprofloxacin tablets and tell your healthcare provider right away if you have yellowing of your skin or white part of your eyes, or if you have dark urine. These can be signs of a serious reaction to ciprofloxacin tablets (a liver problem).
- Aortic aneurysm and dissection. Tell your healthcare provider if you have ever been told that you have an aortic aneurysm, a swelling of the large artery that carries blood from the heart to the body. Get emergency medical help right away if you have sudden chest, stomach, or back pain.
- Intestine infection ( Clostridioides difficile – associated diarrhea) . Clostridioides difficile-associated diarrhea (CDAD) can happen with many antibacterial medicines, including ciprofloxacin tablets. Call your healthcare provider right away if you get watery diarrhea, diarrhea that does not go away, or bloody stools. You may have stomach cramps and a fever. CDAD can happen 2 or more months after you have finished your antibacterial medicine.
-
Serious heart rhythm changes (QT prolongation and torsade de pointes). Tell your healthcare provider right away if you have a change in your heartbeat (a fast or irregular heartbeat), or if you faint. Ciprofloxacin tablets may cause a rare heart problem known as prolongation of the QT interval. This condition can cause an abnormal heartbeat and can be very dangerous. The chances of this event are higher in people:
- who are elderly.
- with a family history of prolonged QT interval.
- with low blood potassium (hypokalemia) or low magnesium (hypomagnesemia).
- who take certain medicines to control heart rhythm (antiarrhythmics)
- Joint Problems. Increased chance of problems with joints and tissues around joints in children under 18 years old can happen. Tell your child's healthcare provider if your child has any joint problems during or after treatment with ciprofloxacin tablets.
- Sensitivity to sunlight (photosensitivity). See " What should I avoid while taking ciprofloxacin tablets?"
- Changes in blood sugar. People who take ciprofloxacin and other fluoroquinolone medicines with oral anti-diabetes medicines or with insulin can get low blood sugar (hypoglycemia) and high blood sugar (hyperglycemia). Follow your healthcare provider's instructions for how often to check your blood sugar. If you have diabetes and you get low blood sugar while taking ciprofloxacin tablets, stop taking ciprofloxacin tablets and call your healthcare provider right away. Your antibiotic medicine may need to be changed.
The most common side effects of ciprofloxacin include:
- nausea
- diarrhea
- changes in liver function tests
- vomiting
- rash
Tell your healthcare provider about any side effect that bothers you or that does not go away.
These are not all the possible side effects of ciprofloxacin tablets. For more information, ask your healthcare provider or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088 or Rising Pharma Holdings, Inc. at 1-844-874-7464.
How should I store ciprofloxacin tablets?
- Store at 20° to 25°C (68° to 77° F); excursions permitted to 15° to 30°C (59° to 86° F).
Keep ciprofloxacin tablets and all medicines out of the reach of children.
General Information about the safe and effective use of ciprofloxacin tablets.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use ciprofloxacin tablets for a condition for which it is not prescribed. Do not give ciprofloxacin tablets to other people, even if they have the same symptoms that you have. It may harm them.
This Medication Guide summarizes the most important information about ciprofloxacin tablets. If you would like more information about ciprofloxacin tablets, talk with your healthcare provider. You can ask your healthcare provider or pharmacist for information about ciprofloxacin tablets that is written for healthcare professionals.
What are the ingredients in ciprofloxacin tablets?
- Active ingredient: ciprofloxacin hydrochloride
- Inactive ingredients: colloidal silicon dioxide, crospovidone, hypromellose, magnesium stearate, microcrystalline cellulose, polyethylene glycol, pregelatinized starch and titanium dioxide.
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Trademarks are the property of their respective owner.
Manufactured by:
Unique Pharmaceutical Laboratories
(A Div. of J. B. Chemicals & Pharmaceuticals Ltd.),
Mumbai 400 030, India
Rx only
Distributed by:
Rising Pharma Holdings, Inc.
East Brunswick, NJ 08816
For more information, call 1-844-874-7464.
132754
Revised: Jan. 2022
