FULL PRESCRIBING INFORMATION
WARNING: ANAPHYLAXIS
-
Anaphylaxis has occurred with MEPSEVII administration, as early as the first dose [see Warnings and Precautions (5.1)], therefore appropriate medical support should be readily available when MEPSEVII is administered.
-
Closely observe patients during and for 60 minutes after MEPSEVII infusion [see Dosage and Administration (2.2), Warnings and Precautions (5.1)].
- Immediately discontinue the MEPSEVII infusion if the patient experiences anaphylaxis [see Dosage and Administration (2.2), Warnings and Precautions (5.1)].
1 INDICATIONS AND USAGE
MEPSEVII is indicated in pediatric and adult patients for the treatment of Mucopolysaccharidosis VII (MPS VII, Sly syndrome).
Limitations of Use
The effect of MEPSEVII on the central nervous system manifestations of MPS VII has not been determined.
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
MEPSEVII should be administered under the supervision of a healthcare professional with the capability to manage anaphylaxis. Premedication is recommended 30 to 60 minutes prior to the start of the infusion [see Dosage and Administration (2.2)].
The recommended dosage of MEPSEVII is 4 mg/kg administered by intravenous infusion every two weeks.
Administer the infusion over approximately 4 hours. Infuse the first 2.5% of the total volume over the first hour. After the first hour, increase the infusion rate as tolerated in order to complete infusion over the following 3 hours according to the recommended rate guidelines in Table 1 [see Dosage and Administration (2.4)].
2.2 Premedication
- Administration of a non-sedating antihistamine with or without an anti-pyretic medication is recommended 30 to 60 minutes prior to the start of the infusion for patient comfort.
- Follow the instructions in Table 1 for the rate of MEPSEVII infusion [see Dosage and Administration (2.4)].
- Observe patients closely during the infusion and following the infusion for a minimum of 60 minutes for the development of anaphylaxis [see Warnings and Precautions (5.1)].
- Discontinue the infusion immediately if the patient experiences a severe systemic reaction, including anaphylaxis [see Warnings and Precautions (5.1)].
2.3 Preparation Instructions
Prepare MEPSEVII according to the following steps using aseptic technique:
1. Determine the number of vials to be diluted based on the patient's actual weight and the recommended dose of 4 mg/kg, using the following calculations (a-b):
a. Total dose (mg) = Patient's weight (kg) x 4 mg/kg (recommended dose)
b. Total number of vials = Total dose (mg) divided by 10 mg/vial
2. Round to the next whole vial and remove the required number of vials from the refrigerator to allow them to reach room temperature. Do not heat, microwave or shake vials.
a. Volume (mL) of calculated dose = Total dose (mg) divided by the 2 mg/mL concentration
3. The final solution will be a 1:1 dilution of MEPSEVII with 0.9% Sodium Chloride Injection, USP. More than 1:1 dilution may be used if the patient can tolerate additional infusion volume, taking into consideration cardiac function and fluid status.
4. For a 1:1 dilution, prepare the solution at room temperature, as follows:
a. Select an empty infusion bag, sized upon the total volume of the final solution.
b. Prior to withdrawing MEPSEVII from the vial, visually inspect the solution for particulate matter and discoloration. Because this is a protein solution, slight flocculation (thin translucent fibers) may occur. The MEPSEVII solution should be colorless to slightly yellow. Discard if the solution is discolored or if there is particulate matter in the solution.
c. Slowly withdraw the volume of the calculated MEPSEVII dose from the appropriate number of vials (step 2a) using caution to avoid excessive agitation and any air or frothing. Use a sufficiently large needle (18 gauge) to minimize bubbles in the solution.
d. Slowly add MEPSEVII to the infusion bag using care to avoid agitation, ensuring liquid to liquid contact without generating bubbles or turbulence.
e. Add 0.9% Sodium Chloride Injection, USP equal to the volume of MEPSEVII to the infusion bag.
f. Gently rock the infusion bag to ensure proper distribution of MEPSEVII. Do not shake the solution.
2.4 Administration Instructions
Administer MEPSEVII as follows:
- The rate of infusion: In the first hour infuse 2.5% of the total volume, and infuse the remaining volume over the subsequent three hours (see Table 1). Account for any dead space in the lines to ensure 2.5% of the total infusion volume is delivered into the patient's bloodstream during the first hour of infusion.
- Use an infusion set equipped with an in-line, low-protein binding 0.2 micron filter to administer the diluted MEPSEVII solution.
- Do not flush the line containing MEPSEVII to avoid a rapid bolus of infused enzyme. Due to the low infusion rate, additional saline may be added through a separate line (piggyback or Y tube) to maintain sufficient intravenous flow to prevent clotting or line blockage.
- Do not infuse with other products in the infusion tubing. Compatibility with other products has not been evaluated.
- Use MEPSEVII immediately after dilution and complete the infusion within 42 hours from the time of dilution. Discard any unused product.
Stability
If immediate use is not possible, the diluted solution may be stored up to 36 hours under refrigeration at 2°C to 8°C (36°F to 46°F) followed by up to 6 hours at room temperature up to a maximum of 25°C (77°F).
| Patient
Weight Range (kg) | Total MEPSEVII
Dose Range (mg) | Total MEPSEVII Volume (rounded)
(mL) | Total
Infusion Volume of Drug and diluent (infused over 4 hours) (mL) | Infusion Rate for 1st Hour (2.5%) (mL/h) | Infusion Rate per Hour for Subsequent 3 Hours (97.5%/3)
(mL/h) |
| 3.5-5.9 | 14-23.6 | 10 | 20 | 0.5 | 6.5 |
| 6-8.4 | 24-33.6 | 15 | 30 | 0.8 | 9.8 |
| 8.5-10.9 | 34-43.6 | 20 | 40 | 1 | 13 |
| 11-13.4 | 44-53.6 | 25 | 50 | 1.3 | 16.3 |
| 13.5-15.9 | 54-63.6 | 30 | 60 | 1.5 | 19.5 |
| 16-18.4 | 64-73.6 | 35 | 70 | 1.8 | 22.8 |
| 18.5-20.9 | 74-83.6 | 40 | 80 | 2 | 26 |
| 21-23.4 | 84-93.6 | 45 | 90 | 2.3 | 29.3 |
| 23.5-25.9 | 94-103.6 | 50 | 100 | 2.5 | 32.5 |
| 26-28.4 | 104-113.6 | 55 | 110 | 2.8 | 35.8 |
| 28.5-30.9 | 114-123.6 | 60 | 120 | 3 | 39 |
| 31-33.4 | 124-133.6 | 65 | 130 | 3.3 | 42.3 |
| 33.5-35.9 | 134-143.6 | 70 | 140 | 3.5 | 45.5 |
| 36-38.4 | 144-153.6 | 75 | 150 | 3.8 | 48.8 |
| 38.5-40.9 | 154-163.6 | 80 | 160 | 4 | 52 |
| 41-43.4 | 164-173.6 | 85 | 170 | 4.3 | 55.3 |
| 43.5-45.9 | 174-183.6 | 90 | 180 | 4.5 | 58.5 |
| 46-48.4 | 184-193.6 | 95 | 190 | 4.8 | 61.8 |
| 48.5-50.9 | 194-203.6 | 100 | 200 | 5 | 65 |
| 51-53.4 | 204-213.6 | 105 | 210 | 5.3 | 68.3 |
| 53.5-55.9 | 214-223.6 | 110 | 220 | 5.5 | 71.5 |
| 56-58.4 | 224-233.6 | 115 | 230 | 5.8 | 74.8 |
| 58.5-60.9 | 234-243.6 | 120 | 240 | 6 | 78 |
| 61-63.4 | 244-253.6 | 125 | 250 | 6.3 | 81.3 |
| 63.5-65.9 | 254-263.6 | 130 | 260 | 6.5 | 84.5 |
| 66-68.4 | 264-273.6 | 135 | 270 | 6.8 | 87.8 |
| 68.5-70.9 | 274-283.6 | 140 | 280 | 7 | 91 |
3 DOSAGE FORMS AND STRENGTHS
Injection: 10 mg/5 mL (2 mg/mL) as a colorless to slightly yellow liquid in a single-dose vial.
5 WARNINGS AND PRECAUTIONS
5.1 Anaphylaxis
Anaphylaxis to MEPSEVII was reported in 2 of 20 patients in the clinical program [see Adverse Reactions (6.1)]. These reactions occurred during MEPSEVII infusion and were observed as early as the first dose of MEPSEVII for one patient. Manifestations included respiratory distress, cyanosis, decreased oxygen saturation, and hypotension. The two patients with anaphylaxis to MEPSEVII during the clinical trials had one occurrence each and tolerated subsequent infusions of MEPSEVII, without recurrence.
Anaphylaxis can be life-threatening. MEPSEVII should be administered under the supervision of a healthcare professional with the capability to manage anaphylaxis. Patients should be observed for 60 minutes after MEPSEVII administration. If severe systemic reactions occur, including anaphylaxis, immediately discontinue the MEPSEVII infusion and provide appropriate medical treatment. Prior to discharge, inform patients of the signs and symptoms of anaphylaxis and instruct them to seek immediate medical care if symptoms occur. Consider the risks and benefits of re-administering MEPSEVII following anaphylaxis.
6 ADVERSE REACTIONS
The following serious adverse reactions are described below and elsewhere in the labeling:
- Anaphylaxis [see Warnings and Precautions (5.1)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The MEPSEVII clinical program included 23 patients aged 5 months to 25 years who received treatment with MEPSEVII at doses up to 4 mg/kg once every two weeks for up to 187 weeks. Nineteen patients were younger than 18 years of age.
Table 2 summarizes the adverse reactions that occurred in Study 301, a randomized start trial in 12 patients with MPS VII between the ages of 8 and 25 years [see Clinical Studies (14)].
Adverse reactions in Table 2 occurred in one or more patients treated with MEPSEVII at a dosage of 4 mg/kg at a higher patient frequency than placebo. Adverse reaction incidence rates are presented in the table below to account for the different duration of exposure to active treatment vs. placebo.
| Adverse Reaction | MEPSEVII
N =12 n (Incidence Rate*) | Placebo
N=9 n (Incidence Rate*) |
| Infusion site extravasation | 4 (0.5) | 1 (0.4) |
| Diarrhea | 3 (0.4) | 0 (0.0) |
| Rash | 3 (0.4) | 2 (0.7) |
| Anaphylaxis | 2 (0.2) | 0 (0.0) |
| Infusion site swelling | 1 (0.1) | 0 (0.0) |
| Peripheral swelling | 1 (0.1) | 0 (0.0) |
| Pruritus | 1 (0.1) | 0 (0.0) |
n = number of reactions
*Adverse reaction incidence rates calculated per 8.3 patient years for exposure to MEPSEVII, and 2.7 years of exposure for placebo
Febrile Convulsion
One patient receiving a dose of 4 mg/kg experienced a febrile convulsion during MEPSEVII treatment at Week 66. The infusion was stopped, the patient received anticonvulsants, antipyretics and antibiotics, and the adverse reaction resolved. The patient subsequently was re-challenged without recurrence and continued on treatment.
6.2 Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies in the studies described below with the incidence of antibodies in other studies to other vestronidase alfa products may be misleading.
Immunogenicity data were available from 23 patients who received MEPSEVII for up to 187 weeks of treatment. Eighteen out of 23 (78%) patients developed anti-vestronidase alfa-vjbk antibodies (ADA). Ten of the 18 (55.6%) ADA-positive patients were tested positive for neutralizing antibodies (NAb). There is no correlation between ADA titer and NAb development.
Six treatment-naïve patients had pre-existing ADA titers at baseline. ADAs were detected in five of these six patients post-treatment. The post-treatment ADA titers were the same as or below the baseline ADA titer values in two patients, but one of these two patients was positive for NAb. ADA titer values after treatment increased 64-fold, 128-fold, and 364-fold, respectively, in the other three patients.
The presence of ADA titer did not appear to affect reduction in urinary glycosaminoglycans (uGAGs).
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no available data on MEPSEVII use in pregnant women to determine a drug-associated risk of adverse developmental outcomes. In embryofetal development studies, vestronidase alfa-vjbk administered intravenously to pregnant rats and rabbits during the period of organogenesis showed no adverse developmental outcomes at doses up to 1.6 and 10 times, respectively for rats and rabbits, the exposure at the recommended human dose. In a pre- and post-natal development study in rats, an increased number of stillbirths were observed at exposures less than the recommended human dose (see Data). The clinical relevance of these animal findings is uncertain.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Animal Data
In embryofetal development studies, vestronidase alfa-vjbk administered intravenously to pregnant rats (once a week) and rabbits (once every 3 days) during the period of organogenesis showed no adverse developmental outcomes at doses up to 20 mg/kg. The 20 mg/kg dose in rats and rabbits provides approximately 1.6 and 10 times the human exposure (AUC) of 57.9 hr*mcg/mL at the 4 mg/kg dose administered once every other week, respectively.
In a pre- and post-natal developmental study in rats, vestronidase alfa-vjbk was administered every 3 days from gestation day 7 through lactation day 20 at doses of 2 mg/kg, 6 mg/kg, and 20 mg/kg. Mortality and adverse clinical signs were observed in the maternal animals at the 20 mg/kg dose (1.6 times the human exposure (AUC) at the recommended human dose of 4 mg/kg). Subsequently, the 20 mg/kg dose was reduced to 12 mg/kg. Maternal toxicity with mortality in one animal was also observed at the 6 mg/kg dose (0.17 times the AUC at the recommended human dose of 4 mg/kg). At the 2 mg/kg dose (0.01 times the AUC at the recommended human dose of 4 mg/kg), no adverse effects were observed in the maternal animals; however, there was a statistically significant decrease in the number of live births and subsequent increase in the number of stillbirths at this dose.
8.2 Lactation
Risk Summary
There are no data on the presence of vestronidase alfa-vjbk in either human or animal milk, the effects on the breastfed infant, or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for MEPSEVII and any potential adverse effects on the breastfed infant from vestronidase alfa-vjbk or from the underlying maternal condition.
11 DESCRIPTION
Vestronidase alfa-vjbk is a recombinant human lysosomal beta glucuronidase which is a purified human enzyme produced by recombinant DNA technology in a Chinese hamster ovary cell line.
Purified vestronidase alfa-vjbk exists as a homotetramer, with each monomer consisting of 629 amino acids. The calculated isotope average molecular mass of each non-glycosylated peptide chain is 72,562 Da.
The amino acid sequence for vestronidase alfa-vjbk is the same as the amino acid sequence for human beta-glucuronidase (GUS).
MEPSEVII (vestronidase alfa-vjbk) injection for intravenous infusion is a sterile, preservative-free, non-pyrogenic, colorless to slightly yellow liquid supplied in a single-dose vial. Each mL of solution contains vestronidase alfa-vjbk (2 mg), L-histidine (3.1 mg), polysorbate 20 (0.1 mg), sodium chloride (7.88 mg) and sodium phosphate monobasic dihydrate (3.12 mg). The pH of the solution is 6.0.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Mucopolysaccharidosis VII (MPS VII or Sly syndrome) is a lysosomal disorder characterized by the deficiency of GUS that results in GAG accumulation in cells throughout the body leading to multisystem tissue and organ damage.
Vestronidase alfa-vjbk is a recombinant form of human GUS and is intended to provide exogenous GUS enzyme for uptake into cellular lysosomes. Mannose-6-phosphate (M6P) residues on the oligosaccharide chains allow binding of the enzyme to cell surface receptors, leading to cellular uptake of the enzyme, targeting to lysosomes and subsequent catabolism of accumulated GAGs in affected tissues.
12.2 Pharmacodynamics
In clinical studies, MEPSEVII treatment resulted in sustained reduction of urinary excretion of GAGs during long-term treatment [see Clinical Studies (14)].
12.3 Pharmacokinetics
The pharmacokinetics of vestronidase alfa-vjbk were evaluated in a total of 23 MPS VII patients including 19 pediatric patients and 4 adults. Serum exposures of vestronidase alfa-vjbk appeared to increase approximately proportionally from 1 mg/kg (0.25 times the approved recommended dosage) to 2 mg/kg (0.5 times the approved recommended dosage), and 4 mg/kg (the recommended dosage). After repeated dosing of 4 mg/kg every other week, the mean ± standard deviation of maximal concentration (Cmax) was 17.3 ± 9.6 mcg/mL (range: 4.7 to 35.7 mcg/mL); and the mean ± standard deviation of area under the concentration-time curve from time zero to the last measurable concentration (AUC0-t) was 50.9 ± 32.2 mcg*hr/mL (range: 17.4 to 153 mcg*hr/mL).
Vestronidase alfa-vjbk concentrations in pediatric patients less than 5 years of age were similar to the concentrations in older children and adults.
Distribution
After repeated dosing of 4 mg/kg every other week in MPS VII patients, the mean ± standard deviation of the total volume of distribution (Vss) was 251 ± 140 mL/kg (range: 97 to 598 mL/kg).
Elimination
After repeated dosing of 4 mg/kg every other week in MPS VII patients, the mean ± standard deviation of the total clearance (CL) was 83.6 ± 43.2 mL/hr/kg (range: 38.3 to 184mL/hr/kg); the mean ± standard deviation of the elimination half-life (t1/2) was 2.33 ± 0.75 hours (range: 0.86 to 3.03 hours). The inter-subject variability (coefficient of variation) in total clearance (CL) was 52%.
Metabolism
Vestronidase alfa-vjbk is a recombinant human enzyme and is therefore eliminated by proteolytic degradation into small peptides and amino acids.
Excretion
No excretion studies have been conducted in humans. Vestronidase alfa-vjbk is not expected to be eliminated through renal or fecal excretion.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term studies in animals to evaluate carcinogenic potential or studies to evaluate the mutagenic potential have not been performed with vestronidase alfa-vjbk.
Vestronidase alfa-vjbk at intravenous doses up to 20 mg/kg administered weekly to rats prior to mating and after mating on gestation days 6, 9, 12, 15 and 18 (females), [approximately up to 4.5 times (male rats) and 1.6 times (female rats) the human AUC0-t of 3440 mcg*min/mL at the 4 mg/kg dose administered once every other week] was found to have no adverse effect on fertility and reproductive performance of male and female rats.
14 CLINICAL STUDIES
The clinical program for MEPSEVII included 23 patients with MPS VII, 17 of whom were evaluable for efficacy, 20 for safety, and 23 for immunogenicity. Patients were enrolled in clinical trials and expanded access protocols receiving treatment at doses up to 4 mg/kg once every two weeks for up to 187 weeks. The patients ranged in age from 5 months to 25 years. Sixteen patients were younger than 18 years of age.
Studies 301 and 202
Study UX003-CL301 (referred to as Study 301, NCT02230566) was a randomized start trial of MEPSEVII 4 mg/kg every two weeks in patients with MPS VII. Twelve patients were randomized to one of four placebo durations before crossing over to active treatment. Three patients received MEPSEVII immediately for a duration of 48 weeks, 3 patients received placebo for 8 weeks then MEPSEVII for 40 weeks, 3 patients received placebo for 16 weeks then MEPSEVII for 32 weeks, and 3 patients received placebo for 24 weeks then MEPSEVII for 24 weeks. Of the 12 patients enrolled in the trial, 4 were male and 8 were female and ranged in age from 8 to 25 years (median 14 years). Nine patients were younger than 18 years of age. The majority of the patients were white (75%), with 50% of Hispanic or Latino ethnicity. Patients who were enrolled in Study 301 were eligible to roll over to Study UX003-CL202 (referred to as Study 202, NCT02432144), an open-label extension trial in which patients received additional doses of MEPSEVII at 4 mg/kg intravenously every other week for up to 144 weeks. Ten patients rolled over directly from the end of Study to Week 0 of Study 202, while 2 patients (17%) had treatment gaps before enrolling in Study 202.
In Study 301, motor function, forced vital capacity, and visual acuity were assessed after 24 weeks of MEPSEVII treatment and measured against pre-specified minimal important differences. The extremely small population of patients with MPS VII globally necessitated the enrollment of all patients able to participate resulting in a highly heterogeneous group. Clinical endpoints were not assessable in some patients due to their extent of disease, age or level of cognition. Repeated assessments of the six minute walk test (6MWT) were feasible in ten of 12 patients and are described further below. Of the three patients who improved on their 6MWT (Figure 1, left panel), two also were noted to have improvement in balance and gross motor proficiency as assessed by the Bruininks-Oseretsky Test of Motor Proficiency (BOT-2).
In this trial, the mean difference in 6MWT distance between MEPSEVII and placebo treatment periods in patients able to perform the test at baseline and subsequent visits through Week 24 is shown in Table 3. The mean difference in 6MWT distance increases with increased treatment duration, however, due to the small size of the trial, standard errors are large.
| Duration of MEPSEVII Treatment | LS mean 6MWT (meters) (± Standard Error)* | Number and Treatment Assignment of Patients Included in Analyis** |
| 8 weeks | -11 (± 24) | 5 placebo period; 8 MEPSEVII period |
| 16 weeks | 13 (± 32) | 5 placebo period; 8 MEPSEVII period |
| 24 weeks | 18 (± 33) | 5 placebo period; 8 MEPSEVII period |
*ANCOVA analysis of change from baseline in least squares (LS) mean between placebo and MEPSEVII for different periods, after adjusting for study cohort, age, and baseline 6MWT distance. Patients who used assistive devices were imputed as zeros in the analysis.
**Number and treatment assignment of patients included in the analysis was based upon a randomized start trial design and patient ability to complete testing. Due to no placebo period for the three patients who received 48 weeks of MEPSEVII in the first cohort of the randomized start design, more data were available for analyses during the treatment period (n=8) than during the placebo period (n=5). While data from 8 participants were available at each time point, due to missing observations, the 8 participants were not the same across all time points.
The observed individual 6MWT distances for the 10 patients who could perform the test in Study 301 and Study 202 through Week 184 are presented in Figure 1. The course of three patients with improvement in distance walked of at least 60 meters during the 301 Study compared to the start of MEPSEVII treatment (Week 0) is shown in the left panel; the relatively stable course in the remaining seven patients, including those who used assistive devices, is shown in the right panel.
Figure 1. 6MWT Distance for MPS VII Patients in Studies 301 and 202
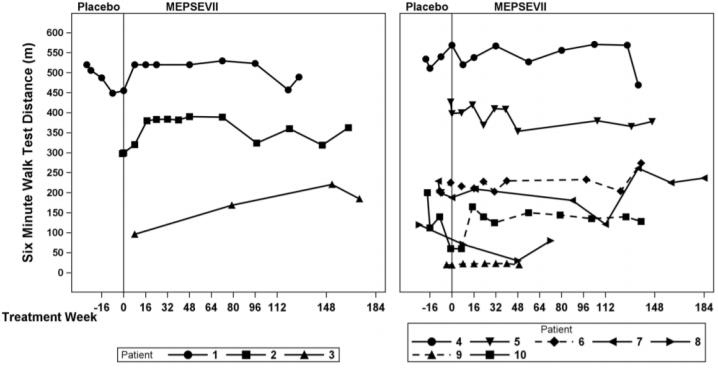
Patient 10 did not use an assistive device at baseline but started using an assistive device post-baseline from Treatment Week 8. Patients 6 and 9 consistently used an assistive device at all visits. A solid line indicates the unassisted assessments and a dotted line indicates the assisted assessments.
Liver and Spleen Volume
In Study 301, imaging by MRI or ultrasound to assess liver and spleen volume was performed in seven of the 12 patients. Most liver volumes were normal or below normal size at baseline (mean 1,591 mL, range 742 to 2,207 mL), and on average were unchanged after treatment (mean 1,459 mL, range 876 to 1,851 mL).
Spleen volumes generally were normal or below normal size at baseline (mean 325 mL, range 131 to 491 mL) and on average were unchanged after treatment (mean 360 mL, range 200 to 582 mL).
Study 203
UX003-CL203 (referred to as Study 203; NCT 02418455 ) was an open-label, uncontrolled single arm study that enrolled 8 patients less than 5 years of age who received MEPSEVII at a dose of 4 mg/kg every two weeks for 48 weeks of treatment and up to an additional 240 weeks during an optional continuation period. The study evaluated urinary GAG excretion, growth and hepatosplenomegaly. With long-term treatment, urinary GAG levels remained decreased upon exposure to MEPSEVII. At baseline, all 8 patients had impaired growth, and height remained near the 5th percentile relative to age-matched gender norms throughout the trial. No significant changes in hepatosplenomegaly were observed.
Other Investigations
Study UX003-CL201 (referred to as Study 201, NCT01856218) was a single arm, open-label, dose exploration trial completed outside the United States that enrolled three MPS VII patients, ranging in age from 5 years to 25 years. Two patients were male; two patients were white and one was Asian. After 120 weeks of exposure to MEPSEVII, one patient demonstrated a 21% improvement over baseline in forced vital capacity (FVC% predicted) on pulmonary function testing in addition to a 105 meter improvement in the 6MWT. Two other patients with baseline hepatosplenomegaly had reduction in liver volume (24% and 53%) and spleen volume (28% and 47%) after 36 weeks of MEPSEVII treatment.
Expanded access to MEPSEVII treatment was provided to a pediatric patient with MPS VII who required continuous ventilatory support at the start of treatment and was subsequently able to tolerate 9 hours daily off ventilator support after 164 weeks of MEPSEVII treatment.
16 HOW SUPPLIED/STORAGE AND HANDLING
MEPSEVII (vestronidase alfa-vjbk) injection is a colorless to slightly yellow liquid supplied as a carton containing one 10 mg/5 mL (2 mg/mL) single-dose vial (NDC 69794-001-01).
Store under refrigeration at 2°C to 8°C (36°F to 46°F). Do not freeze or shake. Protect from light.
17 PATIENT COUNSELING INFORMATION
Anaphylaxis
Advise patients and caregivers that anaphylaxis has occurred with MEPSEVII administration. Inform patients of the signs and symptoms of anaphylaxis, and have them seek immediate medical care should signs and symptoms occur [see Warnings and Precautions (5.1)].
Manufactured by:
Ultragenyx Pharmaceutical Inc.
Novato, CA 94949
U.S. License No. 2040