FULL PRESCRIBING INFORMATION
WARNING: POTENTIAL FOR ABUSE
AMPHETAMINES HAVE A HIGH POTENTIAL FOR ABUSE. ADMINISTRATION OF AMPHETAMINES FOR PROLONGED PERIODS OF TIME MAY LEAD TO DRUG DEPENDENCE. PARTICULAR ATTENTION SHOULD BE PAID TO THE POSSIBILITY OF SUBJECTS OBTAINING AMPHETAMINES FOR NON-THERAPEUTIC USE OR DISTRIBUTION TO OTHERS AND THE DRUGS SHOULD BE PRESCRIBED OR DISPENSED SPARINGLY.
MISUSE OF AMPHETAMINES MAY CAUSE SUDDEN DEATH AND SERIOUS CARDIOVASCULAR ADVERSE EVENTS.
1 INDICATIONS AND USAGE
1.1 Attention Deficit Hyperactivity Disorder
Vyvanse® is indicated for the treatment of Attention Deficit Hyperactivity Disorder (ADHD).
The efficacy of Vyvanse in the treatment of ADHD was established on the basis of two controlled trials in children aged 6 to 12, one controlled trial in adolescents aged 13 to 17, and two controlled trials in adults who met DSM-IV-TR® criteria for ADHD [see CLINICAL STUDIES (14)].
A diagnosis of Attention Deficit Hyperactivity Disorder (ADHD; DSM-IV®) implies the presence of hyperactive-impulsive and/or inattentive symptoms that cause impairment and were present before the age of 7 years. The symptoms must cause clinically significant impairment, e.g. in social, academic, or occupational functioning, and be present in two or more settings, e.g. school (or work) and at home. The symptoms must not be better accounted for by another mental disorder. For the Inattentive Type, at least 6 of the following symptoms must have persisted for at least 6 months: lack of attention to details/careless mistakes; lack of sustained attention; poor listener; failure to follow through on tasks; poor organization; avoids tasks requiring sustained mental effort; loses things; easily distracted; forgetful. For the Hyperactive-Impulsive Type, at least 6 of the following symptoms (or adult equivalent symptoms) must have persisted for at least 6 months: fidgeting/squirming; leaving seat; inappropriate running/climbing; difficulty with quiet activities; "on the go"; excessive talking; blurting answers; can't wait turn; intrusive. The Combined Type requires both inattentive and hyperactive-impulsive criteria to be met.
Special Diagnostic Considerations
Specific etiology of this syndrome is unknown, and there is no single diagnostic test. Adequate diagnosis requires the use not only of medical but also of special psychological, educational, and social resources. Learning may or may not be impaired. The diagnosis must be based upon a complete history and evaluation of the patient and not solely on the presence of the required number of DSM-IV characteristics.
Need for Comprehensive Treatment Program
Vyvanse is indicated as an integral part of a total treatment program for ADHD that may include other measures (psychological, educational, social) for patients with this syndrome. Drug treatment may not be indicated for all patients with this syndrome. Stimulants are not intended for use in patients who exhibit symptoms secondary to environmental factors and/or other primary psychiatric disorders, including psychosis. Appropriate educational/vocational placement is essential and psychosocial intervention is often helpful. When remedial measures alone are insufficient, the decision to prescribe stimulant medication will depend upon the physician's assessment of the chronicity and severity of the patient's symptoms and on the level of functional impairment.
Long-Term Use
The effectiveness of Vyvanse for long-term use, i.e., for more than 4 weeks, has not been systematically evaluated in controlled trials. Therefore, the physician who elects to use Vyvanse for extended periods should periodically re-evaluate the long-term usefulness of the drug for the individual patient.
2 DOSAGE AND ADMINISTRATION
Dosage should be individualized according to the therapeutic needs and response of the patient. Vyvanse should be administered at the lowest effective dosage.
In patients who are either starting treatment for the first time or switching from another medication, 30 mg once daily in the morning is the recommended dose. If the decision is made in the judgment of the clinician to increase the dose beyond 30 mg/day, daily dosage may be adjusted in increments of 10 mg or 20 mg at approximately weekly intervals. The maximum recommended dose is 70 mg/day; doses greater than 70 mg/day of Vyvanse have not been studied. Vyvanse has not been studied in children under 6 years of age.
Vyvanse should be taken in the morning. Afternoon doses should be avoided because of the potential for insomnia.
Vyvanse may be taken with or without food.
Vyvanse capsules may be taken whole, or the capsule may be opened and the entire contents dissolved in a glass of water. The solution should be consumed immediately and should not be stored. The dose of a single capsule should not be divided. The contents of the entire capsule should be taken, and patients should not take anything less than one capsule per day.
Where possible, drug administration should be interrupted occasionally to determine if there is a recurrence of behavioral symptoms sufficient to require continued treatment.
3 DOSAGE FORM AND STRENGTHS
Vyvanse capsules 20 mg: ivory body/ivory cap (imprinted with NRP104 or S489 and 20 mg)
Vyvanse capsules 30 mg: white body/orange cap (imprinted with NRP104 or S489 and 30 mg)
Vyvanse capsules 40 mg: white body/blue green cap (imprinted with NRP104 or S489 and 40 mg)
Vyvanse capsules 50 mg: white body/blue cap (imprinted with NRP104 or S489 and 50 mg)
Vyvanse capsules 60 mg: aqua blue body/aqua blue cap (imprinted with NRP104 or S489 and 60 mg)
Vyvanse capsules 70 mg: blue body/orange cap (imprinted with NRP104 or S489 and 70 mg)
4 CONTRAINDICATIONS
- Advanced arteriosclerosis, symptomatic cardiovascular disease, moderate to severe hypertension, hyperthyroidism, known hypersensitivity or idiosyncratic reaction to sympathomimetic amines, glaucoma
- Agitated states
- Patients with a history of drug abuse
- During or within 14 days following the administration of monoamine oxidase inhibitors (hypertensive crises may result) [See Drug Interactions (7.3)]
5 WARNINGS AND PRECAUTIONS
5.1 Serious Cardiovascular Events
Sudden Death and Pre-existing Structural Cardiac Abnormalities or Other Serious Heart Problems
Children and Adolescents
Sudden death has been reported in association with CNS stimulant treatment at usual doses in children and adolescents with structural cardiac abnormalities or other serious heart problems. Although some serious heart problems alone carry an increased risk of sudden death, stimulant products generally should not be used in children or adolescents with known serious structural cardiac abnormalities, cardiomyopathy, serious heart rhythm abnormalities, or other serious cardiac problems that may place them at increased vulnerability to the sympathomimetic effects of a stimulant drug [see CONTRAINDICATIONS (4)].
Adults
Sudden death, stroke, and myocardial infarction have been reported in adults taking stimulant drugs at usual doses for ADHD. Although the role of stimulants in these adult cases is unknown, adults have a greater likelihood than children of having serious structural cardiac abnormalities, cardiomyopathy, serious heart rhythm abnormalities, coronary artery disease, or other serious cardiac problems. Adults with such abnormalities should also generally not be treated with stimulant drugs [see CONTRAINDICATIONS (4)].
Hypertension and Other Cardiovascular Conditions
Stimulant medications cause a modest increase in average blood pressure (about 2-4 mm Hg) and average heart rate (about 3-6 bpm) and individuals may have larger increases. While the mean changes alone would not be expected to have short-term consequences, all patients should be monitored for larger changes in heart rate and blood pressure. Caution is indicated in treating patients whose underlying medical conditions might be compromised by increases in blood pressure or heart rate, e.g. those with pre-existing hypertension, heart failure, recent myocardial infarction, or ventricular arrhythmia [see CONTRAINDICATIONS (4)].
Assessing Cardiovascular Status in Patients Being Treated with Stimulant Medications
Children, adolescents, or adults who are being considered for treatment with stimulant medications should have a careful history (including assessment for a family history of sudden death or ventricular arrhythmia) and physical exam to assess for the presence of cardiac disease, and should receive further cardiac evaluation if findings suggest such disease (e.g. electrocardiogram and echocardiogram). Patients who develop symptoms such as exertional chest pain, unexplained syncope, or other symptoms suggestive of cardiac disease during stimulant treatment should undergo a prompt cardiac evaluation.
5.2 Psychiatric Adverse Events
Pre-existing Psychosis
Administration of stimulants may exacerbate symptoms of behavior disturbance and thought disorder in patients with a pre-existing psychotic disorder.
Bipolar Illness
Particular care should be taken in using stimulants to treat ADHD in patients with comorbid bipolar disorder because of concern for possible induction of a mixed/manic episode in such patients. Prior to initiating treatment with a stimulant, patients with comorbid depressive symptoms should be adequately screened to determine if they are at risk for bipolar disorder. Such screening should include a detailed psychiatric history, including a family history of suicide, bipolar disorder, and depression.
Emergence of New Psychotic or Manic Symptoms
Treatment-emergent psychotic or manic symptoms, e.g. hallucinations, delusional thinking, or mania in children and adolescents without a prior history of psychotic illness or mania, can be caused by stimulants at usual doses. If such symptoms occur, consideration should be given to a possible causal role of the stimulant, and discontinuation of treatment may be appropriate. In a pooled analysis of multiple short-term, placebo-controlled studies, such symptoms occurred in about 0.1% (4 patients with events out of 3482 exposed to methylphenidate or amphetamine for several weeks at usual doses) of stimulant-treated patients compared to 0 in placebo-treated patients.
Aggression
Aggressive behavior or hostility is often observed in children and adolescents with ADHD, and has been reported in clinical trials and the postmarketing experience of some medications indicated for the treatment of ADHD. Although there is no systematic evidence that stimulants cause aggressive behavior or hostility, patients beginning treatment of ADHD should be monitored for the appearance of, or worsening of, aggressive behavior or hostility.
5.3 Seizures
There is some clinical evidence that stimulants may lower the convulsive threshold in patients with prior history of seizures, in patients with prior EEG abnormalities in absence of seizures, and, very rarely, in patients without a history of seizures and no prior EEG evidence of seizures. In the presence of seizures, the drug should be discontinued.
5.4 Visual Disturbance
Difficulties with accommodation and blurring of vision have been reported with stimulant treatment.
5.5 Tics
Amphetamines have been reported to exacerbate motor and phonic tics and Tourette's syndrome. Therefore, clinical evaluation for tics and Tourette's syndrome should precede use of stimulant medications.
5.6 Long-Term Suppression of Growth
In a controlled trial of Vyvanse in children ages 6 to 12 years, mean weight loss from baseline after 4 weeks of therapy was -0.9, -1.9, and -2.5 lbs., respectively, for patients receiving 30 mg, 50 mg, and 70 mg of Vyvanse, compared to a 1 lb weight gain for patients receiving placebo. Higher doses were associated with greater weight loss with 4 weeks of treatment. Careful follow-up for weight in children ages 6 to 12 years who received Vyvanse over 12 months suggests that consistently medicated children (i.e. treatment for 7 days per week throughout the year) have a slowing in growth rate, measured by body weight as demonstrated by an age- and sex-normalized mean change from baseline in percentile, of -13.4 over 1 year (average percentiles at baseline and 12 months were 60.6 and 47.2, respectively). In a 4-week controlled trial of Vyvanse in adolescents ages 13 to 17 years, mean weight loss from baseline to endpoint was -2.7, -4.3, and -4.8 lbs., respectively, for patients receiving 30 mg, 50 mg, and 70 mg of Vyvanse, compared to a 2.0 lb weight gain for patients receiving placebo.
Careful follow-up of weight and height in children ages 7 to 10 years who were randomized to either methylphenidate or non-medication treatment groups over 14 months, as well as in naturalistic subgroups of newly methylphenidate-treated and non-medication treated children over 36 months (to the ages of 10 to 13 years), suggests that consistently medicated children (i.e. treatment for 7 days per week throughout the year) have a temporary slowing in growth rate (on average, a total of about 2 cm less growth in height and 2.7 kg less growth in weight over 3 years), without evidence of growth rebound during this period of development. In a controlled trial of amphetamine (d- to l-enantiomer ratio of 3:1) in adolescents, mean weight change from baseline within the initial 4 weeks of therapy was -1.1 lbs. and -2.8 lbs., respectively, for patients receiving 10 mg and 20 mg of amphetamine. Higher doses were associated with greater weight loss within the initial 4 weeks of treatment.
Therefore, growth should be monitored during treatment with stimulants, and patients who are not growing or gaining weight as expected may need to have their treatment interrupted.
6 ADVERSE REACTIONS
6.1 Clinical Studies Experience
The premarketing development program for Vyvanse included exposures in a total of 995 participants in clinical trials (348 pediatric patients aged 6 to 12 years, 233 adolescent patients aged 13 to 17 years, 358 adult patients and 56 healthy adult subjects). Of these, 348 pediatric (aged 6 to 12) patients were evaluated in two controlled clinical studies (one parallel-group and one crossover), one open-label extension study, and one single-dose clinical pharmacology study, 233 adolescent (aged 13 to 17) patients were evaluated in one controlled clinical study, and 358 adult patients were evaluated in one controlled clinical study and one open-label extension study. The information included in this section is based on data from the 4-week parallel-group controlled clinical studies in pediatric and adult patients with ADHD. Adverse reactions were assessed by collecting adverse events, results of physical examinations, vital signs, weights, laboratory analyses, and ECGs.
Adverse reactions during exposure were obtained primarily by general inquiry and recorded by clinical investigators using terminology of their own choosing. Consequently, it is not possible to provide a meaningful estimate of the proportion of individuals experiencing adverse reactions without first grouping similar types of reactions into a smaller number of standardized reactions categories. In the tables and listings that follow, MedDRA terminology has been used to classify reported adverse reactions.
The stated frequencies of adverse reactions represent the proportion of individuals who experienced a treatment-emergent adverse reaction of the type listed at least once.
Adverse Reactions Associated with Discontinuation of Treatment in Clinical Trials
In the controlled pediatric (aged 6 to 12) trial, 9% (20/218) of Vyvanse-treated patients discontinued due to adverse reactions compared to 1% (1/72) who received placebo. The most frequent adverse events leading to discontinuation and considered to be drug-related (i.e. leading to discontinuation in at least 1% of Vyvanse-treated patients and at a rate at least twice that of placebo) were ECG voltage criteria for ventricular hypertrophy, tic, vomiting, psychomotor hyperactivity, insomnia, and rash (2/218 each; 1%).
In the controlled adolescent (aged 13 to 17) trial, 4% (10/233) of Vyvanse-treated patients discontinued due to adverse events compared to 1% (1/77) who received placebo. The most frequent adverse reactions leading to discontinuation and considered to be drug-related were irritability (3/233; 1%), decreased appetite (2/233; 1%), and insomnia (2/233; 1%).
In the controlled adult trial, 6% (21/358) of Vyvanse-treated patients discontinued due to adverse events compared to 2% (1/62) who received placebo. The most frequent adverse events leading to discontinuation and considered to be drug-related (i.e. leading to discontinuation in at least 1% of Vyvanse-treated patients and at a rate at least twice that of placebo) were insomnia (8/358; 2%), tachycardia (3/358; 1%), irritability (2/358; 1%), hypertension (4/358; 1%), headache (2/358; 1%), anxiety (2/358; 1%), and dyspnea (3/358; 1%).
Adverse Reactions Occurring at an Incidence of 2% or More Among Vyvanse Treated Patients in Clinical Trials
Adverse reactions reported in the controlled trials in pediatric (aged 6 to 17 years) and adult patients treated with Vyvanse or placebo are presented in Tables 1, 2, and 3 below. The prescriber should be aware that these figures cannot be used to predict the incidence of adverse reactions in the course of usual medical practice where patient characteristics and other factors differ from those which prevailed in the clinical trials. Similarly, the cited frequencies cannot be compared with figures obtained from other clinical investigations involving different treatment uses and investigators. The cited figures, however, do provide the prescribing physician with some basis for estimating the relative contribution of drug and non-drug factors to the adverse reaction incidence rate in the population studied.
Pediatric
Table 1 Adverse Reactions Reported by 2% or More of Children (Aged 6 to 12 Years) Taking Vyvanse in a 4-Week Clinical Trial
| Body System | Preferred Term | Vyvanse (n=218) | Placebo (n=72) |
|---|---|---|---|
| Gastrointestinal Disorders | Abdominal Pain Upper | 12% | 6% |
| Vomiting | 9% | 4% | |
| Nausea | 6% | 3% | |
| Dry Mouth | 5% | 0% | |
| General Disorder and Administration Site Conditions | Pyrexia | 2% | 1% |
| Investigations | Weight Decreased | 9% | 1% |
| Metabolism and Nutrition | Decreased Appetite | 39% | 4% |
| Nervous System Disorders | Dizziness | 5% | 0% |
| Somnolence | 2% | 1% | |
| Psychiatric Disorders | Insomniaa | 23% | 3% |
| Irritability | 10% | 0% | |
| Affect lability | 3% | 0% | |
| Tic | 2% | 0% | |
| Skin and Subcutaneous Tissue Disorders | Rash | 3% | 0% |
a Insomnia includes the following preferred terms reported in the study: Initial Insomnia, Insomnia.
Note: This table includes those reactions for which the incidence in patients taking Vyvanse is at least twice the incidence in patients taking placebo.
| Body System | Preferred Term | Vyvanse (n=233) | Placebo (n=77) |
|---|---|---|---|
| b Insomnia includes the following preferred terms reported in the study: Initial Insomnia, Insomnia. Note: This table includes those reactions for which the incidence in patients taking Vyvanse is at least twice the incidence in patients taking placebo. |
|||
| Gastrointestinal Disorders | Dry Mouth | 4% | 1% |
| Investigations | Weight Decreased | 9% | 0% |
| Metabolism and Nutrition | Decreased Appetite | 34% | 3% |
| Psychiatric Disorders | Insomniab | 13% | 4% |
Adult
Table 3 Adverse Reactions Reported by 2% or More of Adult Patients Taking Vyvanse in a 4-Week Clinical Trial
| Body System | Preferred Term | Vyvanse (n=358) | Placebo (n=62) |
|---|---|---|---|
| Gastrointestinal Disorders | Dry Mouth | 26% | 3% |
| Diarrhea | 7% | 0% | |
| Nausea | 7% | 0% | |
| General Disorder and Administration Site Conditions | Feeling Jittery | 4% | 0% |
| Investigations | Blood Pressure Increased | 3% | 0% |
| Heart Rate Increased | 2% | 0% | |
| Metabolism and Nutrition Disorders | Decreased Appetite | 27% | 3% |
| Anorexia | 5% | 0% | |
| Nervous System Disorders | Tremor | 2% | 0% |
| Psychiatric Disorders | Insomniac | 27% | 8% |
| Anxiety | 6% | 0% | |
| Agitation | 3% | 0% | |
| Restlessness | 3% | 0% | |
| Respiratory, Thoracic, and Mediastinal Disorders | Dyspnea | 2% | 0% |
| Skin and Subcutaneous Tissue Disorders | Hyperhidrosis | 3% | 0% |
c Insomnia includes the following preferred terms reported in the study: Initial Insomnia, Insomnia, Middle Insomnia.
Note: This table includes those events for which the incidence in patients taking Vyvanse is at least twice the incidence in patients taking placebo.
In addition, adverse reactions observed at a rate of less than 2% included decreased libido and erectile dysfunction.
Vital Signs
Weight Loss - In the controlled adult trial, mean weight loss after 4 weeks of therapy was 2.8 lbs, 3.1 lbs, and 4.3 lbs, for patients receiving final doses of 30 mg, 50 mg, and 70 mg of Vyvanse, respectively, compared to a mean weight gain of 0.5 lbs for patients receiving placebo.
6.2 Postmarketing Reports
The following adverse reactions have been identified during post approval use of Vyvanse. Because these reactions are reported voluntarily from a population of uncertain size, it is not possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Cardiac Disorders - Palpitation
Eye Disorders - Vision blurred, mydriasis, diplopia
General Disorders and Administration Site Conditions - Fatigue
Hepatobiliary Disorders - Eosinophilic Hepatitis
Immune System Disorders - Anaphylactic reaction, hypersensitivity
Nervous System Disorders - Somnolence, seizure, dyskinesia
Psychiatric Disorder - Psychotic episodes, mania, hallucination, depression, aggression, dysphoria, euphoria, logorrhoea, dermatillomania
Skin and Subcutaneous Tissue Disorder - Stevens-Johnson Syndrome, angioedema, urticaria
6.3 Adverse Reactions Associated with the Use of Amphetamine
Cardiovascular
Palpitations, tachycardia, elevation of blood pressure, sudden death, myocardial infarction. There have been isolated reports of cardiomyopathy associated with chronic amphetamine use.
Central Nervous System
Psychotic episodes at recommended doses, overstimulation, restlessness, dizziness, insomnia, euphoria, dyskinesia, dysphoria, depression, tremor, headache, exacerbation of motor and phonic tics and Tourette's syndrome, seizures, stroke.
Gastrointestinal
Dryness of the mouth, unpleasant taste, diarrhea, constipation, other gastrointestinal disturbances.
Allergic
Urticaria, rashes, and hypersensitivity reactions, including angioedema and anaphylaxis. Serious skin reactions, including Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis have been reported.
Endocrine
Impotence, changes in libido.
7 DRUG INTERACTIONS
7.1 Agents Whose Blood Levels May be Impacted by Vyvanse
Extended release guanfacine: In a drug interaction study (N=40), administration of an extended release guanfacine (4 mg) in combination with Vyvanse (50 mg) increased guanfacine maximum plasma concentration by 19%, whereas, exposure (area under the curve; AUC) was increased by 7%. These small changes are not expected to be clinically meaningful. In this study, no effect on d-amphetamine exposure was observed following co-administration of extended release guanfacine and Vyvanse.
7.2 Agents that Lower Blood Levels of Amphetamines
Urinary Acidifying Agents
These agents (ammonium chloride, sodium acid phosphate, etc.) increase the concentration of the ionized species of the amphetamine molecule, thereby increasing urinary excretion.
Methenamine Therapy
Urinary excretion of amphetamines is increased, and efficacy is reduced, by acidifying agents used in methenamine therapy.
7.3 Agents that Increase Blood Levels of Amphetamines
Urinary Alkalinizing Agents
These agents (acetazolamide, some thiazides) increase the concentration of the non-ionized species of the amphetamine molecule, thereby decreasing urinary excretion.
Monoamine Oxidase Inhibitors
MAOI antidepressants, as well as a metabolite of furazolidone, slow amphetamine metabolism. This slowing potentiates amphetamines, increasing their effect on the release of norepinephrine and other monoamines from adrenergic nerve endings; this can cause headaches and other signs of hypertensive crisis. A variety of toxic neurological effects and malignant hyperpyrexia can occur, sometimes with fatal results.
7.4 Agents Whose Effects May be Reduced by Amphetamines
Adrenergic Blockers
Adrenergic blockers are inhibited by amphetamines.
Antihistamines
Amphetamines may counteract the sedative effect of antihistamines.
Antihypertensives
Amphetamines may antagonize the hypotensive effects of antihypertensives.
Veratrum Alkaloids
Amphetamines inhibit the hypotensive effect of veratrum alkaloids.
Ethosuximide
Amphetamines may delay intestinal absorption of ethosuximide.
7.5 Agents Whose Effects May be Potentiated by Amphetamines
Antidepressants, Tricyclic
Amphetamines may enhance the activity of tricyclic antidepressants or sympathomimetic agents; d-amphetamine with desipramine or protriptyline and possibly other tricyclics cause striking and sustained increases in the concentration of d-amphetamine in the brain; cardiovascular effects can be potentiated.
Meperidine
Amphetamines potentiate the analgesic effect of meperidine.
Phenobarbital
Amphetamines may delay intestinal absorption of phenobarbital; co-administration of phenobarbital may produce a synergistic anticonvulsant action.
Phenytoin
Amphetamines may delay intestinal absorption of phenytoin; co-administration of phenytoin may produce a synergistic anticonvulsant action.
7.6 Agents that May Reduce the Effects of Amphetamines
Chlorpromazine
Chlorpromazine blocks dopamine and norepinephrine receptors, thus inhibiting the central stimulant effects of amphetamines, and can be used to treat amphetamine poisoning.
Haloperidol
Haloperidol blocks dopamine receptors, thus inhibiting the central stimulant effects of amphetamines.
Lithium Carbonate
The anorectic and stimulatory effects of amphetamines may be inhibited by lithium carbonate.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Animal reproduction studies of lisdexamfetamine dimesylate have not been performed. Studies have been performed with the active metabolite of lisdexamfetamine, d-amphetamine, either alone or in combination with l-amphetamine, as noted below.
Teratogenic Effects
Pregnancy Category C
Amphetamine (d- to l-enantiomer ratio of 3:1) had no apparent effects on embryofetal morphological development or survival when orally administered to pregnant rats and rabbits throughout the period of organogenesis at doses of up to 6 and 16 mg/kg/day, respectively. Fetal malformations and death have been reported in mice following parenteral administration of d-amphetamine doses of 50 mg/kg/day or greater to pregnant animals. Administration of these doses was also associated with severe maternal toxicity.
A number of studies in rodents indicate that prenatal or early postnatal exposure to amphetamine (d- or d,l-) at doses similar to those used clinically can result in longterm neurochemical and behavioral alterations. Reported behavioral effects include learning and memory deficits, altered locomotor activity, and changes in sexual function.
There are no adequate and well-controlled studies in pregnant women. There has been one report of severe congenital bony deformity, tracheo-esophageal fistula, and anal atresia (vater association) in a baby born to a woman who took dextroamphetamine sulfate with lovastatin during the first trimester of pregnancy. Amphetamines should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Nonteratogenic Effects
Infants born to mothers dependent on amphetamines have an increased risk of premature delivery and low birth weight. Also, these infants may experience symptoms of withdrawal as demonstrated by dysphoria, including agitation, and significant lassitude.
8.3 Nursing Mothers
Amphetamines are excreted into human milk. Mothers taking amphetamines should be advised to refrain from nursing.
8.4 Pediatric Use
Vyvanse is indicated for use in pediatric patients with ADHD aged 6 to 17 years. Vyvanse has not been studied in children under 6 years of age. Long-term effects of amphetamines in children have not been well established.
A study was conducted in which juvenile rats received oral doses of 4, 10, or 40 mg/kg/day of lisdexamfetamine dimesylate from day 7 to day 63 of age. These doses are approximately 0.3, 0.7, and 3 times the maximum recommended human daily dose of 70 mg on a mg/m2 basis. Dose-related decreases in food consumption, bodyweight gain, and crown-rump length were seen; after a four-week drug-free recovery period, bodyweights and crown-rump lengths had significantly recovered in females but were still substantially reduced in males. Time to vaginal opening was delayed in females at the highest dose, but there were no drug effects on fertility when the animals were mated beginning on day 85 of age.
In a study in which juvenile dogs received lisdexamfetamine dimesylate for 6 months beginning at 10 weeks of age, decreased bodyweight gain was seen at all doses tested (2, 5, and 12 mg/kg/day, which are approximately 0.5, 1, and 3 times the maximum recommended human daily dose on a mg/m2 basis). This effect partially or fully reversed during a four-week drug-free recovery period.
9 DRUG ABUSE AND DEPENDENCE
9.2 Abuse and Dependence
Amphetamines have been extensively abused. Tolerance, extreme psychological dependence, and severe social disability have occurred. There are reports of patients who have increased the dosage to levels many times higher than recommended. Abrupt cessation following prolonged high-dosage administration results in extreme fatigue and mental depression; changes are also noted on the sleep EEG. Manifestations of chronic intoxication with amphetamines may include severe dermatoses, marked insomnia, irritability, hyperactivity, and personality changes. The most severe manifestation of chronic intoxication is psychosis, often clinically indistinguishable from schizophrenia.
Human Studies
In a human abuse liability study, when equivalent oral doses of 100 mg lisdexamfetamine dimesylate and 40 mg immediate-release d-amphetamine sulfate were administered to individuals with a history of drug abuse, lisdexamfetamine dimesylate 100 mg produced subjective responses on a scale of "Drug Liking Effects" (primary endpoint) that were significantly less than d-amphetamine immediate-release 40 mg. However, oral administration of 150 mg lisdexamfetamine dimesylate produced increases in positive subjective responses on this scale that were statistically indistinguishable from the positive subjective responses produced by 40 mg of oral immediate-release d-amphetamine and 200 mg of diethylpropion (C-IV).1
Intravenous administration of 50 mg lisdexamfetamine dimesylate to individuals with a history of drug abuse produced positive subjective responses on scales measuring "Drug Liking", "Euphoria", "Amphetamine Effects", and "Benzedrine Effects" that were greater than placebo but less than those produced by an equivalent dose (20 mg) of intravenous d-amphetamine.
Animal Studies
In animal studies, lisdexamfetamine dimesylate produced behavioral effects qualitatively similar to those of the CNS stimulant d-amphetamine. In monkeys trained to self-administer cocaine, intravenous lisdexamfetamine dimesylate maintained self-administration at a rate that was statistically less than that for cocaine, but greater than that of placebo.
10 OVERDOSAGE
Individual patient response to amphetamines varies widely. Toxic symptoms may occur idiosyncratically at low doses.
Symptoms: Manifestations of acute overdosage with amphetamines include restlessness, tremor, hyperreflexia, rapid respiration, confusion, assaultiveness, hallucinations, panic states, hyperpyrexia, and rhabdomyolysis. Fatigue and depression usually follow the central nervous system stimulation. Cardiovascular effects include arrhythmias, hypertension or hypotension, and circulatory collapse. Gastrointestinal symptoms include nausea, vomiting, diarrhea, and abdominal cramps. Fatal poisoning is usually preceded by convulsions and coma.
Treatment: Consult with a Certified Poison Control Center for up-to-date guidance and advice. Management of acute amphetamine intoxication is largely symptomatic and includes gastric lavage, administration of activated charcoal, administration of a cathartic, and sedation. Experience with hemodialysis or peritoneal dialysis is inadequate to permit recommendation in this regard. Acidification of the urine increases amphetamine excretion but is believed to increase risk of acute renal failure if myoglobinuria is present. If acute severe hypertension complicates amphetamine overdosage, administration of intravenous phentolamine has been suggested. However, a gradual drop in blood pressure will usually result when sufficient sedation has been achieved. Chlorpromazine antagonizes the central stimulant effects of amphetamines and can be used to treat amphetamine intoxication.
The prolonged release of Vyvanse in the body should be considered when treating patients with overdose.
11 DESCRIPTION
Vyvanse (lisdexamfetamine dimesylate) is designed as a capsule for once-a-day oral administration. The chemical designation for lisdexamfetamine dimesylate is (2S)-2,6-diamino-N-[(1S)-1-methyl-2-phenylethyl] hexanamide dimethanesulfonate. The molecular formula is C15H25N3O•(CH4O3S)2, which corresponds to a molecular weight of 455.60. The chemical structure is:
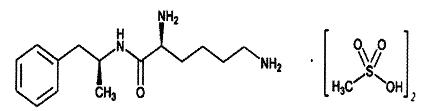
Lisdexamfetamine dimesylate is a white to off-white powder that is soluble in water (792 mg/mL). Vyvanse capsules contain 20 mg, 30 mg, 40 mg, 50 mg, 60 mg, and 70 mg of lisdexamfetamine dimesylate and the following inactive ingredients: microcrystalline cellulose, croscarmellose sodium, and magnesium stearate. The capsule shells (imprinted with NRP104) contain gelatin, titanium dioxide, and one or more of the following: D&C Red #28, D&C Yellow #10, FD&C Blue #1, FD&C Green #3, and FD&C Red #40. The capsule shells (imprinted with S489) contain gelatin, titanium dioxide, and one or more of the following: FD&C Red #3, FD&C Yellow #6, FD&C Blue #1, Black Iron Oxide, and Yellow Iron Oxide.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Lisdexamfetamine is a prodrug of dextroamphetamine. After oral administration, lisdexamfetamine is rapidly absorbed from the gastrointestinal tract and converted primarily in blood due to the hydrolytic activity of red blood cells to dextroamphetamine, which is responsible for the drug's activity. Amphetamines are non-catecholamine sympathomimetic amines with CNS stimulant activity. The mode of therapeutic action in Attention Deficit Hyperactivity Disorder (ADHD) is not known. Amphetamines are thought to block the reuptake of norepinephrine and dopamine into the presynaptic neuron and increase the release of these monoamines into the extraneuronal space. The parent drug, lisdexamfetamine, does not bind to the sites responsible for the reuptake of norepinephrine and dopamine in vitro.
12.3 Pharmacokinetics
Pharmacokinetic studies of dextroamphetamine after oral administration of lisdexamfetamine have been conducted in healthy adult and pediatric (aged 6 to 12) patients with ADHD.
In 18 pediatric patients (aged 6 to 12) with ADHD, the Tmax of dextroamphetamine was approximately 3.5 hours following single-dose oral administration of lisdexamfetamine dimesylate either 30 mg, 50 mg, or 70 mg after an 8-hour overnight fast. The Tmax of lisdexamfetamine was approximately 1 hour. Linear pharmacokinetics of dextroamphetamine after single-dose oral administration of lisdexamfetamine dimesylate was established over the dose range of 30 mg to 70 mg in children aged 6 to 12 years.
There is no unexpected accumulation of dextroamphetamine AUC at steady state in healthy adults and no accumulation of lisdexamfetamine after once-daily dosing for 7 consecutive days.
Food does not affect the observed AUC and Cmax of dextroamphetamine in healthy adults after single-dose oral administration of 70 mg of Vyvanse capsules but prolongs Tmax by approximately 1 hour (from 3.8 hrs at fasted state to 4.7 hrs after a high fat meal). After an 8-hour fast, the AUCs for dextroamphetamine following oral administration of lisdexamfetamine dimesylate in solution and as intact capsules were equivalent.
Weight/Dose normalized AUC and Cmax were 22% and 12% lower, respectively, in adult females than in males on day 7 following a 70 mg/day dose of lisdexamfetamine dimesylate for 7 days. Weight/Dose normalized AUC and Cmax values were the same in girls and boys following single doses of 30-70 mg.
Metabolism and Excretion
After oral administration, lisdexamfetamine is rapidly absorbed from the gastrointestinal tract. Lisdexamfetamine is converted to dextroamphetamine and l-lysine primarily in blood due to the hydrolytic activity of red blood cells. In vitro data demonstrated that red blood cells have a high capacity for metabolism of lisdexamfetamine; substantial hydrolysis occurred even at low hematocrit levels (33% of normal). Lisdexamfetamine is not metabolized by cytochrome P450 enzymes. Following the oral administration of a 70 mg dose of radiolabeled lisdexamfetamine dimesylate to 6 healthy subjects, approximately 96% of the oral dose radioactivity was recovered in the urine and only 0.3% recovered in the feces over a period of 120 hours. Of the radioactivity recovered in the urine, 42% of the dose was related to amphetamine, 25% to hippuric acid, and 2% to intact lisdexamfetamine. Plasma concentrations of unconverted lisdexamfetamine are low and transient, generally becoming non-quantifiable by 8 hours after administration. The plasma elimination half-life of lisdexamfetamine typically averaged less than one hour in studies of lisdexamfetamine dimesylate in volunteers.
Dextroamphetamine is known to inhibit monoamine oxidase. The ability of dextroamphetamine and its metabolites to inhibit various P450 isozymes and other enzymes has not been adequately elucidated. In vitro experiments with human microsomes indicate minor inhibition of CYP2D6 by amphetamine and minor inhibition of CYP1A2, 2D6, and 3A4 by one or more metabolites, but there are no in vivo studies of p450 enzyme inhibition.
Special Populations
Age
The pharmacokinetics of dextroamphetamine is similar in pediatric (aged 6 to 12) and adolescent (aged 13 to 17) ADHD patients, and healthy adult volunteers. Any differences in kinetics seen after oral administration are a result of differences in mg/kg dosing.
Gender
Systemic exposure to dextroamphetamine is similar for men and women given the same mg/kg dose.
Race
Formal pharmacokinetic studies for race have not been conducted.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis/ Mutagenesis and Impairment of Fertility
Carcinogenicity studies of lisdexamfetamine dimesylate have not been performed.
No evidence of carcinogenicity was found in studies in which d-, l-amphetamine (enantiomer ratio of 1:1) was administered to mice and rats in the diet for 2 years at doses of up to 30 mg/kg/day in male mice, 19 mg/kg/day in female mice, and 5 mg/kg/day in male and female rats.
Lisdexamfetamine dimesylate was not clastogenic in the mouse bone marrow micronucleus test in vivo and was negative when tested in the E. coli and S. typhimurium components of the Ames test and in the L5178Y/TK+- mouse lymphoma assay in vitro.
Amphetamine (d- to l-enantiomer ratio of 3:1) did not adversely affect fertility or early embryonic development in the rat at doses of up to 20 mg/kg/day.
14 CLINICAL STUDIES
The efficacy of Vyvanse in the treatment of ADHD was established on the basis of two controlled trials in children aged 6 to 12 years, one controlled trial in adolescents aged 13 to 17 years, and two controlled trials in adults who met Diagnostic and Statistical Manual of Mental Disorders, 4th edition (DSM-IV-TR) criteria for ADHD [see INDICATIONS AND USAGE (1)].
Pediatric
A double-blind, randomized, placebo-controlled, parallel-group study was conducted in children aged 6 to 12 (N=290) who met DSM-IV criteria for ADHD (either the combined type or the hyperactive-impulsive type). Patients were randomized to receive final doses of 30 mg, 50 mg, or 70 mg of Vyvanse or placebo once daily in the morning for a total of four weeks of treatment. All subjects receiving Vyvanse were initiated on 30 mg for the first week of treatment. Subjects assigned to the 50 mg and 70 mg dose groups were titrated by 20 mg per week until they achieved their assigned dose. The primary efficacy outcome was change in Total Score from baseline to endpoint in investigator ratings on the ADHD Rating Scale (ADHD-RS), a measure of the core symptoms of ADHD. Endpoint was defined as the last post-randomization treatment week (i.e. Weeks 1 through 4) for which a valid score was obtained. All Vyvanse dose groups were superior to placebo in the primary efficacy outcome. Mean effects at all doses were fairly similar, although the highest dose (70 mg/day) was numerically superior to both lower doses (30 mg/day and 50 mg/day). The effects were maintained throughout the day based on parent ratings (Conners' Parent Rating Scale) in the morning (approximately 10 am), afternoon (approximately 2 pm), and early evening (approximately 6 pm).
A double-blind, placebo-controlled, randomized, crossover design, analog classroom study was conducted in children aged 6 to 12 (N=52) who met DSM-IV criteria for ADHD (either the combined type or the hyperactive-impulsive type). Following a 3-week open-label dose titration with Adderall XR®, patients were randomly assigned to continue the same dose of Adderall XR (10 mg, 20 mg, or 30 mg), Vyvanse (30 mg, 50 mg, or 70 mg), or placebo once daily in the morning for 1 week each treatment. A significant difference in patient behavior, based upon the average of investigator ratings on the Swanson, Kotkin, Agler, M.Flynn, and Pelham (SKAMP)-Deportment scores across 7 assessments conducted at 2, 3, 4.5, 6, 8, 10, and 12 hours post-dose were observed between patients who received Vyvanse compared to patients who received placebo. The drug effect was similar for all 7 sessions.
A second double-blind, placebo-controlled, randomized, crossover design, analog classroom study was conducted in children aged 6 to 12 (N=129) who met DSM-IV criteria for ADHD (either the combined type or the hyperactive-impulsive type). Following a 4-week open-label dose titration with Vyvanse (30 mg, 50 mg, 70 mg), patients were randomly assigned to continue Vyvanse or placebo once daily in the morning for 1 week each treatment. A significant difference in patient behavior, based upon the average of investigator ratings on the SKAMP-Deportment scores across all 7 assessments conducted at 1.5, 2.5, 5.0, 7.5, 10.0, 12.0, and 13.0 hours post-dose, were observed between patients who received Vyvanse compared to patients who received placebo.
A double-blind, randomized, placebo-controlled, parallel-group study was conducted in adolescents aged 13 to 17 (N=314) who met DSM-IV criteria for ADHD. In this study, patients were randomized in a 1:1:1:1 ratio to a daily morning dose of Vyvanse (30 mg/day, 50 mg/day or 70 mg/day) or placebo for a total of four weeks of treatment. All subjects receiving Vyvanse were initiated on 30 mg for the first week of treatment. Subjects assigned to the 50 mg and 70 mg dose groups were titrated by 20 mg per week until they achieved their assigned dose. The primary efficacy outcome was change in Total Score from baseline to endpoint in investigator ratings on the ADHD Rating Scale (ADHD-RS), a measure of the core symptoms of ADHD. Endpoint was defined as the last post-randomization treatment week (i.e. Weeks 1 through 4) for which a valid score was obtained. All Vyvanse dose groups were superior to placebo in the primary efficacy outcome.
Adult
A double-blind, randomized, placebo-controlled, parallel-group study was conducted in adults (N=420) who met DSM-IV criteria for ADHD. In this study, patients were randomized to receive final doses of 30 mg, 50 mg, or 70 mg of Vyvanse or placebo for a total of four weeks of treatment. All subjects receiving Vyvanse were initiated on 30 mg for the first week of treatment. Subjects assigned to the 50 mg and 70 mg dose groups were titrated by 20 mg per week until they achieved their assigned dose. The primary efficacy outcome was change in Total Score from baseline to endpoint in investigator ratings on the ADHD Rating Scale (ADHD-RS), a measure of the core symptoms of ADHD. Endpoint was defined as the last post-randomization treatment week (i.e. Weeks 1 through 4) for which a valid score was obtained. All Vyvanse dose groups were superior to placebo in the primary efficacy outcome.
The second study was a multi-center, randomized, double-blind, placebo-controlled, crossover design, modified analog classroom study of Vyvanse to simulate a workplace environment in 142 adults who met DSM-IV-TR criteria for ADHD. There was a 4-week open-label, dose optimization phase with Vyvanse (30 mg/day, 50 mg/day, or 70 mg/day in the morning). Subjects were then randomized to one of two treatment sequences: 1) Vyvanse (optimized dose) followed by placebo, each for one week, or 2) placebo followed by Vyvanse, each for one week. Efficacy assessments occurred at the end of each week, using the Permanent Product Measure of Performance (PERMP). The PERMP is a skill-adjusted math test that measures attention in ADHD. Vyvanse treatment, compared to placebo, resulted in a statistically significant improvement in attention across all post-dose time points, as measured by average PERMP total scores over the course of one assessment day, as well as at each time point measured. The PERMP assessments were administered at pre-dose (-0.5 hours) and at 2, 4, 8, 10, 12, and 14 hours post-dose.
15 REFERENCES
1 Jasinski DR, Krishnan S. Abuse liability and safety of oral lisdexamfetamine dimesylate in individuals with a history of stimulant abuse. Journal of Psychopharmacology. 2009; 23(4);419-27.
16 HOW SUPPLIED/STORAGE AND HANDLING
Vyvanse capsules 30 mg: white body/orange cap (imprinted with NRP104 or S489 and 30 mg),
| Bottles of 10 | NDC 54868-5916-0 |
| Bottles of 30 | NDC 54868-5916-1 |
Vyvanse capsules 40 mg: white body/blue green cap (imprinted with NRP104 or S489 and 40 mg),
| Bottles of 10 | NDC 54868-6009-0 |
| Bottles of 30 | NDC 54868-6009-1 |
Vyvanse capsules 50 mg: white body/blue cap (imprinted with NRP104 or S489 and 50 mg),
| Bottles of 10 | NDC 54868-5827-0 |
| Bottles of 30 | NDC 54868-5827-1 |
Vyvanse capsules 70 mg: blue body/orange cap (imprinted with NRP104 or S489 and 70 mg),
| Bottles of 10 | NDC 54868-3655-0 |
| Bottles of 30 | NDC 54868-3655-1 |
Dispense in a tight, light-resistant container as defined in the USP.
Store at 25° C (77° F). Excursions permitted to 15-30° C (59-86° F) [see USP Controlled Room Temperature]
17 PATIENT COUNSELING INFORMATION
See Medication Guide
17.1 Information on Medication Guide
Prescribers or other health professionals should inform patients, their families, and their caregivers about the benefits and risks associated with treatment with Vyvanse and should counsel them in its appropriate use. A patient Medication Guide is available for Vyvanse. The prescriber or health professional should instruct patients, their families, and their caregivers to read the Medication Guide and should assist them in understanding its contents. Patients should be given the opportunity to discuss the contents of the Medication Guide and to obtain answers to any questions they may have. The complete text of the Medication Guide is attached to the package insert.
17.2 Controlled Substance Status/Potential for Abuse, Misuse, and Dependence
Patients should be advised that Vyvanse is a federally controlled substance because it can be abused or lead to dependence. Additionally, it should be emphasized that Vyvanse should be stored in a safe place to prevent misuse and/or abuse. Patient history (including family history) of abuse or dependence on alcohol, prescription medicines, or illicit drugs should be evaluated [see Drug Abuse and Dependence (9)].
17.3 Serious Cardiovascular Risks
Patients should be advised of serious cardiovascular risk (including sudden death, myocardial infarction, stroke, and hypertension) with Vyvanse. Patients who develop symptoms such as exertional chest pain, unexplained syncope, or other symptoms suggestive of cardiac disease during treatment should undergo a prompt cardiac evaluation [see Warnings and Precautions (5.1)].
17.4 Psychiatric Risks
Prior to initiating treatment with a stimulant, patients with comorbid depressive symptoms should be adequately screened to determine if they are at risk for bipolar disorder. Such screening should include a detailed psychiatric history, including a family history of suicide, bipolar disorder, and/or depression. Additionally, stimulant therapy at usual doses may cause treatment-emergent psychotic or manic symptoms in patients without prior history of psychotic symptoms or mania [see Warnings and Precautions (5.2)].
17.5 Growth
Growth should be monitored during treatment with stimulants, and patients who are not growing or gaining weight as expected may need to have their treatment interrupted. [see Warnings and Precautions (5.6)].
17.6 Pregnancy
Patients should be advised to notify their physicians if they become pregnant or intend to become pregnant during treatment [see Dosage and Administration (2) and Use in Specific Populations (8.1)].
17.7 Nursing
Patients should be advised not to breast feed if they are taking Vyvanse [see USE IN SPECIFIC POPULATIONS (8.3)].
17.8 Impairment in Ability to Operate Machinery or Vehicles
Amphetamines may impair the ability of the patient to engage in potentially hazardous activities such as operating machinery or vehicles; the patient should therefore be cautioned accordingly.
Pharmacist: Medication Guide to be dispensed to patients
Manufactured for: Shire US Inc., Wayne, PA 19087
Made in USA
For more information call 1-800-828-2088
Vyvanse® is a trademark of Shire LLC
©2011 Shire US Inc.
US Pat No. 7,105,486 and US Pat No. 7,223,735
Last Modified: 08/2011
Relabeling and Repackaging by:
Physicians Total Care, Inc.
Tulsa, Oklahoma 74146
VYVANSE®
(lisdexamfetamine dimesylate) CII
Read the Medication Guide that comes with Vyvanse before you or your child starts taking it and each time you get a refill. There may be new information. This Medication Guide does not take the place of talking to your doctor about you or your child's treatment with Vyvanse.
| What is the most important information I should know about Vyvanse?
Vyvanse is a stimulant medicine. The following have been reported with use of stimulant medicines. 1. Heart-related problems: • sudden death in patients who have heart problems or heart defects • stroke and heart attack in adults • increased blood pressure and heart rate Tell your doctor if you or your child have any heart problems, heart defects, high blood pressure, or a family history of these problems. Your doctor should check you or your child carefully for heart problems before starting Vyvanse. Your doctor should check your or your child's blood pressure and heart rate regularly during treatment with Vyvanse. Call your doctor right away if you or your child has any signs of heart problems such as chest pain, shortness of breath, or fainting while taking Vyvanse. 2. Mental (Psychiatric) problems: All Patients • new or worse behavior and thought problems • new or worse bipolar illness • new or worse aggressive behavior or hostility Children and Teenagers • new psychotic symptoms (such as hearing voices, believing things that are not true, are suspicious) or new manic symptoms Tell your doctor about any mental problems you or your child have, or about a family history of suicide, bipolar illness, or depression. Call your doctor right away if you or your child have any new or worsening mental symptoms or problems while taking Vyvanse, especially seeing or hearing things that are not real, believing things that are not real, or are suspicious. |
What Is Vyvanse?
Vyvanse is a central nervous system stimulant prescription medicine. It is used for the treatment of Attention-Deficit Hyperactivity Disorder (ADHD). Vyvanse may help increase attention and decrease impulsiveness and hyperactivity in patients with ADHD.
Vyvanse should be used as a part of a total treatment program for ADHD that may include counseling or other therapies.
| Vyvanse is a federally controlled substance (CII) because it can be abused or lead to dependence. Keep Vyvanse in a safe place to prevent misuse and abuse. Selling or giving away Vyvanse may harm others, and is against the law.
Tell your doctor if you or your child have (or have a family history of) ever abused or been dependent on alcohol, prescription medicines or street drugs. |
Who should not take Vyvanse?
Vyvanse should not be taken if you or your child:
- have heart disease or hardening of the arteries
- have moderate to severe high blood pressure
- have hyperthyroidism
- have an eye problem called glaucoma
- are very anxious, tense, or agitated
- have a history of drug abuse
- are taking or have taken within the past 14 days an anti-depression medicine called a monoamine oxidase inhibitor or MAOI.
- is sensitive to, allergic to, or had a reaction to other stimulant medicines
Vyvanse has not been studied in children less than 6 years old.
Vyvanse may not be right for you or your child. Before starting Vyvanse tell your or your child's doctor about all health conditions (or a family history of) including:
- heart problems, heart defects, high blood pressure
- mental problems including psychosis, mania, bipolar illness, or depression
- tics or Tourette's syndrome
- liver or kidney problems
- thyroid problems
- seizures or have had an abnormal brain wave test (EEG)
Tell your doctor if you or your child is pregnant, planning to become pregnant, or breastfeeding.
Can Vyvanse be taken with other medicines?
Tell your doctor about all of the medicines that you or your child take including prescription and non-prescription medicines, vitamins, and herbal supplements. Vyvanse and some medicines may interact with each other and cause serious side effects. Sometimes the doses of other medicines will need to be adjusted while taking Vyvanse.
Your doctor will decide whether Vyvanse can be taken with other medicines.
Especially tell your doctor if you or your child takes:
- anti-depression medicines including MAOIs
- anti-psychotic medicines
- lithium
- blood pressure medicines
- seizure medicines
- narcotic pain medicines
Know the medicines that you or your child takes. Keep a list of your medicines with you to show your doctor and pharmacist.
Do not start any new medicine while taking Vyvanse without talking to your doctor first.
How should Vyvanse be taken?
- Take Vyvanse exactly as prescribed. Vyvanse comes in 6 different strength capsules. Your doctor may adjust the dose until it is right for you or your child.
- Take Vyvanse once a day in the morning.
- Vyvanse can be taken with or without food.
- From time to time, your doctor may stop Vyvanse treatment for a while to check ADHD symptoms.
- Your doctor may do regular checks of the blood, heart, and blood pressure while taking Vyvanse. Children should have their height and weight checked often while taking Vyvanse. Vyvanse treatment may be stopped if a problem is found during these check-ups.
- If you or your child takes too much Vyvanse or overdoses, call your doctor or poison control center right away, or get emergency treatment.
What are possible side effects of Vyvanse?
See "What is the most important information I should know about Vyvanse?" for information on reported heart and mental problems.
Other serious side effects include:
- slowing of growth (height and weight) in children
- seizures, mainly in patients with a history of seizures
- eyesight changes or blurred vision
Common side effects include:
- upper belly pain
- dizziness
- irritability
- nausea
- weight loss
- decreased appetite
- dry mouth
- trouble sleeping
- vomiting
Vyvanse may affect your or your child's ability to drive or do other dangerous activities.
Talk to your doctor if you or your child has side effects that are bothersome or do not go away.
This is not a complete list of possible side effects. Ask your doctor or pharmacist for more information.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store Vyvanse?
- Store Vyvanse in a safe place at room temperature, 59 to 86° F (15 to 30° C). Protect from light.
- Keep Vyvanse and all medicines out of the reach of children.
General information about Vyvanse
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use Vyvanse for a condition for which it was not prescribed. Do not give Vyvanse to other people, even if they have the same condition. It may harm them and it is against the law.
This Medication Guide summarizes the most important information about Vyvanse. If you would like more information, talk with your doctor. You can ask your doctor or pharmacist for information about Vyvanse that was written for healthcare professionals. For more information about Vyvanse, please contact Shire US Inc. at 1-800-828-2088.
What are the ingredients in Vyvanse?
Active Ingredient: lisdexamfetamine dimesylate
Inactive Ingredients: microcrystalline cellulose, croscarmellose sodium, and magnesium stearate. The capsule shells (imprinted with NRP104) contain gelatin, titanium dioxide, and one or more of the following: D&C Red #28, D&C Yellow #10, FD&C Blue #1, FD&C Green #3, and FD&C Red #40. The capsule shells (imprinted with S489) contain gelatin, titanium dioxide, and one or more of the following: FD&C Red #3, FD&C Yellow #6, FD&C Blue #1, Black Iron Oxide, Yellow Iron Oxide.
This Medication Guide has been approved by the U.S. Food and Drug Administration.
©2011 Shire US Inc.
US Pat No. 7,105,486 and US Pat No. 7,223,735
Last Modified: 08/2011



