FULL PRESCRIBING INFORMATION
WARNING: INCREASED RISK OF BLEEDING WITH CONCOMITANT USE OF VITAMIN K ANTAGONISTS
Altered coagulation parameters and/or bleeding, including death, have been reported in patients taking XELODA concomitantly with oral vitamin K antagonists, such as warfarin [see Warnings and Precautions (5.1), Drug Interactions (7.2)].
Clinically significant increases in prothrombin time (PT) and international normalized ratio (INR) have been reported in patients who were on stable doses of a vitamin K antagonist at the time XELODA was introduced. These events occurred within several days and up to several months after initiating XELODA and, in a few cases, within 1 month after stopping XELODA. These events occurred in patients with and without liver metastases.
Monitor INR more frequently and adjust the dose of the vitamin K antagonist as appropriate [see Drug Interactions (7.2)].
1 INDICATIONS AND USAGE
1.1 Colorectal Cancer
XELODA is indicated for the:
- adjuvant treatment of patients with Stage III colon cancer as a single agent or as a component of a combination chemotherapy regimen.
- perioperative treatment of adults with locally advanced rectal cancer as a component of chemoradiotherapy.
- treatment of patients with unresectable or metastatic colorectal cancer as a single agent or as a component of a combination chemotherapy regimen.
1.2 Breast Cancer
XELODA is indicated for the:
- treatment of patients with advanced or metastatic breast cancer as a single agent if an anthracycline- or taxane-containing chemotherapy is not indicated.
- treatment of patients with advanced or metastatic breast cancer in combination with docetaxel after disease progression on prior anthracycline-containing chemotherapy.
1.3 Gastric, Esophageal, or Gastroesophageal Junction Cancer
XELODA is indicated for the:
- treatment of adults with unresectable or metastatic gastric, esophageal, or gastroesophageal junction cancer as a component of a combination chemotherapy regimen.
- treatment of adults with HER2-overexpressing metastatic gastric or gastroesophageal junction adenocarcinoma who have not received prior treatment for metastatic disease as a component of a combination regimen.
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage for Colorectal Cancer
Adjuvant Treatment of Colon Cancer
Single Agent
The recommended dosage of XELODA is 1,250 mg/m2 orally twice daily for the first 14 days of each 21-day cycle for a maximum of 8 cycles.
In Combination with Oxaliplatin-Containing Regimens
The recommended dosage of XELODA is 1,000 mg/m2 orally twice daily for the first 14 days of each 21-day cycle for a maximum of 8 cycles in combination with oxaliplatin 130 mg/m2 administered intravenously on day 1 of each cycle.
Refer to the oxaliplatin prescribing information for additional dosing information as appropriate.
Perioperative Treatment of Rectal Cancer
The recommended dosage of capecitabine is 825 mg/m2 orally twice daily when administered with concomitant radiation therapy and 1,250 mg/m2 orally twice daily when administered without radiation therapy as part of a peri-operative combination regimen.
Unresectable or Metastatic Colorectal Cancer
Single Agent
The recommended dosage of XELODA is 1,250 mg/m2 orally twice daily for the first 14 days of a 21-day cycle until disease progression or unacceptable toxicity.
In Combination with Oxaliplatin
The recommended dosage of XELODA is 1,000 mg/m2 orally twice daily for the first 14 days of each 21-day cycle until disease progression or unacceptable toxicity in combination with oxaliplatin 130 mg/m2 administered intravenously on day 1 of each cycle.
Refer to the Prescribing Information for oxaliplatin for additional dosing information as appropriate.
2.2 Recommended Dosage for Breast Cancer
Advanced or Metastatic Breast Cancer
Single Agent
The recommended dosage of XELODA is 1,000 mg/m2 or 1,250 mg/m2 orally twice daily for the first 14 days of a 21-day cycle until disease progression or unacceptable toxicity. Individualize the dose and dosing schedule of XELODA based on patient risk factors and adverse reactions.
In Combination with Docetaxel
The recommended dosage of XELODA is 1,000 mg/m2 or 1,250 mg/m2 orally twice daily for the first 14 days of a 21-day cycle until disease progression or unacceptable toxicity in combination with docetaxel 75 mg/m2 administered intravenously on day 1 of each cycle.
Refer to the Prescribing Information for docetaxel for additional dosing information as appropriate.
2.3 Recommended Dosage for Gastric, Esophageal, or Gastroesophageal Junction Cancer
The recommended dosage of XELODA for unresectable or metastatic gastric, esophageal, or gastroesophageal junction cancer is:
-
625 mg/m2 orally twice daily on days 1 to 21 of each 21-day cycle for a maximum of 8 cycles in combination with platinum-containing chemotherapy.
OR - 850 mg/m2 or 1,000 mg/m2 orally twice daily for the first 14 days of each 21-day cycle until disease progression or unacceptable toxicity in combination with oxaliplatin 130 mg/m2 administered intravenously on day 1 of each cycle. Individualize the dose and dosing schedule of XELODA based on patient risk factors and adverse reactions.
The recommended dosage of XELODA for HER2-overexpressing metastatic gastric or gastroesophageal junction adenocarcinoma is 1,000 mg/m2 orally twice daily for the first 14 days of each 21-day cycle until disease progression or unacceptable toxicity in combination with cisplatin and trastuzumab.
Refer to the Prescribing Information for agents used in combination for additional dosing information as appropriate.
2.4 Recommended Dosage for Pancreatic Cancer
The recommended dosage of XELODA is 830 mg/m2 orally twice daily for the first 21 days of each 28-day cycle until disease progression, unacceptable toxicity, or for a maximum 6 cycles in combination with gemcitabine 1,000 mg/m2 administered intravenously on days 1, 8, and 15 of each cycle.
Refer to Prescribing Information for gemcitabine for additional dosing information as appropriate.
2.5 Dosage Modifications for Adverse Reactions
Monitor patients for adverse reactions and modify dosages of XELODA as described in Table 1. Do not replace missed doses of XELODA; instead resume XELODA with the next planned dosage.
When XELODA is administered with docetaxel, withhold XELODA and docetaxel until the requirements for resuming both XELODA and docetaxel are met. Refer to the Prescribing Information for docetaxel for additional dosing information as appropriate.
| Severity | Dosage Modification | Resume at Same or Reduced Dose (Percent of Current Dose) |
|---|---|---|
| Grade 2 | ||
| 1st appearance | Withhold until resolved to grade 0-1. | 100% |
| 2nd appearance | 75% | |
| 3rd appearance | 50% | |
| 4th appearance | Permanently discontinue. | - |
| Grade 3 | ||
| 1st appearance | Withhold until resolved to grade 0-1. | 75% |
| 2nd appearance | 50% | |
| 3rd appearance | Permanently discontinue. | - |
| Grade 4 | ||
| 1st appearance | Permanently discontinue OR Withhold until resolved to grade 0-1. | 50% |
Hyperbilirubinemia
Patients with Grade 3-4 hyperbilirubinemia may resume treatment once the event is Grade 2 or less (less than three times the upper limit of normal), using the percent of current dose as shown in column 3 of Table 1 [see Warnings and Precautions (5.10)].
2.6 Dosage Modification For Renal Impairment
Reduce the dose of XELODA by 25% for patients with creatinine clearance (CLcr) of 30 to 50 mL/min as determined by Cockcroft-Gault equation. A dosage has not been established in patients with severe renal impairment (CLcr <30 mL/min) [see Use in Specific Populations (8.6)].
2.7 Administration
Round the recommended dosage for patients to the nearest 150 mg dose to provide whole XELODA tablets.
Swallow XELODA tablets whole with water within 30 minutes after a meal. Do not chew, cut, or crush XELODA tablets [see Warnings and Precautions (5.12)].
Take XELODA at the same time each day approximately 12 hours apart.
Do not take an additional dose after vomiting and continue with the next scheduled dose.
Do not take a missed dose and continue with the next scheduled dose.
XELODA is a hazardous drug. Follow applicable special handling and disposal procedures.1
3 DOSAGE FORMS AND STRENGTHS
Tablets, film-coated:
- 150 mg: biconvex, oblong, light-peach colored, with "XELODA" on one side and "150" on the other
- 500 mg: biconvex, oblong, peach colored, with "XELODA on one side and "500" on the other
4 CONTRAINDICATIONS
XELODA is contraindicated in patients with history of severe hypersensitivity reaction to fluorouracil or capecitabine [see Adverse Reactions (6.1)].
5 WARNINGS AND PRECAUTIONS
5.1 Increased Risk of Bleeding With Concomitant Use of Vitamin K Antagonists
Altered coagulation parameters and/or bleeding, including death, have been reported in patients taking XELODA concomitantly with vitamin K antagonists, such as warfarin.
Clinically significant increases in PT and INR have been reported in patients who were on stable doses of oral vitamin K antagonists at the time XELODA was introduced. These events occurred within several days and up to several months after initiating XELODA and, in a few cases, within 1 month after stopping XELODA. These events occurred in patients with and without liver metastases.
Monitor INR more frequently and adjust the dose of the vitamin K antagonist as appropriate [see Drug Interactions (7.1)].
5.2 Serious Adverse Reactions from Dihydropyrimidine Dehydrogenase (DPD) Deficiency
Patients with certain homozygous or compound heterozygous variants in the DPYD gene known to result in complete or near complete absence of DPD activity (complete DPD deficiency) are at increased risk for acute early-onset toxicity and serious, including fatal, adverse reactions due to XELODA (e.g., mucositis, diarrhea, neutropenia, and neurotoxicity). Patients with partial DPD activity (partial DPD deficiency) may also have increased risk of serious, including fatal, adverse reactions.
XELODA is not recommended for use in patients known to have certain homozygous or compound heterozygous DPYD variants that result in complete DPD deficiency.
Withhold or permanently discontinue XELODA based on clinical assessment of the onset, duration, and severity of the observed adverse reactions in patients with evidence of acute early-onset or unusually severe reactions, which may indicate complete DPD deficiency. No XELODA dose has been proven safe for patients with complete DPD deficiency. There are insufficient data to recommend a specific dose in patients with partial DPD deficiency.
Consider testing for genetic variants of DPYD prior to initiating XELODA to reduce the risk of serious adverse reactions if the patient's clinical status permits and based on clinical judgement [see Clinical Pharmacology (12.5)]. Serious adverse reactions may still occur even if no DPYD variants are identified.
An FDA-authorized test for the detection of genetic variants of DPYD to identify patients at risk of serious adverse reactions due to increased systemic exposure to XELODA is not currently available. Currently available tests used to identify DPYD variants may vary in accuracy and design (e.g., which DPYD variant(s) they identify).
5.3 Cardiotoxicity
Cardiotoxicity can occur with XELODA. Myocardial infarction/ischemia, angina, dysrhythmias, cardiac arrest, cardiac failure, sudden death, electrocardiographic changes, and cardiomyopathy have been reported with XELODA. These adverse reactions may be more common in patients with a prior history of coronary artery disease.
Withhold XELODA for cardiotoxicity as appropriate [see Dosage and Administration (2.5)]. The safety of resumption of XELODA in patients with cardiotoxicity that has resolved have not been established.
5.4 Diarrhea
Diarrhea, sometimes severe, can occur with XELODA. In 875 patients with metastatic breast or colorectal cancer who received XELODA as a single agent, the median time to first occurrence of grade 2 to 4 diarrhea was 34 days (range: 1 day to 1 year). The median duration of grade 3 to 4 diarrhea was 5 days.
Withhold XELODA and then resume at same or reduced dose or permanently discontinue based on severity and occurrence [see Dosage and Administration (2.5)].
5.5 Dehydration
Dehydration can occur with XELODA. Patients with anorexia, asthenia, nausea, vomiting, or diarrhea may be at an increased risk of developing dehydration with XELODA. Optimize hydration before starting XELODA. Monitor hydration status and kidney function at baseline and as clinically indicated. Withhold XELODA and then resume at same or reduced dose, or permanently discontinue, based on severity and occurrence [see Dosage and Administration (2.5)].
5.6 Renal Toxicity
Serious renal failure, sometimes fatal, can occur with XELODA. Renal impairment or coadministration of XELODA with other products known to cause renal toxicity may increase the risk of renal toxicity [see Drug Interactions (7.3)].
Monitor renal function at baseline and as clinically indicated. Optimize hydration before starting XELODA. Withhold XELODA and then resume at same or reduced dose, or permanently discontinue, based on severity and occurrence [see Dosage and Administration (2.5)].
5.7 Serious Skin Toxicities
Severe cutaneous adverse reactions (SCARs), including Stevens-Johnson Syndrome and toxic epidermal necrolysis (TEN), which can be fatal, can occur with XELODA [see Adverse Reactions (6.2)].
Monitor for new or worsening serious skin reactions. Permanently discontinue XELODA for severe cutaneous adverse reactions.
5.8 Palmar-Plantar Erythrodysesthesia Syndrome
Palmar-plantar erythrodysesthesia syndrome (PPES) can occur with XELODA.
In patients with metastatic breast or colorectal cancer who received XELODA as a single agent, the median time to onset of grades 1 to 3 PPES was 2.6 months (range: 11 days to 1 year).
Withhold XELODA and then resume at same or reduced dose or permanently discontinue based on severity and occurrence [see Dosage and Administration (2.5)].
5.9 Myelosuppression
Myelosuppression can occur with XELODA.
In the 875 patients with metastatic breast or colorectal cancer who received XELODA as a single agent, 3.2% had grade 3 or 4 neutropenia, 1.7% had grade 3 or 4 thrombocytopenia, and 2.4% had grade 3 or 4 anemia.
In the 251 patients with metastatic breast cancer who received XELODA with docetaxel, 68% had grade 3 or 4 neutropenia, 2.8% had grade 3 or 4 thrombocytopenia, and 10% had grade 3 or 4 anemia.
Necrotizing enterocolitis (typhlitis) has been reported. Consider typhlitis in patients with fever, neutropenia and abdominal pain.
Monitor complete blood count at baseline and before each cycle. XELODA is not recommended if baseline neutrophil count <1.5 × 109/L or platelet count <100 × 109/L. For grade 3 to 4 myelosuppression, withhold XELODA and then resume at same or reduced dose, or permanently discontinue, based on occurrence [see Dosage and Administration (2.5)].
5.10 Hyperbilirubinemia
Hyperbilirubinemia can occur with XELODA. In the 875 patients with metastatic breast or colorectal cancer who received XELODA as a single agent, grade 3 hyperbilirubinemia occurred in 15% of patients and grade 4 hyperbilirubinemia occurred in 3.9%. Of the 566 patients who had hepatic metastases at baseline and the 309 patients without hepatic metastases at baseline, grade 3 or 4 hyperbilirubinemia occurred in 23% and 12%, respectively. Of these 167 patients with grade 3 or 4 hyperbilirubinemia, 19% had postbaseline increased alkaline phosphatase and 28% had postbaseline increased transaminases at any time (not necessarily concurrent). The majority of these patients with increased transaminases or alkaline phosphatase had liver metastases at baseline. In addition, 58% and 35% of the 167 patients with grade 3 or 4 hyperbilirubinemia had pre- and postbaseline increased alkaline phosphatase or transaminases (grades 1 to 4), respectively. Only 8% (n=13) and 3% (n=5) had grade 3 or 4 increased alkaline phosphatase or transaminases.
In the 596 patients who received XELODA for metastatic colorectal cancer, the incidence of grade 3 or 4 hyperbilirubinemia was similar to that observed for the pooled population of patients with metastatic breast and colorectal cancer. The median time to onset for grade 3 or 4 hyperbilirubinemia was 64 days and median total bilirubin increased from 8 µm/L at baseline to 13 µm/L during treatment with XELODA. Of the 136 patients with grade 3 or 4 hyperbilirubinemia, 49 patients had grade 3 or 4 hyperbilirubinemia as their last measured value, of which 46 had liver metastases at baseline.
In the 251 patients with metastatic breast cancer who received XELODA with docetaxel, grade 3 hyperbilirubinemia occurred in 7% and grade 4 hyperbilirubinemia occurred in 2%.
Withhold XELODA and then resume at a same or reduced dose, or permanently discontinue, based on occurrence [see Dosage and Administration (2.5)]. Patients with Grade 3-4 hyperbilirubinemia may resume treatment once the event is Grade 2 or less than three times the upper limit of normal, using the percent of current dose as shown in Table 1 [see Dosage and Administration (2.5)].
5.11 Embryo-Fetal Toxicity
Based on findings from animal reproduction studies and its mechanism of action, XELODA can cause fetal harm when administered to a pregnant woman. Insufficient data is available on XELODA use in pregnant women to evaluate a drug-associated risk. In animal reproduction studies, administration of capecitabine to pregnant animals during the period of organogenesis caused embryolethality and teratogenicity in mice and embryolethality in monkeys at 0.2 and 0.6 times the human exposure (AUC) in patients who received a dosage of 1,250 mg/m2 twice daily, respectively.
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with XELODA and for 6 months following the last dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with XELODA and for 3 months following the last dose [see Use in Specific Populations (8.1, 8.3)].
5.12 Eye Irritation, Skin Rash, and Other Adverse Reactions from Exposure to Crushed Tablets
In instances of exposure to crushed XELODA tablets, the following adverse reactions have been reported: eye irritation and swelling, skin rash, diarrhea, paresthesia, headache, gastric irritation, vomiting and nausea. Advise patients not to cut or crush tablets.
If XELODA tablets must be cut or crushed, this should be done by a professional trained in safe handling of cytotoxic drugs using appropriate equipment and safety procedures [see Dosage and Administration (2.7)]. The safety and effectiveness have not been established for the administration of crushed XELODA tablets.
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Cardiotoxicity [see Warnings and Precautions (5.3)]
- Diarrhea [see Warnings and Precautions (5.4)]
- Dehydration [see Warnings and Precautions (5.5)]
- Renal Toxicity [see Warnings and Precautions (5.6)]
- Serious Skin Toxicities [see Warnings and Precautions (5.7)]
- Palmar-Plantar Erythrodysesthesia Syndrome [see Warnings and Precautions (5.8)]
- Myelosuppression [see Warnings and Precautions (5.9)]
- Hyperbilirubinemia [see Warnings and Precautions (5.10)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adjuvant Treatment of Colon Cancer
Single Agent
The safety of XELODA as a single agent was evaluated in patients with Stage III colon cancer in X-ACT [see Clinical Studies (14.1)]. Patients received XELODA 1,250 mg/m2 orally twice daily for the first 14 days of a 21-day cycle (N=995) or leucovorin 20 mg/m2 intravenously followed by fluorouracil 425 mg/m2 as an intravenous bolus on days 1 to 5 of each 28-day cycle (N=974). Among patients who received XELODA, the median duration of treatment was 5.4 months.
Deaths due to all causes occurred in 0.8% of patients who received XELODA on study or within 28 days of receiving study drug. Permanent discontinuation due to an adverse reaction occurred in 11% of patients who received XELODA.
Most common adverse reactions (>30%) were palmar-plantar erythrodysesthesia syndrome, diarrhea, and nausea.
Tables 2 and 3 summarize the adverse reactions and laboratory abnormalities in X-ACT.
| Adverse Reaction | XELODA (N=995) | Fluorouracil + Leucovorin (N=974) |
||
|---|---|---|---|---|
| All Grades (%) | Grade 3 or 4 (%) | All Grades (%) | Grade 3 or 4 (%) |
|
| Skin and Subcutaneous Tissue | ||||
| Palmar-plantar erythrodysesthesia syndrome | 60 | 17 | 9 | <1 |
| Gastrointestinal | ||||
| Diarrhea | 47 | 12 | 65 | 14 |
| Nausea | 34 | 2 | 47 | 2 |
| Stomatitis | 22 | 2 | 60 | 14 |
| Vomiting | 15 | 2 | 21 | 2 |
| Abdominal pain | 14 | 3 | 16 | 2 |
| General | ||||
| Fatigue | 16 | <1 | 16 | 1 |
| Asthenia | 10 | <1 | 10 | 1 |
| Lethargy | 10 | <1 | 9 | <1 |
Clinically relevant adverse reactions in <10% of patients are presented below:
Eye: conjunctivitis
Gastrointestinal: constipation, upper abdominal pain, dyspepsia
General: pyrexia
Metabolism and Nutrition: anorexia
Nervous System: dizziness, dysgeusia, headache
Skin & Subcutaneous Tissue: rash, alopecia, erythema
| Laboratory Abnormality | XELODA (N=995) | Fluorouracil + Leucovorin (N=974) |
|---|---|---|
| Grade 3 or 4 | Grade 3 or 4 | |
| (%) | (%) | |
| Bilirubin increased | 20 | 6 |
| Lymphocytes decreased | 13 | 13 |
| Neutrophils/granulocytes decreased | 2.4 | 26 |
| Calcium decreased | 2.3 | 2.2 |
| Neutrophils decreased | 2.2 | 26 |
| ALT increased | 1.6 | 0.6 |
| Calcium increased | 1.1 | 0.7 |
| Hemoglobin decreased | 1 | 1.2 |
| Platelets decreased | 1 | 0.7 |
In Combination with Oxaliplatin-Containing Regimens
The safety of XELODA for the perioperative treatment of adults with Stage III colon cancer as a component of a combination chemotherapy regimen was derived from published literature [see Clinical Studies (14.1)]. The safety of XELODA for the adjuvant treatment of patients with Stage III colon cancer as a component of a combination chemotherapy regimen was similar to those in patients treated with XELODA as a single agent, with the exception of an increased incidence of neurosensory toxicity.
Perioperative Treatment of Rectal Cancer
The safety of XELODA for the perioperative treatment of adults with locally advanced rectal cancer as a component of chemoradiotherapy was derived from published literature [see Clinical Studies (14.1)]. The safety of XELODA for the perioperative treatment of adults with locally advanced rectal cancer as a component of chemoradiotherapy was similar to those in patients treated with XELODA as a single agent, with the exception of an increased incidence of diarrhea.
Metastatic Colorectal Cancer
Single Agent
The safety of XELODA as a single agent was evaluated in a pooled metastatic colorectal cancer population (Study SO14695 and Study SO14796) [see Clinical Studies (14.1)]. Patients received XELODA 1,250 mg/m2 orally twice a day for the first 14 days of a 21-day cycle (N=596) or leucovorin 20 mg/m2 intravenously followed by fluorouracil 425 mg/m2 as an intravenous bolus on days 1 to 5 of each 28-day cycle (N=593). Among the patients who received XELODA, the median duration of treatment was 4.6 months.
Deaths due to all causes occurred in 8% of patients who received XELODA on study or within 28 days of receiving study drug. Permanent discontinuation due to an adverse reaction or intercurrent illness occurred in 13% of patients who received XELODA.
Most common adverse reactions (>30%) were anemia, diarrhea, palmar-plantar erythrodysesthesia syndrome, hyperbilirubinemia, nausea, fatigue, and abdominal pain.
Table 4 shows the adverse reactions occurring in this pooled colorectal cancer population.
| Adverse Reaction | XELODA | Fluorouracil + Leucovorin | ||||
|---|---|---|---|---|---|---|
| (N=596) | (N=593) | |||||
| All Grades (%) | Grade 3 (%) | Grade 4 (%) | All Grades (%) | Grade 3 (%) | Grade 4 (%) |
|
| – Not observed NA = Not Applicable |
||||||
|
||||||
| Blood and Lymphatic System | ||||||
| Anemia | 80 | 2 | <1 | 79 | 1 | <1 |
| Neutropenia | 13 | 1 | 2 | 46 | 8 | 13 |
| Gastrointestinal | ||||||
| Diarrhea | 55 | 13 | 2 | 61 | 10 | 2 |
| Nausea | 43 | 4 | – | 51 | 3 | <1 |
| Abdominal pain | 35 | 9 | <1 | 31 | 5 | – |
| Vomiting | 27 | 4 | <1 | 30 | 4 | <1 |
| Stomatitis | 25 | 2 | <1 | 62 | 14 | 1 |
| Constipation | 14 | 1 | <1 | 17 | 1 | – |
| Gastrointestinal motility disorder | 10 | <1 | – | 7 | <1 | – |
| Oral discomfort | 10 | – | – | 10 | – | – |
| Skin and Subcutaneous Tissue | ||||||
| Palmar-plantar erythrodysesthesia syndrome | 54 | 17 | NA | 6 | 1 | NA |
| Dermatitis | 27 | 1 | – | 26 | 1 | – |
| Hepatobiliary | ||||||
| Hyperbilirubinemia | 48 | 18 | 5 | 17 | 3 | 3 |
| General | ||||||
| Fatigue* | 42 | 4 | – | 46 | 4 | – |
| Pyrexia | 18 | 1 | – | 21 | 2 | – |
| Edema | 15 | 1 | – | 9 | 1 | – |
| Pain | 12 | 1 | – | 10 | 1 | – |
| Metabolism and Nutrition | ||||||
| Decreased appetite | 26 | 3 | <1 | 31 | 2 | <1 |
| Respiratory Thoracic and Mediastinal | ||||||
| Dyspnea | 14 | 1 | – | 10 | <1 | 1 |
| Eye | ||||||
| Eye irritation | 13 | – | – | 10 | <1 | – |
| Nervous System | ||||||
| Peripheral sensory neuropathy | 10 | – | – | 4 | – | – |
| Headache | 10 | 1 | – | 7 | – | – |
| Musculoskeletal | ||||||
| Back pain | 10 | 2 | – | 9 | <1 | – |
Clinically relevant adverse reactions in <10% of patients are presented below:
Eye: abnormal vision
Gastrointestinal: upper gastrointestinal tract inflammatory disorders, gastrointestinal hemorrhage, ileus
General: chest pain
Infections: viral
Metabolism and Nutrition: dehydration
Musculoskeletal: arthralgia
Nervous System: dizziness (excluding vertigo), insomnia, taste disturbance
Psychiatric: mood alteration, depression
Respiratory, Thoracic, and Mediastinal: cough, pharyngeal disorder
Skin and Subcutaneous Tissue: skin discoloration, alopecia
Vascular: venous thrombosis
In Combination with Oxaliplatin
The safety of XELODA for the treatment of patients with unresectable or metastatic colorectal cancer as a component of a combination chemotherapy regimen was derived from published literature [see Clinical Studies (14.1)]. The safety of XELODA for the treatment of patients with unresectable or metastatic colorectal cancer as a component of a combination chemotherapy regimen was similar to those in patients treated with XELODA as a single agent, with the exception of an increased incidence of peripheral neuropathy.
Metastatic Breast Cancer
In Combination with Docetaxel
The safety of XELODA in combination with docetaxel was evaluated in patients with metastatic breast cancer in Study SO14999 [see Clinical Studies (14.2)]. Patients received XELODA 1,250 mg/m2 orally twice daily for the first 14 days of a 21-day cycle with docetaxel 75 mg/m2 as 1-hour intravenous infusion on day 1 of each 21-day cycle for at least 6 weeks or docetaxel 100 mg/m2 as a 1-hour intravenous infusion on day 1 of each 21-day cycle for at least 6 weeks. Among patients who received XELODA, the mean duration of treatment was 4.2 months.
Permanent discontinuation due to an adverse reaction occurred in 26% of patients who received XELODA. Dosage interruptions due to an adverse reaction occurred in 79% of patients who received XELODA and dosage reductions due to an adverse reaction occurred in 65%.
Most common adverse reactions (>30%) were diarrhea, stomatitis, palmar-plantar erythrodysesthesia syndrome, nausea, alopecia, vomiting, edema, and abdominal pain.
Table 5 summarizes the adverse reactions in Study SO14999.
| Adverse Reaction | XELODA with Docetaxel | Docetaxel | ||||
|---|---|---|---|---|---|---|
| (N=251) | (N=255) | |||||
| All Grades (%) | Grade 3 (%) | Grade 4 (%) | All Grades (%) | Grade 3 (%) | Grade 4 (%) |
|
| – Not observed NA = Not Applicable |
||||||
| Gastrointestinal | ||||||
| Diarrhea | 67 | 14 | <1 | 48 | 5 | <1 |
| Stomatitis | 67 | 17 | <1 | 43 | 5 | – |
| Nausea | 45 | 7 | – | 36 | 2 | – |
| Vomiting | 35 | 4 | 1 | 24 | 2 | – |
| Abdominal pain | 30 | 3 | <1 | 24 | 2 | – |
| Constipation | 20 | 2 | – | 18 | – | – |
| Dyspepsia | 14 | – | – | 8 | 1 | – |
| Skin and Subcutaneous Tissue | ||||||
| Palmar-plantar erythrodysesthesia syndrome | 63 | 24 | NA | 8 | 1 | NA |
| Alopecia | 41 | 6 | – | 42 | 7 | – |
| Nail disorder | 14 | 2 | – | 15 | – | – |
| Cardiac | ||||||
| Edema | 33 | <2 | – | 34 | <3 | 1 |
| General | ||||||
| Pyrexia | 28 | 2 | – | 34 | 2 | – |
| Asthenia | 26 | 4 | <1 | 25 | 6 | – |
| Fatigue | 22 | 4 | – | 27 | 6 | – |
| Weakness | 16 | 2 | – | 11 | 2 | – |
| Pain in Limb | 13 | <1 | – | 13 | 2 | – |
| Blood and Lymphatic System | ||||||
| Neutropenic fever | 16 | 3 | 13 | 21 | 5 | 16 |
| Nervous System | ||||||
| Taste disturbance | 16 | <1 | – | 14 | <1 | – |
| Headache | 15 | 3 | – | 15 | 2 | – |
| Paresthesia | 12 | <1 | – | 16 | 1 | – |
| Dizziness | 12 | – | – | 8 | <1 | – |
| Musculoskeletal and Connective Tissue | ||||||
| Arthralgia | 15 | 2 | – | 24 | 3 | – |
| Myalgia | 15 | 2 | – | 25 | 2 | – |
| Back Pain | 12 | <1 | – | 11 | 3 | – |
| Respiratory, Thoracic and Mediastinal | ||||||
| Dyspnea | 14 | 2 | <1 | 16 | 2 | – |
| Cough | 13 | 1 | – | 22 | <1 | – |
| Sore Throat | 12 | 2 | – | 11 | <1 | – |
| Metabolism and Nutrition | ||||||
| Anorexia | 13 | <1 | – | 11 | <1 | – |
| Appetite decreased | 10 | – | – | 5 | – | – |
| Dehydration | 10 | 2 | – | 7 | <1 | <1 |
| Eye | ||||||
| Lacrimation increased | 12 | – | – | 7 | <1 | – |
Clinically relevant adverse reactions in <10% of patients are presented below:
Blood and Lymphatic System: agranulocytosis, prothrombin decreased
Cardiac: supraventricular tachycardia
Eye: conjunctivitis, eye irritation
Gastrointestinal: ileus, necrotizing enterocolitis, esophageal ulcer, hemorrhagic diarrhea, dry mouth
General: chest pain (non-cardiac), lethargy, pain, influenza-like illness
Hepatobiliary: jaundice, abnormal liver function tests, hepatic failure, hepatic coma, hepatotoxicity
Immune System: hypersensitivity
Infection: hypoesthesia, neutropenic sepsis, sepsis, bronchopneumonia, oral candidiasis, urinary tract infection
Metabolism and Nutrition: weight decreased
Musculoskeletal and Connective Tissue: bone pain
Nervous System: insomnia, peripheral neuropathy, ataxia, syncope, taste loss, polyneuropathy, migraine
Psychiatric: depression
Renal and Urinary: renal failure
Respiratory, Thoracic and Mediastinal: upper respiratory tract infection, pleural effusion, epistaxis, rhinorrhea
Skin and Subcutaneous Tissue: pruritis, rash erythematous, dermatitis, nail discoloration, onycholysis
Vascular: lymphedema, hypotension, venous phlebitis and thrombophlebitis, postural hypotension, flushing
Table 6 summarizes the laboratory abnormalities in this trial.
| Laboratory Abnormality | XELODA with Docetaxel | Docetaxel | ||||
|---|---|---|---|---|---|---|
| (N=251) | (N=255) | |||||
| All Grades (%) | Grade 3 (%) | Grade 4 (%) | All Grades (%) | Grade 3 (%) | Grade 4 (%) |
|
| Hematologic | ||||||
| Lymphocytopenia | 99 | 48 | 41 | 98 | 44 | 40 |
| Leukopenia | 91 | 37 | 24 | 88 | 42 | 33 |
| Neutropenia | 86 | 20 | 49 | 87 | 10 | 66 |
| Anemia | 80 | 7 | 3 | 83 | 5 | <1 |
| Thrombocytopenia | 41 | 2 | 1 | 23 | 1 | 2 |
| Hepatobiliary | ||||||
| Hyperbilirubinemia | 20 | 7 | 2 | 6 | 2 | 2 |
Single Agent
The safety of XELODA as a single agent was evaluated in patients with metastatic breast cancer in Study SO14697 [see Clinical Studies (14.2)]. Patients received XELODA 1,250 mg/m2 orally twice daily for the first 14 days of a 21-day cycle. The mean duration of treatment was 3.7 months.
Permanent discontinuation due to an adverse reaction or intercurrent illness occurred in 8% of patients.
Most common adverse reactions (>30%) were lymphopenia, anemia, diarrhea, hand-and-foot syndrome, nausea, fatigue, vomiting, and dermatitis.
Table 7 summarizes the adverse reactions in Study SO14697.
| Adverse Reaction | XELODA (n=162) |
||
|---|---|---|---|
| All Grades (%) | Grade 3 (%) | Grade 4 (%) |
|
| – = Not observed NA = Not Applicable |
|||
| Blood and Lymphatic System | |||
| Lymphopenia | 94 | 44 | 15 |
| Anemia | 72 | 3 | 1 |
| Neutropenia | 26 | 2 | 2 |
| Thrombocytopenia | 24 | 3 | 1 |
| Gastrointestinal | |||
| Diarrhea | 57 | 12 | 3 |
| Nausea | 53 | 4 | – |
| Vomiting | 37 | 4 | – |
| Stomatitis | 24 | 7 | – |
| Abdominal pain | 20 | 4 | – |
| Constipation | 15 | 1 | – |
| Skin and Subcutaneous Tissue | |||
| Hand-and-foot syndrome | 57 | 11 | NA |
| Dermatitis | 37 | 1 | – |
| General | |||
| Fatigue | 41 | 8 | – |
| Pyrexia | 12 | 1 | – |
| Metabolism and Nutrition | |||
| Anorexia | 23 | 3 | – |
| Hepatobiliary | |||
| Hyperbilirubinemia | 22 | 9 | 2 |
| Nervous System | |||
| Paresthesia | 21 | 1 | – |
| Eye | |||
| Eye irritation | 15 | – | – |
Pooled Safety Population
Clinically relevant adverse reactions in <10% of patients who received XELODA as a single agent are presented below.
Blood & Lymphatic System: leukopenia, coagulation disorder, bone marrow depression, pancytopenia
Cardiac: tachycardia, bradycardia, atrial fibrillation, myocarditis, edema
Ear: vertigo
Eye: conjunctivitis
Gastrointestinal: abdominal distension, dysphagia, proctalgia, gastric ulcer, ileus, gastroenteritis, dyspepsia
General: chest pain, influenza-like illness, hot flushes, pain, thirst, fibrosis, hemorrhage, edema, pain in limb
Hepatobiliary: hepatic fibrosis, hepatitis, cholestatic hepatitis, abnormal liver function tests
Immune System: drug hypersensitivity
Infections: bronchitis, pneumonia, keratoconjunctivitis, sepsis, fungal infections
Metabolism and Nutrition: cachexia, hypertriglyceridemia, hypokalemia, hypomagnesemia, dehydration
Musculoskeletal and Connective Tissue: myalgia, arthritis, muscle weakness
Nervous System: insomnia, ataxia, tremor, dysphasia, encephalopathy, dysarthria, impaired balance, headache, dizziness
Psychiatric: depression, confusion
Renal and Urinary: renal impairment
Respiratory, Mediastinal and Thoracic: cough, epistaxis, respiratory distress, dyspnea
Skin and Subcutaneous Tissue: nail disorder, sweating increased, photosensitivity reaction, skin ulceration, pruritus, radiation recall syndrome
Vascular: hypotension, hypertension, lymphedema, pulmonary embolism
Unresectable or Metastatic Gastric, Esophageal, or Gastroesophageal Junction Cancer
The safety of XELODA for the treatment of adults with unresectable or metastatic gastric, esophageal, or gastroesophageal junction cancer as a component of a combination chemotherapy regimen was derived from published literature [see Clinical Studies (14.3)]. The safety of XELODA for the treatment of adults with unresectable or metastatic gastric, esophageal, or gastroesophageal junction cancer as a component of a combination chemotherapy regimen was consistent with the known safety profile of XELODA.
The safety of XELODA for the treatment of patients with HER2-overexpressing metastatic gastric or gastroesophageal junction adenocarcinoma who have not received prior treatment for metastatic disease as a component of a combination regimen was derived from the published literature [see Clinical Studies (14.3)]. The safety of XELODA for the treatment of patients with HER2-overexpressing metastatic gastric or gastroesophageal junction adenocarcinoma was consistent with the known safety profile of XELODA.
Pancreatic Cancer
The safety of XELODA for the adjuvant treatment of adults with pancreatic adenocarcinoma as a component of a combination chemotherapy regimen was derived from the published literature [see Clinical Studies (14.4)]. The safety of XELODA for the adjuvant treatment of adults with pancreatic adenocarcinoma as a component of a combination chemotherapy regimen was consistent with the known safety profile of XELODA.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of XELODA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Eye: lacrimal duct stenosis, corneal disorders including keratitis
Hepatobiliary: hepatic failure
Immune System Disorders: angioedema
Nervous System: toxic leukoencephalopathy
Renal & Urinary: acute renal failure secondary to dehydration including fatal outcome
Skin & Subcutaneous Tissue: cutaneous lupus erythematosus, severe skin reactions such as Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis (TEN), persistent or severe PPES can eventually lead to loss of fingerprints
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on XELODA
Allopurinol
Concomitant use with allopurinol may decrease concentration of capecitabine's active metabolites [see Clinical Pharmacology (12.3)], which may decrease efficacy. Avoid concomitant use of allopurinol with XELODA.
Leucovorin
The concentration of fluorouracil is increased and its toxicity may be enhanced by leucovorin, folic acid, or folate analog products. Deaths from severe enterocolitis, diarrhea, and dehydration have been reported in elderly patients receiving weekly leucovorin and fluorouracil.
Instruct patients not to take products containing folic acid or folate analog products unless directed to do so by their healthcare provider.
7.2 Effect of Xeloda on Other Drugs
CYP2C9 Substrates
XELODA increased exposure of CYP2C9 substrates [see Clinical Pharmacology (12.3)], which may increase the risk of adverse reactions related to these substrates. Closely monitor for adverse reactions of CYP2C9 substrates where minimal concentration changes may lead to serious adverse reactions when used concomitantly with XELODA (e.g., anticoagulants, antidiabetic drugs).
Vitamin K Antagonists
XELODA increases exposure of vitamin K antagonist [see Clinical Pharmacology (12.3)], which may alter coagulation parameters and/or bleeding and could result in death [see Warning and Precautions (5.1)]. These events may occur within days of treatment initiation and up to 1 month after discontinuation of XELODA.
Monitor INR more frequently and refer to the prescribing information of oral vitamin K antagonist for dosage adjustment, as appropriate, when XELODA is used concomitantly with vitamin K antagonist.
Phenytoin
XELODA may increases exposure of phenytoin, which may increase the risk of adverse reactions related to phenytoin. Closely monitor phenytoin levels and refer to the prescribing information of phenytoin for dosage adjustment, as appropriate, when XELODA is used concomitantly with phenytoin.
7.3 Nephrotoxic Drugs
Due of the additive pharmacologic effect, concomitant use of XELODA with other drugs known to cause renal toxicity may increase the risk of renal toxicity [see Warnings and Precautions (5.6)]. Closely monitor for signs of renal toxicity when XELODA is used concomitantly with nephrotoxic drugs (e.g. platinum salts, irinotecan, methotrexate, intravenous bisphosphonates).
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings in animal reproduction studies and its mechanism of action [see Clinical Pharmacology (12.1)], XELODA can cause fetal harm when administered to a pregnant woman. Available human data with XELODA use in pregnant women is not sufficient to inform the drug-associated risk. In animal reproduction studies, administration of capecitabine to pregnant animals during the period of organogenesis caused embryolethality and teratogenicity in mice and embryolethality in monkeys at 0.2 and 0.6 times the exposure (AUC) in patients receiving the recommended dose of 1,250 mg/m2 twice daily, respectively (see Data). Advise pregnant women of the potential risk to a fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
Oral administration of capecitabine to pregnant mice during the period of organogenesis at a dose of 198 mg/kg/day caused malformations and embryo lethality. In separate pharmacokinetic studies, this dose in mice produced 5'-DFUR AUC values that were approximately 0.2 times the AUC values in patients administered the recommended daily dose. Malformations in mice included cleft palate, anophthalmia, microphthalmia, oligodactyly, polydactyly, syndactyly, kinky tail and dilation of cerebral ventricles. Oral administration of capecitabine to pregnant monkeys during the period of organogenesis at a dose of 90 mg/kg/day, caused fetal lethality. This dose produced 5'-DFUR AUC values that were approximately 0.6 times the AUC values in patients administered the recommended daily dose.
8.2 Lactation
Risk Summary
There is no information regarding the presence of capecitabine or its metabolites in human milk, or on its effects on milk production or the breastfed child. Capecitabine metabolites were present in the milk of lactating mice (see Data). Because of the potential for serious adverse reactions in a breastfed child, advise women not to breastfeed during treatment with XELODA and for 1 week after the last dose.
8.3 Females and Males of Reproductive Potential
XELODA can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify pregnancy status in females of reproductive potential prior to initiating XELODA.
Contraception
Females
Advise females of reproductive potential to use effective contraception during treatment with XELODA and for 6 months after the last dose.
Males
Based on genotoxicity findings, advise males with female partners of reproductive potential to use effective contraception during treatment with XELODA and for 3 months after the last dose [see Nonclinical Toxicology (13.1)].
Infertility
Based on animal studies, XELODA may impair fertility in females and males of reproductive potential [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of XELODA in pediatric patients have not been established.
Safety and effectiveness were assessed, but not established in two single arm studies in 56 pediatric patients aged 3 months to <17 years with newly diagnosed gliomas. In both trials, pediatric patients received an investigational pediatric formulation of capecitabine concomitantly with and following completion of radiation therapy (total dose of 5580 cGy in 180 cGy fractions). The relative bioavailability of the investigational formulation to XELODA was similar.
The adverse reaction profile was consistent with that of adults, with the exception of laboratory abnormalities which occurred more commonly in pediatric patients. The most frequently reported laboratory abnormalities (per-patient incidence ≥ 40%) were increased ALT (75%), lymphocytopenia (73%), hypokalemia (68%), thrombocytopenia (57%), hypoalbuminemia (55%), neutropenia (50%), low hematocrit (50%), hypocalcemia (48%), hypophosphatemia (45%) and hyponatremia (45%).
8.5 Geriatric Use
Of 7938 patients with colorectal cancer who were treated with XELODA, 33% were older than 65 years. Of the 4536 patients with metastatic breast cancer who were treated with XELODA, 18% were older than 65 years.
Of 1951 patients with gastric, esophageal, or gastrointestinal junction cancer who were treated with XELODA, 26% were older than 65 years.
Of 364 patients with pancreatic cancer who received adjuvant treatment with XELODA, 47% were 65 years or older.
No overall differences in efficacy were observed comparing older versus younger patients with colorectal cancer, gastric, esophageal or gastrointestinal junction cancer, or pancreatic cancer using the approved recommended dosages and treatment regimens.
Older patients experience increased gastrointestinal toxicity due to XELODA compared to younger patients. Deaths from severe enterocolitis, diarrhea, and dehydration have been reported in elderly patients receiving weekly leucovorin and fluorouracil [see Drug Interactions (7.1)].
8.6 Renal Impairment
The exposure of capecitabine and its inactive metabolites (5-DFUR and FBAL) increases in patients with CLcr <50 mL/min as determined by Cockcroft-Gault [see Clinical Pharmacology (12.3)]. Reduce the dosage for patients with CLcr of 30 to 50 mL/min [see Dosage and Administration (2.6)]. There is limited experience with XELODA in patients with CLcr <30 mL/min, and a dosage has not been established in those patients. If no treatment alternative exists, XELODA could be administered to such patients on an individual basis applying a reduced starting dose, close monitoring of a patient's clinical and biochemical data and dose modifications guided by observed adverse reactions.
8.7 Hepatic Impairment
The exposure of capecitabine increases in patients with mild to moderate hepatic impairment. The effect of severe hepatic impairment on the safety and pharmacokinetics of XELODA is unknown [see Clinical Pharmacology (12.3)]. Monitor patients with hepatic impairment more frequently for adverse reactions.
10 OVERDOSAGE
Administer uridine triacetate within 96 hours for management of XELODA overdose.
Although no clinical experience using dialysis as a treatment for XELODA overdose has been reported, dialysis may be of benefit in reducing circulating concentrations of 5'-DFUR, a low–molecular-weight metabolite of the parent compound.
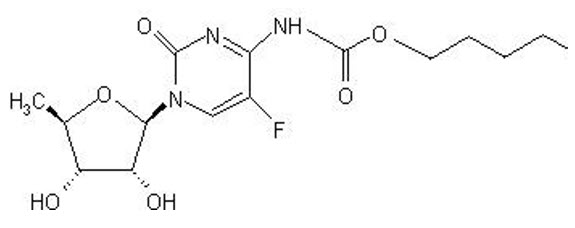
11 DESCRIPTION
Capecitabine is a nucleoside metabolic inhibitor. The chemical name is 5'-deoxy-5-fluoro-N-[(pentyloxy) carbonyl]-cytidine and has a molecular formula of C15H22FN3O6 and a molecular weight of 359.35. Capecitabine has the following structural formula:
Capecitabine is a white to off-white crystalline powder with an aqueous solubility of 26 mg/mL at 20°C.
XELODA (capecitabine) is supplied as biconvex, oblong film-coated tablets for oral use. Each light peach-colored tablet contains 150 mg capecitabine and each peach-colored tablet contains 500 mg capecitabine. The inactive ingredients in XELODA include: anhydrous lactose, croscarmellose sodium, hydroxypropyl methylcellulose, microcrystalline cellulose, magnesium stearate and purified water. The peach or light peach film coating contains hydroxypropyl methylcellulose, talc, titanium dioxide, and synthetic yellow and red iron oxides.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Capecitabine is metabolized to fluorouracil in vivo. Both normal and tumor cells metabolize fluorouracil to 5-fluoro-2'-deoxyuridine monophosphate (FdUMP) and 5-fluorouridine triphosphate (FUTP). These metabolites cause cell injury by two different mechanisms. First, FdUMP and the folate cofactor, N5-10-methylenetetrahydrofolate, bind to thymidylate synthase (TS) to form a covalently bound ternary complex. This binding inhibits the formation of thymidylate from 2'-deoxyuridylate. Thymidylate is the necessary precursor of thymidine triphosphate, which is essential for the synthesis of DNA, so that a deficiency of this compound can inhibit cell division. Second, nuclear transcriptional enzymes can mistakenly incorporate FUTP in place of uridine triphosphate (UTP) during the synthesis of RNA. This metabolic error can interfere with RNA processing and protein synthesis.
12.2 Pharmacodynamics
Population-based exposure-effect analyses demonstrated a positive association between AUC of fluorouracil and grade 3-4 hyperbilirubinemia.
12.3 Pharmacokinetics
The AUC of capecitabine and its metabolite 5'-DFCR increases proportionally over a dosage range of 500 mg/m2/day to 3,500 mg/m2/day (0.2 to 1.4 times the approved recommended dosage). The AUC of capecitabine's metabolites 5'-DFUR and fluorouracil increased greater than proportional to the dose. The interpatient variability in the Cmax and AUC of fluorouracil was greater than 85%.
Absorption
Following oral administration of XELODA 1,255 mg/m2 orally twice daily (the recommended dosage when used as single agent), the median Tmax of capecitabine and its metabolite fluorouracil was approximately 1.5 hours and 2 hours, respectively.
Effect of Food
Following administration of a meal (breakfast medium-rich in fat and carbohydrates), the mean Cmax and AUC0-INF of capecitabine was decreased by 60% and 34%, respectively. The mean Cmax and AUC0-INF of fluorouracil were also decreased by 37 % and 12%, respectively. The Tmax of both capecitabine and fluorouracil was delayed by 1.5 hours.
Distribution
Plasma protein binding of capecitabine and its metabolites is less than 60% and is not concentration-dependent. Capecitabine was primarily bound to human albumin (approximately 35%).
Following oral administration of XELODA 7 days before surgery in patients with colorectal cancer, the median ratio of concentration for the active metabolite fluorouracil in colorectal tumors to adjacent tissues was 2.9 (range: 0.9 to 8.0).
Elimination
The elimination half-lives of capecitabine and fluorouracil were approximately 0.75 hour.
Metabolism
Capecitabine undergoes metabolism by carboxylesterase and is hydrolyzed to 5'-DFCR. 5'-DFCR is subsequently converted to 5'-DFUR by cytidine deaminase. 5'-DFUR is then hydrolized by thymidine phosphorylase (dThdPase) enzymes to the active metabolite fluorouracil.
Fluorouracil is subsequently metabolized by dihydropyrimidine dehydrogenase to 5-fluoro-5, 6-dihydro-fluorouracil (FUH2). The pyrimidine ring of FUH2 is cleaved by dihydropyrimidinase to yield 5-fluoro-ureido-propionic acid (FUPA). Finally, FUPA is cleaved by β-ureido-propionase to α-fluoro-β-alanine (FBAL).
Specific Populations
Following therapeutic doses of XELODA, no clinically meaningful difference in the pharmacokinetics of 5'-DFUR, fluorouracil or FBAL were observed based on sex (202 females and 303 males) and race (455 White, 22 Black, and 28 Other). No clinically meaningful difference on the pharmacokinetics of 5'-DFUR and fluorouracil were observed based on age (range: 27 to 86 years); however, the AUC of FBAL increased by 15% following a 20% increase in age.
Racial or Ethnic Groups
Following administration of XELODA 825 mg/m2 orally twice daily for 14 days (0.66 times the recommended dosage), the Cmax and AUC of capecitabine decreased by 36% and 24%, respectively in Japanese patients (n=18) compared to White patients (n=22). The Cmax and AUC of FBAL decreased by approximately 25% and 34%, respectively in Japanese patients compared to White patients; however, the clinical significance of these differences is unknown. No clinically significant differences in the pharmacokinetics of 5'-DFCR, 5'-DFUR or fluorouracil were observed.
Patients with Renal Impairment
| Renal Impairment * | Changes in AUC † | |||
|---|---|---|---|---|
| Capecitabine | 5'-DFUR‡ | FBAL‡ | 5-FU | |
| CLcr= Creatine Clearance, AUC= Area under the plasma concentration-time curve | ||||
| CLcr 30 to 50 mL/min | Increased by 25% | Increased by 42% | Increased by 85% | No relevant change |
| CLcr <30 mL/min | Increased by 25% | Increased by 71% | Increased by 258% | Increased by 24% |
Patients with Hepatic Impairment
AUC0-INF and Cmax of capecitabine's active principle, fluorouracil, were not affected in patients with mild or moderate hepatic impairment compared to patients with normal hepatic function. The AUC0-INF and Cmax of capecitabine increased by 60%. The effect of severe hepatic impairment on the pharmacokinetics of capecitabine and its metabolites are unknown.
Drug Interaction Studies
Clinical Studies
Effect of Capecitabine on Warfarin: In four patients with cancer, chronic administration of XELODA 1,250 mg/m2 twice daily with a single dose of warfarin 20 mg increased the mean AUC of S-warfarin by 57% and decreased its clearance by 37%. Baseline corrected AUC of INR in these 4 patients increased by 2.8-fold, and the maximum observed mean INR value was increased by 91%.
Effect of Capecitabine on Celecoxib: Concomitant administration of multiple doses of capecitabine (XELODA 1,000 mg/m2 twice daily for 14 days) increased celecoxib (sensitive CYP2C9 substrate) AUC by 28%, Cmax by 24% and Ctrough by 30%.
Effect of Antacids on Capecitabine: When an aluminum hydroxide- and magnesium hydroxide-containing antacid was administered immediately after a XELODA dose of 1,250 mg/m2 in patients with cancer, AUC and Cmax increased by 16% and 35%, respectively, for capecitabine and by 18% and 22%, respectively, for 5'-DFCR. No effect was observed on the other three major metabolites (5'-DFUR, fluorouracil, FBAL) of XELODA.
12.5 Pharmacogenomics
The DPYD gene encodes the enzyme DPD, which is responsible for the catabolism of >80% of fluorouracil. Approximately 3-5% of White populations have partial DPD deficiency and 0.2% of White populations have complete DPD deficiency, which may be due to certain genetic no function or decreased function variants in DPYD resulting in partial to complete or near complete absence of enzyme activity. DPD deficiency is estimated to be more prevalent in Black or African American populations compared to White populations. Insufficient information is available to estimate the prevalence of DPD deficiency in other populations.
Patients who are homozygous or compound heterozygous for no function DPYD variants (i.e., carry two no function DPYD variants) or are compound heterozygous for a no function DPYD variant plus a decreased function DPYD variant have complete DPD deficiency and are at increased risk for acute early-onset of toxicity and serious life-threatening, or fatal adverse reactions due to increased systemic exposure to XELODA. Partial DPD deficiency can result from the presence of either two decreased function DPYD variants or one normal function plus either a decreased function or a no function DPYD variant. Patients with partial DPD deficiency may also be at an increased risk for toxicity from XELODA.
Four DPYD variants have been associated with impaired DPD activity in White populations, especially when present as homozygous or compound heterozygous variants: c.1905+1G>A (DPYD *2A), c.1679T>G (DPYD *13), c.2846A>T, and c.1129-5923C>G (Haplotype B3). DPYD*2A and DPYD*13 are no function variants, and c.2846A>T and c.1129-5923C>G are decreased function variants. The decreased function DPYD variant c.557A>G is observed in individuals of African ancestry. This is not a complete listing of all DPYD variants that may result in DPD deficiency [see Warnings and Precautions (5.2)].
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Adequate studies investigating the carcinogenic potential of capecitabine have not been conducted. Capecitabine was not mutagenic in vitro to bacteria (Ames test) or mammalian cells (Chinese hamster V79/HPRT gene mutation assay). Capecitabine was clastogenic in vitro to human peripheral blood lymphocytes but not clastogenic in vivo to mouse bone marrow (micronucleus test). Fluorouracil causes mutations in bacteria and yeast. Fluorouracil also causes chromosomal abnormalities in the mouse micronucleus test in vivo.
In studies of fertility and general reproductive performance in female mice, oral capecitabine doses of 760 mg/kg/day (about 2,300 mg/m2/day) disturbed estrus and consequently caused a decrease in fertility. In mice that became pregnant, no fetuses survived this dose. The disturbance in estrus was reversible. In males, this dose caused degenerative changes in the testes, including decreases in the number of spermatocytes and spermatids. In separate pharmacokinetic studies, this dose in mice produced 5'-DFUR AUC values about 0.7 times the corresponding values in patients administered the recommended daily dose.
14 CLINICAL STUDIES
14.1 Colorectal Cancer
Adjuvant Treatment of Colon Cancer
Single Agent
The efficacy of XELODA was evaluated in X-ACT (NCT00009737), a multicenter, randomized, controlled clinical trial. Eligible patients were between 18 and 75 years of age with histologically-confirmed Dukes' Stage C colon cancer with at least one positive lymph node and to have undergone (within 8 weeks prior to randomization) complete resection of the primary tumor without macroscopic or microscopic evidence of remaining tumor. Patients were also required to have no prior cytotoxic chemotherapy or immunotherapy (except steroids) and have an ECOG performance status of 0 or 1 (KPS ≥70%), ANC≥1.5×109/L, platelets ≥100×109/L, serum creatinine ≤1.5 ULN, total bilirubin ≤1.5 ULN, AST/ALT ≤2.5 ULN and CEA within normal limits at time of randomization.
Patients (n=1987) were randomized to XELODA 1,250 mg/m2 orally twice daily for the first 14 days of a 21-day cycle for a total of 8 cycles or fluorouracil 425 mg/m2 and leucovorin 20 mg/m2 intravenously on days 1 to 5 of each 28-day cycle for a total of 6 cycles. The XELODA dose was reduced in patients with baseline CLcr of 30 to 50 mL/min. The major efficacy outcome measure was disease-free survival (DFS).
The baseline demographics are shown in Table 9. The baseline characteristics were well-balanced between arms.
| XELODA (N=1004) | Fluorouracil + Leucovorin (N=983) |
|
|---|---|---|
| Age (median, years) | 62 | 63 |
| Range | (25-80) | (22-82) |
| Sex | ||
| Male, % | 54 | 54 |
| Female, % | 46 | 46 |
| ECOG Performance Status | ||
| 0, % | 85 | 85 |
| 1, % | 15 | 15 |
| Staging – Primary Tumor | ||
| PT1, % | 1 | 0.6 |
| PT2, % | 9 | 9 |
| PT3, % | 76 | 76 |
| PT4, % | 14 | 0 |
| Other, % | 0.1 | 14 |
| Staging – Lymph Node | ||
| pN1, % | 69 | 71 |
| pN2, % | 30 | 29 |
| Other, % | 0.4 | 0.1 |
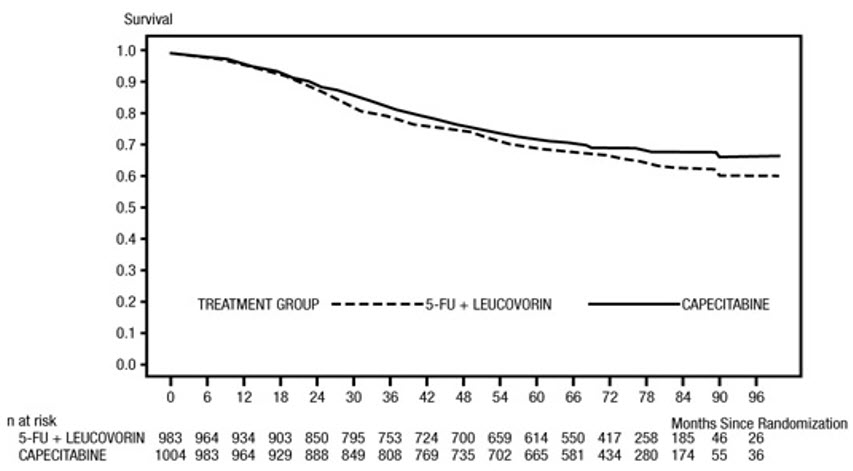
Efficacy results are summarized in Table 10 and Figures 1 and 2. The median follow-up at the time of the analysis was 6.9 years. Because the upper 2-sided 95% confidence limit of hazard ratio for DFS was less than 1.20, XELODA was non-inferior to fluorouracil + leucovorin. The choice of the non-inferiority margin of 1.20 corresponds to the retention of approximately 75% of the fluorouracil + leucovorin effect on DFS. The hazard ratio for XELODA compared to fluorouracil + leucovorin with respect to overall survival was 0.86 (95% CI 0.74, 1.01). The 5-year overall survival rates were 71% for XELODA and 68% for fluorouracil + leucovorin.
| Efficacy Parameters | XELODA (N=1004) | Fluorouracil + Leucovorin (N=983) |
|---|---|---|
| 5-year Disease-free Survival Rate† | 59% | 55% |
| Hazard Ratio | 0.88 | |
| (95% CI) | (0.77, 1.01) | |
| p-value‡ | p = 0.068 | |
Figure 1 Kaplan-Meier Estimates of Disease-Free Survival in X-ACT (All Randomized Population)
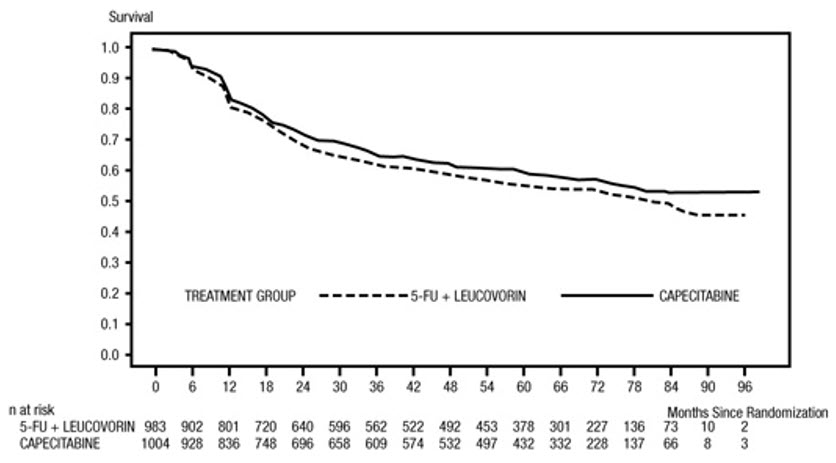
Figure 2 Kaplan-Meier Estimates of Overall Survival in X-ACT (All Randomized Population)
In Combination with Oxaliplatin-Containing Regimens
The efficacy of XELODA in combination with oxaliplatin for the adjuvant treatment of patients with Stage III colon cancer as a component of a combination chemotherapy regimen was derived from studies in the published literature, including NO16968 [NCT00069121], a multicenter, open-label, randomized trial, where the major efficacy outcome measure was disease free survival.
Perioperative Treatment of Rectal Cancer
The efficacy of XELODA for the perioperative treatment of adults with locally advanced rectal cancer as a component of chemoradiotherapy was derived from studies in the published literature, including Rektum-III [NCT01500993], a randomized, open-label, multicenter, non-inferiority trial, where the major efficacy outcome measure was overall survival.
Metastatic Colorectal Cancer
The efficacy of XELODA as a single agent was evaluated in two open-label, multicenter, randomized, controlled clinical trials (Study SO14695 and Study SO14796). Eligible patients received first-line treatment for metastatic colorectal cancer. Patients were randomized to XELODA 1,250 mg/m2 twice daily for first 14 days of a 21-day cycle or leucovorin 20 mg/m2 intravenously followed by fluorouracil 425 mg/m2 as an intravenous bolus on days 1 to 5 of each 28-day cycle.
The efficacy outcome measures were overall survival, time to progression and response rate (complete plus partial responses). Responses were defined by the World Health Organization criteria and submitted to a blinded independent review committee (IRC). Differences in assessments between the investigator and IRC were reconciled by the sponsor, blinded to treatment arm, according to a specified algorithm. Survival was assessed based on a non-inferiority analysis.
The baseline demographics are shown in Table 11.
| Study SO14695 | Study SO14796 | |||
|---|---|---|---|---|
| XELODA (N=302) | Fluorouracil + Leucovorin (N=303) | XELODA (N=301) | Fluorouracil + Leucovorin (N=301) |
|
| Age (median, years) | 64 | 63 | 64 | 64 |
| Range | (23-86) | (24-87) | (29-84) | (36-86) |
| Sex | ||||
| Male, % | 60 | 65 | 57 | 57 |
| Female, % | 40 | 35 | 43 | 43 |
| Karnofsky PS (median) | 90 | 90 | 90 | 90 |
| Range | (70-100) | (70-100) | (70-100) | (70-100) |
| Colon, % | 74 | 77 | 66 | 65 |
| Rectum, % | 26 | 23 | 34 | 35 |
| Prior radiation therapy, % | 17 | 21 | 14 | 14 |
| Prior adjuvant fluorouracil, % | 28 | 36 | 19 | 14 |
Efficacy results for Study SO14695 and Study SO14796 are shown in Table 12 and Table 13.
| XELODA (N=302) | Fluorouracil + Leucovorin (N=303) |
|
|---|---|---|
| Overall Response Rate | ||
| % (95% CI) | 21 (16, 26) | 11 (8, 15) |
| p-value | 0.0014 | |
| Time to Progression | ||
| Median, months (95% CI) | 4.2 (3.9, 4.5) | 4.3 (3.4, 5.0) |
| Hazard Ratio | 0.99 | |
| 95% CI | (0.84, 1.17) | |
| Overall Survival | ||
| Median, months (95% CI) | 12.5 (10.5, 14.3) | 13.4 (12.0, 14.7) |
| Hazard Ratio | 1.00 | |
| 95% CI | (0.84, 1.18) | |
| XELODA (N=301) | Fluorouracil + Leucovorin (N=301) |
|
|---|---|---|
| Overall Response Rate | ||
| % (95% CI) | 21 (16, 26) | 14 (10, 18) |
| p-value | 0.027 | |
| Time to Progression | ||
| Median, months (95% CI) | 4.5 (4.2, 5.5) | 4.3 (3.4, 5.1) |
| Hazard Ratio | 0.97 | |
| 95% CI | (0.82, 1.14) | |
| Overall Survival | ||
| Median, months (95% CI) | 13.3 (12.1, 14.8) | 12.1 (11.1,14.1) |
| Hazard Ratio | 0.92 | |
| 95% CI | (0.78, 1.09) | |
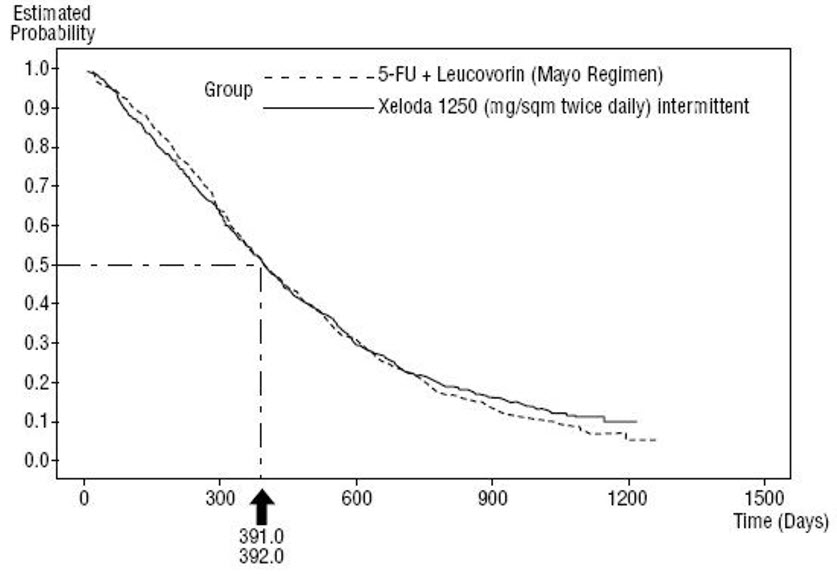
Efficacy results of the pooled population from Study SO14695 and Study SO14796 are shown in Figure 3. Statistical analyses were performed to determine the percent of the survival effect of fluorouracil + leucovorin that was retained by XELODA. The estimate of the survival effect of fluorouracil + leucovorin was derived from a meta-analysis of ten randomized studies from the published literature comparing fluorouracil to regimens of fluorouracil + leucovorin that were similar to the control arms used in these Studies SO14695 and SO14796. The method for comparing the treatments was to examine the worst case (95% confidence upper bound) for the difference between fluorouracil + leucovorin and XELODA, and to show that loss of more than 50% of the fluorouracil + leucovorin survival effect was ruled out. It was demonstrated that the percent of the survival effect of fluorouracil + leucovorin maintained was at least 61% for Study SO14796 and 10% for Study SO14695. The pooled result is consistent with a retention of at least 50% of the effect of fluorouracil + leucovorin. It should be noted that these values for preserved effect are based on the upper bound of the fluorouracil + leucovorin vs XELODA difference.
Figure 3 Kaplan-Meier Curve for Overall Survival of Pooled Data (Studies SO14695 and SO14796)
In Combination with Oxaliplatin
The efficacy of XELODA for the treatment of patients with unresectable or metastatic colorectal cancer as a component of a combination chemotherapy regimen was derived from studies in the published literature, including NO16966 [NCT00069095], a randomized, non-inferiority, 2×2 factorial trial, where the major efficacy outcome measure was progression free survival.
14.2 Metastatic Breast Cancer
In Combination With Docetaxel
The efficacy of XELODA in combination with docetaxel was evaluated in an open-label, multicenter, randomized trial (Study SO14999). Eligible patients had metastatic breast cancer resistant to, or recurring during or after an anthracycline-containing therapy, or relapsing during or recurring within 2 years of completing an anthracycline-containing adjuvant therapy were enrolled. Patients were randomized to XELODA 1,250 mg/m2 twice daily for the first 14 days of a 21-day cycle and docetaxel 75 mg/m2 as a 1-hour intravenous infusion on day 1 of day of a 21-day cycle or docetaxel 100 mg/m2 as a 1-hour intravenous infusion on day 1 of a 21-day cycle. The efficacy outcome measures were time to disease progression, overall survival, and response rate.
Patient demographics are provided in Table 14.
| XELODA + Docetaxel (N=255) | Docetaxel (N=256) |
|
|---|---|---|
|
||
| Age (median, years) | 52 | 51 |
| Karnofsky Performance Status (median) | 90 | 90 |
| Site of Disease | ||
| Lymph nodes, % | 47 | 49 |
| Liver, % | 45 | 48 |
| Bone, % | 42 | 46 |
| Lung, % | 37 | 39 |
| Skin, % | 29 | 29 |
| Prior Chemotherapy | ||
| Anthracycline*, % | 100 | 100 |
| Fluorouracil, % | 77 | 74 |
| Paclitaxel, % | 10 | 9 |
| Resistance to an Anthracycline | ||
| No resistance, % | 7 | 7 |
| Progression on anthracycline therapy, % | 26 | 29 |
| Stable disease after 4 cycles of anthracycline therapy, % | 16 | 16 |
| Relapsed within 2 years of completion of anthracycline-adjuvant therapy, % | 31 | 29 |
| Experienced a brief response to anthracycline therapy, with subsequent progression while on therapy or within 12 months after last dose, % | 20 | 20 |
| No. of Prior Chemotherapy Regimens for Treatment of Metastatic Disease | ||
| 0, % | 35 | 31 |
| 1, % | 48 | 53 |
| 2, % | 17 | 15 |
| 3, % | 0 | 1 |
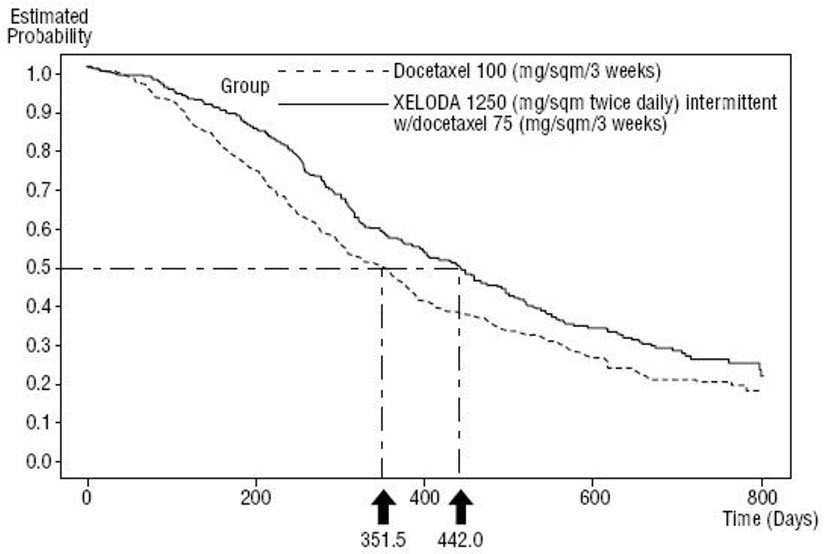
Efficacy results are shown in Table 15, Figure 4 and Figure 5.
| Efficacy Parameter | XELODA + Docetaxel (N=255) | Docetaxel (N=256) |
|---|---|---|
|
||
| Time to Disease Progression | ||
| Median, months | 6.1 | 4.2 |
| 95% CI | (5.4, 6.5) | (3.5, 4.5) |
| Hazard Ratio | 0.643 | |
| p-value | 0.0001 | |
| Overall Survival | ||
| Median, months | 14.5 | 11.6 |
| 95% CI | (12.3, 16.3) | (9.8, 12.7) |
| Hazard Ratio | 0.775 | |
| p-value | 0.0126 | |
| Response Rate* | 32% | 22% |
Figure 4 Kaplan-Meier Estimates for Time to Disease Progression in Metastatic Breast Cancer (Study SO14999)
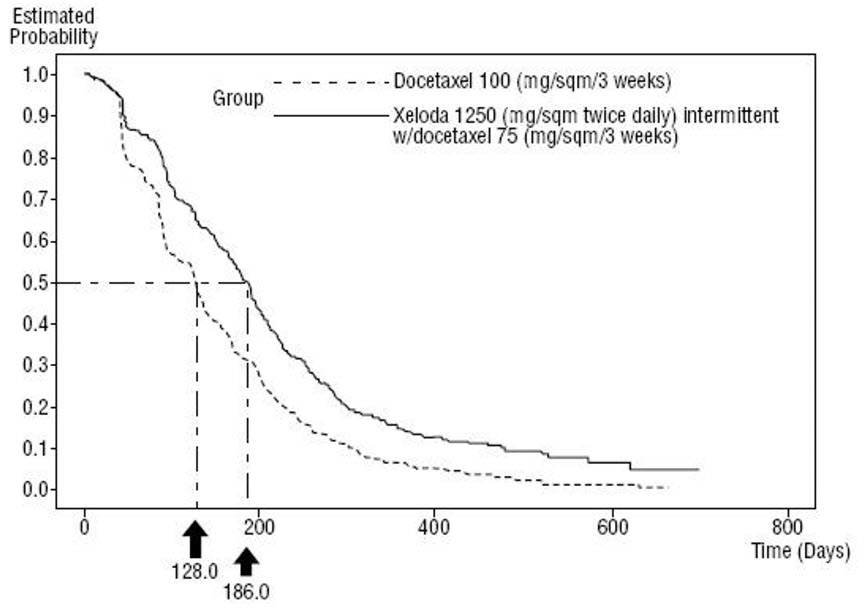
Figure 5 Kaplan-Meier Estimates of Survival in Metastatic Breast Cancer (Study SO14999)
Single Agent
The efficacy of XELODA as a single agent was evaluated in an open-label single-arm trial (Study SO14697). Eligible patients had metastatic breast cancer resistant to both paclitaxel and an anthracycline-containing chemotherapy regimen or resistant to paclitaxel and for whom further anthracycline therapy is not indicated (e.g., patients who have received cumulative doses of 400 mg/m2 of doxorubicin or doxorubicin equivalents). Resistance was defined as progressive disease while on treatment, with or without an initial response, or relapse within 6 months of completing treatment with an anthracycline-containing adjuvant chemotherapy regimen. Patients received XELODA 1,255 mg/m2 orally twice daily for first 14-days of a 21-day treatment cycle. The major efficacy outcome measure was tumor response rate in patients with measurable disease, with response defined as a ≥50% decrease in sum of the products of the perpendicular diameters of bidimensionally measurable disease for at least 1 month.
The baseline demographics are shown in Table 16.
| Patients With Measurable Disease (N=135) | All Patients (N=162) |
|
|---|---|---|
| Age (median, years) | 55 | 56 |
| Karnofsky Performance Status | 90 | 90 |
| No. Disease Sites | ||
| 1-2, % | 32 | 37 |
| 3-4, % | 46 | 43 |
| >5, % | 22 | 21 |
| Dominant Site of Disease | ||
| Visceral*, % | 75 | 68 |
| Soft Tissue, % | 22 | 22 |
| Bone, % | 3 | 10 |
| Prior Chemotherapy | ||
| Paclitaxel, % | 100 | 100 |
| Anthracycline†, % | 90 | 91 |
| Fluorouracil, % | 81 | 82 |
| Resistance to Paclitaxel, % | 76 | 77 |
| Resistance to an Anthracycline†, % | 41 | 41 |
| Resistance to both Paclitaxel and an Anthracycline†, % | 32 | 31 |
Efficacy for Study SO14697 are shown in Table 17.
| Efficacy Parameter | Resistance to Both Paclitaxel and an Anthracycline (N=43) |
|---|---|
| Response Rate* | 25.6% |
| (95% CI) | (13.5, 41.2) |
| Complete Response | 0% |
| Partial Response* | 11% |
| Duration of Response* | |
| Median, months†
(Range) | 5.1 (2.1-7.7) |
For the subgroup of 43 patients who were doubly resistant, the median time to progression was 3.4 months and the median survival was 8.4 months. The objective response rate in this population was supported by a response rate of 18.5% (1 CR, 24 PRs) in the overall population of 135 patients with measurable disease, who were less resistant to chemotherapy (see Table 15). The median time to progression was 3.0 months and the median survival was 10.1 months.
14.3 Gastric, Esophageal, or Gastroesophageal Junction Cancer
The efficacy of XELODA for treatment of adults with unresectable or metastatic gastric, esophageal, or gastroesophageal junction cancer as a component of a combination chemotherapy regimen was derived from studies in the published literature. XELODA was evaluated in REAL-2, a randomized non-inferiority, 2×2 factorial trial, where the major efficacy outcome measure was overall survival, and an additional randomized trial conducted by the North Central Cancer Treatment Group, where the major efficacy outcome measure was objective response rate.
The efficacy of XELODA for the treatment of adults with HER2-overexpressing metastatic gastric or gastroesophageal junction adenocarcinoma who have not received prior treatment for metastatic disease as a component of a combination regimen was derived from studies in the published literature. XELODA was evaluated in the ToGA trial [NCT01041404], an open-label, multicenter, randomized trial where the primary efficacy measure was overall survival.
14.4 Pancreatic Cancer
The efficacy of XELODA for the adjuvant treatment of adults with pancreatic adenocarcinoma as a component of a combination chemotherapy regimen was derived from a study in the published literature. XELODA was evaluated in ESPAC-4 trial, a two-group, open-label, multicenter, randomized trial, where the major efficacy outcome measure was overall survival.
16 HOW SUPPLIED/STORAGE AND HANDLING
XELODA (capecitabine) tablets are supplied as follows:
- 150 mg, biconvex, oblong, film-coated, light peach tablets with "XELODA" on one side and "150" on the other; available in bottles of 60 tablets (NDC 0004-1100-20), individually packaged in a carton.
- 500 mg, biconvex, oblong, film-coated, peach tablets with "XELODA" on one side and "500" on the other; available in bottles of 120 tablets (NDC 0004-1101-50), individually packaged in a carton.
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Increased Risk of Bleeding with Concomitant Use of Vitamin K Antagonists
Advise patients on vitamin K antagonists, such as warfarin, that they are at an increased risk of severe bleeding while taking XELODA. Advise these patients that INR should be monitored more frequently, and dosage modifications of the vitamin K antagonist may be required, while taking and after discontinuation of XELODA. Advise these patients to immediately contact their healthcare provider if signs or symptoms of bleeding occur [see Warnings and Precautions (5.1)].
Serious Adverse Reactions from Dihydropyrimidine Dehydrogenase (DPD) Deficiency
Inform patients of the potential for serious and life-threatening adverse reactions due to DPD deficiency and discuss with your patient whether they should be tested for genetic variants of DPYD that are associated with an increased risk of serious adverse reactions from the use of XELODA. Advise patients to immediately contact their healthcare provider if symptoms of severe mucositis, diarrhea, neutropenia, and neurotoxicity occur [see Warnings and Precautions (5.2) and Clinical Pharmacology (12.5)].
Cardiotoxicity
Advise patients of the risk of cardiotoxicity and to immediately contact their healthcare provider for new onset of chest pain, shortness of breath, dizziness, or lightheadedness [see Warnings and Precautions (5.3)].
Diarrhea
Inform patients experiencing grade 2 diarrhea (an increase of 4 to 6 stools/day or nocturnal stools) or greater or experiencing severe bloody diarrhea with severe abdominal pain and fever to stop taking XELODA. Advise patients on the use of antidiarrheal treatments (e.g., loperamide) to manage diarrhea [see Warnings and Precautions (5.4)].
Dehydration
Instruct patients experiencing grade 2 or higher dehydration to stop taking XELODA immediately and to contact their healthcare provider. Advise patients to not restart XELODA until rehydrated and any precipitating causes have been corrected or controlled [see Warnings and Precautions (5.5)].
Renal Toxicity
Instruct patients experiencing decreased urinary output or other signs and symptoms of renal toxicity to immediately contact their healthcare provider [see Warnings and Precautions (5.6)].
Serious Skin Toxicities
Instruct patients skin rash, blistering, or peeling to immediately contact their healthcare provider [see Warnings and Precautions (5.7)].
Palmar-Plantar Erythrodysesthesia Syndrome
Instruct patients experiencing grade 2 palmar-plantar erythrodysesthesia syndrome or greater to stop taking XELODA immediately and to contact their healthcare provider. Inform patients that initiation of symptomatic treatment is recommended and hand-and-foot syndrome can lead to loss of fingerprints which could impact personal identification [see Warnings and Precautions (5.8)].
Myelosuppression
Inform patients who develop a fever of 100.5°F or greater or other evidence of potential infection to immediately contact their healthcare provider [see Warnings and Precautions (5.9)].
Hyperbilirubinemia
Inform patients who develop jaundice or icterus to immediately contact their healthcare provider [see Warnings and Precautions (5.10)].
Embryo-Fetal Toxicity
Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.11), Use in Specific Populations (8.1)].
Advise females of reproductive potential to use effective contraception during treatment with XELODA and for 6 months after the last dose [see Use in Specific Populations (8.3)].
Advise males with female partners of reproductive potential to use effective contraception during treatment with XELODA and for 3 months after the last dose [see Use in Specific Populations (8.3)].
Lactation
Advise females not to breastfeed during treatment with XELODA and for 1 week after the last dose [see Use in Specific Populations (8.2)].
Infertility
Advise males and females of reproductive potential that XELODA may impair fertility [see Use in Specific Populations (8.3)].
Hypersensitivity and Angioedema
Advise patients that XELODA may cause severe hypersensitivity reactions and angioedema.
Advise patients who have known hypersensitivity to capecitabine or 5-fluorouracil to inform their healthcare provider [see Contraindications (4)]. Instruct patients who develop hypersensitivity reactions or mucocutaneous symptoms (e.g., urticaria, rash, erythema, pruritus, or swelling of the face, lips, tongue or throat which make it difficult to swallow or breathe) to stop taking XELODA and immediately contact their healthcare provider or to go to an emergency room. [see Adverse Reactions (6)].
Nausea and Vomiting
Instruct patients experiencing grade 2 nausea (food intake significantly decreased but able to eat intermittently) or greater to stop taking XELODA and to immediately contact their healthcare provider for management of nausea [see Adverse Reactions (6.1)].
Instruct patients experiencing grade 2 vomiting (2 to 5 episodes in a 24-hour period) or greater to stop taking XELODA immediately and to contact their healthcare provider for management of vomiting [see Adverse Reactions (6.1)].
Stomatitis
Inform patients experiencing grade 2 stomatitis (painful erythema, edema or ulcers of the mouth or tongue, but able to eat) or greater to stop taking XELODA immediately and to contact their healthcare provider [see Adverse Reactions (6.1)].
Important Administration Instructions
Advise patients to swallow XELODA tablets whole with water within 30 minutes after a meal. Advise patients and caregivers not to chew, crush, or cut XELODA tablets. Advise patients if they cannot swallow XELODA tablets whole to inform their healthcare provider [see Dosage and Administration (2.7), Warnings and Precautions (5.12)].
Drug interactions
Instruct patients not to take products containing folic acid or folate analog products (e.g., leucovorin, levoleucovorin) unless directed to do so by their healthcare provider. Advise patients to inform their healthcare provider of all prescription or nonprescription medications, vitamins or herbal products [see Drug Interactions (7.1, 7.2, 7.3)].
Distributed by:
Genentech, Inc.
A Member of the Roche Group
1 DNA Way
South San Francisco, CA 94080-4990
XELODA® is a registered trademark of Hoffmann-La Roche, Inc.
© 2022 Genentech, Inc. All rights reserved.
| This Patient Information has been approved by the U.S. Food and Drug Administration. | Revised: 12/2022 | ||||
|
Patient Information |
|||||
|
What is the most important information I should know about XELODA? XELODA can cause serious side effects, including:
See "What are the possible side effects of XELODA?" for more information about side effects. |
|||||
|
What is XELODA? XELODA is a prescription medicine used to treat:
It is not known if XELODA is safe and effective in children. |
|||||
Talk to your healthcare provider before taking XELODA if you are not sure. |
|||||
|
Before taking XELODA, tell your healthcare provider about all your medical conditions, including if you: See "What is the most important information I should know about XELODA?"
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. XELODA may affect the way other medicines work, and other medicines may affect the way XELODA works. |
|||||
|
|||||
|
What are the possible side effects of XELODA? XELODA can cause serious side effects including:
|
|||||
|
|||||
|
|||||
|
|
|
|||
Your healthcare provider should talk with you about DPYD testing to look for DPD deficiency. |
|||||
|
|||||
|
|
||||
|
|||||
|
|
||||
| Do not chew, cut, or crush XELODA tablets. See "How should I take XELODA tablets." If for any reason your tablets must be cut or crushed, this must be done by your pharmacist or healthcare provider. |
|||||
| Your healthcare provider may decide to decrease your dose, or temporarily or permanently stop XELODA if you have serious side effects with XELODA. | |||||
| The most common side effects in people with colon cancer who take XELODA alone to help prevent it from coming back include: hand and foot syndrome, diarrhea, and nausea. | |||||
| The most common side effects in people with metastatic colorectal carcinoma who take XELODA alone include: | |||||
|
|
||||
| The most common side effects in people with metastatic breast cancer who take XELODA in combination with docetaxel include: | |||||
|
|
||||
| The most common side effects in people with metastatic breast cancer who take XELODA alone include: | |||||
|
|
||||
| Severe allergic reactions can happen with XELODA. See "Do not take XELODA if you:" Stop taking XELODA and call your healthcare provider right away or go to an emergency room if you have any of the following symptoms of a severe allergic reaction to XELODA: | |||||
|
|
|
|||
| XELODA may cause fertility problems in females and males. This may affect the ability to have a child. Talk to your healthcare provider if you have concerns about fertility. | |||||
| These are not all the possible side effects of XELODA. | |||||
| Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. | |||||
|
How should I store XELODA?
Keep XELODA and all medicines out of the reach of children. |
|||||
|
General information about the safe and effective use of XELODA. Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use XELODA for a condition for which it was not prescribed. Do not give XELODA to other people, even if they have the same symptoms you have. It may harm them. You can ask your pharmacist or healthcare provider for information about XELODA that is written for health professionals. |
|||||
|
What are the ingredients in XELODA? Active ingredient: capecitabine Inactive ingredients: anhydrous lactose, croscarmellose sodium, hydroxypropyl methylcellulose, microcrystalline cellulose, magnesium stearate and purified water. The peach or light peach film coating contains hydroxypropyl methylcellulose, talc, titanium dioxide, and synthetic yellow and red iron oxides. Distributed by: Genentech, Inc. A Member of the Roche Group 1 DNA Way South San Francisco, CA 94080-4990 For more information, go to http://www.gene.com/patients/medicines/xeloda or call 1-877-436-3683. |
|||||

