FULL PRESCRIBING INFORMATION
WARNING: EMBRYO-FETAL TOXICITY, HYPERSENSITIVITY REACTIONS AND SEVERE ADVERSE REACTIONS
- Methotrexate tablets can cause embryo-fetal toxicity, including fetal death. For non-neoplastic diseases, methotrexate tablets are contraindicated in pregnancy. For neoplastic diseases, advise females and males of reproductive potential to use effective contraception [see Contraindications (4), Warnings and Precautions (5.1), Use in Specific Populations (8.1, 8.3)] .
- Methotrexate tablets are contraindicated in patients with a history of severe hypersensitivity reactions to methotrexate, including anaphylaxis [see Contraindications (4), Warnings and Precautions (5.2)] .
- Serious adverse reactions, including death, have been reported with methotrexate. Closely monitor for adverse reactions of the bone marrow, gastrointestinal tract, liver, lungs, skin, and kidneys. Withhold or discontinue methotrexate tablets as appropriate [see Warnings and Precautions (5.3, 5.4, 5.5, 5.6, 5.7, 5.8)] .
1 INDICATIONS AND USAGE
1.1 Neoplastic Diseases
Methotrexate tablets are indicated for the:
- treatment of adults and pediatric patients with acute lymphoblastic leukemia (ALL) as part of a combination chemotherapy maintenance regimen.
- treatment of adults with mycosis fungoides (cutaneous T-cell lymphoma) as a single agent or as part of a combination chemotherapy regimen.
- treatment of adults with relapsed or refractory non-Hodgkin lymphomas as part of a metronomic combination chemotherapy regimen.
1.2 Rheumatoid Arthritis
Methotrexate tablets are indicated for the treatment of adults with rheumatoid arthritis.
2 DOSAGE AND ADMINISTRATION
2.1 Important Dosage and Safety Information
Verify pregnancy status in females of reproductive potential before starting methotrexate tablets [see Contraindications (4), Warnings and Precautions (5.1)] .
Instruct patients and caregivers to take the recommended dosage as directed, because medication errors have led to deaths [see Warnings and Precautions (5.9)] .
When switching the dosing regimen from oral administration to intravenous, intramuscular, or subcutaneous administration, an alternative dosing regimen may be necessary.
Do not administer to patients who are unable to swallow a tablet.
Methotrexate tablets are a cytotoxic drug. Follow applicable special handling and disposal procedures. 1
2.2 Recommended Dosage for Neoplastic Diseases
Acute Lymphoblastic Leukemia
The recommended starting dosage of methotrexate tablets is 20 mg/m 2orally once weekly, as part of a combination chemotherapy maintenance regimen. After initiating methotrexate tablets, periodically monitor absolute neutrophil count (ANC) and platelet count and adjust the dose to maintain ANC at a desirable level and for excessive myelosuppression.
2.3 Recommended Dosage for Rheumatoid Arthritis
The recommended starting dosage of methotrexate tablets is 7.5 mg orally once weekly with escalation to achieve optimal response. Dosages of more than 20 mg once weekly result in an increased risk of serious adverse reactions, including myelosuppression. When responses are observed, the majority occurred between 3 and 6 weeks from initiation of treatment; however, responses have occurred up to 12 weeks after treatment initiation.
Administer folic acid or folinic acid to reduce the risk of methotrexate adverse reactions [see Warnings and Precautions (5.10)].
2.4 Recommended Dosage for Polyarticular Juvenile Idiopathic Arthritis
The recommended starting dosage of methotrexate tablets is 10 mg/m 2orally once weekly with escalation to achieve optimal response. Dosages of more than 30 mg/m 2once weekly result in an increased risk of serious adverse reactions, including myelosuppression. When responses are observed, the majority occurred between 3 and 6 weeks from initiation of treatment; however, responses have occurred up to 12 weeks after treatment initiation.
Administer folic acid or folinic acid to reduce the risk of methotrexate adverse reactions [see Warnings and Precautions (5.10)].
2.5 Recommended Dosage for Psoriasis
The recommended dosage of methotrexate tablets is 10 to 25 mg orally once weekly until an adequate response is achieved. Adjust the dose gradually to achieve optimal clinical response; do not exceed a dose of 30 mg per week. Once optimal clinical response has been achieved, reduce the dosage to the lowest possible dosing regimen.
Administer folic acid or folinic acid supplementation to reduce the risk of methotrexate adverse reactions [see Warnings and Precautions (5.10)] .
2.6 Dosage Modifications for Adverse Reactions
Discontinue methotrexate tablets for:
- Anaphylaxis or other severe hypersensitivity reactions [see Warnings and Precautions (5.2)]
- Lymphoproliferative disease [see Warnings and Precautions (5.13)]
Withhold, dose reduce or discontinue methotrexate tablets as appropriate for:
- Myelosuppression [see Warnings and Precautions (5.3)]
Withhold or discontinue methotrexate tablets as appropriate for:
- Severe gastrointestinal toxicity [see Warnings and Precautions (5.4)]
- Hepatotoxicity [see Warnings and Precautions (5.5)]
- Pulmonary toxicity [see Warnings and Precautions (5.6)]
- Severe dermatologic reactions [see Warnings and Precautions (5.7)]
- Severe renal toxicity [see Warnings and Precautions (5.8)]
- Serious infections [see Warnings and Precautions (5.11)]
- Neurotoxicity [see Warnings and Precautions (5.12)]
3 DOSAGE FORMS AND STRENGTHS
Methotrexate Tablets, USP are available containing 2.5 mg methotrexate, USP equivalent to 2.74 mg methotrexate sodium.
- The 2.5 mg tablets are orange, round, scored tablets debossed with Mabove the score and 14below the score on one side of the tablet and blank on the other side of the tablet.
4 CONTRAINDICATIONS
Methotrexate tablets are contraindicated in:
- Pregnant women receiving methotrexate tablets for treatment of non-neoplastic diseases [see Warnings and Precautions (5.1), and Use in Specific Populations (8.1, 8.3)] .
- Patients with a history of a severe hypersensitivity reactions, including anaphylaxis, to methotrexate [see Warnings and Precautions (5.2)] .
5 WARNINGS AND PRECAUTIONS
5.1 Embryo-Fetal Toxicity
Based on published reports and its mechanism of action, methotrexate tablets can cause fetal harm, including fetal death, when administered to a pregnant woman. Methotrexate tablets are contraindicated for use in pregnant women receiving methotrexate tablets for the treatment of non-malignant diseases. Advise pregnant women with neoplastic diseases of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with methotrexate tablets and for 6 months after the final dose. Advise males with female partners of reproductive potential to use effective contraception during methotrexate tablets treatment and for 3 months after the final dose [see Contraindications (4), Use in Specific Populations (8.1, 8.3)].
5.2 Hypersensitivity Reactions
Hypersensitivity reactions, including anaphylaxis, can occur with methotrexate [see Contraindications (4), Adverse Reactions (6.1)] .
If anaphylaxis or other serious hypersensitivity reaction occurs, immediately and permanently discontinue methotrexate tablets [see Dosage and Administration (2.6)].
5.3 Myelosuppression
Methotrexate suppresses hematopoiesis and can cause severe and life-threatening pancytopenia, anemia, leukopenia, neutropenia, and thrombocytopenia [see Adverse Reactions (6.1)] .
Obtain blood counts at baseline, periodically during treatment, and as clinically indicated. Monitor patients for clinical complications of myelosuppression. Withhold, dose reduce, or discontinue methotrexate tablets taking into account the importance of methotrexate tablet treatment in the context of the severity of the disease being treated, the severity of the adverse drug reaction, and availability of alternative therapy [see Dosage and Administration (2.6)].
5.4 Gastrointestinal Toxicity
Diarrhea, vomiting, nausea, and stomatitis occurred in up to 10% of patients receiving methotrexate for treatment of non-neoplastic diseases. Hemorrhagic enteritis and fatal intestinal perforation have been reported [see Adverse Reactions (6.1, 6.2)] . Patients with peptic ulcer disease or ulcerative colitis are at a greater risk of developing severe gastrointestinal adverse reactions [see Drug Interactions (7.1)] .
Withhold or discontinue methotrexate tablets for severe gastrointestinal toxicity taking into account the importance of methotrexate tablet treatment in the context of the severity of the disease being treated, the severity of the adverse drug reaction, and availability of alternative therapy [see Dosage and Administration (2.6)].
5.5 Hepatotoxicity
Methotrexate can cause severe and potentially irreversible hepatotoxicity, including fibrosis, cirrhosis, and fatal liver failure [see Adverse Reactions (6.1)]. The safety of methotrexate tablets in patients with hepatic disease is unknown.
The risk of hepatotoxicity is increased with heavy alcohol consumption. In patients with psoriasis, fibrosis or cirrhosis may occur in the absence of symptoms or abnormal liver tests; the risk of hepatotoxicity appears to increase with total cumulative dose and generally occurs after receipt of a total cumulative dose of 1.5 g or more.
Monitor liver tests at baseline, periodically during treatment and as clinically indicated. Withhold or discontinue methotrexate tablets taking into account the importance of methotrexate tablet treatment in the context of the severity of the disease being treated, the severity of the adverse drug reaction, and availability of alternative therapy [see Dosage and Administration (2.6)].
5.6 Pulmonary Toxicity
Pulmonary toxicity, including acute or chronic interstitial pneumonitis and irreversible or fatal cases, can occur with methotrexate [see Adverse Reactions (6.1, 6.2)] .
Monitor patients for pulmonary toxicity and withhold or discontinue methotrexate tablets taking into account the importance of methotrexate tablet treatment in the context of the severity of the disease being treated, the severity of the adverse drug reaction, and availability of alternative therapy [see Dosage and Administration (2.6)].
5.7 Dermatologic Reactions
Severe, including fatal dermatologic reactions, such as toxic epidermal necrolysis, Stevens-Johnson syndrome, exfoliative dermatitis, skin necrosis, and erythema multiforme, can occur with methotrexate [see Adverse Reactions (6.1, 6.2)] .
Exposure to ultraviolet radiation while taking methotrexate may aggravate psoriasis.
Methotrexate can cause radiation recall dermatitis and photodermatitis (sunburn) reactivation.
Monitor patients for dermatologic toxicity and withhold or permanently discontinue methotrexate tablets for severe dermatologic reactions taking into account the importance of methotrexate tablet treatment in the context of the severity of the disease being treated, the severity of the adverse drug reaction, and availability of alternative therapy [see Dosage and Administration (2.6)] . Advise patients to avoid excessive sun exposure and use sun protection measures.
5.8 Renal Toxicity
Methotrexate can cause renal toxicity, including irreversible acute renal failure [see Adverse Reactions (6.2)] .
Monitor renal function at baseline, periodically during treatment and as clinically indicated. Withhold or discontinue methotrexate tablets for severe renal toxicity taking into account the importance of methotrexate tablet treatment in the context of the severity of the disease being treated, the severity of the adverse drug reaction, and availability of alternative therapy [see Dosage and Administration (2.6)] .
Administer glucarpidase in patients with toxic plasma methotrexate concentrations (> 1 micromole per liter) and delayed methotrexate clearance due to impaired renal function. Refer to the glucarpidase prescribing information for additional information.
5.9 Risk of Serious Adverse Reactions with Medication Error
Deaths occurred in patients as a result of medication errors. Most commonly, these errors occurred in patients who were taking methotrexate daily when a weekly dosing regimen was prescribed.
For patients prescribed a once weekly dosing regimen, instruct patients and caregivers to take the recommended dosage as directed, because medication errors have led to death.
5.10 Folic Acid Supplementation
Neoplastic Diseases
Products containing folic acid or its derivatives may decrease the clinical effectiveness of methotrexate. Therefore, instruct patients not to take products containing folic acid or folinic acid unless directed to do so by their healthcare provider.
Non-neoplastic Diseases
Folate deficiency may increase methotrexate adverse reactions. Administer folic acid or folinic acid for patients with rheumatoid arthritis, pJIA, and psoriasis [see Dosage and Administration (2.3, 2.4, 2.5)].
5.11 Serious Infections
Patients treated with methotrexate are at increased risk for developing life-threatening or fatal bacterial, fungal, or viral infections, including opportunistic infections such as Pneumocystis jirovecipneumonia, invasive fungal infections, hepatitis B reactivation, tuberculosis primary infection or reactivation, and disseminated Herpes zosterand cytomegalovirus infections [see Adverse Reactions (6.2)] .
Monitor patients for infection during and after treatment with methotrexate tablets. Withhold or discontinue methotrexate tablets for serious infections taking into account the importance of methotrexate tablet treatment in the context of the severity of the disease being treated, the severity of the adverse drug reaction, and availability of alternative therapy [see Dosage and Administration (2.6)].
5.12 Neurotoxicity
Methotrexate can cause severe acute and chronic neurotoxicity, which can be progressive, irreversible, and fatal [see Adverse Reactions (6.2)] . The risk of leukoencephalopathy is increased in patients who received prior cranial radiation.
Monitor patients for neurotoxicity and withhold or discontinue methotrexate tablets taking into account the importance of methotrexate tablet treatment in the context of the severity of the disease being treated, the severity of the adverse drug reaction, and availability of alternative therapy [see Dosage and Administration (2.6)].
5.13 Secondary Malignancies
Secondary malignancies can occur with methotrexate [see Adverse Reactions (6.2)] . The risk of cutaneous malignancies is further increased when cyclosporine is administered to patients with psoriasis who received prior methotrexate.
In some cases, lymphoproliferative disease occurring during therapy with low-dose methotrexate regressed completely following withdrawal of methotrexate. If lymphoproliferative disease occurs, discontinue methotrexate tablets [see Dosage and Administration (2.6)].
5.14 Tumor Lysis Syndrome
Methotrexate can induce tumor lysis syndrome in patients with rapidly growing tumors. Institute appropriate prophylactic measures in patients at risk for tumor lysis syndrome prior to initiation of methotrexate tablets.
5.15 Immunization and Risks Associated with Live Vaccines
Disseminated infections following administration of live vaccines have been reported. Immunization with live vaccines is not recommended during treatment. Follow current vaccination practice guidelines for administration of immunizations in patients receiving methotrexate tablets.
Update immunizations according to immunization guidelines prior to initiating methotrexate tablets. The interval between live vaccinations and initiation of methotrexate should be in accordance with current vaccination guidelines regarding immunosuppressive agents.
5.16 Infertility
Based on published reports, methotrexate can cause impairment of fertility, oligospermia, and menstrual dysfunction. It is not known if the infertility may be reversible. Discuss the risk of infertility with females and males of reproductive potential [see Use in Specific Populations (8.3)].
5.17 Increased Risk of Adverse Reactions Due to Third-Space Accumulation
Methotrexate accumulates in third-spaces (e.g., pleural effusions or ascites), which results in prolonged elimination and increases the risk of adverse reactions. Evacuate significant third-space accumulations prior to methotrexate tablets administration taking into account the importance of methotrexate tablet treatment in the context of the severity of the disease being treated, the severity of the adverse drug reaction, and availability of alternative therapy.
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Hypersensitivity Reactions [see Warnings and Precautions (5.2)]
- Myelosuppression [see Warnings and Precautions (5.3)]
- Gastrointestinal Toxicity [see Warnings and Precautions (5.4)]
- Hepatotoxicity [see Warnings and Precautions (5.5)]
- Pulmonary Toxicity [see Warnings and Precautions (5.6)]
- Dermatologic Reactions [see Warnings and Precautions (5.7)]
- Renal Toxicity [see Warnings and Precautions (5.8)]
- Serious Infections [see Warnings and Precautions (5.11)]
- Neurotoxicity [see Warnings and Precautions (5.12)]
- Secondary Malignancies [see Warnings and Precautions (5.13)]
- Tumor Lysis Syndrome [see Warnings and Precautions (5.14)]
- Increased Risk of Adverse Reactions Due to Third-Space Accumulation [see Warnings and Precautions (5.17)]
6.1 Clinical Trials Experience
Because clinical trials and other studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Common adverse reactions were: ulcerative stomatitis, leukopenia, nausea, and abdominal distress. Other clinically relevant adverse reactions were infection, malaise, fatigue, chills, fever, and dizziness.
Rheumatoid Arthritis
The most common adverse reactions of methotrexate that exceeded the rate of placebo in 12- to 18-week double-blind studies in patients (n = 128) with rheumatoid arthritis are listed below. Patients received methotrexate 7.5 to 15 mg orally once weekly. Most patients received concomitant nonsteroidal anti-inflammatory drugs (NSAIDs) and some also received corticosteroids. Hepatic histology was not examined in these short-term studies.
Incidence ≥ 10%: Elevated liver tests 15%, nausea/vomiting 10%
Incidence 3% to < 10%: Stomatitis, thrombocytopenia (platelet count < 100,000/mm 3)
Incidence 1% to < 3%: Rash/pruritus/dermatitis, diarrhea, alopecia, leukopenia (white blood cell count < 3000/mm 3), pancytopenia, dizziness
Two other controlled trials of patients (n = 680) with rheumatoid arthritis who received methotrexate 7.5 mg to 15 mg orally once weekly showed the following serious adverse reaction:
Incidence 1%: Interstitial pneumonitis
Other less common adverse reactions were: anemia, headache, upper respiratory infection, anorexia, arthralgias, chest pain, coughing, dysuria, eye discomfort, epistaxis, fever, infection, sweating, tinnitus, vaginal discharge.
Polyarticular Juvenile Idiopathic Arthritis (pJIA)
The most common adverse reactions reported in patients 2 to 18 years of age with pJIA treated with methotrexate 5 mg/m 2to 20 mg/m 2orally once weekly or 0.1 to 0.65 mg/kg orally once weekly were as follows: elevated liver tests 14%; gastrointestinal reactions (e.g., nausea, vomiting, diarrhea) 11%; stomatitis 2%; leukopenia 2%; headache 1.2%; alopecia 0.5%; dizziness 0.2%; rash 0.2%. Most patients received concomitant NSAIDs and some also received corticosteroids.
Psoriasis
In two published series of adults with psoriasis (n = 204, 248) who received methotrexate up to 25 mg per week for up to 4 years, adverse reaction rates were similar to those in patients with rheumatoid arthritis, except for alopecia, photosensitivity, and “burning of skin lesions” (3% to 10% each). Painful plaque erosions have been reported.
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of methotrexate. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Cardiovascular: Thromboembolic events (including arterial thrombosis, cerebral thrombosis, deep vein thrombosis, retinal vein thrombosis, thrombophlebitis, and pulmonary embolus), pericarditis, pericardial effusion, hypotension, sudden death
Endocrine: Diabetes
Eye: Optic neuropathy, blurred vision, ocular pain, conjunctivitis, xerophthalmia
Gastrointestinal: Hemorrhagic enteritis, intestinal perforation, gingivitis, pancreatitis, pharyngitis, hematemesis, melena, gastrointestinal ulceration
Hematology: Aplastic anemia, lymphadenopathy, hypogammaglobulinemia
Hepatobiliary: Acute hepatitis, decreased serum albumin, fibrosis, cirrhosis
Immune system: Anaphylaxis, anaphylactoid reactions, vasculitis
Metabolism: Hyperglycemia
Musculoskeletal: Stress fracture, soft tissue and bone necrosis, arthralgia, myalgia, osteoporosis
Nervous system: Headaches, drowsiness, blurred vision, speech impairment (including dysarthria and aphasia), transient cognitive dysfunction, mood alteration, unusual cranial sensations, paresis, encephalopathy, and convulsions.
Renal: Azotemia, hematuria, proteinuria, cystitis
Reproductive: Defective oogenesis or spermatogenesis, loss of libido, impotence, gynecomastia, menstrual dysfunction
Respiratory: Pulmonary fibrosis, respiratory failure, chronic interstitial obstructive pulmonary disease, pleuritic pain and thickening, alveolitis
Skin: Toxic epidermal necrolysis, Stevens-Johnson syndrome, exfoliative dermatitis, skin necrosis, and erythema multiforme, erythematous rashes, pruritus, alopecia, skin ulceration, accelerated nodulosis, urticaria, pigmentary changes, ecchymosis, telangiectasia, photosensitivity, acne, furunculosis
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on Methotrexate
Drugs that Increase Methotrexate Exposure
Coadministration of methotrexate with the following products may increase methotrexate plasma concentrations, which may increase the risk of methotrexate severe adverse reactions. In some cases, the coadministration of methotrexate with these products may also subsequently reduce active metabolite formation, which may decrease the clinical effectiveness of methotrexate. Increased organ specific adverse reactions may also occur when methotrexate is coadministered with hepatotoxic or nephrotoxic products.
If coadministration cannot be avoided, monitor closely for methotrexate adverse reactions when coadministered with:
|
|
Nitrous Oxide
Coadministration of methotrexate with nitrous oxide anesthesia potentiates the effect of methotrexate on folate-dependent metabolic pathways, which may increase the risk of severe methotrexate adverse reactions. Avoid nitrous oxide anesthesia in patients receiving methotrexate. Consider alternative therapies in patients who have received prior nitrous oxide anesthesia.
Folic Acid
Coadministration of methotrexate with folic acid or its derivatives decreases the clinical effectiveness of methotrexate in patients with neoplastic diseases. Methotrexate competes with reduced folates for active transport across cell membranes. Instruct patients to take folic or folinic acid only as directed by their healthcare provider [see Warnings and Precautions (5.10)].
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Methotrexate tablets are contraindicated in pregnant women with non-neoplastic diseases [see Contraindications (4)].
Based on published reports and its mechanism of action [see Clinical Pharmacology (12.1)], methotrexate can cause embryo-fetal toxicity and fetal death when administered to a pregnant woman. There are no animal data that meet current standards for nonclinical developmental toxicity studies. Advise pregnant women with neoplastic diseases of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Human Data
Published data from case reports, literature reviews, and observational studies report that methotrexate exposure during pregnancy is associated with an increased risk of embryo-fetal toxicity and fetal death. Methotrexate exposure during the first trimester of pregnancy is associated with an increased incidence of spontaneous abortions and multiple adverse developmental outcomes, including skull anomalies, facial dysmorphism, central nervous system abnormalities, limb abnormalities, and sometimes cardiac anomalies and intellectual impairment. Adverse outcomes associated with exposure during second and third trimesters of pregnancy include intrauterine growth restriction and functional abnormalities. Because methotrexate is widely distributed and persists in the body for a prolonged period, there is a potential risk to the fetus from preconception methotrexate exposure.
A prospective multicenter study evaluated pregnancy outcomes in women taking methotrexate less than or equal to 30 mg/week after conception. The rate of spontaneous abortion and miscarriage in pregnant women exposed to methotrexate was 42% (95% confidence interval [95% CI] 29, 59), which was higher than in unexposed patients with autoimmune disease (22%; 95% CI: 17, 30) and unexposed patients with nonautoimmune disease (17%; 95% CI: 13, 23). Of the live births, the rate of major birth defects in pregnant women exposed to methotrexate after conception was higher than in unexposed patients with autoimmune disease (adjusted odds ratio (OR) 1.8 [95% CI: 0.6, 6]) and unexposed patients with non-autoimmune disease (adjusted OR 3.1 [95% CI: 1, 10]) (2.9%). Major birth defects associated with pregnancies exposed to methotrexate after conception were not always consistent with methotrexate-associated adverse developmental outcomes.
8.2 Lactation
Risk Summary
Limited published literature report the presence of methotrexate in human milk in low amounts, with the highest breast milk to plasma concentration ratio reported to be 0.08:1. There are no data on the effects of methotrexate or its metabolites on the breastfed child or their effects on milk production. Because of the potential for serious adverse reactions in a breastfed child, instruct women not to breastfeed during treatment with methotrexate tablets and for 1 week after the final dose.
8.3 Females and Males of Reproductive Potential
Methotrexate can cause malformations and fetal death at doses less than or equal to the recommended clinical doses [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to initiating methotrexate tablets [see Contraindications (4), Use in Specific Populations (8.1)].
Contraception
Infertility
Females
Based on published reports of female infertility after methotrexate, advise females of reproductive potential that methotrexate can cause impairment of fertility and menstrual dysfunction during treatment with methotrexate tablets and after the final dose. It is not known if the infertility may be reversed in all affected females.
8.4 Pediatric Use
The safety and effectiveness of methotrexate tablets in pediatric patients have been established for the treatment of ALL as part of the combination chemotherapy maintenance regimen and the treatment of pJIA [see Indications and Usage (1), Dosage and Administration (2)]. No new safety signals have been observed in pediatric patients in clinical studies [see Adverse Reactions (6.1)].
The safety and effectiveness of methotrexate tablets have not been established in pediatric patients for the other indications [see Indications and Usage (1)].
8.5 Geriatric Use
Clinical studies of methotrexate did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects.
8.6 Renal Impairment
Methotrexate elimination is reduced in patients with renal impairment [see Clinical Pharmacology (12.3)]. Patients with renal impairment are at increased risk for methotrexate adverse reactions. Closely monitor patients with renal impairment [creatinine clearance (CLcr) less than 90 mL/min, Cockcroft-Gault] for adverse reactions. Reduce the dosage or discontinue methotrexate tablets as appropriate [see Warnings and Precautions (5.8)].
8.7 Hepatic Impairment
The pharmacokinetics and safety of methotrexate in patients with hepatic impairment is unknown. Patients with hepatic impairment may be at increased risk for methotrexate adverse reactions based on the elimination characteristics of methotrexate [see Clinical Pharmacology (12.3)]. Closely monitor patients with hepatic impairment for adverse reactions. Reduce the dosage or discontinue methotrexate tablets as appropriate [see Warnings and Precautions (5.5)] .
10 OVERDOSAGE
Overdosage, including fatal overdosage, has occurred with methotrexate [see Warnings and Precautions (5.9)].
Manifestations: Manifestations of methotrexate overdosage include adverse reactions reported at pharmacologic doses, particularly hematologic and gastrointestinal reactions (e.g., leukopenia, thrombocytopenia, anemia, pancytopenia, myelosuppression, mucositis, stomatitis, oral ulceration, nausea, vomiting, gastrointestinal ulceration, or gastrointestinal bleeding). In some cases, no symptoms were reported; however, sepsis or septic shock, renal failure, and aplastic anemia were also reported.
Management: Leucovorin and levoleucovorin are indicated for diminishing the methotrexate adverse reactions of methotrexate overdosage. Administer leucovorin or levoleucovorin as soon as possible after methotrexate overdosage). Monitor serum creatinine and methotrexate levels to guide leucovorin or levoleucovorin therapy. Refer to the leucovorin or levoleucovorin prescribing information for additional dosage information.
Glucarpidase is indicated for the treatment of toxic plasma methotrexate concentrations (> 1 micromole per liter) in patients with delayed methotrexate clearance due to impaired renal function. Refer to the glucarpidase prescribing information for additional dosage information.
Administer concomitant hydration and urinary alkalinization.
Neither hemodialysis nor peritoneal dialysis has been shown to improve methotrexate elimination; however, methotrexate has been effectively cleared with acute, intermittent hemodialysis using a high-flux dialyzer.
11 DESCRIPTION
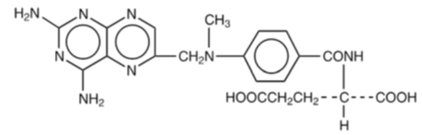
Methotrexate is dihydrofolate reductase inhibitor with the chemical name of N-[4-[[(2,4 diamino-6-pteridinyl) methyl]methylamino]benzoyl]-L glutamic acid. The molecular formula is C 20H 22N 8O 5and the molecular weight is 454.4 g/mol. The structural formula is:
Methotrexate Tablets, USP for oral use are available in bottles of 100 tablets. Each methotrexate tablet contains 2.5 mg methotrexate equivalent to 2.74 mg methotrexate sodium and the following inactive ingredients: colloidal silicon dioxide, FD&C Red No. 40 Aluminum Lake, lactose monohydrate, magnesium stearate, microcrystalline cellulose, pregelatinized starch (corn), sodium carbonate (monohydrate), sodium lauryl sulfate and sodium starch glycolate (potato).
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Methotrexate inhibits dihydrofolic acid reductase. Dihydrofolates must be reduced to tetrahydrofolates by this enzyme before they can be utilized as carriers of one-carbon groups in the synthesis of purine nucleotides and thymidylate. Therefore, methotrexate interferes with DNA synthesis, repair, and cellular replication. Actively proliferating tissues such as malignant cells, bone marrow, fetal cells, buccal and intestinal mucosa, and cells of the urinary bladder are in general more sensitive to this effect of methotrexate.
The mechanism of action in rheumatoid arthritis and in psoriasis is unknown.
12.3 Pharmacokinetics
Absorption
At doses of 30 mg/m 2or less, the mean bioavailability is approximately 60%. Peak plasma concentrations are reached within 0.75 to 6 hours following oral administration. Methotrexate may undergo enterohepatic recirculation; however, this pathway has not been fully characterized.
Distribution
Methotrexate in serum is approximately 50% protein bound.
Methotrexate does not penetrate the blood-cerebrospinal fluid barrier at concentrations achieved with the recommended dosages.
Elimination
The elimination half-life of methotrexate is approximately 3 to 10 hours.
Small amounts of methotrexate polyglutamates may remain in tissues for extended periods. The retention and prolonged drug action of these active metabolites vary among different cells, tissues, and tumors.
Nonlinear elimination due to saturation of renal tubular reabsorption has been observed in studies of patients with psoriasis receiving methotrexate doses between 7.5 mg and 30 mg.
Metabolism
Methotrexate is partially metabolized by intestinal flora after oral administration.
Methotrexate primarily undergoes hepatic and intracellular metabolism to active polyglutamated forms which can be converted back to methotrexate by hydrolase enzymes. Methotrexate also undergoes minor metabolism to active 7-hydroxymethotrexate.
Specific Populations
The effect of hepatic impairment on the pharmacokinetics of methotrexate is unknown.
Pediatric Patients
In pediatric patients with leukemia, oral absorption (23% to 95%) of methotrexate is variable and dose-dependent. The difference between highest and lowest peak methotrexate concentrations (C max0.11 to 2.3 micromolar after a 20 mg/m 2dose) was 20-fold. The time to peak concentration (T max0.67 to 4 hours after a 15 mg/m 2dose) and fraction of dose absorbed is variable. The absorption of doses greater than 40 mg/m 2is significantly less than that of lower doses.
In pediatric patients with pJIA, plasma concentrations of methotrexate are variable. Following oral administration of methotrexate 6.4 mg/m 2/week to 11.2 mg/m 2/week, mean serum concentrations were 0.59 micromolar (0.03 to 1.40) at 1 hour, 0.44 micromolar (0.01 to 1.00) at 2 hours, and 0.29 micromolar (0.06 to 0.58) at 3 hours.
In pediatric patients receiving methotrexate for acute lymphoblastic leukemia (6.3 mg/m 2 to 30 mg/m 2) or for JIA (3.75 mg/m 2 to 26.2 mg/m 2), the terminal half-life has been reported to range from 0.7 to 5.8 hours or from 0.9 to 2.3 hours, respectively.
16 HOW SUPPLIED/STORAGE AND HANDLING
Methotrexate Tablets, USP are available containing 2.5 mg methotrexate, USP equivalent to 2.74 mg methotrexate sodium.
The 2.5 mg tablets are orange, round, scored tablets debossed with Mabove the score and 14below the score on one side of the tablet and blank on the other side of the tablet. They are available as follows:
NDC 51079-670-05 – Unit dose blister packages of 20 (2 cards of 10 tablets each).
Store at 20°C to 25°C (68°F to 77°F). [See USP Controlled Room Temperature.]
Protect from light.
Methotrexate tablets are a cytotoxic drug. Follow applicable special handling and disposal procedures. 1
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling ( Patient Information).
Embryo-Fetal Toxicity
- Advise females of reproductive potential of the potential risk to a fetus and to inform their healthcare provider of a known or suspected pregnancy [see Contraindications (4), Warnings and Precautions (5.1), Use in Specific Populations (8.1)].
- Advise females of reproductive potential to use effective contraception during treatment with methotrexate tablets and for 6 months after the final dose [see Use in Specific Populations (8.3)].
- Advise males of reproductive potential to use effective contraception during treatment with methotrexate tablets and for 3 months after the final dose [see Use in Specific Populations (8.3)].
Hypersensitivity Reactions: Advise patients and their caregivers of the potential risk of hypersensitivity and that methotrexate tablets are contraindicated in patients with a history of hypersensitivity reactions to methotrexate. Instruct patients to seek immediate medical attention for signs of a hypersensitivity reaction [see Warnings and Precautions (5.2)].
Myelosuppression and Serious Infections: Inform patients and their caregivers that methotrexate tablets can cause myelosuppression and the need for frequent monitoring of blood cell counts. Advise patients and their caregivers to immediately report new onset fever, symptoms of infection, easy bruising or persistent bleeding to their healthcare provider [see Warnings and Precautions (5.3, 5.11)].
Gastrointestinal Toxicity: Advise patients and their caregivers to report new or worsening diarrhea, vomiting, or stomatitis to their healthcare provider. Advise patients to immediately contact their healthcare provider for high fever, rigors, persistent or severe abdominal pain, severe constipation, hematemesis, or melena [see Warnings and Precautions (5.4)].
Hepatotoxicity: Advise patients and their caregivers to report signs or symptoms of hepatic toxicity to their healthcare provider [see Warnings and Precautions (5.5)].
Pulmonary Toxicity: Advise patients and their caregivers to report new or worsening cough, fever, or dyspnea to their healthcare provider [see Warnings and Precautions (5.6)].
Dermatologic Reactions: Advise patients and their caregivers that methotrexate tablets can cause serious skin rash and to immediately contact their healthcare provider for new or worsening skin rash. Advise patients and their caregivers to avoid excessive sun exposure and use sun protection measures [see Warnings and Precautions (5.7)].
Renal Toxicity: Advise patients and their caregivers to immediately contact their healthcare provider for signs or symptoms of renal toxicity, such as marked increases or decreases in urinary output [see Warnings and Precautions (5.8)].
Risk of Serious Adverse Reactions with Medication Error: For patients who are prescribed a once weekly dosing regimen, advise patients and caregivers that the recommended dosage is to be taken once weekly as a single dose and that mistakenly taking the recommended weekly dosage once daily has led to fatal adverse reactions [see Warnings and Precautions (5.9)].
Neurotoxicity: Advise patients and their caregivers to report new neurological signs or symptoms to their healthcare provider [see Warnings and Precautions (5.12)].
Secondary Malignancies: Advise patients on the risk of second primary malignancies during treatment with methotrexate tablets [see Warnings and Precautions (5.13)].
Lactation: Instruct women not to breastfeed during treatment with methotrexate tablets and for 1 week after the final dose [see Use in Specific Populations (8.2)].
Infertility: Advise females and males of reproductive potential that methotrexate may impair fertility [see Warnings and Precautions (5.16), Use in Specific Populations (8.3)].
Drug Interactions: Advise patients and caregivers to inform their healthcare provider of all concomitant medications, including prescription medicines, over-the-counter drugs, vitamins, and herbal products [see Drug Interactions (7)].
Patient Information
|
Methotrexate Tablets, USP (meth" oh trex' ate) |
||
|
What is the most important information I should know about methotrexate tablets?
Methotrexate tablets can cause serious side effects that may be severe and lead to death,including:
Harm to an unborn baby, including birth defects or death of an unborn baby.
Females who can become pregnant:
Males with female partners who are able to become pregnant:
Severe allergic reactions. Severe allergic reactions can happen with methotrexate tablets. Signs and symptoms of a severe allergic reaction may include: |
||
|
|
|
|
Do nottake methotrexate tablets if you have had a severe allergic reaction to methotrexate in the past.
Get medical help right awayif you develop any of the signs or symptoms of a severe allergic reaction listed above.
Decreased blood cell counts.Methotrexate tablets can affect your bone marrow and cause decreases in red blood counts, white blood cell counts, and platelets that can be severe and life-threatening.
Call your healthcare provider right away if you develop any of the following: |
||
|
|
|
|
Severe stomach and intestine problems (gastrointestinal) problems.
Tell your healthcare provider if you develop new or worsening diarrhea, vomiting, or mouth sores during treatment with methotrexate tablets.
Tell your healthcare provider right away if you develop high fever, shaking chills (rigors), pain in your stomach-area (abdomen) that will not go away or is severe, severe constipation, if you are vomiting blood or have blood in your stools.
Liver problems.Methotrexate tablets can cause severe liver problems including liver scarring (fibrosis), cirrhosis, and liver failure that may not get better (possibly irreversible) and can cause death.
Tell your healthcare provider if you have any signs or symptoms of liver problemsduring treatment with methotrexate tablets, including: |
||
|
|
|
|
Lung problems.Lung problems can happen suddenly (acute) with methotrexate tablets or they can develop over a long period-of-time (chronic). Lung problems may not get better (possibly irreversible) and can cause death.
Tell your healthcare provider if you have any new or worsening symptoms including: cough (especially a dry cough), fever, or trouble breathing.
Severe skin reactions.Severe skin reactions can happen with methotrexate tablets and can lead to death.
Limit sunlight exposure during treatment with methotrexate tablets. Use sunscreen and wear protective clothing when you will be exposed to sunlight during treatment with methotrexate tablets.
Tell your healthcare provider right away about any new or worsening skin rash during treatment with methotrexate tablets.
Kidney problems. Kidney problems can happen with methotrexate tablets, including kidney failure which can happen suddenly (acute) and may not go away (irreversible).
Your healthcare provider will check your kidney function before you start and during treatment with methotrexate tablets.
Tell your healthcare provider right awayif you have any signs or symptoms of kidney problems, including: |
||
|
|
|
|
See “What are the possible side effects of methotrexate tablets”for more information about side effects. |
||
|
What are methotrexate tablets? Methotrexate tablets are a prescription medicine used:
It is not known if methotrexate tablets are safe and effective in treating children with any disease other than ALL as part of a combination regimen used for maintenance therapy of their cancer, and for the treatment of pJIA.
It is not known if methotrexate tablets are safe in people with liver problems. |
||
|
Do not take methotrexate tablets if you:
|
||
|
Before taking methotrexate tablets tell your healthcare provider about all of your medical conditions, including if you:
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins and herbal supplements. Methotrexate tablets and certain other medicines can affect each other and cause serious side effects. Do not start or change any medicines unless you have talked to your doctor and your doctor has told you it is safe. Know all the medicines that you take and keep a list of them with you at all times to show doctors and pharmacists. |
||
|
How should I take methotrexate tablets?
If you are taking methotrexate tablets for treatment of severe psoriasis, rheumatoid arthritis, or polyarticular juvenile idiopathic arthritis:
If you are taking methotrexate tablets to treat your cancer:
|
||
|
What are the possible side effects of methotrexate tablets? Methotrexate tablets can cause serious side effects that may be severe and lead to death including:
The most common side effects of methotrexate tablets include:
These are not all the side effects of methotrexate tablets. Ask your healthcare provider or pharmacist for more information. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800- FDA-1088. |
||
|
How should I store methotrexate tablets?
Keep methotrexate tablets and all medicines out of the reach of children. |
||
|
General information about the safe and effective use of methotrexate tablets. Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use methotrexate tablets for a condition for which they were not prescribed. Do not give methotrexate tablets to other people, even if they have the same symptoms that you have. They may harm them. This leaflet summarizes the most important information about methotrexate tablets. If you would like more information, talk with your healthcare provider. You can ask your pharmacist or healthcare provider for information about methotrexate tablets that is written for healthcare professionals. |
||
|
What are the ingredients in methotrexate tablets? Active Ingredient:methotrexate Inactive Ingredients:colloidal silicon dioxide, FD&C Red No. 40 Aluminum Lake, lactose monohydrate, magnesium stearate, microcrystalline cellulose, pregelatinized starch (corn), sodium carbonate (monohydrate), sodium lauryl sulfate and sodium starch glycolate (potato).
Manufactured for: Mylan Pharmaceuticals Inc., Morgantown, WV 26505 U.S.A. For additional information contact Mylan at 1-877-446-3679 (1-877-4-INFO-RX). |
||
This Patient Information has been approved by the U.S. Food and Drug Administration.
Manufactured for:
Mylan Pharmaceuticals Inc.
Morgantown, WV 26505 U.S.A.
Manufactured by:
Auro PR Inc.
RD 156 Caguas West Industrial Park, Lot 24
Caguas, PR 00725 U.S.A.
Distributed by:
Mylan Institutional Inc.
Rockford, IL 61103 U.S.A.
S-12021 R5
10/22
PACKAGE/LABEL PRINCIPAL DISPLAY PANEL
NDC 51079-670-05
Methotrexate
Tablets, USP
2.5 mg
20 Tablets (2 x 10)
Caution: Cytotoxic Agent
Each tablet contains 2.5 mg
methotrexate, USP equivalent to
2.74 mg methotrexate sodium.
Store at 20°C to 25°C (68°F to 77°F). [See USP Controlled Room Temperature.]
Protect from light.
Recommended Dosage:See accompanying prescribing information
and Patient Information Leaflet.
Manufactured for:
Mylan Pharmaceuticals Inc.
Morgantown, WV 26505 U.S.A.
Made in Puerto Rico
Rx only
S-10757 R6
Distributed by:
Mylan Institutional Inc.
Rockford, IL 61103 U.S.A.
This unit dose package is not child resistant.
For institutional use only.
Keep this and all drugs out of the reach of children.
This container provides light-resistance.
See window for lot number and expiration date.

