To reduce the development of drug-resistant bacteria and maintain the effectiveness of Metronidazole Injection, USP and other antibacterial drugs, Metronidazole Injection, USP should be used only to treat or prevent infections that are proven or strongly suspected to be caused by bacteria.
|
WARNING Metronidazole has been shown to be carcinogenic in mice and rats (see PRECAUTIONS). Unnecessary use of the drug should be avoided. Its use should be reserved for the conditions described in the INDICATIONS AND USAGE section below. |
DESCRIPTION
Metronidazole Injection, USP, is a parenteral formulation of the synthetic nitroimidazole antibacterial agent 2-methyl-5-nitro-1H-imidazole-1-ethanol.
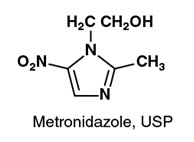
Metronidazole Injection, USP, in 100 mL VIAFLEX Plus single dose plastic container, is a sterile, nonpyrogenic, iso-osmotic, buffered solution of 500 mg Metronidazole, USP, 790 mg Sodium Chloride, USP, 47.6 mg Dibasic Sodium Phosphate Dried, USP and 22.9 mg Citric Acid Anhydrous, USP. Metronidazole Injection, USP has an osmolarity of 310 mOsmol/L (calc) and a pH of 5.5 (4.5 to 7.0). Each container contains 14 mEq of sodium.
The plastic container is fabricated from a specially formulated polyvinyl chloride plastic. Water can permeate from inside the container into the overwrap in amounts insufficient to affect the solution significantly. Solutions in contact with the plastic container can leach out certain of its chemical components in very small amounts within the expiration period, e.g., di-2-ethylhexyl phthalate (DEHP), up to 5 parts per million. However, the safety of the plastic has been confirmed in tests in animals according to USP biological tests for plastic containers as well as by tissue culture toxicity studies.
CLINICAL PHARMACOLOGY
In patients treated with intravenous metronidazole, using a dosage regimen of 15 mg/kg loading dose followed 6 hours later by 7.5 mg/kg every 6 hours, the average peak steady-state plasma concentrations (Cmax) and trough concentrations (Cmin) were 25 mcg/mL and 18 mcg/mL, respectively. Plasma concentrations of metronidazole are proportional to the administered dose. An eight-hour intravenous infusion of 100 mg to 4,000 mg of metronidazole in normal subjects showed a linear relationship between dose and peak plasma concentration. The average elimination half-life of metronidazole in healthy subjects is eight hours.
Distribution
Metronidazole is the major component appearing in the plasma, with lesser quantities of metabolites also being present. Less than 20% of the circulating metronidazole is bound to plasma proteins. Metronidazole appears in cerebrospinal fluid, saliva and breast milk in concentrations similar to those found in plasma. Bactericidal concentrations of metronidazole have also been detected in pus from hepatic abscesses.
Following a single intravenous dose of metronidazole 500 mg, 4 healthy subjects who underwent gastrointestinal endoscopy had peak gastric juice metronidazole concentrations of 5 to 6 mcg/mL at one hour post-dose. In patients receiving intravenous metronidazole in whom gastric secretions are continuously removed by nasogastric aspiration, sufficient metronidazole may be removed in the aspirate to cause a reduction in serum levels.
Metabolism
The metabolites of metronidazole result primarily from side-chain oxidation [1-(ß-hydroxyethyl)-2-hydroxymethyl-5-nitroimidazole and 2-methyl-5-nitroimidazole-1-yl-acetic acid] and glucuronide conjugation. Both the parent compound and the hydroxyl metabolite possess in vitro antimicrobial activity.
Excretion
The major route of elimination of metronidazole and its metabolites is via the urine (60-80% of the dose), with approximately 20% of the amount excreted appearing as unchanged metronidazole. Renal clearance of metronidazole is approximately 10 mL/min/1.73 m2. Fecal excretion accounts for 6-15% of the dose.
Renal Impairment
Decreased renal function does not alter the single-dose pharmacokinetics of metronidazole.
Subjects with end-stage renal disease (ESRD; CLCR= 8.1±9.1 mL/min) and who received a single intravenous infusion of metronidazole 500 mg had no significant change in metronidazole pharmacokinetics but had 2-fold higher Cmax of hydroxy-metronidazole and 5-fold higher Cmax of metronidazole acetate, compared to healthy subjects with normal renal function (CLCR= 126 ± 16 mL/min). Thus, on account of the potential accumulation of metronidazole metabolites in ESRD patients, monitoring for metronidazole associated adverse events is recommended (see PRECAUTIONS).
Effect of Dialysis
Following a single intravenous infusion or oral dose of metronidazole 500 mg, the clearance of metronidazole was investigated in ESRD subjects undergoing hemodialysis or continuous ambulatory peritoneal dialysis (CAPD). A hemodialysis session lasting for 4 to 8 hours removed 40% to 65% of the administered metronidazole dose, depending on the type of the dialyzer membrane used and the duration of the dialysis session. If the administration of metronidazole cannot be separated from the dialysis session, supplementation of metronidazole dose following hemodialysis should be considered (see DOSAGE AND ADMINISTRATION). A peritoneal dialysis session lasting for 7.5 hours removed approximately 10% of the administered metronidazole dose. No adjustment in metronidazole dose is needed in ESRD patients undergoing CAPD.
Hepatic Impairment
Following a single intravenous infusion of 500 mg metronidazole, the mean AUC24 of metronidazole was higher by 114% in patients with severe (Child-Pugh C) hepatic impairment, and by 54% and 53% in patients with a mild (Child-Pugh A) and moderate (Child-Pugh B) hepatic impairment, respectively, compared to healthy control subjects. There were no significant changes in the AUC24 of hydroxy-metronidazole in these hepatically impaired patients. A reduction in metronidazole dosage by 50% is recommended in patients with severe (Child-Pugh C) hepatic impairment (see DOSAGE AND ADMINISTRATION). No dosage adjustment is needed for patients with mild to moderate hepatic impairment. Patients with mild to moderate hepatic impairment should be monitored for metronidazole associated adverse events (see PRECAUTIONS and DOSAGE AND ADMINISTRATION).
Geriatric Patients
Following a single 500 mg oral or IV dose of metronidazole, subjects >70 years old with no apparent renal or hepatic dysfunction had a 40% to 80% higher mean AUC of hydroxy-metronidazole (active metabolite), with no apparent increase in the mean AUC of metronidazole (parent compound), compared to young healthy controls < 40 years old. In geriatric patients, monitoring for metronidazole associated adverse events is recommended (see PRECAUTIONS).
Pediatric Patients
In one study newborn infants appeared to demonstrate diminished capacity to eliminate metronidazole. The elimination half-life, measured during the first three days of life, was inversely related to gestational age. In infants whose gestational ages were between 28 and 40 weeks, the corresponding elimination half-lives ranged from 109 to 22.5 hours.
Microbiology
Mechanism of Action
Metronidazole, a nitroimidazole, exerts antibacterial effects in an anaerobic environment against most obligate anaerobes. Once metronidazole enters the organism by passive diffusion and is activated in the cytoplasm of susceptible anaerobic bacteria, it is reduced; this process includes intra-cellular electron transport proteins such as ferredoxin, transfer of an electron to the nitro group of the metronidazole, and formation of a short-lived nitroso free radical. Because of this alteration of the metronidazole molecule, a concentration gradient is created and maintained which promotes the drug’s intracellular transport. The reduced form of metronidazole and free radicals can interact with DNA leading to inhibition of DNA synthesis and DNA degradation leading to death of bacteria. The precise mechanism of action of metronidazole is unclear.
Drug Resistance
A potential for development of resistance exists against metronidazole.
Resistance may be due to multiple mechanisms that include decreased uptake of the drug, altered reduction efficiency, overexpression of the efflux pumps, inactivation of the drug, and/or increased DNA damage repair.
Metronidazole does not possess any clinically relevant activity against facultative anaerobes or obligate aerobes.
Activity In Vitro and in Clinical Infections
Metronidazole has been shown to be active against most isolates of the following bacteria both in vitro and in clinical infections as described in the INDICATIONS AND USAGE section.
Gram-positive anaerobes
Clostridium species
Eubacterium species
Peptococcus species
Peptostreptococcus species
Gram-negative anaerobes
Bacteroides fragilis group (B. fragilis, B. distasonis, B. ovatus, B. thetaiotaomicron, B.vulgatus)
Fusobacterium species
The following in vitro data are available, but their clinical significance is unknown.
Metronidazole exhibits in vitro minimal inhibitory concentrations (MIC’s) of 8 mcg/mL or less against most (≥ 90%) isolates of the following bacteria; however, the safety and effectiveness of metronidazole in treating clinical infections due to these bacteria have not been established in adequate and well-controlled clinical trials.
Gram-negative anaerobes
Bacteroides fragilis group (B. caccae, B. uniformis)
Prevotella species (P. bivia, P. buccae, P. disiens)
Susceptibility Tests
For specific information regarding susceptibility test interpretive criteria and associated test methods and quality control standards recognized by FDA for this drug, please see: http://www.fda.gov/STIC.
INDICATIONS AND USAGE
Treatment of Anaerobic Bacterial Infections
Metronidazole Injection, USP is indicated in the treatment of serious infections caused by susceptible anaerobic bacteria. Indicated surgical procedures should be performed in conjunction with Metronidazole Injection, USP therapy. In a mixed aerobic and anaerobic infection, antibiotics appropriate for the treatment of the aerobic infection should be used in addition to Metronidazole Injection, USP.
Metronidazole Injection, USP is effective in Bacteroides fragilis infections resistant to clindamycin, chloramphenicol and penicillin.
Intra-Abdominal Infections, including peritonitis, intra-abdominal abscess and liver abscess, caused by Bacteroides species including the B. fragilis group (B. fragilis, B. distasonis, B. ovatus, B. thetaiotaomicron, B. vulgatus), Clostridium species, Eubacterium species, Peptococcus species and Peptostreptococcus species.
Skin and Skin Structure Infections caused by Bacteroides species including the B. fragilis group, Clostridium species, Peptococcus species, Peptostreptococcus species and Fusobacterium species.
Gynecologic Infections, including endometritis, endomyometritis, tubo-ovarian abscess and postsurgical vaginal cuff infection, caused by Bacteroides species including the B. fragilis group, Clostridium species, Peptococcus species, Peptostreptococcus species and Fusobacterium species.
Bacterial Septicemia caused by Bacteroides species including the B. fragilis group and Clostridium species.
Bone and Joint Infections, as adjunctive therapy, caused by Bacteroides species including the B. fragilis group.
Central Nervous System (CNS) Infections, including meningitis and brain abscess, caused by Bacteroides species including the B. fragilis group.
Lower Respiratory Tract Infections, including pneumonia, empyema and lung abscess, caused by Bacteroides species including the B. fragilis group.
Endocarditis caused by Bacteroides species including the B. fragilis group.
Prophylaxis
The prophylactic administration of Metronidazole Injection, USP preoperatively, intraoperatively and postoperatively may reduce the incidence of postoperative infection in patients undergoing elective colorectal surgery which is classified as contaminated or potentially contaminated. Prophylactic use of Metronidazole Injection, USP should be discontinued within 12 hours after surgery. If there are signs of infection, specimens for cultures should be obtained for the identification of the causative organism(s) so that appropriate therapy may be given (see DOSAGE AND ADMINISTRATION).
To reduce the development of drug-resistant bacteria and maintain the effectiveness of Metronidazole Injection, USP and other antibacterial drugs, Metronidazole Injection, USP should be used only to treat or prevent infections that are proven or strongly suspected to be caused by susceptible bacteria. When culture and susceptibility information are available, they should be considered in selecting or modifying antibacterial therapy. In the absence of such data, local epidemiology and susceptibility patterns may contribute to the empiric selection of therapy.
CONTRAINDICATIONS
Hypersensitivity
Metronidazole Injection, USP is contraindicated in patients with a prior history of hypersensitivity to metronidazole or other nitroimidazole derivatives.
Psychotic Reaction with Disulfiram
Use of oral metronidazole is associated with psychotic reactions in alcoholic patients who were using disulfiram concurrently. Do not administer metronidazole to patients who have taken disulfiram within the last two weeks (see PRECAUTIONS-Drug Interactions).
Interaction with Alcohol
Use of oral metronidazole is associated with a disulfiram-like reaction to alcohol, including abdominal cramps, nausea, vomiting, headaches, and flushing. Discontinue consumption of alcohol or products containing propylene glycol during and for at least three days after therapy with metronidazole (see PRECAUTIONS-Drug Interactions).
Cockayne Syndrome
Metronidazole Injection is contraindicated in patients with Cockayne syndrome. Severe irreversible hepatotoxicity/acute liver failure with fatal outcomes have been reported after initiation of metronidazole in patients with Cockayne syndrome (see ADVERSE REACTIONS).
WARNINGS
Central and Peripheral Nervous System Effects
Severe neurological disturbances, including encephalopathy, cerebellar symptoms, convulsive seizures, peripheral neuropathy, optic neuropathy, and aseptic meningitis, have been reported in patients treated with metronidazole.
Encephalopathy associated with metronidazole may manifest as confusion or decreased level of consciousness, and is associated with widespread lesions on magnetic resonance imaging (MRI) of the brain. Cerebellar toxicity associated with metronidazole may manifest as ataxia, dizziness, dysarthria, nystagmus and saccadic pursuit and is accompanied by T2 flair lesions within the dentate nuclei seen on MRI. Cerebellar toxicity may concurrently occur with encephalopathy, peripheral neuropathy or seizures. CNS symptoms and CNS lesions are generally reversible within days to weeks upon discontinuation of Metronidazole Injection, USP. Peripheral neuropathy, usually symmetric and mainly of sensory type has been reported and is characterized by numbness or paresthesia of an extremity. Symptoms may be prolonged after drug discontinuation. Aseptic meningitis may occur within hours of dose administration and generally resolve after metronidazole therapy is discontinued (see ADVERSE REACTIONS).
Advise patients to report neurologic symptoms that occur during metronidazole administration. Discontinue metronidazole treatment if any abnormal neurologic symptoms occur such as ataxia, dizziness, confusion or any other CNS adverse reaction (see ADVERSE REACTIONS).
PRECAUTIONS
General
Hepatic Impairment
Patients with hepatic impairment metabolize metronidazole slowly, with resultant accumulation of metronidazole and increase the plasma concentrations. Reduce the dose of Metronidazole Injection, USP by 50% in patients with severe hepatic impairment (Child-Pugh C). For patients with mild to moderate hepatic impairment, no dosage adjustment is needed but these patients should be monitored for metronidazole associated adverse events (see CLINICAL PHARMACOLOGY and DOSAGE AND ADMINISTRATION).
Patients with severe hepatic encephalopathy metabolize metronidazole slowly, with resultant accumulation of metronidazole. This may cause exacerbation of CNS adverse effects. Reduce the dose of Metronidazole Injection, USP as necessary.
Renal Impairment
For patients with mild to moderate renal impairment does adjustment is not considered necessary as elimination half-life is not significantly altered. In patients with severe renal impairment or end stage of renal disease, metronidazole and metronidazole metabolites may accumulate significantly because of reduced urinary excretion in those patients. Monitoring for metronidazole associated adverse events is recommended when metronidazole is administered in patients with severe renal impairment or end stage of renal disease who are not undergoing hemodialysis (see CLINICAL PHARMACOLOGY).
Hemodialysis removes significant amounts of metronidazole and its metabolites from systemic circulation. Therefore, supplementation of metronidazole following a hemodialysis session may be necessary.
Patients receiving peritoneal dialysis should be monitored for signs of toxicity due to the potential accumulation of metronidazole metabolites.
Fungal Superinfections
Known or previously unrecognized candidiasis may present more prominent symptoms during therapy with Metronidazole Injection, USP and requires treatment with a candicidal agent.
Use in Patients with Blood Dyscrasias
Metronidazole is a nitroimidazole, and should be used with care in patients with evidence of or history of blood dyscrasia, Agranulocytosis, leukopenia and neutropenia have associated with metronidazole administration. Monitor complete blood count in these patients.
Monitoring for Leukopenia
Monitoring of complete blood count (CBC) is recommended before, during, and after prolonged or repeated courses of metronidazole therapy.
Information for Patients
Interaction with Alcohol
Discontinue consumption of alcoholic beverages or products containing propylene glycol while taking Metronidazole Injection, USP and for at least three days afterward because abdominal cramps, nausea, vomiting, headaches, and flushing may occur (see CONTRAINDICATIONS, PRECAUTIONS-Drug Interactions).
Treatment of Bacterial Infections
Patients should be counseled that antibacterial drugs including Metronidazole Injection, USP should only be used to treat bacterial infections. They do not treat viral infections (e.g., the common cold). When Metronidazole Injection, USP is prescribed to treat a bacterial infection, patients should be told that although it is common to feel better early in the course of therapy, the medication should be taken exactly as directed. Skipping doses or not completing the full course of therapy may (1) decrease the effectiveness of the immediate treatment and (2) increase the likelihood that bacteria will develop resistance and will not be treatable by Metronidazole Injection, USP or other antibacterial drugs in the future.
Drug Interactions
Disulfiram
Psychotic reactions and confusion have been reported in alcoholic patients who are using metronidazole and disulfiram concurrently. Do not administer Metronidazole Injection, USP to patients who have taken disulfiram within the last two weeks (see CONTRAINDICATIONS).
Alcoholic Beverages
Abdominal cramps, nausea, vomiting, headaches, tachycardia and flushing may occur if alcoholic beverages or products containing propylene glycol are consumed during or following metronidazole therapy. Discontinue consumption of alcohol or products containing propylene glycol before, during and up to 72 hours after therapy with Metronidazole Injection, USP (see CONTRAINDICATIONS).
Warfarin and other Oral Anticoagulants
Metronidazole has been reported to potentiate the anticoagulant effect of warfarin and other oral coumarin anticoagulants, resulting in a prolongation of prothrombin time and increased risk of hemorrhages. When Metronidazole Injection, USP is prescribed for patients on this type of anticoagulant therapy, prothrombin time and international normalized ratio (INR) should be carefully monitored and their anticoagulant dose adjusted accordingly. Monitor patients for signs and symptoms of bleeding.
Lithium
In patients stabilized on relatively high doses of lithium, short-term metronidazole therapy has been associated with elevation of serum lithium and, in a few cases, signs of lithium toxicity. Lithium toxicity may lead to renal damage. Frequent monitoring of serum lithium and serum creatinine levels is necessary.
Busulfan
Metronidazole has been reported to increase plasma concentrations of busulfan, which can result in an increased risk for serious busulfan toxicity such as sinusoidal obstruction syndrome, gastrointestinal mucositis, and hepatic veno-occlusive disease. Metronidazole Injection, USP should not be administered concomitantly with busulfan unless the benefit outweighs the risk. If no therapeutic alternatives to metronidazole are available, and concomitant administration with busulfan is medically needed, frequent monitoring of busulfan plasma concentration should be performed and the busulfan dose should be adjusted accordingly.
Drugs that Inhibit CYP450 Enzymes
The simultaneous administration of drugs that decrease microsomal liver enzyme activity, such as cimetidine, may decrease metabolism and reduce plasma clearance of metronidazole which may result in metronidazole toxicity.
Drugs that Induce CYP450 Enzymes
The simultaneous administration of drugs that induce microsomal liver enzyme activity, such as phenytoin or phenobarbital, may accelerate the elimination of metronidazole and therefore decrease its efficacy.
Cytochrome P450 3A4 (CYP3A4) substrates
Concomitant use of Metronidazole Injection, USP and CYP3A4 substrates (e.g., amiodarone, tacrolimus, cyclosporine, carbamazepine, phenytoin, and quinidine) may increase respective CYP3A4-substrate plasma levels. Monitoring of plasma concentrations of CYP3A4 substrates may be necessary.
Drug/Laboratory Test Interactions
Metronidazole may interfere with certain types of determinations of serum chemistry values, such as aspartate aminotransferase (AST, SGOT), alanine aminotransferase (ALT, SGPT), lactate dehydrogenase (LDH), triglycerides and glucose hexokinase. Metronidazole causes an increase in ultraviolet absorbance at 340 nm resulting in falsely decreased values.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Tumors affecting the liver, lung, mammary and lymphatic tissues have been detected in several studies of metronidazole in rats and mice, but not hamsters.
Pulmonary tumors have been observed in all six reported studies in the mouse, including one study in which the animals were dosed on an intermittent schedule (administration during every fourth week only). Malignant tumors were increased in male mice treated at approximately 1500 mg/m2 (similar to the maximum recommended daily dose, based on body surface area comparisons). Malignant lymphomas and pulmonary neoplasms were also increased with lifetime feeding of the drug to mice. Mammary and hepatic tumors were increased among female rats administered oral metronidazole compared to concurrent controls. Two lifetime tumorigenicity studies in hamsters have been performed and reported to be negative.
Metronidazole has shown mutagenic activity in in vitro assay systems including the Ames test. Studies in mammals in vivo have failed to demonstrate a potential for genetic damage.
Metronidazole failed to produce any adverse effects on fertility or testicular function in male rats at doses up to 400 mg/kg/day (approximately 2 times the maximum recommended daily dose based on body surface area comparison) for 28 days. However, rats treated at the same dose for 6 weeks, or longer were infertile and showed severe degeneration of the seminiferous epithelium in the testes as well as marked decreases in testicular spermatid counts and epididymal sperm counts. Fertility was restored in most rats after an eight week, drug-free recovery period.
Fertility studies have been performed in male mice at doses up to six times the maximum recommended human dose based on mg/m2 and have revealed no evidence of impaired fertility. However, metronidazole was associated with reversible adverse effects on the male reproductive system (significantly decreased testes and epididymides weight, decreased sperm viability, and increased the incidence of abnormal sperm).
Pregnancy
Teratogenic Effects
There are no adequate and well-controlled studies of Metronidazole Injection, USP in pregnant women. There are published data from case-control studies, cohort studies, and 2 meta-analyses that include more than 5000 pregnant women who used metronidazole during pregnancy. Many studies included first trimester exposures. One study showed an increased risk of cleft lip, with or without cleft palate, in infants exposed to metronidazole in utero; however, these findings were not confirmed. In addition, more than ten randomized, placebo-controlled clinical trials enrolled more than 5000 pregnant women to assess the use of antibiotic treatment (including metronidazole) for bacterial vaginosis on the incidence of preterm delivery. Most studies did not show an increased risk for congenital anomalies or other adverse fetal outcomes following metronidazole exposure during pregnancy. Three studies conducted to assess the risk of infant cancer following metronidazole exposure during pregnancy did not show an increased risk; however, the ability of these studies to detect such a signal was limited.
Metronidazole crosses the placental barrier and its effects on the human fetal organogenesis are not known. Reproduction studies have been performed in rats, rabbits and mice at doses similar to the maximum recommended daily dose based on body surface area comparisons. There was no evidence of harm to the fetus due to metronidazole.
Healthcare provider should carefully consider the potential risks and benefits for each specific patient before prescribing Metronidazole Injection, USP.
Nursing Mothers
Metronidazole is present in human milk at concentrations similar to maternal serum levels, and infant serum levels can be close to or comparable to infant therapeutic levels. Because of the potential for tumorigenicity shown for metronidazole in mouse and rat studies, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother. Alternatively, a nursing mother may choose to pump and discard human milk for the duration of metronidazole therapy, and for 24 hours after therapy ends and feed her infant stored human milk or formula.
Geriatric Use
In general, dose selection for an elderly patient should be cautious, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
In geriatric patients, monitoring for metronidazole associated adverse events is recommended (see CLINICAL PHARMACOLOGY, PRECAUTIONS). Decreased liver function in geriatric patients can result in increased concentrations of metronidazole that may necessitate adjustment of metronidazole dosage (see DOSAGE AND ADMINISTRATION).
ADVERSE REACTIONS
The following reactions have been reported during treatment with metronidazole formulations.
INFECTIONS AND INFESTATIONS: Vaginal candidiasis
BLOOD AND LYMPHATIC SYSTEM DISORDERS: Agranulocytosis, Leukopenia, Neutropenia, Thrombocytopenia, Eosinophilia
IMMUNE SYSTEM DISORDERS: Anaphylactic reaction, Hypersensitivity
METABOLISM AND NUTRITION DISORDERS: Decreased appetite
PSYCHIATRIC DISORDERS: Confusional state, Depression, Insomnia, Decreased libido
NERVOUS SYSTEM DISORDERS: Encephalopathy, Seizure, Neuropathy peripheral, Ataxia, Dizziness, Hypoesthesia, Paresthesia, Dysgeusia, Headache, Nystagmus, Aseptic meningitis, Somonlence, Dysarthria, Numbness, Syncope
EYE DISORDERS: Optic neuropathy, Saccadic eye movement
EAR AND LABYRINTH DISORDERS: Vertigo
CARDIAC DISORDERS: QT prolongation has been reported, particularly when metronidazole was administered with drugs with the potential for prolonging the QT interval. Flattening of the T-wave may be seen in electrocardiographic tracings, Tachycardia, Palpitation
RESPIRATORY, THORACIC AND MEDIASTINAL DISORDERS: Dyspnea
GASTROINTESTINAL DISORDERS: Pancreatitis, Abdominal pain, Diarrhea, Nausea, Vomiting, Asthenia, Proctitis
HEPATOBILIARY DISORDERS: Hepatotoxicity/Liver Failure in patients with Cockayne syndrome (see CONTRAINDICATIONS), Jaundice
SKIN AND SUBCUTANEOUS DISORDERS: Toxic epidermal necrolysis, Swelling face, Pruritus, Urticaria, Hyperhidrosis, Erythema, Rash; Stevens-Johnson syndrome, Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS)
MUSCULOSKELETAL AND CONNECTIVE TISSUE DISORDERS: Muscle spasms, Arthralgia, Myalgia
RENAL AND URINARY DISORDERS: Chromaturia, Dysuria
Hepatic: Cases of severe irreversible hepatotoxicity/acute liver failure, including cases with fatal outcomes with very rapid onset after initiation of systemic use of metronidazole, have been reported in patients with Cockayne Syndrome (latency from drug start to signs of liver failure as short as 2 days) (see CONTRAINDICATIONS)
REPRODUCTIVE: Dyspareunia
GENERAL DISORDERS AND ADMINISTRATION SITE CONDITIONS: Injection site reaction, Malaise, Face edema, Edema peripheral, Chest pain, Chills,
INVESTIGATIONS: Hepatic enzyme increased
Patients with Crohn's disease are known to have an increased incidence of gastrointestinal and certain extraintestinal cancers. There have been some reports in the medical literature of breast and colon cancer in Crohn's disease patients who have been treated with metronidazole at high doses for extended periods of time. A cause and effect relationship has not been established. Crohn's disease is not an approved indication for Metronidazole Injection, USP.
OVERDOSAGE
Signs and symptoms of an overdose may include nausea, vomiting, and neurotoxic effects, including ataxia, confusion, disorientation, seizures, and peripheral neuropathy.
Treatment of Overdosage
Symptomatic treatment is recommended. There is no specific antidote for an overdose of metronidazole. Discontinue metronidazole administration in the event of an overdose. Hemodialysis removes significant amounts of metronidazole and its metabolites from systemic circulation.
DOSAGE AND ADMINISTRATION
Dosage, rate of administration, and duration of treatment are to be individualized and depend upon the indication for use, the patient’s age, weight, clinical condition and concomitant treatment, and on the patient’s clinical and laboratory response to the treatment.
Treatment of Anaerobic Bacterial Infections
The recommended dosage schedule for adults is:
|
Loading Dose |
15 mg/kg infused intravenously over one hour (approximately 1 gram for a 70-kg adult). |
|
Maintenance Dose |
7.5 mg/kg infused intravenously over one hour every six hours (approximately 500 mg for a 70-kg adult). The first maintenance dose should be instituted six hours following the initiation of the loading dose. |
Parenteral therapy may be changed to oral metronidazole when conditions warrant, based upon the severity of the disease and the response of the patient to Metronidazole Injection, USP treatment. The usual adult oral dosage is 7.5 mg/kg every six hours (approximately 500 mg for a 70-kg adult).
A maximum of 4 grams should not be exceeded during a 24-hour period.
The usual duration of therapy is 7 to 10 days; however, infections of the bone and joint, lower respiratory tract and endocardium may require longer treatment.
Dosage Adjustments
Patients with Severe Hepatic Impairment
For patients with severe hepatic impairment (Child-Pugh C), the metronidazole dose should be reduced by 50% (see CLINICAL PHARMACOLOGY and PRECAUTIONS).
Patients Undergoing Hemodialysis
Hemodialysis removes significant amounts of metronidazole and its metabolites from systemic circulation. The clearance of metronidazole will depend on the type of dialysis membrane used, the duration of the dialysis session, and other factors. If the administration of metronidazole cannot be separated from a hemodialysis session, supplementation of metronidazole dosage following a hemodialysis session should be considered, depending on the patient’s clinical situation (see CLINICAL PHARMACOLOGY).
Prophylaxis
For surgical prophylactic use, to prevent postoperative infection in contaminated or potentially contaminated colorectal surgery, the recommended dosage schedule for adults is:
- a.
- 15 mg/kg infused over 30 to 60 minutes and completed approximately one hour before surgery; followed by
- b.
- 7.5 mg/kg infused over 30 to 60 minutes at 6 and 12 hours after the initial dose.
It is important that (1) administration of the initial preoperative dose be completed approximately one hour before surgery so that adequate drug levels are present in the serum and tissues at the time of initial incision, and (2) Metronidazole Injection, USP be administered, if necessary, at 6-hour intervals to maintain effective drug levels. Prophylactic use of Metronidazole Injection, USP should be limited to the day of surgery only, following the above guidelines.
Caution: Metronidazole Injection, USP is to be administered by slow intravenous drip infusion only, either as a continuous or intermittent infusion. Additives should not be introduced into Metronidazole Injection, USP, unless compatibility is known. If used with a primary intravenous fluid system, the primary solution should be discontinued during metronidazole infusion. DO NOT USE EQUIPMENT CONTAINING ALUMINUM (e.g., NEEDLES, CANNULAE) THAT WOULD COME IN CONTACT WITH THE DRUG SOLUTION AS PRECIPITATES MAY FORM.
Metronidazole Injection, USP is incompatible with (includes but is not limited to): Aztreonam, Cefamandole nafate, Cefoxitin, Penicillin G.
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal products (see DIRECTIONS FOR USE OF VIAFLEX PLUS PLASTIC CONTAINER).
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Do not administer unless the solution is clear and the seal is intact.
HOW SUPPLIED
Metronidazole Injection, USP is supplied in 100 mL single dose plastic containers, each containing an iso-osmotic, buffered solution of 500 mg metronidazole as follows:
|
2B3422 |
NDC 0338-9541-24 |
500 mg/100 mL |
Store at controlled room temperature (77°F or 25°C) and protect from light during storage. Do not remove unit from overwrap until ready for use. The overwrap is a moisture barrier. The inner bag maintains the sterility of the product. After removing overwrap, check for minute leaks by squeezing inner bag firmly. If leaks are found, discard solution as sterility may be impaired.
DIRECTIONS FOR USE OF VIAFLEX PLUS PLASTIC CONTAINER
Metronidazole Injection, USP is a ready-to-use iso-osmotic solution. No dilution or buffering is required. Do not refrigerate. Each container of Metronidazole Injection, USP contains 14 mEq of sodium.
Do not connect flexible plastic containers in series in order to avoid air embolism due to possible residual air contained in the primary container.
Pressurizing intravenous solutions contained in flexible plastic containers to increase flow rates can result in air embolism if the residual air in the container is not fully evacuated prior to administration.
Use of a vented intravenous administration set with the vent in the open position could result in air embolism. Vented intravenous administration sets with the vent in the open position should not be used with flexible plastic containers.
Discard any unused portion.
For single dose only.
To open
Tear overwrap down side at slit and remove solution container. Visually inspect the container. If the outlet port protector is damaged, detached, or not present, discard container as solution path sterility may be impaired. Some opacity of the plastic due to moisture absorption during the sterilization process may be observed. This is normal and does not affect the solution quality or safety. The opacity will diminish gradually. Check for leaks. Do not add supplementary medication.
Manufactured by:
Baxter Healthcare Corporation
Deerfield, IL 60015 USA
Printed in USA
PREMIERPro™Rx
Baxter and Viaflex are registered trademarks of Baxter International Inc.
PremierPro Rx is a trademark of Premier, Inc. used under license
07-19-00-3753
Rev. 12/2021
PACKAGE/LABEL PRINCIPAL DISPLAY PANEL

LOT
EXP
NDC 0338-9541-24
Metronidazole
Metronidazole Injection USP
500 mg per 100 mL
(5 mg / mL)
100 mL STERILE SINGLE DOSE
CONTAINER EACH 100 mL CONTAINS
500 mg METRONIDAZOLE USP 790 mg
SODIUM CHLORIDE USP 47.6 mg DIBASIC
SODIUM PHOSPHATE DRIED USP 22.9 mg
CITRIC ACID ANHYDROUS USP pH 5.5
(4.5 TO 7.0) OSMOLARITY 310 mOsmol/L
(CALC) USUAL DOSAGE SEE INSERT DO
NOT ADD SUPPLEMENTARY MEDICATION
STORE IN MOISTURE BARRIER OVERWRAP
AT ROOM TEMPERATURE (77°F OR 25°C)
AND PROTECT FROM LIGHT DURING STORAGE
RX ONLY
Manufactured By
Baxter Logo
PREMIER Pro ™ Rx
USA

Lift here
to open
Baxter Logo
Baxter Logo
Store at room temperature (77°F or 25°C) and protect contents
from light during storage.
24/100 mL single dose containers
Store at room temperature (77°F or 25°C) and protect contents
from light during storage.
24/100 mL single dose containers
Use no sharp instruments
Baxter Healthcare Corporation
Deerfield, IL 60015
Made in USA
Baxter Healthcare Corporation
Deerfield, IL 60015
Made in USA
Mfg. Stamp
07-05-70-265
24PK – 100 mL
Corrugated
Recycles