FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
1.1 Monotherapy
AVODART (dutasteride) soft gelatin capsules are indicated for the treatment of symptomatic benign prostatic hyperplasia (BPH) in men with an enlarged prostate to:
- •
- improve symptoms,
- •
- reduce the risk of acute urinary retention (AUR), and
- •
- reduce the risk of the need for BPH-related surgery.
2 DOSAGE AND ADMINISTRATION
The capsules should be swallowed whole and not chewed or opened, as contact with the capsule contents may result in irritation of the oropharyngeal mucosa. AVODART may be administered with or without food.
3 DOSAGE FORMS AND STRENGTHS
0.5-mg, opaque, dull yellow, gelatin capsules imprinted with “GX CE2” in red ink on one side.
4 CONTRAINDICATIONS
AVODART is contraindicated for use in:
- •
- Pregnancy. Dutasteride use is contraindicated in women who are pregnant. In animal reproduction and developmental toxicity studies, dutasteride inhibited development of male fetus external genitalia. Therefore, AVODART may cause fetal harm when administered to a pregnant woman [see Warnings and Precautions (5.4), Use in Specific Populations (8.1)].
- •
- Patients with previously demonstrated clinically significant hypersensitivity (e.g., serious skin reactions, angioedema) to AVODART or other 5 alpha-reductase inhibitors [see Adverse Reactions (6.2)].
5 WARNINGS AND PRECAUTIONS
5.1 Effects on Prostate-Specific Antigen (PSA) and the Use of PSA in Prostate Cancer Detection
In clinical trials, AVODART reduced serum PSA concentration by approximately 50% within 3 to 6 months of treatment. This decrease was predictable over the entire range of PSA values in subjects with symptomatic BPH, although it may vary in individuals. AVODART may also cause decreases in serum PSA in the presence of prostate cancer. To interpret serial PSAs in men taking AVODART, a new PSA baseline should be established at least 3 months after starting treatment and PSA monitored periodically thereafter. Any confirmed increase from the lowest PSA value while on AVODART may signal the presence of prostate cancer and should be evaluated, even if PSA levels are still within the normal range for men not taking a 5 alpha-reductase inhibitor. Noncompliance with AVODART may also affect PSA test results.
To interpret an isolated PSA value in a man treated with AVODART for 3 months or more, the PSA value should be doubled for comparison with normal values in untreated men. The free-to-total PSA ratio (percent free PSA) remains constant, even under the influence of AVODART. If clinicians elect to use percent free PSA as an aid in the detection of prostate cancer in men receiving AVODART, no adjustment to its value appears necessary.
Coadministration of dutasteride and tamsulosin resulted in similar changes to serum PSA as dutasteride monotherapy.
5.2 Increased Risk of High-grade Prostate Cancer
In men aged 50 to 75 years with a prior negative biopsy for prostate cancer and a baseline PSA between 2.5 ng/mL and 10.0 ng/mL taking AVODART in the 4-year Reduction by Dutasteride of Prostate Cancer Events (REDUCE) trial, there was an increased incidence of Gleason score 8-10 prostate cancer compared with men taking placebo (AVODART 1.0% versus placebo 0.5%) [see Indications and Usage (1.3), Adverse Reactions (6.1)]. In a 7-year placebo-controlled clinical trial with another 5 alpha-reductase inhibitor (finasteride 5 mg, PROSCAR), similar results for Gleason score 8-10 prostate cancer were observed (finasteride 1.8% versus placebo 1.1%).
5 alpha-reductase inhibitors may increase the risk of development of high-grade prostate cancer. Whether the effect of 5 alpha-reductase inhibitors to reduce prostate volume or trial-related factors impacted the results of these trials has not been established.
5.3 Evaluation for Other Urological Diseases
Prior to initiating treatment with AVODART, consideration should be given to other urological conditions that may cause similar symptoms. In addition, BPH and prostate cancer may coexist.
5.4 Transdermal Exposure of AVODART in Pregnant Women—Risk to Male Fetus
AVODART capsules should not be handled by women who are pregnant or may be pregnant. Dutasteride can be absorbed through the skin and could result in unintended fetal exposure and potential risk to a male fetus. If a pregnant woman comes in contact with leaking dutasteride capsules, the contact area should be washed immediately with soap and water [see Use in Specific Populations (8.1)]. Dutasteride can be absorbed through the skin based on animal studies [see Nonclinical Toxicology (13.2)].
5.5 Blood Donation
Men being treated with AVODART should not donate blood until at least 6 months have passed following their last dose. The purpose of this deferred period is to prevent administration of dutasteride to a pregnant female transfusion recipient.
5.6 Effect on Semen Characteristics
The effects of dutasteride 0.5 mg/day on semen characteristics were evaluated in healthy men throughout 52 weeks of treatment and 24 weeks of post‑treatment follow‑up. At 52 weeks, compared with placebo, dutasteride treatment resulted in mean reduction in total sperm count, semen volume, and sperm motility; the effects on total sperm count were not reversible after 24 weeks of follow-up. Sperm concentration and sperm morphology were unaffected and mean values for all semen parameters remained within the normal range at all timepoints. The clinical significance of the effect of dutasteride on semen characteristics for an individual patient’s fertility is not known [see Use in Specific Populations (8.3)].
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trial of another drug and may not reflect the rates observed in practice.
From clinical trials with AVODART as monotherapy or in combination with tamsulosin:
- •
- The most common adverse reactions reported in subjects receiving AVODART were impotence, decreased libido, breast disorders (including breast enlargement and tenderness), and ejaculation disorders. The most common adverse reactions reported in subjects receiving combination therapy (AVODART plus tamsulosin) were impotence, decreased libido, breast disorders (including breast enlargement and tenderness), ejaculation disorders, and dizziness. Ejaculation disorders occurred significantly more in subjects receiving combination therapy (11%) compared with those receiving AVODART (2%) or tamsulosin (4%) as monotherapy.
- •
- Trial withdrawal due to adverse reactions occurred in 4% of subjects receiving AVODART and 3% of subjects receiving placebo in placebo-controlled trials with AVODART. The most common adverse reaction leading to trial withdrawal was impotence (1%).
- •
- In the clinical trial evaluating the combination therapy, trial withdrawal due to adverse reactions occurred in 6% of subjects receiving combination therapy (AVODART plus tamsulosin) and 4% of subjects receiving AVODART or tamsulosin as monotherapy. The most common adverse reaction in all treatment arms leading to trial withdrawal was erectile dysfunction (1% to 1.5%).
Monotherapy
Over 4,300 male subjects with BPH were randomly assigned to receive placebo or 0.5-mg daily doses of AVODART in 3 identical 2-year, placebo-controlled, double-blind, Phase 3 treatment trials, each followed by a 2-year open-label extension. During the double-blind treatment period, 2,167 male subjects were exposed to AVODART, including 1,772 exposed for 1 year and 1,510 exposed for 2 years. When including the open-label extensions, 1,009 male subjects were exposed to AVODART for 3 years and 812 were exposed for 4 years. The population was aged 47 to 94 years (mean age: 66 years) and greater than 90% were white. Table 1 summarizes clinical adverse reactions reported in at least 1% of subjects receiving AVODART and at a higher incidence than subjects receiving placebo.
| a These sexual adverse reactions are associated with dutasteride treatment (including monotherapy and combination with tamsulosin). These adverse reactions may persist after treatment discontinuation. The role of dutasteride in this persistence is unknown. b Includes breast tenderness and breast enlargement. |
||||
|
Adverse Reaction |
Adverse Reaction Time of Onset |
|||
|
Months 0‑6 |
Months 7‑12 |
Months 13‑18 |
Months 19‑24 |
|
|
AVODART (n) |
(n = 2,167) |
(n = 1,901) |
(n = 1,725) |
(n = 1,605) |
|
Placebo (n) |
(n = 2,158) |
(n = 1,922) |
(n = 1,714) |
(n = 1,555) |
|
Impotencea | ||||
|
AVODART |
4.7% |
1.4% |
1.0% |
0.8% |
|
Placebo |
1.7% |
1.5% |
0.5% |
0.9% |
|
Decreased libidoa | ||||
|
AVODART |
3.0% |
0.7% |
0.3% |
0.3% |
|
Placebo |
1.4% |
0.6% |
0.2% |
0.1% |
|
Ejaculation disordersa | ||||
|
AVODART |
1.4% |
0.5% |
0.5% |
0.1% |
|
Placebo |
0.5% |
0.3% |
0.1% |
0.0% |
|
Breast disordersb | ||||
|
AVODART |
0.5% |
0.8% |
1.1% |
0.6% |
|
Placebo |
0.2% |
0.3% |
0.3% |
0.1% |
Long-term Treatment (Up to 4 Years)
High-grade Prostate Cancer: The REDUCE trial was a randomized, double-blind, placebo-controlled trial that enrolled 8,231 men aged 50 to 75 years with a serum PSA of 2.5 ng/mL to 10 ng/mL and a negative prostate biopsy within the previous 6 months. Subjects were randomized to receive placebo (n = 4,126) or 0.5-mg daily doses of AVODART (n = 4,105) for up to 4 years. The mean age was 63 years and 91% were white. Subjects underwent protocol-mandated scheduled prostate biopsies at 2 and 4 years of treatment or had “for-cause biopsies” at non-scheduled times if clinically indicated. There was a higher incidence of Gleason score 8-10 prostate cancer in men receiving AVODART (1.0%) compared with men on placebo (0.5%) [see Indications and Usage (1.3), Warnings and Precautions (5.2)]. In a 7-year placebo-controlled clinical trial with another 5 alpha-reductase inhibitor (finasteride 5 mg, PROSCAR), similar results for Gleason score 8-10 prostate cancer were observed (finasteride 1.8% versus placebo 1.1%).
No clinical benefit has been demonstrated in patients with prostate cancer treated with AVODART.
Reproductive and Breast Disorders
In the 3 pivotal placebo-controlled BPH trials with AVODART, each 4 years in duration, there was no evidence of increased sexual adverse reactions (impotence, decreased libido, and ejaculation disorder) or breast disorders with increased duration of treatment. Among these 3 trials, there was 1 case of breast cancer in the dutasteride group and 1 case in the placebo group. No cases of breast cancer were reported in any treatment group in the 4-year CombAT trial or the 4-year REDUCE trial.
The relationship between long-term use of dutasteride and male breast neoplasia is currently unknown.
Combination with Alpha-blocker Therapy (CombAT)
Over 4,800 male subjects with BPH were randomly assigned to receive 0.5-mg AVODART, 0.4-mg tamsulosin, or combination therapy (0.5-mg AVODART plus 0.4-mg tamsulosin) administered once daily in a 4-year double-blind trial. Overall, 1,623 subjects received monotherapy with AVODART; 1,611 subjects received monotherapy with tamsulosin; and 1,610 subjects received combination therapy. The population was aged 49 to 88 years (mean age: 66 years) and 88% were white. Table 2 summarizes adverse reactions reported in at least 1% of subjects in the combination group and at a higher incidence than subjects receiving monotherapy with AVODART or tamsulosin.
| a Combination = AVODART 0.5 mg once daily plus tamsulosin 0.4 mg once daily. b Includes anorgasmia, retrograde ejaculation, semen volume decreased, orgasmic sensation decreased, orgasm abnormal, ejaculation delayed, ejaculation disorder, ejaculation failure, and premature ejaculation. c These sexual adverse reactions are associated with dutasteride treatment (including monotherapy and combination with tamsulosin). These adverse reactions may persist after treatment discontinuation. The role of dutasteride in this persistence is unknown. d Includes erectile dysfunction and disturbance in sexual arousal. e Includes libido decreased, libido disorder, loss of libido, sexual dysfunction, and male sexual dysfunction. f Includes breast enlargement, gynecomastia, breast swelling, breast pain, breast tenderness, nipple pain, and nipple swelling. |
||||||||||
|
Adverse Reaction |
Adverse Reaction Time of Onset |
|||||||||
|
Year 1 | ||||||||||
|
Months 0‑6 |
Months 7‑12 |
Year 2 |
Year 3 |
Year 4 |
||||||
|
Combinationa |
(n = 1,610) |
(n = 1,527) |
(n = 1,428) |
(n = 1,283) |
(n = 1,200) |
|||||
|
AVODART |
(n = 1,623) |
(n = 1,548) |
(n = 1,464) |
(n = 1,325) |
(n = 1,200) |
|||||
|
Tamsulosin |
(n = 1,611) |
(n = 1,545) |
(n = 1,468) |
(n = 1,281) |
(n = 1,112) |
|||||
|
Ejaculation disordersb,c | ||||||||||
|
Combination |
7.8% |
1.6% |
1.0% |
0.5% |
<0.1% |
|||||
|
AVODART |
1.0% |
0.5% |
0.5% |
0.2% |
0.3% |
|||||
|
Tamsulosin |
2.2% |
0.5% |
0.5% |
0.2% |
0.3% |
|||||
|
Impotencec,d | ||||||||||
|
Combination |
5.4% |
1.1% |
1.8% |
0.9% |
0.4% |
|||||
|
AVODART |
4.0% |
1.1% |
1.6% |
0.6% |
0.3% |
|||||
|
Tamsulosin |
2.6% |
0.8% |
1.0% |
0.6% |
1.1% |
|||||
|
Decreased libidoc,e | ||||||||||
|
Combination |
4.5% |
0.9% |
0.8% |
0.2% |
0.0% |
|||||
|
AVODART |
3.1% |
0.7% |
1.0% |
0.2% |
0.0% |
|||||
|
Tamsulosin |
2.0% |
0.6% |
0.7% |
0.2% |
<0.1% |
|||||
|
Breast disordersf | ||||||||||
|
Combination |
1.1% |
1.1% |
0.8% |
0.9% |
0.6% |
|||||
|
AVODART |
0.9% |
0.9% |
1.2% |
0.5% |
0.7% |
|||||
|
Tamsulosin |
0.4% |
0.4% |
0.4% |
0.2% |
0.0% |
|||||
|
Dizziness | ||||||||||
|
Combination |
1.1% |
0.4% |
0.1% |
<0.1% |
0.2% |
|||||
|
AVODART |
0.5% |
0.3% |
0.1% |
<0.1% |
<0.1% |
|||||
|
Tamsulosin |
0.9% |
0.5% |
0.4% |
<0.1% |
0.0% |
|||||
Cardiac Failure: In CombAT, after 4 years of treatment, the incidence of the composite term cardiac failure in the combination therapy group (12/1,610; 0.7%) was higher than in either monotherapy group: AVODART, 2/1,623 (0.1%) and tamsulosin, 9/1,611 (0.6%). Composite cardiac failure was also examined in a separate 4-year placebo-controlled trial evaluating AVODART in men at risk for development of prostate cancer. The incidence of cardiac failure in subjects taking AVODART was 0.6% (26/4,105) compared with 0.4% (15/4,126) in subjects on placebo. A majority of subjects with cardiac failure in both trials had comorbidities associated with an increased risk of cardiac failure. Therefore, the clinical significance of the numerical imbalances in cardiac failure is unknown. No causal relationship between AVODART alone or in combination with tamsulosin and cardiac failure has been established. No imbalance was observed in the incidence of overall cardiovascular adverse events in either trial.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of AVODART. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure. These reactions have been chosen for inclusion due to a combination of their seriousness, frequency of reporting, or potential causal connection to AVODART.
Immune System Disorders
Hypersensitivity reactions, including rash, pruritus, urticaria, localized edema, serious skin reactions, and angioedema.
Neoplasms
Male breast cancer.
Psychiatric Disorders
Depressed mood.
Reproductive System and Breast Disorders
Testicular pain and testicular swelling.
7 DRUG INTERACTIONS
7.1 Cytochrome P450 3A Inhibitors
Dutasteride is extensively metabolized in humans by the cytochrome P450 (CYP)3A4 and CYP3A5 isoenzymes. The effect of potent CYP3A4 inhibitors on dutasteride has not been studied. Because of the potential for drug‑drug interactions, use caution when prescribing AVODART to patients taking potent, chronic CYP3A4 enzyme inhibitors (e.g., ritonavir) [see Clinical Pharmacology (12.3)].
7.2 Alpha-adrenergic Antagonists
The administration of AVODART in combination with tamsulosin or terazosin has no effect on the steady-state pharmacokinetics of either alpha-adrenergic antagonist. The effect of administration of tamsulosin or terazosin on dutasteride pharmacokinetic parameters has not been evaluated.
7.3 Calcium Channel Antagonists
Coadministration of verapamil or diltiazem decreases dutasteride clearance and leads to increased exposure to dutasteride. The change in dutasteride exposure is not considered to be clinically significant. No dose adjustment is recommended [see Clinical Pharmacology (12.3)].
7.4 Cholestyramine
Administration of a single 5-mg dose of AVODART followed 1 hour later by 12 g of cholestyramine does not affect the relative bioavailability of dutasteride [see Clinical Pharmacology (12.3)].
7.5 Digoxin
AVODART does not alter the steady-state pharmacokinetics of digoxin when administered concomitantly at a dose of 0.5 mg/day for 3 weeks [see Clinical Pharmacology (12.3)].
7.6 Warfarin
Concomitant administration of AVODART 0.5 mg/day for 3 weeks with warfarin does not alter the steady-state pharmacokinetics of the S- or R-warfarin isomers or alter the effect of warfarin on prothrombin time [see Clinical Pharmacology (12.3)].
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
AVODART is contraindicated for use in pregnancy because it may cause harm to the male fetus [see Contraindications (4)]. AVODART is not indicated for use in women.
AVODART is a 5 alpha‑reductase inhibitor that prevents conversion of testosterone to dihydrotestosterone (DHT), a hormone necessary for normal development of male genitalia. Abnormalities in the genitalia of male fetuses is an expected physiological consequence of inhibition of this conversion. These results are similar to observations in male infants with genetic 5 alpha‑reductase deficiency.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
In animal reproduction studies, dutasteride inhibited normal development of external genitalia in male offspring when given to rats or rabbits during organogenesis at less than the maximum recommended human dose (MRHD) of 0.5 mg daily, in the absence of maternal toxicity. At 15 times the MRHD, prolonged pregnancy, decreased reproductive organ weights, and delayed puberty in male offspring were observed in rats, with no-effect levels less than the MRHD of 0.5 mg daily. Increased placental weights in rabbits were also observed, with no-effect levels less than the MRHD of 0.5 mg daily (see Data).
Although dutasteride is secreted into human semen, the drug concentration in the human female partner is approximately 100 times less than concentrations producing abnormalities of male genitalia in animal studies (see Data). In monkeys dosed during organogenesis at blood concentrations comparable to or above levels to which a human female partner is estimated to be exposed, male offspring external genitalia was not adversely affected. No feminization occurred in male offspring of untreated female rats mated to treated male rats even though detectable blood levels of dutasteride were observed in the female rats [see Nonclinical Toxicology (13.1)].
Data
Human Data: The highest measured semen concentration of dutasteride in treated men was 14 ng/mL. Although dutasteride is detected in semen, assuming exposure of a 50‑kg woman to 5 mL of semen and 100% absorption, the woman’s expected dutasteride blood concentration through semen would be about 0.0175 ng/mL. This concentration is approximately 100 times less than blood concentrations producing abnormalities of male genitalia in animal studies. Dutasteride is highly protein bound in human semen (greater than 96%), which may reduce the amount of dutasteride available for vaginal absorption.
Animal Data: In an embryo‑fetal development study in rats, oral administration of dutasteride at 10 times less than the MRHD of 0.5 mg daily (based on average blood levels in men) resulted in feminization of male genitalia in the fetus (decreased anogenital distance at 0.05 mg/kg/day, with a lack of a no-effect level) in the absence of maternal toxicity. In addition, nipple development, hypospadias, and distended preputial glands occurred in fetuses of dams treated at doses of 2.5 mg/kg/day or greater (approximately 15 times the MRHD). Reduced fetal body weight and associated delayed ossification in the presence of maternal toxicity (decreased body weight gain) were observed at maternal exposure approximately 15 times the MRHD (dose of 2.5 mg/kg/day or greater). An increase in stillborn pups was observed in dams treated at 30 mg/kg/day (approximately 111 times the MRHD), with a no-effect level of 12.5 mg/kg/day.
In a rabbit embryo-fetal development study, doses 28 times the MRHD (doses of 30 mg/kg/day or greater), based on average blood levels in men, were administered orally on Gestation Days 7 to 29 (during organogenesis and the late period of external genitalia development). Histological evaluation of the genital papilla of fetuses revealed evidence of feminization of the male fetus as well as fused skull bones and increased placental weights at all doses in the absence of maternal toxicity. A second embryo-fetal development study in rabbits dosed throughout pregnancy (organogenesis and later period of external genitalia development [Gestation Days 6 to 29]) at 0.3 times the MRHD (doses of 0.05 mg/kg/day or greater, with no no-effect level), also produced evidence of feminization of the genitalia in male fetuses and increased placental weights at all doses in the absence of maternal toxicity.
In an embryo-fetal development study, pregnant rhesus monkeys were exposed intravenously during organogenesis (Gestation Days 20 to 100) to a dutasteride blood level comparable to or above the estimated dutasteride exposure of a human female partner. Dutasteride was administered on Gestation Days 20 to 100 (during organogenesis) at doses of 400, 780, 1,325, or 2,010 ng/day (12 monkeys/group). No feminization of male external genitalia of monkey offspring was observed. Reduction of fetal adrenal weights, reduction in fetal prostate weights, and increases in fetal ovarian and testis weights were observed at the highest dose tested. Based on the highest measured semen concentration of dutasteride in treated men (14 ng/mL), these doses in the monkey represent up to 16 times the potential maximum exposure of a 50‑kg human female to 5 mL of semen daily from a dutasteride-treated male, assuming 100% absorption. The dose levels (on a ng/kg basis) administered to monkeys in this study are 32 to 186 times the nominal (ng/kg) dose to which a female would potentially be exposed via the semen. It is not known whether rabbits or rhesus monkeys produce any of the major human metabolites.
In an oral pre- and post-natal development study in rats, feminization of the male genitalia was observed. Decreased anogenital distance was observed at 0.05 times the MRHD and greater (0.05 mg/kg/day and greater), with a lack of a no-effect level, based on average blood levels in men as an estimation of AUC. Hypospadias and nipple development were observed at 2.5 mg/kg/day or greater (14 times the MRHD or greater, with a no-effect level at 0.05 mg/kg/day). Doses of 2.5 mg/kg/day and greater also resulted in prolonged gestation in the parental females, an increase in time to balano-preputial separation in male offspring, a decrease in time to vaginal patency for female offspring, and a decrease in prostate and seminal vesicle weights in male offspring. Increased stillbirths and decreased neonatal viability in offspring were noted at 30 mg/kg/day (102 times the MRHD in the presence of maternal toxicity [decreased body weights]).
8.2 Lactation
Risk Summary
AVODART is not indicated for use in women. There is no information available on the presence of dutasteride in human milk, the effects on the breastfed child, or the effects on milk production.
8.3 Females and Males of Reproductive Potential
Infertility
Males: The effects of dutasteride 0.5 mg/day on semen characteristics were evaluated in normal volunteers aged 18 to 52 years (n = 27 dutasteride, n = 23 placebo) throughout 52 weeks of treatment and 24 weeks of post-treatment follow-up. At 52 weeks, the mean percent reductions from baseline in total sperm count, semen volume, and sperm motility were 23%, 26%, and 18%, respectively, in the dutasteride group when adjusted for changes from baseline in the placebo group. Sperm concentration and sperm morphology were unaffected. After 24 weeks of follow-up, the mean percent change in total sperm count in the dutasteride group remained 23% lower than baseline. While mean values for all semen parameters at all timepoints remained within the normal ranges and did not meet predefined criteria for a clinically significant change (30%), 2 subjects in the dutasteride group had decreases in sperm count of greater than 90% from baseline at 52 weeks, with partial recovery at the 24-week follow-up. The clinical significance of dutasteride’s effect on semen characteristics for an individual patient’s fertility is not known [see Warnings and Precautions (5.6)].
8.4 Pediatric Use
AVODART is not indicated for use in pediatric patients. Safety and effectiveness in pediatric patients have not been established.
8.5 Geriatric Use
Of 2,167 male subjects treated with AVODART in 3 clinical trials, 60% were aged 65 years and older and 15% were aged 75 years and older. No overall differences in safety or efficacy were observed between these subjects and younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out [see Clinical Pharmacology (12.3)].
8.6 Renal Impairment
No dose adjustment is necessary for AVODART in patients with renal impairment [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
The effect of hepatic impairment on dutasteride pharmacokinetics has not been studied. Because dutasteride is extensively metabolized, exposure could be higher in hepatically impaired patients. However, in a clinical trial where 60 subjects received 5 mg (10 times the therapeutic dose) daily for 24 weeks, no additional adverse events were observed compared with those observed at the therapeutic dose of 0.5 mg [see Clinical Pharmacology (12.3)].
10 OVERDOSAGE
In volunteer trials, single doses of dutasteride up to 40 mg (80 times the therapeutic dose) for 7 days have been administered without significant safety concerns. In a clinical trial, daily doses of 5 mg (10 times the therapeutic dose) were administered to 60 subjects for 6 months with no additional adverse effects to those seen at therapeutic doses of 0.5 mg.
There is no specific antidote for dutasteride. Therefore, in cases of suspected overdosage, symptomatic and supportive treatment should be given as appropriate, taking the long half-life of dutasteride into consideration.
11 DESCRIPTION
AVODART is a synthetic 4-azasteroid compound that is a selective inhibitor of both the type 1 and type 2 isoforms of steroid 5 alpha-reductase, an intracellular enzyme that converts testosterone to DHT.
Dutasteride is chemically designated as (5α,17β)-N-{2,5 bis(trifluoromethyl)phenyl}-3-oxo-4-azaandrost-1-ene-17-carboxamide. The empirical formula of dutasteride is C27H30F6N2O2, representing a molecular weight of 528.5 with the following structural formula:
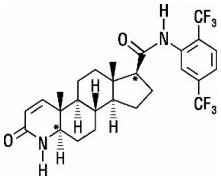
Dutasteride is a white to pale yellow powder with a melting point of 242° to 250°C. It is soluble in ethanol (44 mg/mL), methanol (64 mg/mL), and polyethylene glycol 400 (3 mg/mL), but it is insoluble in water.
Each AVODART soft gelatin capsule, administered orally, contains 0.5 mg of dutasteride dissolved in a mixture of mono-di-glycerides of caprylic/capric acid and butylated hydroxytoluene. The inactive excipients in the capsule shell are ferric oxide (yellow), gelatin (from certified BSE-free bovine sources), glycerin, and titanium dioxide. The soft gelatin capsules are printed with edible red ink.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Dutasteride inhibits the conversion of testosterone to DHT. DHT is the androgen primarily responsible for the initial development and subsequent enlargement of the prostate gland. Testosterone is converted to DHT by the enzyme 5 alpha-reductase, which exists as 2 isoforms, type 1 and type 2. The type 2 isoenzyme is primarily active in the reproductive tissues, while the type 1 isoenzyme is also responsible for testosterone conversion in the skin and liver.
Dutasteride is a competitive and specific inhibitor of both type 1 and type 2 5 alpha-reductase isoenzymes, with which it forms a stable enzyme complex. Dissociation from this complex has been evaluated under in vitro and in vivo conditions and is extremely slow. Dutasteride does not bind to the human androgen receptor.
12.2 Pharmacodynamics
Effect on 5 Alpha-Dihydrotestosterone and Testosterone
The maximum effect of daily doses of dutasteride on the reduction of DHT is dose dependent and is observed within 1 to 2 weeks. After 1 and 2 weeks of daily dosing with dutasteride 0.5 mg, median serum DHT concentrations were reduced by 85% and 90%, respectively. In patients with BPH treated with dutasteride 0.5 mg/day for 4 years, the median decrease in serum DHT was 94% at 1 year, 93% at 2 years, and 95% at both 3 and 4 years. The median increase in serum testosterone was 19% at both 1 and 2 years, 26% at 3 years, and 22% at 4 years, but the mean and median levels remained within the physiologic range.
In patients with BPH treated with 5 mg/day of dutasteride or placebo for up to 12 weeks prior to transurethral resection of the prostate, mean DHT concentrations in prostatic tissue were significantly lower in the dutasteride group compared with placebo (784 and 5,793 pg/g, respectively, P <0.001). Mean prostatic tissue concentrations of testosterone were significantly higher in the dutasteride group compared with placebo (2,073 and 93 pg/g, respectively, P <0.001).
Adult males with genetically inherited type 2 5 alpha-reductase deficiency also have decreased DHT levels. These 5 alpha-reductase-deficient males have a small prostate gland throughout life and do not develop BPH. Except for the associated urogenital defects present at birth, no other clinical abnormalities related to 5 alpha-reductase deficiency have been observed in these individuals.
Effects on Other Hormones
In healthy volunteers, 52 weeks of treatment with dutasteride 0.5 mg/day (n = 26) resulted in no clinically significant change compared with placebo (n = 23) in sex hormone-binding globulin, estradiol, luteinizing hormone, follicle-stimulating hormone, thyroxine (free T4), and dehydroepiandrosterone. Statistically significant, baseline-adjusted mean increases compared with placebo were observed for total testosterone at 8 weeks (97.1 ng/dL, P <0.003) and thyroid-stimulating hormone at 52 weeks (0.4 mcIU/mL, P <0.05). The median percentage changes from baseline within the dutasteride group were 17.9% for testosterone at 8 weeks and 12.4% for thyroid-stimulating hormone at 52 weeks. After stopping dutasteride for 24 weeks, the mean levels of testosterone and thyroid-stimulating hormone had returned to baseline in the group of subjects with available data at the visit. In subjects with BPH treated with dutasteride in a large randomized, double-blind, placebo-controlled trial, there was a median percent increase in luteinizing hormone of 12% at 6 months and 19% at both 12 and 24 months.
Other Effects
Plasma lipid panel and bone mineral density were evaluated following 52 weeks of dutasteride 0.5 mg once daily in healthy volunteers. There was no change in bone mineral density as measured by dual energy x-ray absorptiometry compared with either placebo or baseline. In addition, the plasma lipid profile (i.e., total cholesterol, low density lipoproteins, high density lipoproteins, triglycerides) was unaffected by dutasteride. No clinically significant changes in adrenal hormone responses to adrenocorticotropic hormone (ACTH) stimulation were observed in a subset population (n = 13) of the 1-year healthy volunteer trial.
12.3 Pharmacokinetics
Absorption
Following administration of a single 0.5-mg dose of a soft gelatin capsule, time to peak serum concentrations (Tmax) of dutasteride occurs within 2 to 3 hours. Absolute bioavailability in 5 healthy subjects is approximately 60% (range: 40% to 94%). When the drug is administered with food, the maximum serum concentrations were reduced by 10% to 15%. This reduction is of no clinical significance.
Distribution
Pharmacokinetic data following single and repeat oral doses show that dutasteride has a large volume of distribution (300 to 500 L). Dutasteride is highly bound to plasma albumin (99.0%) and alpha-1 acid glycoprotein (96.6%).
In a trial of healthy subjects (n = 26) receiving dutasteride 0.5 mg/day for 12 months, semen dutasteride concentrations averaged 3.4 ng/mL (range: 0.4 to 14 ng/mL) at 12 months and, similar to serum, achieved steady-state concentrations at 6 months. On average, at 12 months 11.5% of serum dutasteride concentrations partitioned into semen.
Metabolism and Elimination
Dutasteride is extensively metabolized in humans. In vitro studies showed that dutasteride is metabolized by the CYP3A4 and CYP3A5 isoenzymes. Both of these isoenzymes produced the 4′-hydroxydutasteride, 6-hydroxydutasteride, and the 6,4′-dihydroxydutasteride metabolites. In addition, the 15-hydroxydutasteride metabolite was formed by CYP3A4. Dutasteride is not metabolized in vitro by human cytochrome P450 isoenzymes CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP2E1. In human serum following dosing to steady state, unchanged dutasteride, 3 major metabolites (4′-hydroxydutasteride, 1,2-dihydrodutasteride, and 6-hydroxydutasteride), and 2 minor metabolites (6,4′-dihydroxydutasteride and 15-hydroxydutasteride), as assessed by mass spectrometric response, have been detected. The absolute stereochemistry of the hydroxyl additions in the 6 and 15 positions is not known. In vitro, the 4′-hydroxydutasteride and 1,2-dihydrodutasteride metabolites are much less potent than dutasteride against both isoforms of human 5 alpha-reductase. The activity of 6β-hydroxydutasteride is comparable to that of dutasteride.
Dutasteride and its metabolites were excreted mainly in feces. As a percent of dose, there was approximately 5% unchanged dutasteride (~1% to ~15%) and 40% as dutasteride-related metabolites (~2% to ~90%). Only trace amounts of unchanged dutasteride were found in urine (<1%). Therefore, on average, the dose unaccounted for approximated 55% (range: 5% to 97%).
The terminal elimination half-life of dutasteride is approximately 5 weeks at steady state. The average steady-state serum dutasteride concentration was 40 ng/mL following 0.5 mg/day for 1 year. Following daily dosing, dutasteride serum concentrations achieve 65% of steady-state concentration after 1 month and approximately 90% after 3 months. Due to the long half-life of dutasteride, serum concentrations remain detectable (greater than 0.1 ng/mL) for up to 4 to 6 months after discontinuation of treatment.
Specific Populations
Pediatric Patients: Dutasteride pharmacokinetics have not been investigated in subjects younger than 18 years.
Geriatric Patients: No dose adjustment is necessary in the elderly. The pharmacokinetics and pharmacodynamics of dutasteride were evaluated in 36 healthy male subjects aged between 24 and 87 years following administration of a single 5‑mg dose of dutasteride. In this single‑dose trial, dutasteride half‑life increased with age (approximately 170 hours in men aged 20 to 49 years, approximately 260 hours in men aged 50 to 69 years, and approximately 300 hours in men older than 70 years). Of 2,167 men treated with dutasteride in the 3 pivotal trials, 60% were age 65 and over and 15% were age 75 and over. No overall differences in safety or efficacy were observed between these patients and younger patients.
Male and Female Patients: AVODART is contraindicated in pregnancy and is not indicated for use in women [see Contraindications (4), Warnings and Precautions (5.1)]. The pharmacokinetics of dutasteride in women have not been studied.
Racial and Ethnic Groups: The effect of race on dutasteride pharmacokinetics has not been studied.
Patients with Renal Impairment: The effect of renal impairment on dutasteride pharmacokinetics has not been studied. However, less than 0.1% of a steady‑state 0.5‑mg dose of dutasteride is recovered in human urine, so no adjustment in dosage is anticipated for patients with renal impairment.
Patients with Hepatic Impairment: The effect of hepatic impairment on dutasteride pharmacokinetics has not been studied. Because dutasteride is extensively metabolized, exposure could be higher in hepatically impaired patients.
Drug Interaction Studies
Cytochrome P450 Inhibitors: No clinical drug interaction trials have been performed to evaluate the impact of CYP3A enzyme inhibitors on dutasteride pharmacokinetics. However, based on in vitro data, blood concentrations of dutasteride may increase in the presence of inhibitors of CYP3A4/5 such as ritonavir, ketoconazole, verapamil, diltiazem, cimetidine, troleandomycin, and ciprofloxacin.
Dutasteride does not inhibit the in vitro metabolism of model substrates for the major human cytochrome P450 isoenzymes (CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4) at a concentration of 1,000 ng/mL, 25 times greater than steady-state serum concentrations in humans.
Alpha-adrenergic Antagonists: In a single-sequence, crossover trial in healthy volunteers, the administration of tamsulosin or terazosin in combination with AVODART had no effect on the steady-state pharmacokinetics of either alpha-adrenergic antagonist. Although the effect of administration of tamsulosin or terazosin on dutasteride pharmacokinetic parameters was not evaluated, the percent change in DHT concentrations was similar for AVODART alone compared with the combination treatment.
Calcium Channel Antagonists: In a population pharmacokinetics analysis, a decrease in clearance of dutasteride was noted when coadministered with the CYP3A4 inhibitors verapamil (-37%, n = 6) and diltiazem (-44%, n = 5). In contrast, no decrease in clearance was seen when amlodipine, another calcium channel antagonist that is not a CYP3A4 inhibitor, was coadministered with dutasteride (+7%, n = 4).
The decrease in clearance and subsequent increase in exposure to dutasteride in the presence of verapamil and diltiazem is not considered to be clinically significant. No dose adjustment is recommended.
Cholestyramine: Administration of a single 5-mg dose of AVODART followed 1 hour later by 12 g cholestyramine did not affect the relative bioavailability of dutasteride in 12 normal volunteers.
Digoxin: In a trial of 20 healthy volunteers, AVODART did not alter the steady-state pharmacokinetics of digoxin when administered concomitantly at a dose of 0.5 mg/day for 3 weeks.
Warfarin: In a trial of 23 healthy volunteers, 3 weeks of treatment with AVODART 0.5 mg/day did not alter the steady-state pharmacokinetics of the S- or R-warfarin isomers or alter the effect of warfarin on prothrombin time when administered with warfarin.
Other Concomitant Therapy: Although specific interaction trials were not performed with other compounds, approximately 90% of the subjects in the 3 randomized, double-blind, placebo-controlled safety and efficacy trials receiving AVODART were taking other medications concomitantly. No clinically significant adverse interactions could be attributed to the combination of AVODART and concurrent therapy when AVODART was coadministered with anti-hyperlipidemics, angiotensin-converting enzyme (ACE) inhibitors, beta-adrenergic blocking agents, calcium channel blockers, corticosteroids, diuretics, nonsteroidal anti-inflammatory drugs (NSAIDs), phosphodiesterase Type V inhibitors, and quinolone antibiotics.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
A 2-year carcinogenicity study was conducted in B6C3F1 mice at doses of 3, 35, 250, and 500 mg/kg/day for males and 3, 35, and 250 mg/kg/day for females; an increased incidence of benign hepatocellular adenomas was noted at 250 mg/kg/day (290-fold the MRHD of a 0.5-mg daily dose) in female mice only. Two of the 3 major human metabolites have been detected in mice. The exposure to these metabolites in mice is either lower than in humans or is not known.
In a 2-year carcinogenicity study in Han Wistar rats, at doses of 1.5, 7.5, and 53 mg/kg/day in males and 0.8, 6.3, and 15 mg/kg/day in females, there was an increase in Leydig cell adenomas in the testes at 135-fold the MRHD (53 mg/kg/day and greater). An increased incidence of Leydig cell hyperplasia was present at 52-fold the MRHD (male rat doses of 7.5 mg/kg/day and greater). A positive correlation between proliferative changes in the Leydig cells and an increase in circulating luteinizing hormone levels has been demonstrated with 5 alpha-reductase inhibitors and is consistent with an effect on the hypothalamic-pituitary-testicular axis following 5 alpha-reductase inhibition. At tumorigenic doses, luteinizing hormone levels in rats were increased by 167%. In this study, the major human metabolites were tested for carcinogenicity at approximately 1 to 3 times the expected clinical exposure.
Mutagenesis
Dutasteride was tested for genotoxicity in a bacterial mutagenesis assay (Ames test), a chromosomal aberration assay in Chinese hamster ovary cells, and a micronucleus assay in rats. The results did not indicate any genotoxic potential of the parent drug. Two major human metabolites were also negative in either the Ames test or an abbreviated Ames test.
Impairment of Fertility
Treatment of sexually mature male rats with dutasteride at 0.1 times the MRHD (animal doses of 0.05 mg/kg/day or greater for up to 31 weeks) based on mean serum concentration resulted in dose- and time‑dependent decreases in fertility at all doses; reduced cauda epididymal (absolute) sperm counts but not sperm concentration (at 50 and 500 mg/kg/day); reduced weights of the epididymis, prostate, and seminal vesicles; and microscopic changes (cytoplasmic vacuolation of tubular epithelium in the epididymides and/or decreased cytoplasmic content of epithelium, consistent with decreased secretory activity in the prostate and seminal vesicles) in the reproductive organs at all doses in the absence of paternal toxicity. The fertility effects were reversed by Recovery Week 6 at all doses, and sperm counts were normal at the end of a 14‑week recovery period. The microscopic changes were no longer present at Recovery Week 14 at 0.1 times the MRHD and were partly recovered in the remaining treatment groups. Low levels of dutasteride (0.6 to 17 ng/mL) were detected in the serum of untreated female rats mated to treated males (10 to 500 mg/kg/day for 29 to 30 weeks) which are 16 to 110 times the MRHD based on mean serum concentration. No feminization occurred in male offspring of untreated female rats mated to treated male rats even though detectable blood levels of dutasteride were observed in the female rats.
In a fertility study in female rats with dosing 4 weeks prior to mating through early gestation, oral administration of dutasteride at doses of 0.05, 2.5, 12.5, and 30 mg/kg/day resulted in reduced litter size due to increased resorptions and in feminization of male fetuses (decreased anogenital distance) at 2- to 10 times the MRHD (animal doses of 2.5 mg/kg/day or greater) based on mean serum concentration, in the presence of maternal toxicity (decreased body weight gain). Fetal body weights were also reduced at approximately 0.02 times the MRHD (rat dose of 0.05 mg/kg/day or greater) based on mean serum concentration, with no no-effect level, in the absence of maternal toxicity.
13.2 Animal Toxicology and/or Pharmacology
Central Nervous System Toxicology Studies
In rats and dogs, repeated oral administration of dutasteride resulted in some animals showing signs of non-specific, reversible, centrally-mediated toxicity without associated histopathological changes at exposures 425- and 315-fold the expected clinical exposure (of parent drug), respectively.
Rabbit Dermal Absorption
In a rabbit dermal pharmacokinetics study, dermal absorption of dutasteride in CAPMUL (glyceryl oleate) in rabbits resulted in serum concentrations of 2.7 to 40.5 mcg/h/mL for doses of 1 to 20 mg/mL, respectively, or 56% to 100% of applied dutasteride to be absorbed under occluded and prolonged conditions. AVODART soft gelatin capsules administered orally contain 0.5 mg dutasteride dissolved in a mixture of mono‑di‑glycerides of caprylic/capric acid and butylated hydroxytoluene. Dutasteride in water was minimally absorbed in rabbits (2,000 mg/kg).
14 CLINICAL STUDIES
14.1 Monotherapy
AVODART 0.5 mg/day (n = 2,167) or placebo (n = 2,158) was evaluated in male subjects with BPH in three 2‑year multicenter, placebo‑controlled, double‑blind trials, each with 2‑year open‑label extensions (n = 2,340). More than 90% of the trial population was white. Subjects were aged at least 50 years with a serum PSA ≥1.5 ng/mL and <10 ng/mL and BPH diagnosed by medical history and physical examination, including enlarged prostate (≥30 cc) and BPH symptoms that were moderate to severe according to the American Urological Association Symptom Index (AUA‑SI). Most of the 4,325 subjects randomly assigned to receive either dutasteride or placebo completed 2 years of double‑blind treatment (70% and 67%, respectively). Most of the 2,340 subjects in the trial extensions completed 2 additional years of open‑label treatment (71%).
Effect on Symptom Scores
Symptoms were quantified using the AUA-SI, a questionnaire that evaluates urinary symptoms (incomplete emptying, frequency, intermittency, urgency, weak stream, straining, and nocturia) by rating on a 0 to 5 scale for a total possible score of 35, with higher numerical total symptom scores representing greater severity of symptoms. The baseline AUA-SI score across the 3 trials was approximately 17 units in both treatment groups.
Subjects receiving dutasteride achieved statistically significant improvement in symptoms versus placebo by Month 3 in 1 trial and by Month 12 in the other 2 pivotal trials. At Month 12, the mean decrease from baseline in AUA‑SI total symptom scores across the 3 trials pooled was ‑3.3 units for dutasteride and ‑2.0 units for placebo with a mean difference between the 2 treatment groups of ‑1.3 (range: ‑1.1 to ‑1.5 units in each of the 3 trials, P <0.001) and was consistent across the 3 trials. At Month 24, the mean decrease from baseline was ‑3.8 units for dutasteride and ‑1.7 units for placebo with a mean difference of -2.1 (range: ‑1.9 to ‑2.2 units in each of the 3 trials, P <0.001). See Figure 1. The improvement in BPH symptoms seen during the first 2 years of double‑blind treatment was maintained throughout an additional 2 years of open‑label extension trials.
These trials were prospectively designed to evaluate effects on symptoms based on prostate size at baseline. In men with prostate volumes ≥40 cc, the mean decrease was -3.8 units for dutasteride and -1.6 units for placebo, with a mean difference between the 2 treatment groups of -2.2 at Month 24. In men with prostate volumes <40 cc, the mean decrease was -3.7 units for dutasteride and -2.2 units for placebo, with a mean difference between the 2 treatment groups of -1.5 at Month 24.
Figure 1. AUA-SI Scorea Change from Baseline (Randomized, Double-blind, Placebo-Controlled Trials Pooled)
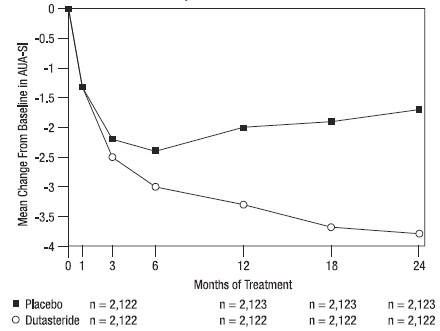
a AUA-SI score ranges from 0 to 35.
Effect on Acute Urinary Retention and the Need for BPH-Related Surgery
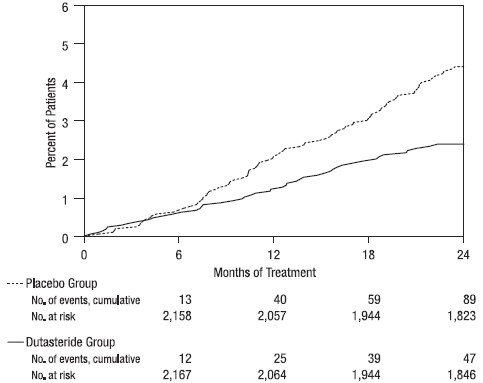
Efficacy was also assessed after 2 years of treatment by the incidence of AUR requiring catheterization and BPH‑related urological surgical intervention. Compared with placebo, AVODART was associated with a statistically significantly lower incidence of AUR (1.8% for AVODART versus 4.2% for placebo, P <0.001; 57% reduction in risk, [95% CI: 38% to 71%]) and with a statistically significantly lower incidence of surgery (2.2% for AVODART versus 4.1% for placebo, P <0.001; 48% reduction in risk, [95% CI: 26% to 63%]). See Figures 2 and 3.
Figure 2. Percent of Subjects Developing Acute Urinary Retention over a 24-Month Period (Randomized, Double-blind, Placebo-Controlled Trials Pooled)
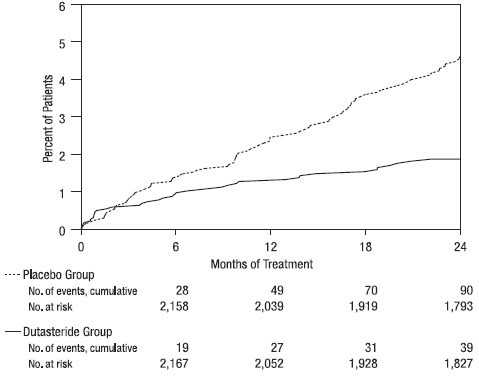
Figure 3. Percent of Subjects Having Surgery for Benign Prostatic Hyperplasia over a 24-Month Period (Randomized, Double-blind, Placebo-Controlled Trials Pooled)
Effect on Prostate Volume
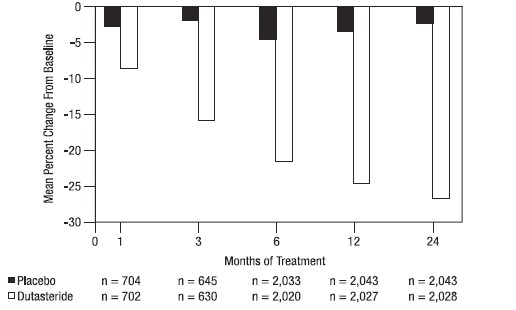
A prostate volume of at least 30 cc measured by transrectal ultrasound was required for trial entry. The mean prostate volume at trial entry was approximately 54 cc.
Statistically significant differences (AVODART versus placebo) were noted at the earliest post-treatment prostate volume measurement in each trial (Month 1, Month 3, or Month 6) and continued through Month 24. At Month 12, the mean percent change in prostate volume across the 3 trials pooled was -24.7% for dutasteride and -3.4% for placebo; the mean difference (dutasteride minus placebo) was -21.3% (range: -21.0% to -21.6% in each of the 3 trials, P <0.001). At Month 24, the mean percent change in prostate volume across the 3 trials pooled was -26.7% for dutasteride and -2.2% for placebo with a mean difference of -24.5% (range: ‑24.0% to ‑25.1% in each of the 3 trials, P <0.001). See Figure 4. The reduction in prostate volume seen during the first 2 years of double-blind treatment was maintained throughout an additional 2 years of open‑label extension trials.
Figure 4. Prostate Volume Percent Change from Baseline (Randomized, Double-blind, Placebo-Controlled Trials Pooled)
Effect on Maximum Urine Flow Rate
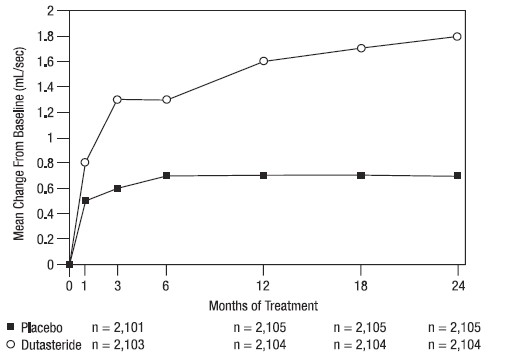
A mean peak urine flow rate (Qmax) of ≤15 mL/sec was required for trial entry. Qmax was approximately 10 mL/sec at baseline across the 3 pivotal trials.
Differences between the 2 groups were statistically significant from baseline at Month 3 in all 3 trials and were maintained through Month 12. At Month 12, the mean increase in Qmax across the 3 trials pooled was 1.6 mL/sec for AVODART and 0.7 mL/sec for placebo; the mean difference (dutasteride minus placebo) was 0.8 mL/sec (range: 0.7 to 1.0 mL/sec in each of the 3 trials, P <0.001). At Month 24, the mean increase in Qmax was 1.8 mL/sec for dutasteride and 0.7 mL/sec for placebo, with a mean difference of 1.1 mL/sec (range: 1.0 to 1.2 mL/sec in each of the 3 trials, P <0.001). See Figure 5. The increase in maximum urine flow rate seen during the first 2 years of double‑blind treatment was maintained throughout an additional 2 years of open-label extension trials.
Figure 5. Qmax Change from Baseline (Randomized, Double-blind, Placebo-Controlled Trials Pooled)
Summary of Clinical Trials
Data from 3 large, well-controlled efficacy trials demonstrate that treatment with AVODART (0.5 mg once daily) reduces the risk of both AUR and BPH-related surgical intervention relative to placebo, improves BPH-related symptoms, decreases prostate volume, and increases maximum urinary flow rates. These data suggest that AVODART arrests the disease process of BPH in men with an enlarged prostate.
14.2 Combination with Alpha-blocker Therapy (CombAT)
The efficacy of combination therapy (AVODART 0.5 mg/day plus tamsulosin 0.4 mg/day, n = 1,610) was compared with AVODART alone (n = 1,623) or tamsulosin alone (n = 1,611) in a 4‑year multicenter, randomized, double‑blind trial. Trial entry criteria were similar to the double‑blind, placebo‑controlled monotherapy efficacy trials described in Section 14.1. Eighty‑eight percent (88%) of the enrolled trial population was white. Approximately 52% of subjects had previous exposure to 5 alpha‑reductase–inhibitor or alpha-adrenergic–antagonist treatment. Of the 4,844 subjects randomly assigned to receive treatment, 69% of subjects in the combination group, 67% in the group receiving AVODART, and 61% in the tamsulosin group completed 4 years of double‑blind treatment.
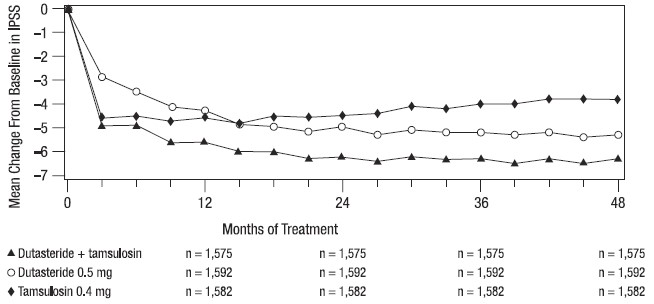
Effect on Symptom Score
Symptoms were quantified using the first 7 questions of the International Prostate Symptom Score (IPSS) (identical to the AUA‑SI). The baseline score was approximately 16.4 units for each treatment group. Combination therapy was statistically superior to each of the monotherapy treatments in decreasing symptom score at Month 24, the primary time point for this endpoint. At Month 24 the mean changes from baseline (±SD) in IPSS total symptom scores were -6.2 (±7.14) for combination, -4.9 (±6.81) for AVODART, and -4.3 (±7.01) for tamsulosin, with a mean difference between combination and AVODART of ‑1.3 units (P <0.001; [95% CI: ‑1.69, ‑0.86]), and between combination and tamsulosin of ‑1.8 units (P <0.001; [95% CI: ‑2.23, ‑1.40]). A significant difference was seen by Month 9 and continued through Month 48. At Month 48 the mean changes from baseline (±SD) in IPSS total symptom scores were -6.3 (±7.40) for combination, -5.3 (±7.14) for AVODART, and -3.8 (±7.74) for tamsulosin, with a mean difference between combination and AVODART of ‑0.96 units (P <0.001; [95% CI: ‑1.40, ‑0.52]), and between combination and tamsulosin of ‑2.5 units (P <0.001; [95% CI: ‑2.96, ‑2.07]). See Figure 6.
Figure 6. International Prostate Symptom Score Change from Baseline over a 48-Month Period (Randomized, Double-blind, Parallel-Group Trial [CombAT Trial])
Effect on Acute Urinary Retention or the Need for BPH-Related Surgery
After 4 years of treatment, combination therapy with AVODART and tamsulosin did not provide benefit over monotherapy with AVODART in reducing the incidence of AUR or BPH-related surgery.
Effect on Maximum Urine Flow Rate
The baseline Qmax was approximately 10.7 mL/sec for each treatment group. Combination therapy was statistically superior to each of the monotherapy treatments in increasing Qmax at Month 24, the primary time point for this endpoint. At Month 24, the mean increases from baseline (±SD) in Qmax were 2.4 (±5.26) mL/sec for combination, 1.9 (±5.10) mL/sec for AVODART, and 0.9 (±4.57) mL/sec for tamsulosin, with a mean difference between combination and AVODART of 0.5 mL/sec (P = 0.003; [95% CI: 0.17, 0.84]), and between combination and tamsulosin of 1.5 mL/sec (P <0.001; [95% CI: 1.19, 1.86]). This difference was seen by Month 6 and continued through Month 24. See Figure 7.
The additional improvement in Qmax of combination therapy over monotherapy with AVODART was no longer statistically significant at Month 48.
Figure 7. Qmax Change from Baseline over a 24-Month Period (Randomized, Double-blind, Parallel-Group Trial [CombAT Trial])
![Figure 7. Q-max Change Ffrom Baseline Over a 24-Month Period (Randomized, Double-Blind, Parallel Group Study [CombAT Study])](/dailymed/image.cfm?name=avodart-spl-image-08.jpg&id=749719)
Effect on Prostate Volume
The mean prostate volume at trial entry was approximately 55 cc. At Month 24, the primary time point for this endpoint, the mean percent changes from baseline (±SD) in prostate volume were -26.9% (±22.57) for combination therapy, -28.0% (±24.88) for AVODART, and 0% (±31.14) for tamsulosin, with a mean difference between combination and AVODART of 1.1% (P = NS; [95% CI: -0.6, 2.8]), and between combination and tamsulosin of -26.9% (P <0.001; [95% CI: -28.9, -24.9]). Similar changes were seen at Month 48: -27.3% (±24.91) for combination therapy, -28.0% (±25.74) for AVODART, and +4.6% (±35.45) for tamsulosin.
16 HOW SUPPLIED/STORAGE AND HANDLING
AVODART soft gelatin capsules 0.5 mg are oblong, opaque, dull yellow, gelatin capsules imprinted with “GX CE2” with red edible ink on one side, packaged in bottles of 30 (NDC 80725-712-15) and 90 (NDC 80725-712-04) with child-resistant closures.
Store at 25°C (77°F); excursions permitted to 15° to 30°C (59° to 86°F) [see USP Controlled Room Temperature].
Dutasteride is absorbed through the skin. AVODART capsules should not be handled by women who are pregnant or who could become pregnant because of the potential for absorption of dutasteride and the subsequent potential risk to a developing male fetus [see Warnings and Precautions (5.4)].
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
PSA Monitoring
Inform patients that AVODART reduces serum PSA levels by approximately 50% within 3 to 6 months of therapy, although it may vary for each individual. For patients undergoing PSA screening, increases in PSA levels while on treatment with AVODART may signal the presence of prostate cancer and should be evaluated by a healthcare provider [see Warnings and Precautions (5.1)].
Increased Risk of High-grade Prostate Cancer
Inform patients that there was an increase in high-grade prostate cancer in men treated with 5 alpha-reductase inhibitors (which are indicated for BPH treatment), including AVODART, compared with those treated with placebo in trials looking at the use of these drugs to reduce the risk of prostate cancer [see Indications and Usage (1.3), Warnings and Precautions (5.2), Adverse Reactions (6.1)].
Transdermal Exposure of AVODART in Pregnant or Potentially Pregnant Women—Risk to Male Fetus
Inform patients that AVODART capsules should not be handled by women who are pregnant or may potentially be pregnant because of the potential for absorption of dutasteride and the subsequent potential risk to a developing male fetus. Dutasteride can be absorbed through the skin and could result in unintended fetal exposure. If a pregnant or potentially pregnant woman comes in contact with leaking AVODART capsules, the contact area should be washed immediately with soap and water [see Warnings and Precautions (5.4), Use in Specific Populations (8.1)].
Effects on Semen Parameters
Advise men that AVODART may affect sperm characteristics but the effect on fertility is unknown [see Warnings and Precautions (5.6), Use in Specific Populations (8.3)].
Blood Donation
Inform men treated with AVODART that they should not donate blood until at least 6 months following their last dose to prevent pregnant women from receiving dutasteride through blood transfusion [see Warnings and Precautions (5.5)]. Serum levels of dutasteride are detectable for 4 to 6 months after treatment ends [see Clinical Pharmacology (12.3)].
AVODART is a trademark used under license by Waylis Therapeutics LLC.
The other brands listed are trademarks owned by or licensed to their respective owners and are not owned by or licensed to Waylis Therapeutics LLC. The makers of these brands are not affiliated with and do not endorse Waylis Therapeutics LLC or its products.
Manufactured for:
Waylis Therapeutics LLC
Wixom, MI 48393
PHARMACIST-DETACH HERE AND GIVE INSTRUCTIONS TO PATIENT
----------------------------------------------------------------------------------------------------------
|
PATIENT INFORMATION |
|
|
AVODART (av’ ō dart) (dutasteride) capsules |
|
|
AVODART is for use by men only. |
|
|
What is AVODART? AVODART is a prescription medicine that contains dutasteride. AVODART is used to treat the symptoms of benign prostatic hyperplasia (BPH) in men with an enlarged prostate to:
Prostate growth is caused by a hormone in the blood called dihydrotestosterone (DHT). AVODART lowers DHT production in the body, leading to shrinkage of the enlarged prostate in most men. While some men have fewer problems and symptoms after 3 months of treatment with AVODART, a treatment period of at least 6 months is usually necessary to see if AVODART will work for you. |
|
|
Do not take AVODART if you are:
|
|
|
Before you take AVODART, tell your healthcare provider about all of your medical conditions, including if you:
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. AVODART and other medicines may affect each other, causing side effects. AVODART may affect the way other medicines work, and other medicines may affect how AVODART works. Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine. |
|
|
How should I take AVODART?
|
|
|
What should I avoid while taking AVODART?
|
|
|
What are the possible side effects of AVODART? AVODART may cause serious side effects, including:
The most common side effects of AVODART include:
*Some of these events may continue after you stop taking AVODART. Depressed mood has been reported in patients receiving AVODART. AVODART has been shown to reduce sperm count, semen volume, and sperm movement. However, the effect of AVODART on male fertility is not known. Prostate-Specific Antigen (PSA) Test: Your healthcare provider may check you for other prostate problems, including prostate cancer, before you start and while you take AVODART. A blood test called PSA (prostate-specific antigen) is sometimes used to see if you might have prostate cancer. AVODART will reduce the amount of PSA measured in your blood. Your healthcare provider is aware of this effect and can still use PSA to see if you might have prostate cancer. Increases in your PSA levels while on treatment with AVODART (even if the PSA levels are in the normal range) should be evaluated by your healthcare provider. These are not all the possible side effects of AVODART. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1‑800‑FDA‑1088. |
|
|
How should I store AVODART?
Keep AVODART and all medicines out of the reach of children. |
|
|
General information about the safe and effective use of AVODART. Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use AVODART for a condition for which it was not prescribed. Do not give AVODART to other people, even if they have the same symptoms that you have. It may harm them. You can ask your healthcare provider or pharmacist for information about AVODART that is written for health professionals. For more information call 1-888-825-5249. |
|
|
What are the ingredients in AVODART? Active ingredient: dutasteride Inactive ingredients: butylated hydroxytoluene, ferric oxide (yellow), gelatin (from certified BSE‑free bovine sources), glycerin, mono‑di‑glycerides of caprylic/capric acid, titanium dioxide, and edible red ink. |
|
|
Manufactured for: AVODART is a trademark under license by Waylis Therapeutics LLC. The other brand listed is a trademark owned by or licensed to its respective owner and is not a trademark owned by or licensed to Waylis Therapeutics LLC. The maker of this brand is not affiliated with and does not endorse Waylis Therapeutics LLC or its products. |
|
|
This Patient Information has been approved by the U.S. Food and Drug Administration. |
Revised: 10/2023 |
PRINCIPAL DISPLAY PANEL
NDC 80725-712-04
AVODART®
(dutasteride)
Soft Gelatin Capsules
0.5 mg
RX only
GX CE2
90 Capsules
WARNING: AVODART should not be used by women or children. Women who are or may potentially be pregnant should not use or handle AVODART Soft Gelatin Capsules (see prescribing information). If contact is made with leaking capsule, wash immediately with soap and water.
Each capsule contains 0.5 mg dutasteride.
Usual Dosage: 0.5 mg once a day.
Capsules should be swallowed whole and not chewed or opened. See prescribing information for further dosing information.
Store at 25oC (77oF); excursions permitted to 15-30oC (59-86oF) [see USP Controlled Room Temperature].
Dispense in a well-closed container as defined in the USP.
Do not use if printed safety seal under cap is broken or missing.
Manufactured for:
Waylis Therapeutics LLC
Wixom, MI 48393
Rev. 10/23