FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
OBIZUR, Antihemophilic Factor (Recombinant), Porcine Sequence, is a recombinant DNA derived, antihemophilic factor indicated for the on-demand treatment and control of bleeding episodes in adults with acquired hemophilia A.
2 DOSAGE AND ADMINISTRATION
For intravenous use after reconstitution only.
2.1 Dose
- Dose, dosing frequency, and duration of treatment with OBIZUR depend on the location and severity of bleeding episode, target factor VIII levels, and the patient's clinical condition. Monitor replacement therapy in cases of major surgery or life-threatening bleeding episodes.
- Each vial of OBIZUR has the recombinant porcine factor VIII potency in units stated on the vial.
- Patients may vary in their pharmacokinetic (e.g., half-life, in vivo recovery) and clinical responses. Titrate dose and frequency based on factor VIII recovery levels and individual clinical response.
- Initial dosing below the recommended 200 U/kg has been associated with lack of efficacy.
A guide for dosing OBIZUR for the on-demand treatment and control of bleeding episodes is provided in Table 1. Maintain the factor VIII activity within the target range. Plasma levels of factor VIII should not exceed 200% of normal or 200 units per dL.
| Type of Bleeding | Factor VIII Level Required (Units per dL or % of normal) | Initial Dose (Units per kg) | Subsequent Dose | Frequency and Duration of Subsequent Dosing |
|---|---|---|---|---|
| Minor and Moderate
Superficial muscle/no neurovascular compromise, and joint | 50 to 100 | 200 | Titrate subsequent doses to maintain recommended factor VIII trough levels and individual clinical response | Dose every 4 to 12 hours, frequency may be adjusted based on clinical response and measured factor VIII levels |
| Major
Moderate to severe intramuscular bleeding, retroperitoneal, gastrointestinal, intracranial | 100 to 200 (To treat an acute bleed) 50 to 100 (After acute bleed is controlled, if required) | 200 | Titrate subsequent doses to maintain recommended factor VIII trough levels and individual clinical response | Dose every 4 to 12 hours, frequency may be adjusted based on clinical response and measured factor VIII levels |
2.2 Reconstitution
- Use aseptic technique during the reconstitution procedure.
- If the patient needs more than one vial of OBIZUR per injection, reconstitute each vial according to the following instructions:
- 1.
- Bring the OBIZUR vial and the pre-filled diluent syringe to room temperature.
- 2.
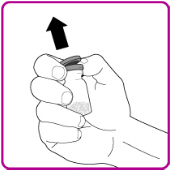
- Remove the plastic cap from the OBIZUR vial (Figure A).
| Figure A |
 |
- 3.
- Wipe the rubber stopper with an alcohol swab (not supplied) and allow it to dry prior to use.
- 4.
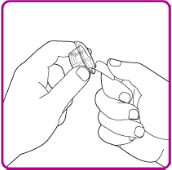
- Peel back the cover of the vial adapter package. (Figure B). Do not touch the luer-lock (tip) in the center of the vial adapter. Do not remove the vial adapter from the plastic package.
| Figure B |
 |
- 5.
- Place the vial adapter package on a clean surface with the luer-lock pointing up.
- 6.
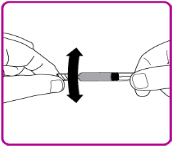
- Snap off the tamper resistant cap of the pre-filled syringe (Figure C).
| Figure C |
 |
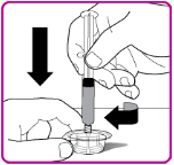
- 7.
- While firmly holding the vial adapter package, connect the pre-filled syringe to the vial adapter by pushing the syringe tip down onto the luer lock in the center of the vial adapter, and turning it clockwise until the syringe is secured. Do not over tighten (Figure D).
| Figure D |
 |
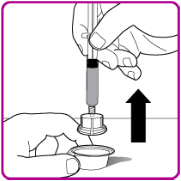
- 8.
- Remove the vial adapter from the plastic package (Figure E).
| Figure E |
 |
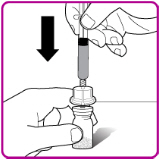
- 9.
- Place the OBIZUR vial on a clean, flat, hard surface. Place the vial adapter over the OBIZUR vial and firmly push the filter spike of the vial adapter through the center of the OBIZUR vial's rubber circle until the clear plastic cap snaps onto the vial (Figure F).
| Figure F |
 |
- 10.
- Push the plunger down to slowly inject all of the diluent from the syringe into the OBIZUR vial.
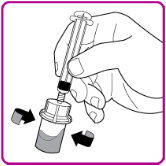
- 11.
- Gently swirl (in a circular motion) the OBIZUR vial without removing the syringe until all of the powder is fully dissolved (Figure G). The reconstituted solution should be inspected visually for particulate matter before administration. Do not use if particulate matter or discoloration is observed.
| Figure G |
 |
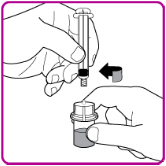
- 12.
- With one hand hold the vial and vial adapter, and with the other hand firmly grasp the barrel of the pre-filled syringe and in a counterclockwise motion unscrew the syringe from the vial adapter (Figure H).
| Figure H |
 |
- 13.
- Use OBIZUR within 3 hours after reconstitution when stored at room temperature.
2.3 Administration
For intravenous use after reconstitution only.
- Inspect the reconstituted OBIZUR solution for particulate matter and discoloration prior to administration. The solution should be clear and colorless in appearance. Do not administer if particulate matter or discoloration is observed.
- Do not administer OBIZUR in the same tubing or container with other medicinal products for infusion.
- 1.
- Once all vials have been reconstituted, connect a large syringe to the vial adapter by gently pushing the syringe tip down onto the luer lock in the center of the vial adapter, and turning clockwise until the syringe is secured.
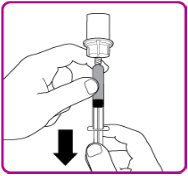
- 2.
- Invert the vial; push the air in the syringe into the vial and withdraw the reconstituted OBIZUR into the syringe (Figure I).
| Figure I |
 |
- 3.
- Unscrew the large syringe counterclockwise from the vial adapter and repeat this process for all reconstituted vials of OBIZUR until the total volume to be administered is reached.
- 4.
- Administer the reconstituted OBIZUR intravenously at a rate of 1 to 2 mL per minute.
3 DOSAGE FORMS AND STRENGTHS
OBIZUR is available as a white lyophilized powder in single-dose glass vials containing nominally 500 units per vial.
4 CONTRAINDICATIONS
OBIZUR is contraindicated in patients:
- Who have had life-threatening hypersensitivity reactions to OBIZUR or its components (including traces of hamster proteins).
- With congenital hemophilia A with inhibitors (CHAWI) due to the high incidence of anamnestic reactions to human factor VIII (hFVIII) and porcine factor VIII (pFVIII) [see Adverse Reactions (6.1)].
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions
Hypersensitivity reactions can occur with OBIZUR. OBIZUR contains trace amounts of hamster proteins. Early signs of allergic reactions, which can progress to anaphylaxis, include angioedema, chest-tightness, dyspnea, hypotension, wheezing, urticaria, and pruritus. Immediately discontinue administration and initiate appropriate treatment if allergic or anaphylactic-type reactions occur.
5.2 Inhibitory Antibodies
Inhibitory antibodies to OBIZUR have occurred in patients treated with OBIZUR. Lack of efficacy could be due to inhibitory antibodies to OBIZUR.
Anamnestic reactions with rise in human factor VIII and/or recombinant factor VIII, porcine sequence inhibitors have also been reported in patients treated with OBIZUR. These anamnestic rises also may result in a lack of efficacy.
Consider evaluating for presence of anti-recombinant porcine factor VIII (anti-rpFVIII) antibodies prior to initiation of treatment with OBIZUR. In instances where positive anti-rpFVIII antibody results are detected after treatment with OBIZUR is initiated, factor VIII levels may be used to decide whether treatment with OBIZUR should be continued [see Warnings and Precautions (5.3)].
Monitor patients for the development of antibodies to OBIZUR by appropriate assays [see Warnings and Precautions (5.3)]. If the plasma factor VIII level fails to increase as expected, or if bleeding is not controlled after OBIZUR administration, suspect the presence of an anti-porcine factor VIII antibody.
If such inhibitory antibodies are suspected and there is a lack of efficacy, consider management options such as discontinuing OBIZUR and initiating other therapeutics such as a Factor VIII bypassing agent.
5.3 Monitoring Laboratory Tests
- Perform one-stage clotting assay to confirm that adequate factor VIII levels have been achieved and maintained [see Dosage and Administration (2)].
- Monitor factor VIII activity 30 minutes and 3 hours after initial dose.
- Monitor factor VIII activity 30 minutes after subsequent doses.
- Monitor the development of inhibitory antibodies to OBIZUR. Perform a Nijmegen Bethesda inhibitor assay with recombinant porcine factor VIII as substrate for recombinant porcine factor VIII antibodies if expected plasma factor VIII activity levels are not attained or if bleeding is not controlled with the expected dose of OBIZUR. Use Bethesda Units (BU) to report inhibitor levels. Contact Takeda Pharmaceuticals U.S.A., Inc. at 1-877-TAKEDA-7 (1-877-825-3327) if additional information is needed regarding testing.
6 ADVERSE REACTIONS
Common adverse reactions observed in greater than 5% of subjects in the clinical trial were development of inhibitors to porcine factor VIII.
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction (AR) rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The safety and efficacy of OBIZUR was evaluated in a multi-center, prospective, open-label, clinical trial that investigated adult patients with acquired hemophilia A. Twenty-nine adult subjects were enrolled in the study, received at least one dose of OBIZUR and were evaluable for safety [see Clinical Studies (14)]. Of the 29 adult subjects, 10 were between the ages of 42 and 65, and 19 were 65 years of age or older (18 Caucasian, 6 African-American, and 5 Asian). Ten (34%) subjects were female.
Immunogenicity: Anti-Drug Antibody-Associated Adverse Reactions
All subjects were monitored for development of inhibitory antibodies to OBIZUR using the Nijmegen modification of the Bethesda inhibitor assay. A subject was considered to have developed an OBIZUR inhibitor if the titer was ≥0.6 Bethesda Units (BU)/mL.
Acquired Hemophilia A
Of the 29 subjects treated with OBIZUR, 19 subjects were negative for anti-porcine factor VIII antibodies at baseline. Five of the 19 (26%) developed anti-porcine factor VIII antibodies following exposure to OBIZUR. Serious adverse drug reactions (ADRs) occurred in 9 subjects. Two subjects (6.9%) developed anti-porcine FVIII inhibitors (≥0.6 Bethesda Units). Seven subjects (24.1%) developed anamnestic reactions with a rise ≥10 BU in human factor VIII and/or recombinant factor VIII, porcine sequence inhibitors. At 24 hours after initial dose, OBIZUR was effective in 9/10 patients with baseline anti-porcine antibodies (and partially effective in 1/10 patients) and was effective in all 5 with de novo anti-porcine antibodies.
All subjects were also monitored for development of binding antibodies to baby hamster kidney (BHK) protein by a validated sequential ELISA (enzyme-linked immunosorbent assay). No patients developed de novo anti-BHK antibodies.
The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors, including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to OBIZUR with the incidence of antibodies to other products may be misleading.
Congenital Hemophilia A with Inhibitors
In the clinical study of OBIZUR in patients with congenital hemophilia A with FVIII inhibitors (CHAWI) undergoing surgery, out of 8 adult patients evaluable for safety analysis a total of 5 subjects experienced anamnestic reactions [see Contraindications (4)].
A total of 5 (62.5%) subjects who underwent major surgeries reported anamnestic reactions with a rise ≥10 BU in human factor VIII and/or recombinant factor VIII, porcine sequence inhibitors (see Table 2). 3 (37.5%) subjects developed anamnestic reactions to human Factor VIII (hFVIII) and porcine Factor VIII (pFVIII), 2 (25.0%) subjects developed anamnestic reactions to hFVIII. The highest increases in antibody levels reported are from 15.4 at baseline to 1039.6 BU for hFVIII and from 2.6 to 335.8 BU for pFVIII. Inhibitory antibodies may result in lack of efficacy.
| Subjects | hFVIII Inhibitors Titers (BU/mL) | Obizur pFVIII Inhibitors Titers (BU/mL) |
||||
|---|---|---|---|---|---|---|
| Baseline | Day 14 Visit | End of the Study Visit | Baseline | Day 14 Visit | End of the Study Visit | |
| Subject 1 | 1.3 | 1.2 (D3) | 38.1/39.9 | <0.6 | <0.6 (D3) | 29.3/38.8 |
| Subject 2 | 15.4 | 1039.6 /807.9 | 2.6 | 0.7 (D5) | 327.3/335.8 | |
| Subject 3 | 3.3 | 19.9 (D20) | 23.3 | 5.6 | 36.1 (D20) | 30.5 |
| Subject 4 | 2.6 | 16.6 | 12.1 | <0.6 | 1 | 0.9 |
| Subject 5 | 4.6 | 3.8 (D3)/488 (D16) | 336.3 | 1.7 | <0.6 (D3)/285.9 (D16) | 280.5 |
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no data with OBIZUR use in pregnant women to inform a drug-associated risk. There are no adequate and well-controlled studies in pregnant women. Animal reproduction studies have not been conducted with OBIZUR. It is also not known whether OBIZUR can cause fetal harm when administered to a pregnant woman or can affect reproduction capacity.
In the U.S. general population, the estimated background risk for major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
8.2 Lactation
Risk Summary
There is no information regarding the presence of OBIZUR in human milk, the effect on the breastfed child, or the effects on milk production.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for OBIZUR and any potential adverse effects on the breastfed child from OBIZUR or from the underlying maternal condition.
8.4 Pediatric Use
The safety and efficacy of OBIZUR have not been established in pediatric patients.
8.5 Geriatric Use
Of the 29 subjects within the trial, the average age was 70 years of age. Nineteen subjects were 65 years of age or older. Clinical studies suggest that OBIZUR is safe and effective in the adult population [see Adverse Reactions (6) and Clinical Studies (14)]. While no differences were observed between geriatric and adult responses to OBIZUR, these findings are inconclusive given the small number of subjects enrolled in either group.
Dose adjustments in the geriatric population have not been studied. Specific hazards associated with the concomitant use of OBIZUR with other drugs in the elderly population have not been studied in the clinical trial.
11 DESCRIPTION
The active ingredient in OBIZUR is a recombinant (r) analogue of porcine factor VIII (pFVIII) with an approximate molecular weight of 170 kDa. The rpFVIII molecule in OBIZUR is a glycoprotein containing a 90 kDa heavy chain and a 80 kDa light chain. The B-domain normally present in naturally occurring porcine factor VIII has been replaced with a twenty-four amino acid linker. Once activated, the resulting rpFVIIIa has a comparable activity to the endogenous human FVIIIa.
OBIZUR is expressed in a genetically engineered baby hamster kidney (BHK) cell line which secretes rpFVIII into the cell culture medium, and the rpFVIII protein is purified using a series of chromatography and filtration steps. The production process includes two dedicated viral clearance steps - a solvent/detergent treatment step for viral inactivation and a nanofiltration step through a series of two 15-nm filters for removal of viruses. No additives of human or animal origin are used in the formulation of OBIZUR.
OBIZUR is formulated as a sterile, non-pyrogenic, lyophilized powder for intravenous injection after reconstitution with the diluent (Sterile Water for Injection). OBIZUR is available in single-dose vials that nominally contain 500 units (U) per vial. When reconstituted with the diluent, the product contains the following components per mL: 8.8 mg sodium chloride, 0.04 mg Tris-base, 0.73 mg Tris-HCl, 1.47 mg tri-sodium citrate dehydrate, 0.15 mg calcium chloride dehydrate, 1.9 mg sucrose, and 0.05 mg polysorbate 80.
Each vial of OBIZUR is labeled with the actual rpFVIII activity expressed in units determined by a one-stage clotting assay, using a reference rpFVIII material calibrated against the World Health Organization (WHO) 8th International Standard for human FVIII concentrates. The specific activity of OBIZUR is in the range of 11000 to 18000 U per milligram of protein. The potency values of OBIZUR determined by the chromogenic assay vary and are approximately 20 to 50% lower than those of the one-stage clotting assay.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
OBIZUR temporarily replaces the inhibited endogenous factor VIII that is needed for effective hemostasis in patients with acquired hemophilia A.
12.2 Pharmacodynamics
Patients with acquired hemophilia A (AHA) have normal factor VIII genes but develop autoantibodies against their own factor VIII (i.e., inhibitors). These autoantibodies neutralize circulating human factor VIII and create a functional deficiency of this procoagulant protein. AHA results in a prolonged clotting time as measured by the activated partial thromboplastin time (aPTT) assay, a conventional in vitro test for biological activity of factor VIII. Treatment with OBIZUR should normalize the aPTT during treatment; however, aPTT normalization should not be used as a measure of efficacy.
14 CLINICAL STUDIES
The efficacy of OBIZUR for the treatment of serious bleeding episodes in subjects with acquired hemophilia A was investigated in a prospective, open-label trial (N=29). The trial was conducted in 18 Caucasian, 6 African-American, and 5 Asian subjects diagnosed with acquired hemophilia A (AHA), having auto-immune inhibitory antibodies to human factor VIII, and experiencing serious bleeding episodes that required hospitalization. Subjects with a prior history of bleeding disorders other than AHA, anti-porcine factor VIII antibody titer >20 Bethesda Units (BU), or in whom the bleeding episode was judged likely to resolve on its own were excluded. One subject was considered evaluable at study entry; however, it was later determined that this subject did not have AHA, leaving 28 subjects evaluable for efficacy.
An initial dose of 200 units per kg OBIZUR was administered to subjects for the treatment of life- or limb-threatening initial bleeding episodes. Patients were treated with OBIZUR until resolution of bleeding or dosing was continued at the physician's discretion according to the clinical assessment. These bleeding episodes included 19 intramuscular or joint bleeding episodes, 4 post-surgical bleeding episodes, 2 intracranial episodes, 2 surgeries, 1 retroperitoneal hemorrhage, and 1 periorbital bleed. Hemostatic response was assessed by the study site investigator at specified time points after initiation of OBIZUR treatment using a pre-specified rating scale that was based on subjective clinical assessments combined with objective factor VIII activity levels achieved. An assessment of effective or partially effective was considered as a positive response (see Table 3 for definitions).
| Assessment of efficacy | Control of bleeding | Clinical Assessment | Factor VIII levels | Response |
|---|---|---|---|---|
| Effective | bleeding stopped | clinical control | ≥50% | positive |
| Partially effective | bleeding reduced | clinical stabilization or improvement; or alternative reason for bleeding | ≥20% | positive |
| Poorly effective | bleeding slightly reduced or unchanged | not clinically stable | <50% | negative |
| Not effective | bleeding worsening | Clinically deteriorating | <20% | negative |
Of the 28 subjects evaluable for efficacy, all subjects had a positive response to treatment for the initial bleeding episodes at 24 hours after dosing. A positive response was observed in 95% (19/20) of subjects evaluated at 8 hours and 100% (18/18) at 16 hours.
In addition to response to treatment, the overall treatment success was determined by the investigator based on his/her ability to discontinue or reduce the dose and/or dosing frequency of OBIZUR. A total of 24/28 (86%) had successful treatment of the initial bleeding episode. Of those subjects treated with OBIZUR as first-line therapy, defined as no immediate previous use of anti-hemorrhagic agents prior to the first OBIZUR treatment, 16/17 (94%) had eventual treatment success reported. Eleven subjects were reported to have received anti-hemorrhagics (e.g., rFVIIa, activated prothrombin-complex concentrate, tranexamic acid) prior to first treatment with OBIZUR. Of these 11 subjects, eight had eventual successful treatment (73%).
The median dose per infusion to successfully treat the primary bleeding episode was 133 units per kg and a median total dose of 1523 units per kg. In the initial 24-hour period, a median of 3 infusions (median dose 200 U/kg) were utilized in the clinical study. When treatment was required beyond 24 hours, a median of 10.5 infusions (median dose 100 U/kg) were given for a median of 6 days to control a bleeding episode.
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
OBIZUR is supplied as a white lyophilized powder in single-dose vials in the following package size:
| Nominal Strength | Package Size | Kit NDC |
|---|---|---|
| 500 units | 1-vial package | 0944-5001-01 |
Each package contains one package insert and one of each of the components listed below:
- Single-dose vial of OBIZUR [NDC 0944-5011-01]
- Pre-filled syringe with 1 mL Sterile Water for Injection [NDC 0944-0011-01]
- Vial adapter with filter
The actual amount of OBIZUR in units is stated on the label of each vial.
Not made with natural rubber latex.
Storage and Handling
- Store OBIZUR at refrigeration temperature of 2° to 8°C [36° to 46°F]. Do not freeze.
- Store vials in the original package to protect from light.
- Do not use beyond the expiration date printed on the carton or vial.
- Use OBIZUR within 3 hours after reconstitution. Discard any unused reconstituted product if not used within 3 hours after reconstitution.
- Do not use OBIZUR if the reconstituted solution is cloudy or has particulate matter.
17 PATIENT COUNSELING INFORMATION
Inform patients:
- of early signs and symptoms of hypersensitivity reactions (including angioedema, chest tightness, dyspnea, hypotension, wheezing, urticaria and pruritus) and anaphylaxis. Instruct patients to discontinue use and contact their physician.
- that new or increase in existing inhibitory antibodies to OBIZUR may develop and this may result in a lack of control of their bleeding.
- to report any adverse reactions or problems following OBIZUR administration to their physician or healthcare provider.