WARNING
Paclitaxel should be administered under the supervision of a physician experienced in the use of cancer chemotherapeutic agents. Appropriate management of complications is possible only when adequate diagnostic and treatment facilities are readily available.
Anaphylaxis and severe hypersensitivity reactions characterized by dyspnea and hypotension requiring treatment, angioedema, and generalized urticaria have occurred in 2 to 4% of patients receiving paclitaxel in clinical trials. Fatal reactions have occurred in patients despite premedication. All patients should be pretreated with corticosteroids, diphenhydramine, and H2 antagonists (see DOSAGE AND ADMINISTRATION). Patients who experience severe hypersensitivity reactions to paclitaxel should not be rechallenged with the drug.
Paclitaxel therapy should not be given to patients with solid tumors who have baseline neutrophil counts of less than 1,500 cells/mm3 and should not be given to patients with AIDS-related Kaposi's sarcoma if the baseline neutrophil count is less than 1,000 cells/mm3. In order to monitor the occurrence of bone marrow suppression, primarily neutropenia, which may be severe and result in infection, it is recommended that frequent peripheral blood cell counts be performed on all patients receiving paclitaxel.
DESCRIPTION
Paclitaxel Injection, USP is a clear, colorless to slightly yellow viscous solution. It is supplied as a nonaqueous solution intended for dilution with a suitable parenteral fluid prior to intravenous infusion. Paclitaxel Injection, USP is available in 30 mg (5 mL), 100 mg (16.7 mL), and 300 mg (50 mL) multidose vials. Each mL of sterile nonpyrogenic solution contains 6 mg paclitaxel, USP, 527 mg of polyoxyl 35 castor oil, NF, and 49.7% (v/v) dehydrated alcohol, USP.
Paclitaxel is a natural product with antitumor activity. Paclitaxel is obtained via a semi-synthetic process from Taxus baccata. The chemical name for paclitaxel is (2aR,4S,4aS,6R,9S,11S,12S,12aR,12bS)-1,2a,3,4,4a,6,9,10,11,12, 12a,12b-Dodecahydro-4,6,9,11,12,12b-hexahydroxy-4a,8,13, 13-tetramethyl-7,11-methano-5H-cyclodeca[3,4]-benz[1,2-b] oxet-5-one 6,12b-diacetate, 12-benzoate, 9-ester with (2R,3S)- N-benzoyl-3-phenylisoserine.
Paclitaxel has the following structural formula:
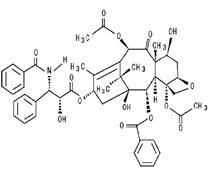
Paclitaxel, USP is a white to off-white powder with the empirical formula C47H51NO14 and a molecular weight of 853.9. It is insoluble in water, soluble in alcohol and melts at around 212°C to 217°C.
CLINICAL PHARMACOLOGY
Paclitaxel is a novel antimicrotubule agent that promotes the assembly of microtubules from tubulin dimers and stabilizes microtubules by preventing depolymerization. This stability results in the inhibition of the normal dynamic reorganization of the microtubule network that is essential for vital interphase and mitotic cellular functions. In addition, paclitaxel induces abnormal arrays or “bundles” of microtubules throughout the cell cycle and multiple asters of microtubules during mitosis.
Following intravenous administration of paclitaxel, paclitaxel plasma concentrations declined in a biphasic manner. The initial rapid decline represents distribution to the peripheral compartment and elimination of the drug. The later phase is due, in part, to a relatively slow efflux of paclitaxel from the peripheral compartment.
Pharmacokinetic parameters of paclitaxel following 3- and 24-hour infusions of paclitaxel at dose levels of 135 and 175 mg/m2 were determined in a Phase 3 randomized study in ovarian cancer patients and are summarized in the following table:
TABLE 1. SUMMARY OF PHARMACOKINETIC PARAMETERS – MEAN VALUES
| Dose
(mg/m2) | Infusion
Duration (h) | N
(patients) | Cmax
(ng/mL) | AUC(0-∞)
(ng•h/mL) | T-HALF
(h) | CLT
(L/h/m2) |
| 135 | 24 | 2 | 195 | 6,300 | 52.7 | 21.7 |
| 175 | 24 | 4 | 365 | 7,993 | 15.7 | 23.8 |
| 135 | 3 | 7 | 2,170 | 7,952 | 13.1 | 17.7 |
| 175 | 3 | 5 | 3,650 | 15,007 | 20.2 | 12.2 |
| Cmax = Maximum plasma concentration AUC (0-∞) = Area under the plasma concentration-time curve from time 0 to infinity CLT = Total body clearance |
||||||
It appeared that with the 24-hour infusion of paclitaxel, a 30% increase in dose (135 mg/m2 vs 175 mg/m2) increased the Cmax by 87%, whereas the AUC (0-∞) remained proportional. However, with a 3-hour infusion, for a 30% increase in dose, the Cmax and AUC (0-∞) were increased by 68% and 89%, respectively. The mean apparent volume of distribution at steady state, with the 24-hour infusion of paclitaxel, ranged from 227 to 688 L/m2, indicating extensive extravascular distribution and/or tissue binding of paclitaxel.
The pharmacokinetics of paclitaxel were also evaluated in adult cancer patients who received single doses of 15 to 135 mg/m2 given by 1-hour infusions (n=15), 30 to 275 mg/m2 given by 6-hour infusions (n=36), and 200 to 275 mg/m2 given by 24-hour infusions (n=54) in Phase 1 and 2 studies. Values for CLT and volume of distribution were consistent with the findings in the Phase 3 study. The pharmacokinetics of paclitaxel in patients with AIDS-related Kaposi's sarcoma have not been studied.
In vitro studies of binding to human serum proteins, using paclitaxel concentrations ranging from 0.1 to 50 mcg/mL, indicate that between 89 to 98% of drug is bound; the presence of cimetidine, ranitidine, dexamethasone, or diphenhydramine did not affect protein binding of paclitaxel.
After intravenous administration of 15 to 275 mg/m2 doses of paclitaxel as 1-, 6-, or 24-hour infusions, mean values for cumulative urinary recovery of unchanged drug ranged from 1.3% to 12.6% of the dose, indicating extensive non-renal clearance. In 5 patients administered a 225 or 250 mg/m2 dose of radiolabeled paclitaxel as a 3-hour infusion, a mean of 71% of the radioactivity was excreted in the feces in 120 hours, and 14% was recovered in the urine. Total recovery of radioactivity ranged from 56% to 101% of the dose. Paclitaxel represented a mean of 5% of the administered radioactivity recovered in the feces, while metabolites, primarily 6α-hydroxypaclitaxel, accounted for the balance. In vitro studies with human liver microsomes and tissue slices showed that paclitaxel was metabolized primarily to 6α-hydroxypaclitaxel by the cytochrome P450 isozyme CYP2C8; and to 2 minor metabolites, 3’-p-hydroxypaclitaxel and 6α, 3’-p-dihydroxypaclitaxel, by CYP3A4. In vitro, the metabolism of paclitaxel to 6α-hydroxypaclitaxel was inhibited by a number of agents (ketoconazole, verapamil, diazepam, quinidine, dexamethasone, cyclosporin, teniposide, etoposide, and vincristine), but the concentrations used exceeded those found in vivo following normal therapeutic doses. Testosterone, 17α-ethinyl estradiol, retinoic acid, and quercetin, a specific inhibitor of CYP2C8, also inhibited the formation of 6α-hydroxypaclitaxel in vitro. The pharmacokinetics of paclitaxel may also be altered in vivo as a result of interactions with compounds that are substrates, inducers, or inhibitors of CYP2C8 and/or CYP3A4 (see PRECAUTIONS, Drug Interactions).
The disposition and toxicity of paclitaxel 3-hour infusion were evaluated in 35 patients with varying degrees of hepatic function. Relative to patients with normal bilirubin, plasma paclitaxel exposure in patients with abnormal serum bilirubin ≤2 times upper limit of normal (ULN) administered 175 mg/m2 was increased, but with no apparent increase in the frequency or severity of toxicity. In 5 patients with serum total bilirubin >2 times ULN, there was a statistically nonsignificant higher incidence of severe myelosuppression, even at a reduced dose (110 mg/m2), but no observed increase in plasma exposure (see PRECAUTIONS, Hepatic and DOSAGE AND ADMINISTRATION). The effect of renal or hepatic dysfunction on the disposition of paclitaxel has not been investigated.
Possible interactions of paclitaxel with concomitantly administered medications have not been formally investigated.
CLINICAL STUDIES
Ovarian Carcinoma
First-Line Data
The safety and efficacy of paclitaxel followed by cisplatin in patients with advanced ovarian cancer and no prior chemotherapy were evaluated in 2, Phase 3 multicenter, randomized, controlled trials. In an Intergroup study led by the European Organization for Research and Treatment of Cancer involving the Scandinavian Group NOCOVA, the National Cancer Institute of Canada, and the Scottish Group, 680 patients with Stage IIB–C, III, or IV disease (optimally or non-optimally debulked) received either paclitaxel 175 mg/m2 infused over 3 hours followed by cisplatin 75 mg/m2 (Tc) or cyclophosphamide 750 mg/m2 followed by cisplatin 75 mg/m2 (Cc) for a median of 6 courses. Although the protocol allowed further therapy, only 15% received both drugs for 9 or more courses. In a study conducted by the Gynecological Oncology Group (GOG), 410 patients with Stage III or IV disease (>1 cm residual disease after staging laparotomy or distant metastases) received either paclitaxel 135 mg/m2 infused over 24 hours followed by cisplatin 75 mg/m2 or cyclophosphamide 750 mg/m2 followed by cisplatin 75 mg/m2 for 6 courses.
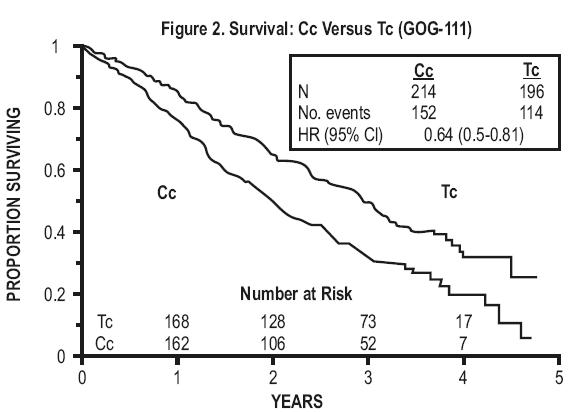
In both studies, patients treated with paclitaxel in combination with cisplatin had significantly higher response rate, longer time to progression, and longer survival time compared with standard therapy. These differences were also significant for the subset of patients in the Intergroup study with non-optimally debulked disease, although the study was not fully powered for subset analyses (TABLES 2A and 2B). Kaplan- Meier survival curves for each study are shown in FIGURES 1 and 2.
TABLE 2A. EFFICACY IN THE PHASE 3 FIRST-LINE OVARIAN CARCINOMA STUDIES
| Intergroup (non-optimally debulked subset) | GOG-111 | |||||
| T175/3a
c75 (n=218) | C750a
c75 (n=227) | T135/24a
c75 (n=196) | C750a
c75 (n=214) |
|||
| • Clinical Responseb
- rate (percent) - p-valuec | (n=153) 58 | 0.016 | (n=153) 43 | (n=113) 62 | 0.04 | (n=127) 48 |
| • Time to Progression - median (months) - p-valuec - hazard ratio (HR)c - 95% CIc | 13.2 | 0.0060 0.76 0.62 to 0.92 | 9.9 | 16.6 | 0.0008 0.70 0.56 to 0.86 | 13.0 |
| • Survival - median (months) - p-valuec - hazard ratio (HR)c - 95% CIc | 29.5 | 0.0057 73 0.58 to 0.91 | 21.9 | 35.5 | 0.0002 0.64 0.50 to 0.81 | 24.2 |
| a Paclitaxel dose in mg/m2/infusion duration in hours; cyclophosphamide and cisplatin doses in mg/m2. b Among patients with measurable disease only. c Unstratified for the Intergroup Study, Stratified for Study GOG-111. |
TABLE 2B. EFFICACY IN THE PHASE 3 FIRST-LINE OVARIAN CARCINOMA INTERGROUP STUDY
| T175/3a
c75 (n=342) | C750a
c75 (n=338) |
||
| • Clinical Responseb
- rate (percent) - p-valuec | (n=162) 59 | 0.014 | (n=161) 45 |
| • Time to Progression - median (months) - p-valuec - hazard ratio (HR)c - 95% CIc | 15.3 | 0.0005 0.74 0.63 to 0.88 | 11.5 |
| • Survival
- median (months) - p-valuec - hazard ratio (HR)c - 95% CIc | 35.6 | 0.0016 0.73 0.60 to 0.89 | 25.9 |
| a Paclitaxel dose in mg/m2/infusion duration in hours; cyclophosphamide and cisplatin doses in mg/m2. b Among patients with measurable disease only. c Unstratified. |
Figure 1. Survival: Cc Versus Tc (Intergroup)
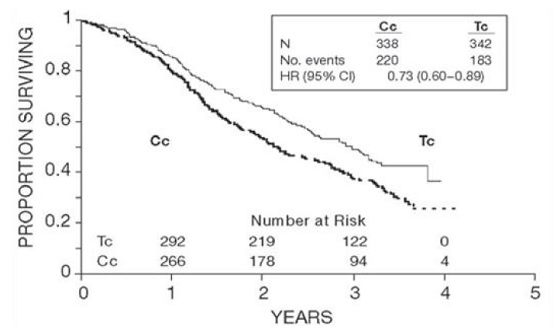
Figure 2. Survival: Cc Versus Tc (GOG-111)
The adverse event profile for patients receiving paclitaxel in combination with cisplatin in these studies was qualitatively consistent with that seen for the pooled analysis of data from 812 patients treated with single-agent paclitaxel in 10 clinical studies. These adverse events and adverse events from the Phase 3 first-line ovarian carcinoma studies are described in the ADVERSE REACTIONSsection in tabular (TABLES 10 and 11) and narrative form.
Second-Line Data
Data from 5, Phase 1 and 2 clinical studies (189 patients), a multicenter randomized Phase 3 study (407 patients), as well as an interim analysis of data from more than 300 patients enrolled in a treatment referral center program were used in support of the use of paclitaxel in patients who have failed initial or subsequent chemotherapy for metastatic carcinoma of the ovary. Two of the Phase 2 studies (92 patients) utilized an initial dose of 135 to 170 mg/m2 in most patients (>90%) administered over 24 hours by continuous infusion. Response rates in these 2 studies were 22% (95% CI, 11 to 37%) and 30% (95% CI, 18 to 46%) with a total of 6 complete and 18 partial responses in 92 patients. The median duration of overall response in these 2 studies measured from the first day of treatment was 7.2 months (range, 3.5 to 15.8 months) and 7.5 months (range, 5.3 to 17.4 months), respectively. The median survival was 8.1 months (range, 0.2 to 36.7 months) and 15.9 months (range, 1.8 to 34.5+ months).
The Phase 3 study had a bifactorial design and compared the efficacy and safety of paclitaxel, administered at 2 different doses (135 or 175 mg/m2) and schedules (3- or 24-hour infusion). The overall response rate for the 407 patients was 16.2% (95% CI, 12.8 to 20.2%), with 6 complete and 60 partial responses. Duration of response, measured from the first day of treatment was 8.3 months (range, 3.2 to 21.6 months). Median time to progression was 3.7 months (range, 0.1+ to 25.1+ months). Median survival was 11.5 months (range, 0.2 to 26.3+ months).
Response rates, median survival, and median time to progression for the 4 arms are given in the following table.
TABLE 3. EFFICACY IN THE PHASE 3 SECOND-LINE OVARIAN CARCINOMA STUDY
|
|
175/3 (n=96) |
175/24 (n=106) |
135/3 (n=99) |
135/24 (n=106) |
|
• Response - rate (percent) - 95% Confidence Interval |
14.6 (8.5 to 23.6) |
21.7 (14.5 to 31) |
15.2 (9 to 24.1) |
13.2 (7.7 to 21.5) |
|
• Time to Progression - median (months) - 95% Confidence Interval |
4.4 (3 to 5.6) |
4.2 (3.5 to 5.1) |
3.4 (2.8 to 4.2) |
2.8 (1.9 to 4) |
|
• Survival - median (months) - 95% Confidence Interval |
11.5 (8.4 to 14.4) |
11.8 (8.9 to 14.6) |
13.1 (9.1 to 14.6) |
10.7 (8.1 to 13.6) |
Analyses were performed as planned by the bifactorial study design described in the protocol, by comparing the 2 doses (135 or 175 mg/m2) irrespective of the schedule (3 or 24 hours) and the 2 schedules irrespective of dose. Patients receiving the 175 mg/m2 dose had a response rate similar to that for those receiving the 135 mg/m2 dose: 18% versus 14% (p=0.28). No difference in response rate was detected when comparing the 3-hour with the 24-hour infusion: 15% versus 17% (p=0.50). Patients receiving the 175 mg/m2 dose of paclitaxel had a longer time to progression than those receiving the 135 mg/m2 dose: median 4.2 versus 3.1 months (p=0.03). The median time to progression for patients receiving the 3-hour versus the 24-hour infusion was 4 months versus 3.7 months, respectively. Median survival was 11.6 months in patients receiving the 175 mg/m2 dose of paclitaxel and 11 months in patients receiving the 135 mg/m2 dose (p=0.92). Median survival was 11.7 months for patients receiving the 3-hour infusion of paclitaxel and 11.2 months for patients receiving the 24-hour infusion (p=0.91). These statistical analyses should be viewed with caution because of the multiple comparisons made.
Paclitaxel remained active in patients who had developed resistance to platinum-containing therapy (defined as tumor progression while on, or tumor relapse within 6 months from completion of, a platinum-containing regimen) with response rates of 14% in the Phase 3 study and 31% in the Phase 1 and 2 clinical studies.
The adverse event profile in this Phase 3 study was consistent with that seen for the pooled analysis of data from 812 patients treated in 10 clinical studies. These adverse events and adverse events from the Phase 3 second-line ovarian carcinoma study are described in the ADVERSE REACTIONSsection in tabular (TABLES 10 and 12) and narrative form.
The results of this randomized study support the use of paclitaxel at doses of 135 to 175 mg/m2, administered by a 3-hour intravenous infusion. The same doses administered by 24-hour infusion were more toxic. However, the study had insufficient power to determine whether a particular dose and schedule produced superior efficacy.
Breast Carcinoma
Adjuvant Therapy
A Phase 3 Intergroup study (Cancer and Leukemia Group B [CALGB], Eastern Cooperative Oncology Group [ECOG], North Central Cancer Treatment Group [NCCTG], and Southwest Oncology Group [SWOG]) randomized 3,170 patients with node-positive breast carcinoma to adjuvant therapy with paclitaxel or to no further chemotherapy following 4 courses of doxorubicin and cyclophosphamide (AC). This multicenter trial was conducted in women with histologically positive lymph nodes following either a mastectomy or segmental mastectomy and nodal dissections. The 3 x 2 factorial study was designed to assess the efficacy and safety of 3 different dose levels of doxorubicin (A) and to evaluate the effect of the addition of paclitaxel administered following the completion of AC therapy. After stratification for the number of positive lymph nodes (1 to 3, 4 to 9, or 10+), patients were randomized to receive cyclophosphamide at a dose of 600 mg/m2 and doxorubicin at doses of either 60 mg/m2 (on day 1), 75 mg/m2 (in 2 divided doses on days 1 and 2), or 90 mg/m2 (in 2 divided doses on days 1 and 2 with prophylactic G-CSF support and ciprofloxacin) every 3 weeks for 4 courses and either paclitaxel 175 mg/m2 as a 3-hour infusion every 3 weeks for 4 additional courses or no additional chemotherapy. Patients whose tumors were positive were to receive subsequent tamoxifen treatment (20 mg daily for 5 years); patients who received segmental mastectomies prior to study were to receive breast irradiation after recovery from treatment-related toxicities.
At the time of the current analysis, median follow-up was 30.1 months. Of the 2,066 patients who were hormone receptor positive, 93% received tamoxifen. The primary analyses of disease-free survival and overall survival used multivariate Cox models, which included paclitaxel administration, doxorubicin dose, number of positive lymph nodes, tumor size, menopausal status, and estrogen receptor status as factors. Based on the model for disease-free survival, patients receiving AC followed by paclitaxel had a 22% reduction in the risk of disease recurrence compared to patients randomized to AC alone (Hazard Ratio [HR]=0.78, 95% CI, 0.67 to 0.91, p=0.0022). They also had a 26% reduction in the risk of death (HR=0.74, 95% CI, 0.60 to 0.92, p=0.0065). For disease-free survival and overall survival, p-values were not adjusted for interim analyses. Kaplan-Meier curves are shown in FIGURES 3 and 4. Increasing the dose of doxorubicin higher than 60 mg/m2 had no effect on either disease-free are shown in FIGURE 3 survival or overall survival.
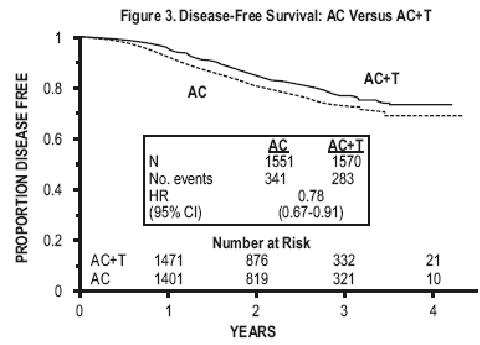
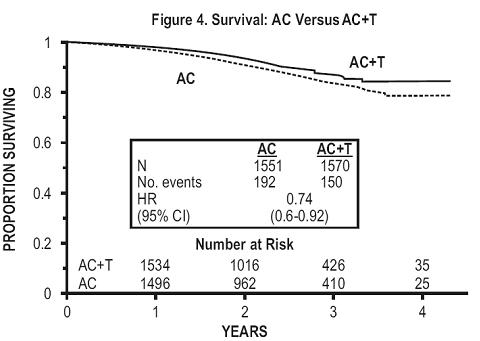
Subset Analyses
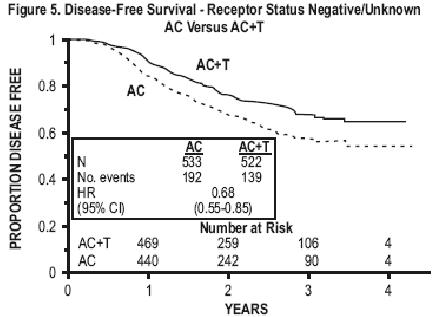
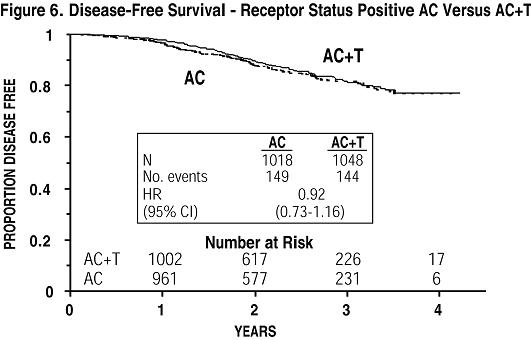
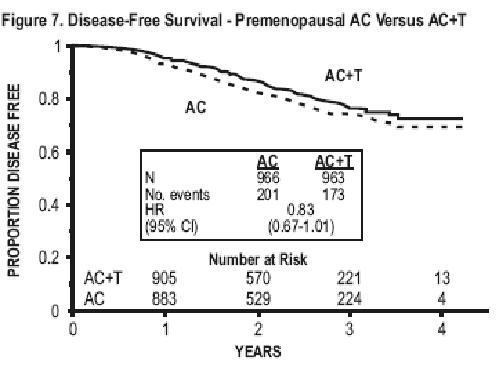
Subsets defined by variables of known prognostic importance in adjuvant breast carcinoma were examined, including number of positive lymph nodes, tumor size, hormone receptor status, and menopausal status. Such analyses must be interpreted with care, as the most secure finding is the overall study result. In general, a reduction in hazard similar to the overall reduction was seen with paclitaxel for both disease-free and overall survival in all of the larger subsets with one exception; patients with receptor-positive tumors had a smaller reduction in hazard (HR=0.92) for disease-free survival with paclitaxel than other groups. Results of subset analyses are shown in TABLE 4.
TABLE 4. SUBSET ANALYSES — ADJUVANT BREAST CARCINOMA STUDY
| Disease-Free Survival | Overall Survival | ||||
| Patient Subset | No. of Patients | No. of Recurrences | Hazard Ratio (95% CI) | No. of Deaths | Hazard Ratio (95% CI) |
| • No. of Positive Nodes 1 to 3 4 to 9 10+ | 1,4491 1,310 360 | 221 274 129 | 0.72 (0.55 to 0.94) 0.78 (0.61 to 0.99) 0.93 (0.66 to 1.31) | 107 148 87 | 0.76 (0.52-1.12) 0.66 (0.47-0.91) 0.90 (0.59-1.36) |
| • Tumor Size (cm) ≤2 >2 and ≤5 >5 | 1,096 1,611 397 | 153 358 111 | 0.79 (0.57 to 1.08) 0.79 (0.64 to 0.97) 0.75 (0.51 to 1.08) | 67 201 72 | 0.73 (0.45-1.18) 0.74 (0.56-0.98) 0.73 (0.46-1.16) |
| • Menopausal Status Pre Post | 1,929 1,183 | 374 250 | 0.83 (0.67 to 1.01) 0.73 (0.57 to 0.93) | 187 155 | 0.72 (0.54-0.97) 0.77 (0.56-1.06) |
| • Receptor Status Positivea Negative/Unknownb | 2,066 1,055 | 293 331 | 0.92 (0.73 to 1.16) 0.68 (0.55 to 0.85) | 126 216 | 0.83 (0.59-1.18) 0.71 (0.54-0.93) |
| a Positive for either estrogen or progesterone receptors. b Negative or missing for both estrogen and progesterone receptors (both missing: n=15). |
These retrospective subgroup analyses suggest that the beneficial effect of paclitaxel is clearly established in the receptor-negative subgroup, but the benefit in receptor-positive patients is not yet clear. With respect to menopausal status, the benefit of paclitaxel is consistent (see TABLE 4 and FIGURES 5 to 8).

The adverse event profile for the patients who received paclitaxel subsequent to AC was consistent with that seen in the pooled analysis of data from 812 patients (TABLE 10) treated with single-agent paclitaxel in 10 clinical studies. These adverse events are described in the ADVERSE REACTIONSsection in tabular (TABLES 10 and 13) and narrative form.
After Failure of Initial Chemotherapy
Data from 83 patients accrued in three Phase 2 open label studies and from 471 patients enrolled in a Phase 3 randomized study were available to support the use of paclitaxel in patients with metastatic breast carcinoma.
Phase 2 Open Label Studies
Two studies were conducted in 53 patients previously treated with a maximum of one prior chemotherapeutic regimen. Paclitaxel was administered in these two trials as a 24-hour infusion at initial doses of 250 mg/m2 (with G-CSF support) or 200 mg/m2. The response rates were 57% (95% CI: 37% to 75%) and 52% (95% CI: 32% to 72%), respectively. The third Phase 2 study was conducted in extensively pretreated patients who had failed anthracycline therapy and who had received a minimum of two chemotherapy regimens for the treatment of metastatic disease. The dose of paclitaxel was 200 mg/m2 as a 24-hour infusion with G-CSF support. Nine of 30 patients achieved a partial response, for a response rate of 30% (95% CI: 15% to 50%).
Phase 3 Randomized Study
This multicenter trial was conducted in patients previously treated with one or two regimens of chemotherapy. Patients were randomized to receive paclitaxel at a dose of either 175 mg/m2 or 135 mg/m2 given as a 3-hour infusion. In the 471 patients enrolled, 60% had symptomatic disease with impaired performance status at study entry, and 73% had visceral metastases. These patients had failed prior chemotherapy either in the adjuvant setting (30%), the metastatic setting (39%), or both (31%). Sixty-seven percent of the patients had been previously exposed to anthracyclines and 23% of them had disease considered resistant to this class of agents.
The overall response rate for the 454 evaluable patients was 26% (95% CI: 22% to 30%), with 17 complete and 99 partial responses. The median duration of response, measured from the first day of treatment, was 8.1 months (range: 3.4 to 18.1+ months). Overall for the 471 patients, the median time to progression was 3.5 months (range: 0.03 to 17.1 months). Median survival was 11.7 months (range: 0 to 18.9 months).
Response rates, median survival and median time to progression for the 2 arms are given in the following table.
TABLE 5: EFFICACY IN BREAST CANCER AFTER FAILURE OF INITIAL CHEMOTHERAPY OR WITHIN 6 MONTHS OF ADJUVANT CHEMOTHERAPY
| 175/3 (n=235) | 135/3 (n=236) | ||
| • Response - rate (percent) - p-value | 28 | 0.135 | 22 |
| • Time to Progression
- median (months) - p-value | 4.2 | 0.027 | 3 |
| • Survival - median (months) - p-value | 11.7 | 0.321 | 10.5 |
The adverse event profile of the patients who received single-agent Paclitaxel Injection, USP, in the Phase 3 study was consistent with that seen for the pooled analysis of data from 812 patients treated in 10 clinical studies. These adverse events and adverse events from the Phase 3 breast carcinoma study are described in the ADVERSE REACTIONSsection in tabular (TABLES 10 and 14) and narrative form.
Non-Small Cell Lung Carcinoma (NSCLC)
In a Phase 3 open-label randomized study conducted by the ECOG, 599 patients were randomized to either paclitaxel (T) 135 mg/m2 as a 24-hour infusion in combination with cisplatin (c) 75 mg/m2, paclitaxel (T) 250 mg/m2 as a 24-hour infusion in combination with cisplatin (c) 75 mg/m2 with G-CSF support, or cisplatin (c) 75 mg/m2 on day 1, followed by etoposide (VP) 100 mg/m2 on days 1, 2, and 3 (control). Response rates, median time to progression, median survival, and 1-year survival rates are given in the following table. The reported p-values have not been adjusted for multiple comparisons. There were statistically significant differences favoring each of the paclitaxel plus cisplatin arms for response rate and time to tumor progression. There was no statistically significant difference in survival between either paclitaxel plus cisplatin arm and the cisplatin plus etoposide arm.
TABLE 6: EFFICACY PARAMETERS IN THE PHASE 3 FIRST-LINE NSCLC STUDY
| T135/24 c75 (n=198) | T250/24 c75 (n=201) | VP100a
c75 (n=200) |
|
| • Response
- rate (percent) - p-valueb | 25 0.001 | 23 <0.001 | 12 |
| • Time to Progression - median (months) - p-valueb | 4.3 0.05 | 4.9 0.004 | 2.7 |
| • Survival - median (months) - p-valueb | 9.3 0.12 | 10 0.08 | 7.4 |
| • 1-Year Survival - percent of patients | 36 | 40 | 32 |
| a Etoposide (VP) 100 mg/m2 was administered IV on days 1, 2, and 3. b Compared to cisplatin/etoposide. |
In the ECOG study, the Functional Assessment of Cancer Therapy-Lung (FACT-L) questionnaire had 7 subscales that measured subjective assessment of treatment. Of the 7, the Lung Cancer Specific Symptoms subscale favored the paclitaxel 135 mg/m2/24 hour plus cisplatin arm compared to the cisplatin/etoposide arm. For all other factors, there was no difference in the treatment groups.
The adverse event profile for patients who received paclitaxel in combination with cisplatin in this study was generally consistent with that seen for the pooled analysis of data from 812 patients treated with single-agent paclitaxel in 10 clinical studies. These adverse events and adverse events from the Phase 3 first-line NSCLC study are described in the ADVERSE REACTIONSsection in tabular (TABLES 10 and 15) and narrative form.
Data from 2, Phase 2 open-label studies support the use of paclitaxel as second-line therapy in patients with AIDS-related Kaposi's sarcoma. Fifty-nine of the 85 patients enrolled in these studies had previously received systemic therapy, including interferon alpha (32%), Dauno Xome® (31%), DOXIL® (2%), and doxorubicin containing chemotherapy (42%), with 64% having received prior anthracyclines. Eighty-five percent of the pretreated patients had progressed on, or could not tolerate, prior systemic therapy.1
In Study CA139-174, patients received paclitaxel at 135 mg/m2 as a 3-hour infusion every 3 weeks (intended dose intensity 45 mg/m2/week). If no dose-limiting toxicity was observed, patients were to receive 155 mg/m2 and 175 mg/m2 in subsequent courses. Hematopoietic growth factors were not to be used initially. In Study CA139-281, patients received paclitaxel at 100 mg/m2 as a 3-hour infusion every 2 weeks (intended dose intensity 50 mg/m2/week). In this study patients could be receiving hematopoietic growth factors before the start of paclitaxel therapy, or this support was to be initiated as indicated; the dose of paclitaxel was not increased. The dose intensity of paclitaxel used in this patient population was lower than the dose intensity recommended for other solid tumors.
All patients had widespread and poor-risk disease. Applying the ACTG staging criteria to patients with prior systemic therapy, 93% were poor risk for extent of disease (T1), 88% had a CD4 count <200 cells/mm3 (I1), and 97% had poor risk considering their systemic illness (S1).
All patients in Study CA139-174 had a Karnofsky performance status of 80 or 90 at baseline; in Study CA139-281, there were 26 (46%) patients with a Karnofsky performance status of 70 or worse at baseline.
TABLE 7. EXTENT OF DISEASE AT STUDY ENTRY PERCENT OF PATIENTS
| Prior Systemic Therapy (n=59) |
|
| Visceral ± edema ± oral ± cutaneous | 42 |
| Edema or lymph nodes ± oral ± cutaneous | 41 |
| Oral ± cutaneous | 10 |
| Cutaneous only | 7 |
Although the planned dose intensity in the 2 studies was slightly different (45 mg/m2/week in Study CA139-174 and 50 mg/m2/week in Study CA139-281), delivered dose intensity was 38 to 39 mg/m2/week in both studies, with a similar range (20 to 24 to 51 to 61).
Efficacy
The efficacy of paclitaxel was evaluated by assessing cutaneous tumor response according to the amended ACTG criteria and by seeking evidence of clinical benefit in patients in 6 domains of symptoms and/or conditions that are commonly related to AIDS-related Kaposi's sarcoma.
Cutaneous Tumor Response (Amended ACTG Criteria)
The objective response rate was 59% (95% CI, 46 to 72%) (35 of 59 patients) in patients with prior systemic therapy. Cutaneous responses were primarily defined as flattening of more than 50% of previously raised lesions.
TABLE 8. OVERALL BEST RESPONSE (AMENDED ACTG CRITERIA) PERCENT OF PATIENTS
| Prior Systemic Therapy (n=59) |
|
| Complete response | 3 |
| Partial response | 56 |
| Stable disease | 29 |
| Progression | 8 |
| Early death/toxicity | 3 |
The median time to response was 8.1 weeks and the median duration of response measured from the first day of treatment was 10.4 months (95% CI, 7 to 11 months) for the patients who had previously received systemic therapy. The median time to progression was 6.2 months (95% CI, 4.6 to 8.7 months).
Additional Clinical Benefit
Most data on patient benefit were assessed retrospectively (plans for such analyses were not included in the study protocols). Nonetheless, clinical descriptions and photographs indicated clear benefit in some patients, including instances of improved pulmonary function in patients with pulmonary involvement, improved ambulation, resolution of ulcers, and decreased analgesic requirements in patients with Kaposi’s sarcoma (KS) involving the feet and resolution of facial lesions and edema in patients with KS involving the face, extremities, and genitalia.
Safety
The adverse event profile of paclitaxel administered to patients with advanced HIV disease and poor-risk AIDS-related Kaposi's sarcoma was generally similar to that seen in the pooled analysis of data from 812 patients with solid tumors. These adverse events and adverse events from the Phase 2 second-line Kaposi's sarcoma studies are described in the ADVERSE REACTIONSsection in tabular (TABLES 10 and 16) and narrative form. In this immunosuppressed patient population, however, a lower dose intensity of paclitaxel and supportive therapy including hematopoietic growth factors in patients with severe neutropenia are recommended. Patients with AIDS-related Kaposi’s sarcoma may have more severe hematologic toxicities than patients with solid tumors.
INDICATIONS AND USAGE
Paclitaxel Injection, USP is indicated as subsequent therapy for the treatment of advanced carcinoma of the ovary. As first-line therapy, Paclitaxel Injection, USP is indicated in combination with cisplatin.
Paclitaxel Injection, USP is indicated for the adjuvant treatment of node-positive breast cancer administered sequentially to standard doxorubicin-containing combination chemotherapy. In the clinical trial, there was an overall favorable effect on disease-free and overall survival in the total population of patients with receptor-positive and receptor-negative tumors, but the benefit has been specifically demonstrated by available data (median follow-up 30 months) only in the patients with estrogen and progesterone receptor-negative tumors (see CLINICAL STUDIES, Breast Carcinoma).
Paclitaxel Injection, USP is indicated for the treatment of breast cancer after failure of combination chemotherapy for metastatic disease or relapse within 6 months of adjuvant chemotherapy. Prior therapy should have included an anthracycline unless clinically contraindicated.
Paclitaxel Injection, USP, in combination with cisplatin, is indicated for the first-line treatment of non-small cell lung cancer in patients who are not candidates for potentially curative surgery and/or radiation therapy.
Paclitaxel Injection, USP is indicated for the second-line treatment of AIDS-related Kaposi's sarcoma.
CONTRAINDICATIONS
Paclitaxel is contraindicated in patients who have a history of hypersensitivity reactions to paclitaxel or other drugs formulated in polyoxyl 35 castor oil.
Paclitaxel should not be used in patients with solid tumors who have baseline neutrophil counts of <1,500 cells/mm3 or in patients with AIDS-related Kaposi's sarcoma with baseline neutrophil counts of <1,000 cells/mm3.
WARNINGS
Anaphylaxis and severe hypersensitivity reactions characterized by dyspnea and hypotension requiring treatment, angioedema, and generalized urticaria have occurred in 2 to 4% of patients receiving paclitaxel in clinical trials. Fatal reactions have occurred in patients despite premedication. All patients should be pretreated with corticosteroids, diphenhydramine, and H2 antagonists (see DOSAGE AND ADMINISTRATION). Patients who experience severe hypersensitivity reactions to paclitaxel should not be rechallenged with the drug.
Bone marrow suppression (primarily neutropenia) is dose-dependent and is the dose-limiting toxicity. Neutrophil nadirs occurred at a median of 11 days. Paclitaxel should not be administered to patients with baseline neutrophil counts of less than 1,500 cells/mm3 (<1,000 cells/mm3 for patients with KS). Frequent monitoring of blood counts should be instituted during paclitaxel treatment. Patients should not be re-treated with subsequent cycles of paclitaxel until neutrophils recover to a level >1,500 cells/mm3 (>1,000 cells/mm3 for patients with KS) and platelets recover to a level >100,000 cells/mm3.
Severe conduction abnormalities have been documented in <1% of patients during paclitaxel therapy and in some cases requiring pacemaker placement. If patients develop significant conduction abnormalities during paclitaxel infusion, appropriate therapy should be administered and continuous cardiac monitoring should be performed during subsequent therapy with paclitaxel.
Pregnancy
Paclitaxel can cause fetal harm when administered to a pregnant woman. Administration of paclitaxel during the period of organogenesis to rabbits at doses of 3 mg/kg/day (about 0.2 the daily maximum recommended human dose on a mg/m2 basis) caused embryo-and fetotoxicity, as indicated by intrauterine mortality, increased resorptions, and increased fetal deaths. Maternal toxicity was also observed at this dose. No teratogenic effects were observed at 1 mg/kg/day (about 1/15 the daily maximum recommended human dose on a mg/m2 basis); teratogenic potential could not be assessed at higher doses due to extensive fetal mortality.
There are no adequate and well-controlled studies in pregnant women. If paclitaxel is used during pregnancy, or if the patient becomes pregnant while receiving this drug, the patient should be apprised of the potential hazard to the fetus. Women of child-bearing potential should be advised to avoid becoming pregnant.
PRECAUTIONS
Contact of the undiluted concentrate with plasticized polyvinyl chloride (PVC) equipment or devices used to prepare solutions for infusion is not recommended. In order to minimize patient exposure to the plasticizer DEHP [di-(2-ethylhexyl)phthalate], which may be leached from PVC infusion bags or sets, diluted paclitaxel solutions should preferably be stored in bottles (glass, polypropylene) or plastic bags (polypropylene, polyolefin) and administered through polyethylene-lined administration sets.
Paclitaxel should be administered through an in-line filter with a microporous membrane not greater than 0.22 microns. Use of filter devices such as IVEX-2® filters which incorporate short inlet and outlet PVC-coated tubing has not resulted in significant filter devices such leaching of DEHP.
Drug Interactions
In a Phase I trial using escalating doses of paclitaxel (110 to 200 mg/m2) and cisplatin (50 or 75 mg/m2) given as sequential infusions, myelosuppression was more profound when paclitaxel was given after cisplatin than with the alternate sequence (i.e., paclitaxel before cisplatin). Pharmacokinetic data from these patients demonstrated a decrease in paclitaxel clearance of approximately 33% when paclitaxel was administered following cisplatin.
The metabolism of paclitaxel is catalyzed by cytochrome P450 isoenzymes CYP2C8 and CYP3A4. Caution should be exercised when administering paclitaxel concomitantly with known substrates or inhibitors of the cytochrome P450 isoenzymes CYP2C8 and CYP3A4. Caution should be exercised when paclitaxel is concomitantly administered with known substrates (e.g., midazolam, buspirone, felodipine, lovastatin, eletriptan, sildenafil, simvastatin, and triazolam), inhibitors (e.g., atazanavir, clarithromycin, indinavir, itraconazole, ketoconazole, nefazodone, nelfinavir, ritonavir, saquinavir, and telithromycin), and inducers (e.g., rifampin and carbamazepine) of CYP3A4 (see CLINICAL PHARMACOLOGY).
Caution should also be exercised when paclitaxel is concomitantly administered with known substrates (e.g., repaglinide and rosiglitazone), inhibitors (e.g., gemfibrozil), and inducers (e.g., rifampin) of CYP2C8 (see CLINICAL PHARMACOLOGY).
Potential interactions between paclitaxel, a substrate of CYP3A4, and protease inhibitors (ritonavir, saquinavir, indinavir, and nelfinavir), which are substrates and/or inhibitors of CYP3A4, have not been evaluated in clinical trials.
Reports in the literature suggest that plasma levels of doxorubicin (and its active metabolite doxorubicinol) may be increased when paclitaxel and doxorubicin are used in combination.
Hematology
Paclitaxel therapy should not be administered to patients with baseline neutrophil counts of less than 1,500 cells/mm3. In order to monitor the occurrence of myelotoxicity, it is recommended that frequent peripheral blood cell counts be performed on all patients receiving paclitaxel. Patients should not be re-treated with subsequent cycles of paclitaxel until neutrophils recover to a level >1,500 cells/mm3 and platelets recover to a level >100,000 cells/mm3. In the case of severe neutropenia (<500 cells/mm3 for 7 days or more) during a course of paclitaxel therapy, a 20% reduction in dose for subsequent courses of therapy is recommended.
For patients with advanced HIV disease and poor-risk AIDS-related Kaposi's sarcoma, paclitaxel, at the recommended dose for this disease, can be initiated and repeated if the neutrophil count is at least 1,000 cells/mm3.
Hypersensitivity Reactions
Patients with a history of severe hypersensitivity reactions to products containing polyoxyl 35 castor oil (e.g., cyclosporin for injection concentrate and teniposide for injection concentrate) should not be treated with paclitaxel. In order to avoid the occurrence of severe hypersensitivity reactions, all patients treated with paclitaxel should be premedicated with corticosteroids (such as dexamethasone), diphenhydramine and H antagonists (such as cimetidine or ranitidine). Minor symptoms such as flushing, skin reactions, dyspnea, hypotension, or tachycardia do not require interruption of therapy. However, severe reactions, such as hypotension requiring treatment, dyspnea requiring bronchodilators, angioedema, or generalized urticaria require immediate discontinuation of paclitaxel and aggressive symptomatic therapy. Patients who have developed severe hypersensitivity reactions should not be rechallenged with paclitaxel.
Cardiovascular
Hypotension, bradycardia, and hypertension have been observed during administration of paclitaxel, but generally do not require treatment. Occasionally paclitaxel infusions must be interrupted or discontinued because of initial or recurrent hypertension. Frequent vital sign monitoring, particularly during the first hour of paclitaxel infusion, is recommended. Continuous cardiac monitoring is not required except for patients with serious conduction abnormalities (see WARNINGS). When paclitaxel is used in combination with doxorubicin for treatment of metastatic breast cancer, monitoring of cardiac function is recommended (see ADVERSE REACTIONS).
Nervous System
Although the occurrence of peripheral neuropathy is frequent, the development of severe symptomatology is unusual and requires a dose reduction of 20% for all subsequent courses of paclitaxel.
Paclitaxel contains dehydrated alcohol USP, 396 mg/mL; consideration should be given to possible CNS and other effects of alcohol (see PRECAUTIONS, Pediatric Use).
Hepatic
There is limited evidence that the myelotoxicity of paclitaxel may be exacerbated in patients with serum total bilirubin >2 times ULN (see CLINICAL PHARMACOLOGY). Extreme caution should be exercised when administering paclitaxel to such patients, with dose reduction as recommended in DOSAGE AND ADMINISTRATION, TABLE 17.
Injection Site Reaction
Injection site reactions, including reactions secondary to extravasation, were usually mild and consisted of erythema, tenderness, skin discoloration, or swelling at the injection site. These reactions have been observed more frequently with the 24-hour infusion than with the 3-hour infusion. Recurrence of skin reactions at a site of previous extravasation following administration of paclitaxel at a different site, i.e., “recall”, has been reported.
More severe events such as phlebitis, cellulitis, induration, skin exfoliation, necrosis, and fibrosis have been reported. In some cases, the onset of the injection site reaction either occurred during a prolonged infusion or was delayed by a week to 10 days.
A specific treatment for extravasation reactions is unknown at this time. Given the possibility of extravasation, it is advisable to closely monitor the infusion site for possible infiltration during drug administration.
Carcinogenesis, Mutagenesis, Impairment of Fertility
The carcinogenic potential of paclitaxel has not been studied.
Paclitaxel has been shown to be clastogenic in vitro (chromosome aberrations in human lymphocytes) and in vivo (micronucleus test in mice). Paclitaxel was not mutagenic in the Ames test or the CHO/HGPRT gene mutation assay.
Administration of paclitaxel prior to and during mating produced impairment of fertility in male and female rats at doses equal to or greater than 1 mg/kg/day (about 0.04 the daily maximum recommended human dose on a mg/m2 basis). At this dose, to or greater than 1 mg/kg/day (about 0.04 the daily maximum recommended human dose on a mg/m2 basis). At this paclitaxel caused reduced fertility and reproductive indices, and increased embryo- and fetotoxicity (see WARNINGS).
Nursing Mothers
It is not known whether the drug is excreted in human milk. Following intravenous administration of carbon-14 labeled paclitaxel to rats on days 9 to 10 postpartum, concentrations of radioactivity in milk were higher than in plasma and declined in parallel with the plasma concentrations. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants, it is recommended that nursing be discontinued when receiving paclitaxel therapy.
Pediatric Use
The safety and effectiveness of paclitaxel in pediatric patients have not been established.
There have been reports of central nervous system (CNS) toxicity (rarely associated with death) in a clinical trial in pediatric patients in which paclitaxel was infused intravenously over 3 hours at doses ranging from 350 mg/m2 to 420 mg/m2. The toxicity is most likely attributable to the high dose of the ethanol component of the paclitaxel vehicle given over a short infusion time. The use of concomitant antihistamines may intensify this effect. Although a direct effect of the paclitaxel itself cannot be discounted, the high doses used in this study (over twice the recommended adult dosage) must be considered in assessing the discounted, the high doses used in this study safety of paclitaxel for use in this population.
Geriatric Use
Of 2,228 patients who received paclitaxel in 8 clinical studies evaluating its safety and effectiveness in the treatment of advanced ovarian cancer, breast carcinoma, or NSCLC, and 1,570 patients who were randomized to receive paclitaxel in the adjuvant breast cancer study, 649 patients (17%) were 65 years or older and 49 patients (1%) were 75 years or older. In most studies, severe myelosuppression was more frequent in elderly patients; in some studies, severe neuropathy was more common in elderly patients. In 2 clinical studies in NSCLC, the elderly patients treated with paclitaxel had a higher incidence of cardiovascular events. Estimates of efficacy appeared similar in elderly patients and in younger patients; however, comparative efficacy cannot be determined with confidence due to the small number of elderly patients studied. In a study of first-line treatment of ovarian cancer, elderly patients had a lower median survival than younger patients, but no other efficacy parameters favored the younger group. TABLE 9 presents the incidences of Grade IV neutropenia and severe neuropathy in clinical studies according to age.
TABLE 9: SELECTED ADVERSE EVENTS IN GERIATRIC PATIENTS RECEIVING PACLITAXEL IN CLINICAL STUDIES
|
| Patients [n/total (%)]
|
|||
|---|---|---|---|---|
|
| Neutropenia
(Grade IV) | Peripheral Neuropathy
(Grades III/IV) |
||
| INDICATION
| Age (y)
| Age (y)
|
||
| (Study/Regimen)
| ≥65
| <65
| ≥65
| <65
|
| • OVARIAN Cancer
| | | | |
| (Intergroup First-Line/T175/3 c75a) | 34/83 (41) | 78/252 (31) | 24/84 (29)*b
| 46/255 (18)b
|
| (GOG-111 First-Line/T135/24 c75a) | 48/61 (79) | 106/129 (82) | 3/62 (5) | 2/134 (1) |
| (Phase 3 Second-Line/T175/3c) | 5/19 (26) | 21/76 (28) | 1/19 (5) | 0/76 (0) |
| (Phase 3 Second-Line/T175/24c) | 21/25 (84) | 57/79 (72) | 0/25 (0) | 2/80 (3) |
| (Phase 3 Second-Line/T135/3c) | 4/16 (25) | 10/81 (12) | 0/17 (0) | 0/81 (0) |
| (Phase 3 Second-Line/T135/24c) | 17/22 (77) | 53/83 (64) | 0/22 (0) | 0/83 (0) |
| (Phase 3 Second-Line Pooled) | 47/82 (57)*
| 141/319 (44) | 1/83 (1) | 2/320 (1) |
| •Adjuvant BREAST Cancer
| | | | |
| (Intergroup/AC followed by Td) | 56/102 (55) | 734/1,468 (50) | 5/102 (5)e
| 46/1,468 (3)e
|
| BREAST Cancer After Failure of Initial Therapy
| | | | |
| (Phase 3/T175/3c) | 7/24 (29) | 56/200 (28) | 3/25 (12) | 12/204 (6) |
| (Phase 3/T135/3c) | 7/20 (35) | 37/207 (18) | 0/20 (0) | 6/209 (3) |
| • Non-Small Cell LUNG Cancer
| | | | |
| (ECOG/T135/24 c75a) | 58/71 (82) | 86/124 (69) | 9/71 (13)f
| 16/124 (13)f
|
| (Phase 3/T175/3 c80a) | 37/89 (42)*
| 56/267 (21) | 11/91 (12)*
| 11/271 (4) |
| * p<0.05 a Paclitaxel dose in mg/m2/infusion duration in hours; cisplatin doses in mg/m2. b Peripheral neuropathy was included within the neurotoxicity category in the Intergroup First-Line Ovarian Cancer study (see TABLE 11). c Paclitaxel dose in mg/m2/infusion duration in hours. d Paclitaxel (T) following 4 courses of doxorubicin and cyclophosphamide (AC) at a dose of 175 mg/m2/3 hours every 3 weeks for 4 courses. e Peripheral neuropathy reported as neurosensory toxicity in the Intergroup Adjuvant Breast Cancer study (see TABLE 13). f Peripheral neuropathy reported as neurosensory toxicity in the ECOG NSCLC study (see TABLE 15). |
||||
Information for Patients
(seePatient Information Leaflet).
ADVERSE REACTIONS
Pooled Analysis of Adverse Event Experiences from Single-Agent Studies
Data in the following table are based on the experience of 812 patients (493 with ovarian carcinoma and 319 with breast carcinoma) enrolled in 10 studies who received single-agent paclitaxel injection. Two hundred and seventy-five patients were treated in 8, Phase 2 studies with paclitaxel doses ranging from 135 to 300 mg/m2 administered over 24 hours (in 4 of these studies, G-CSF was administered as hematopoietic support). Three hundred and one patients were treated in the randomized Phase 3 ovarian carcinoma study which compared 2 doses (135 or 175 mg/m2) and 2 schedules (3 or 24 hours) of paclitaxel.
Two hundred and thirty-six patients with breast carcinoma received paclitaxel (135 or 175 mg/m2) administered over 3 hours in a controlled study.
TABLE10. SUMMARYa OF ADVERSE EVENTS IN PATIENTS WITH SOLID TUMORS RECEIVING SINGLE-AGENT PACLITAXEL
| Percent of Patients (n=812) |
||
| Bone Marrow | ||
| Neutropenia | < 2,000/mm3 | 90 |
| < 500/mm3 | 52 | |
| Leukopenia | < 4,000/mm3 | 90 |
| < 1,000/mm3 | 17 | |
| Thrombocytopenia | < 100,000/mm3 | 20 |
| < 50,000/mm3 | 7 | |
| Anemia | < 11 g/dL | 78 |
| < 8 g/dL | 16 | |
| Infections | 30 | |
| Bleeding | 14 | |
| Red Cell Transfusions | 25 | |
| Platelet Transfusions | 2 | |
| Hypersensitivity Reactionb | ||
| All | 41 | |
| Severe† | 2 | |
| Cardiovascular | ||
| Vital Sign Changesc | ||
| Bradycardia (n=537) | 3 | |
| Hypotension (n=532) | 12 | |
| Significant Cardiovascular Events | 1 | |
| Abnormal ECG | ||
| All Pts | 23 | |
| Pts with normal baseline (n=559) | 14 | |
| Peripheral Neuropathy | ||
| Any symptoms | 60 | |
| Severe symptoms† | 3 | |
| Myalgia/Arthralgia | ||
| Any symptoms | 60 | |
| Severe symptoms† | 8 | |
| Gastrointestinal | ||
| Nausea and vomiting | 52 | |
| Diarrhea | 38 | |
| Mucositis | 31 | |
| Alopecia | 87 | |
| Hepatic (Pts with normal baseline and on study data) | ||
| Bilirubin elevations (n=765) | 7 | |
| Alkaline phosphatase elevations (n=575) | 22 | |
| AST (SGOT) elevations (n=591) | 19 | |
| Injection Site Reaction | 13 | |
| a Based on worst course analysis. b All patients received premedication. c During the first 3 hours of infusion. † Severe events are defined as at least Grade III toxicity. |
None of the observed toxicities were clearly influenced by age.
Disease-Specific Adverse Event Experiences
First-Line Ovary in Combination
For the 1,084 patients who were evaluable for safety in the Phase 3 first-line ovary combination therapy studies, TABLE 11 shows the incidence of important adverse events. For both studies, the analysis of safety was based on all courses of therapy (6 courses for the GOG-111 study and up to 9 courses for the Intergroup study).
TABLE 11: FREQUENCYa OF IMPORTANT ADVERSE EVENTS IN THE PHASE 3 FIRST-LINE OVARIAN CARCINOMA STUDIES
| Percent of Patients | ||||
| Intergroup | GOG-111 | |||
| T175/3b
c75c (n=339) | C750c
c75c (n=336) | T135/24b
c75c (n=196) | C750c
c75c (n=213) |
|
| • Bone Marrow | ||||
| - Neutropenia < 2,000/mm3 | 91d | 95d | 96 | 92 |
| < 500/mm3 | 33d | 43d | 81d | 58d |
| - Thrombocytopenia < 100,000/mm3e | 21d | 33d | 26 | 30 |
| < 50,000/mm3 | 3d | 7d | 10 | 9 |
| - Anemia < 11 g/dLf | 96 | 97 | 88 | 86 |
| < 8 g/dL | 3d | 8d | 13 | 9 |
| - Infections | 25 | 27 | 21 | 15 |
| - Febrile Neutropenia | 4 | 7 | 15d | 4d |
| • Hypersensitivity Reaction | ||||
| - All | 11d | 6d | 8d,g | 1d,g |
| - Severe† | 1 | 1 | 3d,g | --d,g |
| • Neurotoxicityh | ||||
| - Any symptoms | 87d | 52d | 25 | 20 |
| - Severe symptoms† | 21d | 2d | 3d | --d |
| • Nausea and Vomiting | ||||
| - Any symptoms | 88 | 93 | 65 | 69 |
| - Severe symptoms† | 18 | 24 | 10 | 11 |
| • Myalgia/Arthralgia | ||||
| - Any symptoms | 60d | 27d | 9d | 2d |
| - Severe symptoms† | 6d | 1d | 1 | -- |
| • Diarrhea | ||||
| - Any symptoms | 37d | 29d | 16d | 8d |
| - Severe symptoms† | 2 | 3 | 4 | 1 |
| • Asthenia | ||||
| - Any symptoms | NC | NC | 17d | 10d |
| - Severe symptoms† | NC | NC | 1 | 1 |
| • Alopecia | ||||
| - Any symptoms | 96d | 89d | 55d | 37d |
| - Severe symptoms† | 51d | 21d | 6 | 8 |
| a Based on worst course analysis. b Paclitaxel (T) dose in mg/m2/infusion duration in hours. c Cyclophosphamide (C) or cisplatin (c) dose in mg/m2. d p<0.05 by Fisher exact test. e <130,000/mm3 in the Intergroup study. f <12 g/dL in the Intergroup study. g All patients received premedication. h In the GOG-111 study, neurotoxicity was collected as peripheral neuropathy and in the Intergroup study, neurotoxicity was collected as either neuromotor or neurosensory symptoms. † Severe events are defined as at least Grade III toxicity. NC Not Collected. |
Second-Line Ovary
For the 403 patients who received single-agent paclitaxel injection in the Phase 3 second-line ovarian carcinoma study, the following table shows the incidence of important adverse events.
TABLE 12. FREQUENCYa OF IMPORTANT ADVERSE EVENTS IN THE PHASE 3 SECOND-LINE OVARIAN CARCINOMA STUDY
| Percent of Patients | ||||
| 175/3b
(n=95) | 175/24b
(n=105) | 135/3b
(n=98) | 135/24b
(n=105) |
|
| • Bone Marrow | ||||
| -Neutropenia <2,000/mm3 | 78 | 98 | 78 | 98 |
| <500/mm3 | 27 | 75 | 14 | 67 |
| -Thrombocytopenia <100,000/mm3 | 4 | 18 | 8 | 6 |
| <50,000/mm3 | 1 | 7 | 2 | 1 |
| - Anemia <11 g/dL | 84 | 90 | 68 | 88 |
| <8 g/dL | 11 | 12 | 6 | 10 |
| - Infections | 26 | 29 | 20 | 18 |
| • Hypersensitivity ReactionC | ||||
| - All | 41 | 45 | 38 | 45 |
| - Severe† | 2 | 0 | 2 | 1 |
| • Peripheral Neuropathy | ||||
| - Any symptoms | 63 | 60 | 55 | 42 |
| - Severe symptoms† | 1 | 2 | 0 | 0 |
| • Mucositis | ||||
| - Any symptoms | 17 | 35 | 21 | 25 |
| - Severe symptoms† | 0 | 3 | 0 | 2 |
| a Based on worst course analysis. b Paclitaxel dose in mg/m2/infusion duration in hours. c All patients received premedication. † Severe events are defined as at least Grade III toxicity. |
Myelosuppression was dose and schedule related, with the schedule effect being more prominent. The development of severe hypersensitivity reactions (HSRs) was rare; 1% of the patients and 0.2% of the courses overall. There was no apparent dose or schedule effect seen for the HSRs. Peripheral neuropathy was clearly dose related, but schedule did not appear to affect the incidence.
Adjuvant Breast
For the Phase 3 adjuvant breast carcinoma study, the following table shows the incidence of important severe adverse events for the 3,121 patients (total population) who were evaluable for safety as well as for a group of 325 patients (early population) who, per the study protocol, were monitored more intensively than other patients.
TABLE 13. FREQUENCYa OF IMPORTANT SEVEREb ADVERSE EVENTS IN THE PHASE 3 ADJUVANT BREAST CARCINOMA STUDY
| Percent of Patients | ||||
| Early Population | Total Population | |||
| ACc
(n=166) | ACc followed by Td
(n=159) | ACc
(n=1,551) | ACc followed by Td
(n=1,570) |
|
| • Bone Marrowe | ||||
| - Neutropenia < 500/mm3 | 79 | 76 | 48 | 50 |
| -Thrombocytopenia < 50,000/mm3 | 27 | 25 | 11 | 11 |
| - Anemia < 8 g/dL | 17 | 21 | 8 | 8 |
| - Infections | 6 | 14 | 5 | 6 |
| - Fever without Infection | - | 3 | <1 | 1 |
| • Hypersensitivity Reactionf | 1 | 4 | 1 | 2 |
| • Cardiovascular Events | 1 | 2 | 1 | 2 |
| • Neuromotor Toxicity | 1 | 1 | <1 | 1 |
| • Neurosensory Toxicity | - | 3 | <1 | 3 |
| • Myalgia/Arthralgia | - | 2 | <1 | 2 |
| • Nausea/Vomiting | 13 | 18 | 8 | 9 |
| • Mucositis | 13 | 4 | 6 | 5 |
| a Based on worst course analysis. b Severe events are defined as at least Grade III toxicity. c Patients received 600 mg/m2 cyclophosphamide and doxorubicin (AC) at doses of either 60 mg/m2, 75 mg/m2, or 90 mg/m2 (with prophylactic G-CSF support and ciprofloxacin), every 3 weeks for 4 courses. d Paclitaxel (T) following 4 courses of AC at a dose of 175 mg/m2/3 hours every 3 weeks for 4 courses. e The incidence of febrile neutropenia was not reported in this study. f All patients were to receive premedication. |
The incidence of an adverse event for the total population likely represents an underestimation of the actual incidence given that safety data were collected differently based on enrollment cohort. However, since safety data were collected consistently across regimens, the safety of the sequential addition of paclitaxel following AC therapy may be compared with AC therapy alone. Compared to patients who received AC alone, patients who received AC followed by paclitaxel experienced more Grade III/IV neurosensory toxicity, more Grade III/IV myalgia/arthralgia, more Grade III/IV neurologic pain (5% vs 1%), more Grade III/IV flu-like symptoms (5% vs 3%), and more Grade III/IV hyperglycemia (3% vs 1%). During the additional 4 courses of treatment with paclitaxel, 2 deaths (0.1%) were attributed to treatment. During paclitaxel treatment, Grade IV neutropenia was reported for 15% of patients, Grade II/III neurosensory toxicity for 15%, Grade II/III myalgias for 23%, and alopecia for 46%.
The incidences of severe hematologic toxicities, infections, mucositis, and cardiovascular events increased with higher doses of doxorubicin.
Breast Cancer After Failure of Initial Chemotherapy
For the 458 patients who received single-agent paclitaxel in the Phase 3 breast carcinoma study, the following table shows the incidence of important adverse events by treatment arm (each arm was administered by a 3-hour infusion).
TABLE 14: FREQUENCYa OF IMPORTANT ADVERSE EVENTS IN THE PHASE 3 STUDY OF BREAST CANCER AFTER FAILURE OF INITIAL CHEMOTHERAPY OR WITHIN 6 MONTHS OF ADJUVANT CHEMOTHERAPY
| Percent of Patients | ||
| 175/3b
(n=229) | 135/3b
(n=229) |
|
| • Bone Marrow | ||
| - Neutropenia < 2,000/mm3 | 90 | 81 |
| < 500/mm3 | 28 | 19 |
| - Thrombocytopenia < 100,000/mm3 | 11 | 7 |
| < 50,000/mm3 | 3 | 2 |
| - Anemia < 11 g/dL | 55 | 47 |
| < 8 g/dL | 4 | 2 |
| - Infections | 23 | 15 |
| - Febrile Neutropenia | 2 | 2 |
| • Hypersensitivity Reactionc | ||
| - All | 36 | 31 |
| - Severe† | 0 | <1 |
| • Peripheral Neuropathy | ||
| - Any symptoms | 70 | 46 |
| - Severe symptoms† | 7 | 3 |
| • Mucositis | ||
| - Any symptoms | 23 | 17 |
| - Severe symptoms† | 3 | <1 |
| a Based on worst course analysis. b Paclitaxel dose in mg/m2/infusion duration in hours. c All patients received premedication. † Severe events are defined as at least Grade III toxicity. |
Myelosuppression and peripheral neuropathy were dose related. There was one severe hypersensitivity reaction (HSR) observed at the dose of 135 mg/m2.
First-Line NSCLC in Combination
In the study conducted by the Eastern Cooperative Oncology Group (ECOG), patients were randomized to either paclitaxel (T) 135 mg/m2 as a 24-hour infusion in combination with cisplatin (c) 75 mg/m2, paclitaxel (T) 250 mg/m2 as a 24-hour infusion in combination with cisplatin (c) 75 mg/m2 with G-CSF support, or cisplatin (c) 75 mg/m2 on day 1, followed by etoposide (VP) 100 mg/m2 on days 1, 2, and 3 (control).
The following table shows the incidence of important adverse events.
TABLE 15. FREQUENCYa OF IMPORTANT ADVERSE EVENTS IN THE PHASE 3 STUDY FOR FIRST-LINE NSCLC
| Percent of Patients | |||
| T135/24b
c75 (n=195) | T250/24c
c75 (n=197) | VP100d
c75 (n=196) |
|
| • Bone Marrow | |||
| - Neutropenia < 2,000/mm3 | 89 | 86 | 84 |
| < 500/mm3 | 74e | 65 | 55 |
| - Thrombocytopenia < normal | 48 | 68 | 62 |
| < 50,000/mm3 | 6 | 12 | 16 |
| - Anemia < normal | 94 | 96 | 95 |
| < 8 g/dL | 22 | 19 | 28 |
| - Infections | 38 | 31 | 35 |
| • Hypersensitivity Reactionf | |||
| - All | 16 | 27 | 13 |
| - Severe† | 1 | 4e | 1 |
| • Arthralgia/Myalgia | |||
| - Any symptoms | 21e | 42e | 9 |
| - Severe symptoms† | 3 | 11 | 1 |
| • Nausea/Vomiting | |||
| - Any symptoms | 85 | 87 | 81 |
| - Severe symptoms† | 27 | 29 | 22 |
| • Mucositis | |||
| - Any symptoms | 18 | 28 | 16 |
| - Severe symptoms† | 1 | 4 | 2 |
| • Neuromotor Toxicity | |||
| - Any symptoms | 37 | 47 | 44 |
| - Severe symptoms† | 6 | 12 | 7 |
| • Neurosensory Toxicity | |||
| - Any symptoms | 48 | 61 | 25 |
| - Severe symptoms† | 13 | 28e | 8 |
| • Cardiovascular Events | |||
| - Any symptoms | 33 | 39 | 24 |
| - Severe symptoms† | 13 | 12 | 8 |
| a Based on worst course analysis. b Paclitaxel (T) dose in mg/m2/infusion duration in hours; cisplatin (c) dose in mg/m2. c Paclitaxel dose in mg/m2/infusion duration in hours with G-CSF support; cisplatin dose in mg/m2. d Etoposide (VP) dose in mg/m2 was administered IV on days 1, 2, and 3; cisplatin dose in mg/m2. e p<0.05. f All patients received premedication. † Severe events are defined as at least Grade III toxicity. |
Toxicity was generally more severe in the high-dose paclitaxel treatment arm (T250/c75) than in the low-dose paclitaxel arm (T135/c75). Compared to the cisplatin/etoposide arm, patients in the low-dose paclitaxel arm experienced more arthralgia/myalgia of any grade and more severe neutropenia. The incidence of febrile neutropenia was not reported in this study.
Kaposi's Sarcoma
The following table shows the frequency of important adverse events in the 85 patients with KS treated with 2 different single-agent paclitaxel regimens.
TABLE 16. FREQUENCYa OF IMPORTANT ADVERSE EVENTS IN THE AIDS -RELATED KAPOSI'S SARCOMA STUDIES
| Percent of Patients | ||
| Study CA139-174 Paclitaxel 135/3b q 3 wk (n=29) | Study CA139-281 Paclitaxel 100/3b q 2 wk (n=56) |
|
| • Bone Marrow | ||
| - Neutropenia < 2,000/mm3 | 100 | 95 |
| < 500/mm3 | 76 | 35 |
| - Thrombocytopenia < 100,000/mm3 | 52 | 27 |
| < 50,000/mm3 | 17 | 5 |
| - Anemia < 11 g/dL | 86 | 73 |
| < 8 g/dL | 34 | 25 |
| - Febrile Neutropenia | 55 | 9 |
| • Opportunistic Infection | ||
| - Any | 76 | 54 |
| - Cytomegalovirus | 45 | 27 |
| - Herpes Simplex | 38 | 11 |
| - Pneumocystis carinii | 14 | 21 |
| - M. avium intracellulare | 24 | 4 |
| - Candidiasis, esophageal | 7 | 9 |
| - Cryptosporidiosis | 7 | 7 |
| - Cryptococcal meningitis | 3 | 2 |
| - Leukoencephalopathy | - | 2 |
| • Hypersensitivity Reactionc | ||
| - All | 14 | 9 |
| • Cardiovascular | ||
| - Hypotension | 17 | 9 |
| - Bradycardia | 3 | - |
| • Peripheral Neuropathy | ||
| - Any | 79 | 46 |
| - Severe† | 10 | 2 |
| • Myalgia/Arthralgia | ||
| - Any | 93 | 48 |
| - Severe† | 14 | 16 |
| • Gastrointestinal | ||
| - Nausea and Vomiting | 69 | 70 |
| - Diarrhea | 90 | 73 |
| - Mucositis | 45 | 20 |
| • Renal (creatinine elevation) | ||
| - Any | 34 | 18 |
| - Severe† | 7 | 5 |
| • Discontinuation for drug toxicity | 7 | 16 |
| a Based on worst course analysis. b Paclitaxel dose in mg/m2/infusion duration in hours. c All patients received premedication. † Severe events are defined as at least Grade III toxicity. |
Adverse Event Experiences by Body System
The following discussion refers to the overall safety database of 812 patients with solid tumors treated with single-agent paclitaxel in clinical studies. Toxicities that occurred with greater severity or frequency in previously untreated patients with ovarian carcinoma or NSCLC who received paclitaxel in combination with cisplatin or in patients with breast cancer who received paclitaxel after doxorubicin/cyclophosphamide in the adjuvant setting and that occurred with a difference that was clinically significant in these populations are also described.
The frequency and severity of important adverse events for the Phase 3 ovarian carcinoma, breast carcinoma, NSCLC, and the Phase 2 Kaposi’s sarcoma carcinoma studies are presented above in tabular form by treatment arm. In addition, rare events have been reported from postmarketing experience or from other clinical studies. The frequency and severity of adverse events have been generally similar for patients receiving paclitaxel for the treatment of ovarian, breast, or lung carcinoma or Kaposi’s sarcoma, but patients with AIDS-related Kaposi’s sarcoma may have more frequent and severe hematologic toxicity, infections (including opportunistic infections, see TABLE 16), and febrile neutropenia. These patients require a lower dose intensity and supportive care (see CLINICAL STUDIES, AIDS-Related Kaposi's Sarcoma). Toxicities that were observed only in or were noted to have occurred with greater severity in the population with Kaposi's sarcoma and that occurred with a difference that was clinically significant in this population are described. Elevated liver function tests and renal toxicity have a higher incidence in KS patients as compared to patients with solid tumors.
Hematologic
Bone marrow suppression was the major dose-limiting toxicity of paclitaxel. Neutropenia, the most important hematologic toxicity, was dose and schedule dependent and was generally rapidly reversible. Among patients treated in the Phase 3 second line ovarian study with a 3- hour infusion, neutrophil counts declined below 500 cells/mm3 in 14% of the patients treated with a dose of 135 mg/m2 compared to 27% at a dose of 175 mg/m2 (p=0.05). In the same study, severe neutropenia (<500 cells/mm3) was more frequent with the 24-hour than with the 3-hour infusion; infusion duration had a greater impact on myelosuppression than dose. Neutropenia did not appear to increase with cumulative exposure and did not appear to be more frequent nor more severe for patients previously treated with radiation therapy.
In the study where paclitaxel was administered to patients with ovarian carcinoma at a dose of 135 mg/m2/24 hours in combination with cisplatin versus the control arm of cyclophosphamide plus cisplatin, the incidences of grade IV neutropenia and of febrile neutropenia were significantly greater in the paclitaxel plus cisplatin arm than in the control arm. Grade IV neutropenia occurred in 81% on the paclitaxel plus cisplatin arm versus 58% on the cyclophosphamide plus cisplatin arm, and febrile neutropenia occurred in 15% and 4% respectively. On the paclitaxel/cisplatin arm, there were 35/1,074 (3%) courses with fever in which Grade IV neutropenia was reported at some time during the course. When paclitaxel followed by cisplatin was administered to patients with advanced NSCLC in the ECOG study, the incidences of Grade IV neutropenia were 74% (paclitaxel 135 mg/m2/24 hours followed by cisplatin) and 65% (paclitaxel 250 mg/m2/24 hours followed by cisplatin and G-CSF) compared with 55% in patients who received cisplatin/etoposide.
Fever was frequent (12% of all treatment courses). Infectious episodes occurred in 30% of all patients and 9% of all courses; these episodes were fatal in 1% of all patients, and included sepsis, pneumonia and peritonitis. In the Phase 3 second-line ovarian study, infectious episodes were reported in 20% and 26% of the patients treated with a dose of 135 mg/m2 or 175 mg/m2 given as a 3-hour infusions respectively. Urinary tract infections and upper respiratory tract infections were the most frequently reported infectious complications. In the immunosuppressed patient population with advanced HIV disease and poor-risk AIDS-related Kaposi’s sarcoma, 61% of the patients reported at least one opportunistic infection (see CLINICAL STUDIES, AIDS-Related Kaposi's Sarcoma). The use of supportive therapy, including G-CSF, is recommended for patients who have experienced severe neutropenia (see DOSAGE AND ADMINISTRATION).
Thrombocytopenia was reported. Twenty percent of the patients experienced a drop in their platelet count below 100,000 cells/mm3 at least once while on treatment; 7% had a platelet count <50,000 cells/mm3 at the time of their worst nadir. Bleeding episodes were reported in 4% of all courses and by 14% of all patients but most of the hemorrhagic episodes were localized and the frequency of these events was unrelated to the paclitaxel dose and schedule. In the Phase 3 second-line ovarian study, bleeding episodes were reported in 10% of the patients; no patients treated with the 3- hour infusion received platelet transfusions. In the adjuvant breast carcinoma trial, the incidence of severe thrombocytopenia and platelet transfusions increased with higher doses of doxorubicin.
Anemia (Hb <11 g/dL) was observed in 78% of all patients and was severe (Hb <8 g/dL) in 16% of the cases. No consistent relationship between dose or schedule and the frequency of anemia was observed. Among all patients with normal baseline hemoglobin, 69% became anemic on study but only 7% had severe anemia. Red cell transfusions were required in 25% of all hemoglobin, 69% became anemic on study but only 7% had severe anemia. Red cell transfusions were required in 25% of all patients and in 12% of those with normal baseline hemoglobin levels.
Hypersensitivity Reactions (HSRs)
All patients received premedication prior to paclitaxel administration (see WARNINGSand PRECAUTIONS: Hypersensitivity Reactions). The frequency and severity of HSRs were not affected by the dose or schedule of paclitaxel administration. In the Phase 3 second-line ovarian study, the 3-hour infusion was not associated with a greater increase in HSRs when compared to the 24-hour infusion. Hypersensitivity reactions were observed in 20% of all courses and in 41% of all patients. These reactions were severe in less than 2% of the patients and 1% of the courses. No severe reactions were observed after course 3 and severe symptoms occurred generally within the first hour of paclitaxel infusion. The most frequent symptoms observed during these severe reactions were dyspnea, flushing, chest pain, and tachycardia. Abdominal pain, pain in the extremities, diaphoresis, and hypertension were also noted.
The minor hypersensitivity reactions consisted mostly of flushing (28%), rash (12%), hypotension (4%), dyspnea (2%), tachycardia (2%), and hypertension (1%). The frequency of hypersensitivity reactions remained relatively stable during the entire treatment period.
Chills, shock, and back pain in association with hypersensitivity reactions have been reported.
Cardiovascular
Hypotension, during the first 3 hours of infusion, occurred in 12% of all patients and 3% of all courses administered. Bradycardia, during the first 3 hours of infusion, occurred in 3% of all patients and 1% of all courses. In the Phase 3 second-line ovarian study, neither dose nor schedule had an effect on the frequency of hypotension and bradycardia. These vital sign changes most often caused no symptoms and required neither specific therapy nor treatment discontinuation. The frequency of hypotension and bradycardia were not influenced by prior anthracycline therapy.
Significant cardiovascular events possibly related to single-agent paclitaxel occurred in approximately 1% of all patients. These events included syncope, rhythm abnormalities, hypertension, and venous thrombosis. One of the patients with syncope treated with paclitaxel at 175 mg/m2 over 24 hours had progressive hypotension and died. The arrhythmias included asymptomatic ventricular tachycardia, bigeminy and complete AV block requiring pacemaker placement. Among patients with NSCLC treated with paclitaxel in combination with cisplatin in the Phase 3 study, significant cardiovascular events occurred in 12 to 13%. This apparent increase in cardiovascular events is possibly due to an increase in cardiovascular risk factors in patients with lung cancer.
Electrocardiogram (ECG) abnormalities were common among patients at baseline. ECG abnormalities on study did not usually result in symptoms, were not dose-limiting, and required no intervention. ECG abnormalities were noted in 23% of all patients. Among patients with a normal ECG prior to study entry, 14% of all patients developed an abnormal tracing while on study. The most frequently reported ECG modifications were non-specific repolarization abnormalities, sinus bradycardia, sinus tachycardia, and premature beats. Among patients with normal ECGs at baseline, prior therapy with anthracyclines did not influence the frequency of ECG abnormalities.
Cases of myocardial infarction have been reported. Congestive heart failure, including cardiac dysfunction and reduction of left ventricular ejection fraction or ventricular failure, has been reported typically in patients who have received other chemotherapy, notably anthracyclines (see PRECAUTIONS, Drug Interactions).
Atrial fibrillation and supraventricular tachycardia have been reported.
Respiratory
Interstitial pneumonia, lung fibrosis, and pulmonary embolism have been reported.
Radiation pneumonitis has been reported in patients receiving concurrent radiotherapy.
Pleural effusion and respiratory failure have been reported.
Neurologic
The assessment of neurologic toxicity was conducted differently among the studies as evident from the data reported in each individual study (see TABLES 10 to 16). Moreover, the frequency and severity of neurologic manifestations were influenced by prior and/or concomitant therapy with neurotoxic agents.
In general, the frequency and severity of neurologic manifestations were dose-dependent in patients receiving single-agent paclitaxel. Peripheral neuropathy was observed in 60% of all patients (3% severe) and in 52% (2% severe) of the patients without pre-existing neuropathy. The frequency of peripheral neuropathy increased with cumulative dose. Paresthesia commonly occurs in the form of hyperesthesia. Neurologic symptoms were observed in 27% of the patients after the first course of treatment and in 34 to 51% from course 2 to 10. Peripheral neuropathy was the cause of paclitaxel discontinuation in 1% of all patients. Sensory symptoms have usually improved or resolved within several months of paclitaxel discontinuation. Pre-existing neuropathies resulting from prior therapies are not a contraindication for paclitaxel therapy.
In the Intergroup first-line ovarian carcinoma study (see TABLE 11), neurotoxicity included reports of neuromotor and neurosensory events. The regimen with paclitaxel 175 mg/m2 given by 3-hour infusion plus cisplatin 75 mg/m2 resulted in greater incidence and severity of neurotoxicity than the regimen containing cyclophosphamide and cisplatin, 87% (21% severe) versus 52% (2% severe), respectively. The duration of grade III or IV neurotoxicity cannot be determined with precision for the Intergroup study since the resolution dates of adverse events were not collected in the case report forms for this trial and complete follow-up documentation was available only in a minority of these patients. In the GOG first-line ovarian carcinoma study, neurotoxicity was reported as peripheral neuropathy. The regimen with paclitaxel 135 mg/m2 given by 24-hour infusion plus cisplatin 75 mg/m2 resulted in an incidence of neurotoxicity that was similar to the regimen containing cyclophosphamide plus cisplatin, 25% (3% severe) versus 20% (0% severe), respectively. Cross-study comparison of neurotoxicity in the Intergroup and GOG trials suggests that when paclitaxel is given in combination with cisplatin 75 mg/m2, the incidence of severe neurotoxicity is more common at a paclitaxel dose of 175 mg/m2 given by 3-hour infusion (21%) than at a dose of 135 mg/m2 given by 24-hour infusion (3%).
In patients with NSCLC, administration of paclitaxel followed by cisplatin resulted in a greater incidence of severe neurotoxicity compared to the incidence in patients with ovarian or breast cancer treated with single-agent paclitaxel. Severe neurosensory symptoms were noted in 13% of NSCLC patients receiving paclitaxel 135 mg/m2 by 24-hour infusion followed by cisplatin 75 mg/m2 and 8% of NSCLC patients receiving cisplatin/etoposide (see TABLE 15).
Other than peripheral neuropathy, serious neurologic events following paclitaxel administration have been rare (<1%) and have included grand mal seizures, syncope, ataxia, and neuroencephalopathy.
Autonomic neuropathy resulting in paralytic ileus has been reported. Optic nerve and/or visual disturbances (scintillating scotomata) have also been reported, particularly in patients who have received higher doses than those recommended. These effects generally have been reversible.
However, reports in the literature of abnormal visual evoked potentials in patients have suggested persistent optic nerve damage. Postmarketing reports of ototoxicity (hearing loss and tinnitus) have also been received.
Convulsions, dizziness, and headache have been reported.
Arthralgia/Myalgia
There was no consistent relationship between dose or schedule of paclitaxel and the frequency or severity of arthralgia/myalgia. Sixty percent of all patients treated experienced arthralgia/myalgia; 8% experienced severe symptoms. The symptoms were usually transient, occurred 2 or 3 days after paclitaxel administration, and resolved within a few days. The frequency and severity of musculoskeletal symptoms remained unchanged throughout the treatment period.
Hepatic
No relationship was observed between liver function abnormalities and either dose or schedule of paclitaxel administration. Among patients with normal baseline liver function 7%, 22%, and 19% had elevations in bilirubin, alkaline phosphatase, and AST (SGOT), respectively. Prolonged exposure to paclitaxel was not associated with cumulative hepatic toxicity.
Hepatic necrosis and hepatic encephalopathy leading to death have been reported.
Renal
Among the patients treated for Kaposi’s sarcoma with paclitaxel, 5 patients had renal toxicity of grade III or IV severity. One patient with suspected HIV nephropathy of grade IV severity had to discontinue therapy. The other 4 patients had renal insufficiency with reversible elevations of serum creatinine.
Patients with gynecological cancers treated with paclitaxel and cisplatin may have an increased risk of renal failure with the combination therapy of paclitaxel and cisplatin in gynecological cancers as compared to cisplatin alone.
Gastrointestinal (GI)
Nausea/vomiting, diarrhea, and mucositis were reported by 52%, 38%, and 31% of all patients, respectively. These manifestations were usually mild to moderate. Mucositis was schedule dependent and occurred more frequently with the 24-hour than with the 3-hour infusion.
In patients with poor-risk AIDS-related Kaposi’s sarcoma, nausea/vomiting, diarrhea, and mucositis were reported by 69%, 79%, and 28% of patients, respectively. One-third of patients with Kaposi’s sarcoma complained of diarrhea prior to study start (see CLINICAL STUDIES: AIDS-Related Kaposi's Sarcoma).
In the first-line Phase 3 ovarian carcinoma studies, the incidence of nausea and vomiting when paclitaxel was administered in combination with cisplatin appeared to be greater compared with the database for single-agent paclitaxel in ovarian and breast carcinoma. In addition, diarrhea of any grade was reported more frequently compared to the control arm, but there was no difference for severe diarrhea in these studies.
Intestinal obstruction, intestinal perforation, pancreatitis, ischemic colitis, dehydration, esophagitis, constipation, and ascites have been reported. Neutropenic enterocolitis (typhlitis), despite the coadministration of G-CSF, was observed in patients treated with paclitaxel alone and in combination with other chemotherapeutic agents.
Injection Site Reaction
Injection site reactions, including reactions secondary to extravasation, were usually mild and consisted of erythema, tenderness, skin discoloration, or swelling at the injection site. These reactions have been observed more frequently with the 24-hour infusion than with the 3-hour infusion. Recurrence of skin reactions at a site of previous extravasation following administration of paclitaxel at a different site, i.e., “recall”, has been reported.
More severe events such as phlebitis, cellulitis, induration, skin exfoliation, necrosis, and fibrosis have been reported. In some cases the onset of the injection site reaction either occurred during a prolonged infusion or was delayed by a week to 10 days.
A specific treatment for extravasation reactions is unknown at this time. Given the possibility of extravasation, it is advisable to closely monitor the infusion site for possible infiltration during drug administration.
Other Clinical Events
Alopecia was observed in almost all (87%) of the patients. Transient skin changes due to paclitaxel-related hypersensitivity reactions have been observed, but no other skin toxicities were significantly associated with paclitaxel administration. Nail changes (changes in pigmentation or discoloration of nail bed) were uncommon (2%). Edema was reported in 21% of all patients (17% of those without baseline edema); only 1% had severe edema and none of these patients required treatment discontinuation. Edema was most commonly focal and disease-related. Edema was observed in 5% of all courses for patients with normal baseline and did not increase with time on study.
Skin abnormalities related to radiation recall as well as maculopapular rash, pruritus, Stevens-Johnson syndrome, and toxic epidermal necrolysis have been reported. In postmarketing experience, diffuse edema, thickening, and sclerosing of the skin have been reported following paclitaxel administration. Paclitaxel has been reported to exacerbate signs and symptoms of scleroderma.
Reports of asthenia and malaise have been received as part of the continuing surveillance of paclitaxel safety. In the Phase 3 trial of paclitaxel 135 mg/m2 over 24 hours in combination with cisplatin as first-line therapy of ovarian cancer, asthenia was reported in 17% of the patients, significantly greater than the 10% incidence observed in the control arm of cyclophosphamide/cisplatin.
Conjunctivitis, increased lacrimation, anorexia, confusional state, photopsia, visual floaters, vertigo, and increase in blood creatinine have been reported.
Accidental Exposure
Upon inhalation, dyspnea, chest pain, burning eyes, sore throat, and nausea have been reported. Following topical exposure, events have included tingling, burning, and redness.
To report SUSPECTED ADVERSE REACTIONS, contact Fresenius Kabi USA, LLC at 1-800-551-7176 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
OVERDOSAGE
There is no known antidote for paclitaxel overdosage. The primary anticipated complications of overdosage would consist of bone marrow suppression, peripheral neurotoxicity, and mucositis.
Overdoses in pediatric patients may be associated with acute ethanol toxicity (see PRECAUTIONS, Pediatric Use).
DOSAGE AND ADMINISTRATION
NOTE: Contact of the undiluted concentrate with plasticized PVC equipment or devices used to prepare solutions for infusion is not recommended. In order to minimize patient exposure to the plasticizer DEHP [di-(2-ethylhexyl)phthalate], which may be leached from PVC infusion bags or sets, diluted paclitaxel solutions should be stored in bottles (glass, polypropylene) or plastic bags (polypropylene, polyolefin) and administered through polyethylene-lined administration sets.
All patients should be premedicated prior to paclitaxel administration in order to prevent severe hypersensitivity reactions. Such premedication may consist of dexamethasone 20 mg PO administered approximately 12 and 6 hours before paclitaxel, diphenhydramine (or its equivalent) 50 mg I.V. 30 to 60 minutes prior to paclitaxel, and cimetidine (300 mg) or ranitidine (50 mg) I.V. 30 to 60 minutes before paclitaxel.
For patients with carcinoma of the ovary, the following regimens are recommended: (see CLINICAL STUDIES, Ovarian Carcinoma):
1. For previously untreated patients with carcinoma of the ovary, one of the following recommended regimens may be given every 3 weeks. In selecting the appropriate regimen, differences in toxicities should be considered (see TABLE 11 in ADVERSE REACTIONS, Disease-Specific Adverse Event Experiences).
1. Paclitaxel administered intravenously over 3 hours at a dose of 175 mg/m2 followed by cisplatin at a dose of 75 mg/m2; or
2. Paclitaxel administered intravenously over 24 hours at a dose of 135 mg/m2 followed by cisplatin at a dose of 75 mg/m2.
2. In patients previously treated with chemotherapy for carcinoma of the ovary, paclitaxel has been used at several doses and schedules; however, the optimal regimen is not yet clear. (see CLINICAL STUDIES, Ovarian Carcinoma). The recommended regimen is paclitaxel 135 mg/m2 or 175 mg/m2 administered intravenously over 3 hours every 3 weeks.
For patients with carcinoma of the breast, the following is recommended (see CLINICAL STUDIES, Breast Carcinoma):
1. For the adjuvant treatment of node-positive breast cancer, the recommended regimen is paclitaxel, at a dose of 175 mg/m2 intravenously over 3 hours every 3 weeks for 4 courses administered sequentially to doxorubicin-containing combination chemotherapy. The clinical trial used 4 courses of doxorubicin and cyclophosphamide (see CLINICAL STUDIES, Breast Carcinoma).
2. After failure of initial chemotherapy for metastatic disease or relapse within 6 months of adjuvant chemotherapy, paclitaxel at a dose of 175 mg/m2 administered intravenously over 3 hours every 3 weeks has been shown to be effective.
For patients with non-small cell lung carcinoma, the recommended regimen, given every 3 weeks, is paclitaxel administered intravenously over 24 hours at a dose of 135 mg/m2 followed by cisplatin, 75 mg/m2.
For patients with AIDS-related Kaposi’s sarcoma, paclitaxel administered at a dose of 135 mg/m2 given intravenously over 3 hours every 3 weeks or at a dose of 100 mg/m2 given intravenously over 3 hours every 2 weeks is recommended (dose intensity 45 to 50 mg/m2/week). In the 2 clinical trials evaluating these schedules (see CLINICAL STUDIES, AIDS-Related Kaposi'sSarcoma), the former schedule (135 mg/m2 every 3 weeks) was more toxic than the latter. In addition, all patients with low performance status were treated with the latter schedule (100 mg/m2 every 2 weeks).
Based upon the immunosuppression in patients with advanced HIV disease, the following modifications are recommended in these patients:
1. Reduce the dose of dexamethasone as 1 of the 3 premedication drugs to 10 mg PO (instead of 20 mg PO);
2. Initiate or repeat treatment with paclitaxel only if the neutrophil count is at least 1,000 cells/mm3;
3. Reduce the dose of subsequent courses of paclitaxel by 20% for patients who experience severe neutropenia (neutrophil <500 cells/mm3 for a week or longer); and
4. Initiate concomitant hematopoietic growth factor (G-CSF) as clinically indicated.
For the therapy of patients with solid tumors (ovary, breast and NSCLC), courses of paclitaxel should not be repeated until the neutrophil count is at least 1,500 cells/mm3 and the platelet count is at least 100,000 cells/mm3. Paclitaxel should not be given to patients with AIDS-related Kaposi’s sarcoma if the baseline or subsequent neutrophil count is less than 1,000 cells/mm3. Patients who experience severe neutropenia (neutrophil <500 cells/mm3 for a week or longer) or severe peripheral neuropathy during paclitaxel therapy should have dosage reduced by 20% for subsequent courses of paclitaxel. The incidence of neurotoxicity and the severity of neutropenia increase with dose.
Preparation and Administration Precautions:
Paclitaxel is a cytotoxic anticancer drug and, as with other potentially toxic compounds, caution should be exercised in handling paclitaxel. Adequate puncture technique should be used to reduce the probability of coring. Injection needles or cannulas larger than 21G in diameter tends to promote coring. Stoppers should be punctured slowly in the center of vial stopper. Multiple puncturing at same site, rapid puncturing, puncturing with blunt spiking devices and puncturing near the edge of the stopper increases the probability of coring. The use of gloves is recommended. If paclitaxel solution contacts the skin, wash the skin immediately and thoroughly with soap and water. Following topical exposure, events have included tingling, burning and redness. If paclitaxel contacts mucous membranes, the membranes should be flushed thoroughly with water. Upon inhalation, dyspnea, chest pain, burning eyes, sore throat, and nausea have been reported.
Hepatic Impairment
Patients with hepatic impairment may be at increased risk of toxicity, particularly grade III–IV myelosuppression (see CLINICAL PHARMACOLOGY and PRECAUTIONS, Hepatic). Recommendations for dosage adjustment for the first course of therapy are shown in TABLE 17 for both 3- and 24-hour infusions. Further dose reduction in subsequent courses should be based on individual tolerance. Patients should be monitored closely for the development of profound myelosuppression.
TABLE 17. RECOMMENDATIONS FOR DOSING IN PATIENTS WITH HEPATIC IMPAIRMENT BASED ON CLINICAL TRIAL DATAa
| Degree of Hepatic Impairment | |||
| Transaminase Levels | Bilirubin Levelsb | Recommended Paclitaxel Dosec | |
| 24-Hour Infusion | |||
| <2 x ULN | and | ≤1.5 mg/dL | 135 mg/m2 |
| 2 to <10 x ULN | and | ≤1.5 mg/dL | 100 mg/m2 |
| <10 x ULN | and | 1.6 to 7.5 mg/dL | 50 mg/m2 |
| ≥10 x ULN | or | > 7.5 mg/dL | Not recommended |
| 3-Hour Infusion | |||
| <10 x ULN | and | ≤1.25 x ULN | 175 mg/m2 |
| <10 x ULN | and | 1.26 to 2.0 x ULN | 135 mg/m2 |
| <10 x ULN | and | 2.01 to 5.0 x ULN | 90 mg/m2 |
| <10 x ULN | or | > 5.0 x ULN | Not recommended |
| a These recommendations are based on dosages for patients without hepatic impairment of 135 mg/m2 over 24 hours or 175 mg/m2 over 3 hours; data are not available to make dose adjustment recommendations for other regimens (eg, for AIDS-related Kaposi’s sarcoma). b Differences in criteria for bilirubin levels between the 3- and 24-hour infusion are due to differences in clinical trial design. c Dosage recommendations are for the first course of therapy; further dose reduction in subsequent courses should be based on individual tolerance. |
Preparation and Administration Precautions
Procedures for proper handling and disposal of anticancer drugs should be considered. Several guidelines on this subject have been published1-4. To minimize the risk of dermal exposure, always wear impervious gloves when handling vials containing paclitaxel injection. If paclitaxel solution contacts the skin, wash the skin immediately and thoroughly with soap and water. Following topical exposure, events have included tingling, burning, and redness. If paclitaxel contacts mucous membranes, the membranes should be flushed thoroughly with water. Upon inhalation, dyspnea, chest pain, burning eyes, sore throat, and nausea have been reported.
Given the possibility of extravasation, it is advisable to closely monitor the infusion site for possible infiltration during drug administration (see PRECAUTIONS, Injection Site Reaction).
Preparation for Intravenous Administration
Paclitaxel must be diluted prior to infusion. Paclitaxel should be diluted in 0.9% Sodium Chloride Injection, USP; 5% Dextrose Injection, USP; 5% Dextrose and 0.9% Sodium Chloride Injection, USP or 5% Dextrose in Ringer’s Injection to a final concentration of 0.3 to 1.2 mg/mL. The solutions are physically and chemically stable for up to 27 hours at ambient temperature (approximately 25°C) and room lighting conditions. Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration whenever solution and container permit.
Upon preparation, solutions may show haziness, which is attributed to the formulation vehicle. No significant losses in potency have been noted following simulated delivery of the solution through I.V. tubing containing an in-line (0.22 micron) filter.
Data collected for the presence of the extractable plasticizer DEHP [di-(2-ethylhexyl)phthalate] show that levels increase with time and concentration when dilutions are prepared in PVC containers. Consequently, the use of plasticized PVC containers and administration sets is not recommended. Paclitaxel solutions should be prepared and stored in glass, polypropylene, or polyolefin containers. Non-PVC containing administration sets, such as those which are polyethylene-lined, should be used.
Paclitaxel should be administered through an in-line filter with a microporous membrane not greater than 0.22 microns. Use of filter devices such as IVEX-2® filters which incorporate short inlet and outlet PVC-coated tubing has not resulted in significant leaching of DEHP.
The Chemo Dispensing Pin™ device or similar devices with spikes should not be used with vials of paclitaxel since they can cause the stopper to collapse resulting in loss of sterile integrity of the paclitaxel solution.
Stability
Unopened vials of paclitaxel are stable until the date indicated on the package when stored between 20° to 25°C (68° to 77°F), in the original package. Neither freezing nor refrigeration adversely affects the stability of the product. Upon refrigeration, components in the paclitaxel vial may precipitate, but will redissolve upon reaching room temperature with little or no agitation. There is no impact on product quality under these circumstances. If the solution remains cloudy or if an insoluble precipitate is noted, the vial should be discarded. Solutions for infusion prepared as recommended are stable at ambient temperature (approximately 25°C) and lighting conditions for up to 27 hours.
HOW SUPPLIED
Paclitaxel Injection, USP (6 mg per mL) is supplied in the following:
| Product Code | Unit of Sale | Strength |
|---|---|---|
| 760305 | NDC 63323-763-05 Multiple dose vial, packaged individually | 30 mg per 5 mL (6 mg per mL) |
| 760316 | NDC 63323-763-16 Multiple dose vial, packaged individually | 100 mg per 16.7 mL (6 mg per mL) |
| 760350 | NDC 63323-763-50 Multiple dose vial, packaged individually | 300 mg per 50 mL (6 mg per mL) |
The container closure is not made with natural rubber latex.
Storage
Store the vials in original cartons between 20° to 25°C (68° to 77°F) [see USP Controlled Room Temperature]. Retain in the original package to protect from light.
Handling and Disposal
See DOSAGE AND ADMINISTRATION, Preparation and Administration Precautions.
REFERENCES
1. NIOSH Alert: Preventing occupational exposures to antineoplastic and other hazardous drugs in healthcare settings. 2004. U.S. Department of Health and Human Services, Public Health Service, Centers for Disease Control and Prevention, National Institute for Occupational Safety and Health, DHHS (NIOSH) Publication No. 2004-165.
2. OSHA Technical Manual, TED 1-0.15A, Section VI: Chapter 2. Controlling occupational exposure to hazardous drugs. OSHA, 1999. http://www.osha.gov/dts/osta/otm/otm_vi/otm_vi_2.html.
3. American Society of Health-System Pharmacists. ASHP guidelines on handling hazardous drugs. Am J Health-Syst Pharm. 2006;63:1172-1193.
4. Polovich M, White JM, Kelleher LO, eds. 2005. Chemotherapy and biotherapy guidelines and recommendations for practice. 2nd ed. Pittsburgh, PA: Oncology Nursing Society.
*DaunoXome® is a registered trademark of Gilead Sciences, Inc.
DOXIL® is a registered trademark of ALZA Corporation.
IVEX-2® is a registered trademark of the Millipore Corporation.
Chemo Dispensing Pin™ is a trademark of B. Braun Medical Incorporated.
PATIENT INFORMATION
Paclitaxel (pak-li tax-el) Injection, USP
Rx only
Read this patient information leaflet before you start taking paclitaxel. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or your treatment.
What is the most important information I should know about paclitaxel?
Paclitaxel can cause serious side effects including death.
Serious allergic reactions (anaphylaxis) can happen in people who receive paclitaxel injection.
Anaphylaxis is a serious medical emergency that can lead to death and must be treated right away.
Tell your healthcare provider right away if you have any of these signs of an allergic reaction:
• trouble breathing
• sudden swelling of your face, lips, tongue, throat, or trouble swallowing
• hives (raised bumps) or rash
Your healthcare provider will give you medicines to lessen your chance of having an allergic reaction.
What is paclitaxel?
Paclitaxel is a prescription medicine used to treat some forms of:
• ovarian cancer
• breast cancer
• lung cancer
• Kaposi’s sarcoma
It is not known if paclitaxel is safe or effective in children.
Who should not receive paclitaxel?
Do not receive paclitaxel if:
• you are allergic to any of the ingredients in paclitaxel. See the end of this leaflet for a complete list of ingredients in paclitaxel.
• are allergic to medicines containing polyoxyl 35 castor oil.
• you have low white blood cell counts.
What should I tell my healthcare provider before receiving paclitaxel?
Before receiving paclitaxel, tell your healthcare provider about all your medical conditions, including if you:
• have liver problems
• have heart problems
• are pregnant or plan to become pregnant. Paclitaxel can harm your unborn baby. Talk to your healthcare provider if you are pregnant or plan to become pregnant.
• are breast-feeding or plan to breast-feed. It is not known if paclitaxel passes into your breast milk. You and your healthcare provider should decide if you will receive paclitaxel or breast-feed.
Tell your healthcare provider about all the medicines you take, including prescription and non-prescription medicines, vitamins, and herbal supplements.
Know the medicines you take. Keep a list of them and show it to your healthcare provider and pharmacist when you get a new medicine.
How will I receive paclitaxel?
• Paclitaxel is injected into a vein (intravenous [IV] infusion) by your healthcare provider.
Your healthcare provider will do certain tests while you receive paclitaxel.
What are the possible side effects of paclitaxel?
Tell your healthcare provider right away if you have:
• severe stomach pain
• severe diarrhea
The most common side effects of paclitaxel Injection, USP include:
• low red blood cell count (anemia) feeling weak or tired
• hair loss
• numbness, tingling, or burning in your hands or feet (neuropathy)
• joint and muscle pain
• nausea and vomiting
• hypersensitivity reaction - trouble breathing; sudden swelling of your face, lips, tongue, throat, or trouble swallowing; hives (raised bumps) or rash
• diarrhea
• mouth or lip sores (mucositis)
• infections - if you have a fever (temperature above 100.4°F) or other sign of infection, tell your healthcare provider right away
• swelling of your hands, face, or feet
• bleeding events
• irritation at the injection site
• low blood pressure (hypotension)
Tell your healthcare provider if you have any side effect that bothers you or that does not go away.
These are not all the possible side effects of paclitaxel. For more information, ask your healthcare provider or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800- FDA-1088 or go to www.fresenius-kabi.com/us or call 1-800-551-7176.
General information about the safe and effective use of paclitaxel.
Medicines are sometimes prescribed for purposes other than those listed in a patient information leaflet. Do not use paclitaxel for a condition for which it was not prescribed. Do not give paclitaxel to other people, even if they have the same symptoms that you have. It may harm them.
This patient information leaflet summarizes the most important information about paclitaxel. If you would like more information, talk with your healthcare provider. You can ask your pharmacist or healthcare provider for information about paclitaxel that is written for health professionals. For more information go to www.fresenius-kabi.com/us or call 1-800-551-7176.
What are the ingredients in paclitaxel?
Active ingredient: paclitaxel, USP.
Inactive ingredients include: polyoxyl 35 castor oil, NF and dehydrated alcohol, USP.
What is cancer?
Under normal conditions, the cells in your body divide and grow in an orderly, controlled way. Cell division and growth are necessary for the human body to perform its functions and to repair itself, when necessary. Cancer cells are different from normal cells because they are not able to control their own growth. The reasons for this abnormal growth are not yet fully understood.
A tumor is a mass of unhealthy cells that are dividing and growing fast and in an uncontrolled way. When a tumor invades surrounding healthy body tissue, it is known as a malignant tumor. A malignant tumor can spread (metastasize) from its original site to other parts of the body if not found and treated early.

Lake Zurich, IL 60047
For Product Inquiry:
1-800-551-7176 or www.fresenius-kabi.com/us
451224E/Revised: November 2018
PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
Paclitaxel 30 mg per 5 mL- Container Label
NDC 63323-763-05 760305
PACLITAXEL
INJECTION, USP
(semisynthetic)
30 mg per 5 mL
(6 mg/mL)
MUST BE DILUTED PRIOR
TO INTRAVENOUS USE
Cytotoxic Agent
Rx only
Multiple Dose Vial

Paclitaxel 30 mg per 5 mL- Carton Label
NDC 63323-763-05 760305
PACLITAXEL
INJECTION, USP
(semisynthetic)
30 mg per 5 mL
(6 mg per mL)
MUST BE DILUTED PRIOR
TO INTRAVENOUS USE
Cytotoxic Agent
Rx only
Multiple Dose Vial

PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
Paclitaxel 100 mg per 16.7 mL- Container Label
NDC 63323-763-16 760316
PACLITAXEL
INJECTION, USP
(semisynthetic)
100 mg per 16.7 mL
(6 mg/mL)
MUST BE DILUTED PRIOR
TO INTRAVENOUS USE
Cytotoxic Agent
Rx only
Multiple Dose Vial
Paclitaxel 100 mg per 16.7 mL- Carton Label
NDC 63323-763-16 760316
PACLITAXEL
INJECTION, USP
(semisynthetic)
100 mg per 16.7 mL
(6 mg/mL)
MUST BE DILUTED PRIOR
TO INTRAVENOUS USE
Cytotoxic Agent
Rx only
Multiple Dose Vial

PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
Paclitaxel 300 mg per 50 mL- Container Label
NDC 63323-763-50 760350
PACLITAXEL
INJECTION, USP
(semisynthetic)
300 mg per 50 mL
(6 mg/mL)
MUST BE DILUTED PRIOR
TO INTRAVENOUS USE
Cytotoxic Agent
Rx only
Multiple Dose Vial

Paclitaxel 300 mg per 50 mL- Carton Label
NDC 63323-763-50 760350
PACLITAXEL
INJECTION, USP
(semisynthetic)
300 mg per 50 mL
(6 mg/mL)
MUST BE DILUTED PRIOR
TO INTRAVENOUS USE
Cytotoxic Agent
Rx only
Multiple Dose Vial
