DESCRIPTION
Buspirone hydrochloride is an antianxiety agent that is not chemically or pharmacologically related to the benzodiazepines, barbiturates, or other sedative/anxiolytic drugs.
Buspirone hydrochloride is a white crystalline, water soluble compound with a molecular weight of 422.0. Chemically, buspirone hydrochloride is 8-[4-[4-(2-pyrimidinyl)-1-piperazinyl]butyl]-8-azaspiro[4.5]decane-7,9-dione monohydrochloride. The molecular formula C21H31N5O2•HCl is represented by the following structural formula:
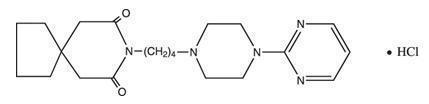
Each tablet, for oral administration, contains 5 mg, 10 mg, 15 mg or 30 mg of buspirone hydrochloride, USP (equivalent to 4.6 mg, 9.1 mg, 13.7 mg and 27.4 mg of buspirone free base, respectively). The 5 mg and 10 mg tablets are scored so they can be bisected. Thus, the 5 mg tablet can also provide a 2.5 mg dose, and the 10 mg tablet can provide a 5 mg dose. The 15 mg and 30 mg tablets are provided in a multi-scored tablet design. These tablets are scored so they can be either bisected or trisected. Thus, a single 15 mg tablet can provide the following doses: 15 mg (entire tablet), 10 mg (two-thirds of a tablet), 7.5 mg (one-half of a tablet), or 5 mg (one-third of a tablet). A single 30 mg tablet can provide the following doses: 30 mg (entire tablet), 20 mg (two-thirds of a tablet), 15 mg (one-half of a tablet), or 10 mg (one-third of a tablet). Buspirone hydrochloride tablets contain the following inactive ingredients: anhydrous lactose, colloidal silicon dioxide, magnesium stearate, microcrystalline cellulose, sodium lauryl sulfate, and sodium starch glycolate.
CLINICAL PHARMACOLOGY
The mechanism of action of buspirone is unknown. Buspirone differs from typical benzodiazepine anxiolytics in that it does not exert anticonvulsant or muscle relaxant effects. It also lacks the prominent sedative effect that is associated with more typical anxiolytics. In vitro preclinical studies have shown that buspirone has a high affinity for serotonin (5-HT1A) receptors. Buspirone has no significant affinity for benzodiazepine receptors and does not affect GABA binding in vitro or in vivo when tested in preclinical models.
Buspirone has moderate affinity for brain D2-dopamine receptors. Some studies do suggest that buspirone may have indirect effects on other neurotransmitter systems.
Buspirone is rapidly absorbed in man and undergoes extensive first-pass metabolism. In a radiolabeled study, unchanged buspirone in the plasma accounted for only about 1% of the radioactivity in the plasma. Following oral administration, plasma concentrations of unchanged buspirone are very low and variable between subjects. Peak plasma levels of 1 ng/mL to 6 ng/mL have been observed 40 to 90 minutes after single oral doses of 20 mg. The single-dose bioavailability of unchanged buspirone when taken as a tablet is on the average about 90% of an equivalent dose of solution, but there is large variability.
The effects of food upon the bioavailability of buspirone have been studied in eight subjects. They were given a 20 mg dose with and without food; the area under the plasma concentration-time curve (AUC) and peak plasma concentration (Cmax) of unchanged buspirone increased by 84% and 116% respectively, but the total amount of buspirone immunoreactive material did not change. This suggests that food may decrease the extent of presystemic clearance of buspirone (see DOSAGE AND ADMINISTRATION).
A multiple-dose study conducted in 15 subjects suggests that buspirone has nonlinear pharmacokinetics. Thus, dose increases and repeated dosing may lead to somewhat higher blood levels of unchanged buspirone than would be predicted from results of single-dose studies.
An in vitro protein binding study indicated that approximately 86% of buspirone is bound to plasma proteins. It was also observed that aspirin increased the plasma levels of free buspirone by 23%, while flurazepam decreased the plasma levels of free buspirone by 20%. However, it is not known whether these drugs cause similar effects on plasma levels of free buspirone in vivo, or whether such changes, if they do occur, cause clinically significant differences in treatment outcome. An in vitro study indicated that buspirone did not displace highly protein bound drugs such as phenytoin, warfarin, and propranolol from plasma protein, and that buspirone may displace digoxin.
Buspirone is metabolized primarily by oxidation, which in vitro has been shown to be mediated by cytochrome P450 3A4 (CYP3A4). (See PRECAUTIONS: Drug Interactions.) Several hydroxylated derivatives and a pharmacologically active metabolite, 1-pyrimidinylpiperazine (1-PP), are produced. In animal models predictive of anxiolytic potential, 1-PP has about one quarter of the activity of buspirone, but is present in up to 20-fold greater amounts. However, this is probably not important in humans; blood samples from humans chronically exposed to buspirone do not exhibit high levels of 1-PP; mean values are approximately 3 ng/mL and the highest human blood level recorded among 108 chronically dosed patients was 17 ng/mL, less than 1/200th of 1-PP levels found in animals given large doses of buspirone without signs of toxicity.
In a single-dose study using 14C-labeled buspirone, 29% to 63% of the dose was excreted in the urine within 24 hours, primarily as metabolites; fecal excretion accounted for 18% to 38% of the dose. The average elimination half-life of unchanged buspirone after single doses of 10 mg to 40 mg is about 2 to 3 hours.
Special Populations
Age and Gender Effects
After single or multiple doses in adults, no significant differences in buspirone pharmacokinetics (AUC and Cmax) were observed between elderly and younger subjects or between men and women.
Hepatic Impairment
After multiple-dose administration of buspirone to patients with hepatic impairment, steady-state AUC of buspirone increased 13-fold compared with healthy subjects (see PRECAUTIONS).
Renal Impairment
After multiple-dose administration of buspirone to renally impaired (CIcr = 10 to 70 mL/min/1.73 m2) patients, steady-state AUC of buspirone increased 4-fold compared with healthy (CIcr ≥ 80 mL/min/1.73 m2) subjects (see PRECAUTIONS).
INDICATIONS AND USAGE
Buspirone hydrochloride tablets are indicated for the management of anxiety disorders or the short-term relief of the symptoms of anxiety. Anxiety or tension associated with the stress of everyday life usually does not require treatment with an anxiolytic.
The efficacy of buspirone hydrochloride has been demonstrated in controlled clinical trials of outpatients whose diagnosis roughly corresponds to Generalized Anxiety Disorder (GAD). Many of the patients enrolled in these studies also had coexisting depressive symptoms and buspirone relieved anxiety in the presence of these coexisting depressive symptoms. The patients evaluated in these studies had experienced symptoms for periods of 1 month to over 1 year prior to the study, with an average symptom duration of 6 months. Generalized Anxiety Disorder (300.02) is described in the American Psychiatric Association’s Diagnostic and Statistical Manual, III1 as follows:
Generalized, persistent anxiety (of at least 1 month continual duration), manifested by symptoms from three of the four following categories:
- Motor tension: shakiness, jitteriness, jumpiness, trembling, tension, muscle aches, fatigability, inability to relax, eyelid twitch, furrowed brow, strained face, fidgeting, restlessness, easy startle.
- Autonomic hyperactivity: sweating, heart pounding or racing, cold, clammy hands, dry mouth, dizziness, lightheadedness, paresthesias (tingling in hands or feet), upset stomach, hot or cold spells, frequent urination, diarrhea, discomfort in the pit of the stomach, lump in the throat, flushing, pallor, high resting pulse and respiration rate.
- Apprehensive expectation: anxiety, worry, fear, rumination, and anticipation of misfortune to self or others.
- Vigilance and scanning: hyperattentiveness resulting in distractibility, difficulty in concentrating, insomnia, feeling “on edge,” irritability, impatience.
The above symptoms would not be due to another mental disorder, such as a depressive disorder or schizophrenia. However, mild depressive symptoms are common in GAD.
The effectiveness of buspirone in long-term use, that is, for more than 3 to 4 weeks, has not been demonstrated in controlled trials. There is no body of evidence available that systematically addresses the appropriate duration of treatment for GAD. However, in a study of long-term use, 264 patients were treated with buspirone for 1 year without ill effect. Therefore, the physician who elects to use buspirone for extended periods should periodically reassess the usefulness of the drug for the individual patient.
CONTRAINDICATIONS
Buspirone is contraindicated in patients hypersensitive to buspirone hydrochloride.
WARNINGS
The administration of buspirone to a patient taking a monoamine oxidase inhibitor (MAOI) may pose a hazard. There have been reports of the occurrence of elevated blood pressure when buspirone has been added to a regimen including an MAOI. Therefore, it is recommended that buspirone not be used concomitantly with an MAOI.
Because buspirone has no established antipsychotic activity, it should not be employed in lieu of appropriate antipsychotic treatment.
PRECAUTIONS
General
Interference with Cognitive and Motor Performance
Studies indicate that buspirone is less sedating than other anxiolytics and that it does not produce significant functional impairment. However, its CNS effects in any individual patient may not be predictable. Therefore, patients should be cautioned about operating an automobile or using complex machinery until they are reasonably certain that buspirone treatment does not affect them adversely.
While formal studies of the interaction of buspirone with alcohol indicate that buspirone does not increase alcohol-induced impairment in motor and mental performance, it is prudent to avoid concomitant use of alcohol and buspirone.
Potential for Withdrawal Reactions in Sedative/Hypnotic/Anxiolytic Drug-Dependent Patients
Because buspirone does not exhibit cross-tolerance with benzodiazepines and other common sedative/hypnotic drugs, it will not block the withdrawal syndrome often seen with cessation of therapy with these drugs. Therefore, before starting therapy with buspirone, it is advisable to withdraw patients gradually, especially patients who have been using a CNS-depressant drug chronically, from their prior treatment. Rebound or withdrawal symptoms may occur over varying time periods, depending in part on the type of drug, and its effective half-life of elimination.
The syndrome of withdrawal from sedative/hypnotic/anxiolytic drugs can appear as any combination of irritability, anxiety, agitation, insomnia, tremor, abdominal cramps, muscle cramps, vomiting, sweating, flu-like symptoms without fever, and occasionally, even as seizures.
Possible Concerns Related to Buspirone’s Binding to Dopamine Receptors
Because buspirone can bind to central dopamine receptors, a question has been raised about its potential to cause acute and chronic changes in dopamine-mediated neurological function (e.g., dystonia, pseudo-parkinsonism, akathisia, and tardive dyskinesia). Clinical experience in controlled trials has failed to identify any significant neuroleptic-like activity; however, a syndrome of restlessness, appearing shortly after initiation of treatment, has been reported in some small fraction of buspirone-treated patients. The syndrome may be explained in several ways. For example, buspirone may increase central noradrenergic activity; alternatively, the effect may be attributable to dopaminergic effects (i.e., represent akathisia). See ADVERSE REACTIONS: Post-Marketing Experience.
Information for Patients
To assure safe and effective use of buspirone hydrochloride tablets, the following information and instructions should be given to patients:
- Inform your physician about any medications, prescription or nonprescription, alcohol, or drugs that you are now taking or plan to take during your treatment with buspirone.
- Inform your physician if you are pregnant, or if you are planning to become pregnant, or if you become pregnant while you are taking buspirone.
- Inform your physician if you are breast-feeding an infant.
- Until you experience how this medication affects you, do not drive a car or operate potentially dangerous machinery.
- You should take buspirone consistently, either always with or always without food.
- During your treatment with buspirone, avoid drinking large amounts of grapefruit juice.
Drug Interactions
Psychotropic Agents
MAO inhibitors
It is recommended that buspirone not be used concomitantly with MAO inhibitors (see WARNINGS).
Amitriptyline
After addition of buspirone to the amitriptyline dose regimen, no statistically significant differences in the steady-state pharmacokinetic parameters (Cmax, AUC, and Cmin) of amitriptyline or its metabolite nortriptyline were observed.
Diazepam
After addition of buspirone to the diazepam dose regimen, no statistically significant differences in the steady-state pharmacokinetic parameters (Cmax, AUC, and Cmin) were observed for diazepam, but increases of about 15% were seen for nordiazepam, and minor adverse clinical effects (dizziness, headache, and nausea) were observed.
Haloperidol
In a study in normal volunteers, concomitant administration of buspirone and haloperidol resulted in increased serum haloperidol concentrations. The clinical significance of this finding is not clear.
Trazodone
There is one report suggesting that the concomitant use of trazodone and buspirone may have caused 3-fold to 6-fold elevations of SGPT (ALT) in a few patients. In a similar study attempting to replicate this finding, no interactive effect on hepatic transaminases was identified.
Inhibitors and Inducers of Cytochrome P450 3A4 (CYP3A4)
Buspirone has been shown in vitro to be metabolized by CYP3A4. This finding is consistent with the in vivo interactions observed between buspirone and the following:
Diltiazem and Verapamil
In a study of nine healthy volunteers, coadministration of buspirone (10 mg as a single dose) with verapamil (80 mg t.i.d.) or diltiazem (60 mg t.i.d.) increased plasma buspirone concentrations (verapamil increased AUC and Cmax of buspirone 3.4-fold while diltiazem increased AUC and Cmax 5.5-fold and 4-fold, respectively). Adverse events attributable to buspirone may be more likely during concomitant administration with either diltiazem or verapamil. Subsequent dose adjustment may be necessary and should be based on clinical assessment.
Erythromycin
In a study in healthy volunteers, coadministration of buspirone (10 mg as a single dose) with erythromycin (1.5 g/day for 4 days) increased plasma buspirone concentrations (5-fold increase in Cmax and 6-fold increase in AUC). These pharmacokinetic interactions were accompanied by an increased incidence of side effects attributable to buspirone. If the two drugs are to be used in combination, a low dose of buspirone (e.g., 2.5 mg b.i.d.) is recommended. Subsequent dose adjustment of either drug should be based on clinical assessment.
Grapefruit Juice
In a study in healthy volunteers, coadministration of buspirone (10 mg as a single dose) with grapefruit juice (200 mL double-strength t.i.d. for 2 days) increased plasma buspirone concentrations (4.3-fold increase in Cmax; 9.2-fold increase in AUC). Patients receiving buspirone should be advised to avoid drinking such large amounts of grapefruit juice.
Itraconazole
In a study in healthy volunteers, coadministration of buspirone (10 mg as a single dose) with itraconazole (200 mg/day for 4 days) increased plasma buspirone concentrations (13-fold increase in Cmax and 19-fold increase in AUC). These pharmacokinetic interactions were accompanied by an increased incidence of side effects attributable to buspirone. If the two drugs are to be used in combination, a low dose of buspirone (e.g., 2.5 mg q.d.) is recommended. Subsequent dose adjustment of either drug should be based on clinical assessment.
Nefazodone
In a study of steady-state pharmacokinetics in healthy volunteers, coadministration of buspirone (2.5 mg or 5 mg b.i.d.) with nefazodone (250 mg b.i.d.) resulted in marked increases in plasma buspirone concentrations (increases up to 20-fold in Cmax and up to 50-fold in AUC) and statistically significant decreases (about 50%) in plasma concentrations of the buspirone metabolite 1-PP. With 5 mg b.i.d. doses of buspirone, slight increases in AUC were observed for nefazodone (23%) and its metabolites hydroxynefazodone (HO-NEF) (17%) and meta-chlorophenylpiperazine (9%). Slight increases in Cmax were observed for nefazodone (8%) and its metabolite HO-NEF (11%). Subjects receiving buspirone 5 mg b.i.d. and nefazodone 250 mg b.i.d. experienced lightheadedness, asthenia, dizziness, and somnolence, adverse events also observed with either drug alone. If the two drugs are to be used in combination, a low dose of buspirone (e.g., 2.5 mg q.d.) is recommended. Subsequent dose adjustment of either drug should be based on clinical assessment.
Rifampin
In a study in healthy volunteers, coadministration of buspirone (30 mg as a single dose) with rifampin (600 mg/day for 5 days) decreased the plasma concentrations (83.7% decrease in Cmax; 89.6% decrease in AUC) and pharmacodynamic effects of buspirone. If the two drugs are to be used in combination, the dosage of buspirone may need adjusting to maintain anxiolytic effect.
Other Inhibitors and Inducers of CYP3A4
Substances that inhibit CYP3A4, such as ketoconazole or ritonavir, may inhibit buspirone metabolism and increase plasma concentrations of buspirone while substances that induce CYP3A4, such as dexamethasone or certain anticonvulsants (phenytoin, phenobarbital, carbamazepine), may increase the rate of buspirone metabolism. If a patient has been titrated to a stable dosage on buspirone, a dose adjustment of buspirone may be necessary to avoid adverse events attributable to buspirone or diminished anxiolytic activity. Consequently, when administered with a potent inhibitor of CYP3A4, a low dose of buspirone used cautiously is recommended. When used in combination with a potent inducer of CYP3A4 the dosage of buspirone may need adjusting to maintain anxiolytic effect.
Protein Binding
In vitro, buspirone does not displace tightly bound drugs like phenytoin, propranolol, and warfarin from serum proteins. However, there has been one report of prolonged prothrombin time when buspirone was added to the regimen of a patient treated with warfarin. The patient was also chronically receiving phenytoin, phenobarbital, digoxin, and levothyroxine sodium. In vitro, buspirone may displace less firmly bound drugs like digoxin. The clinical significance of this property is unknown.
Therapeutic levels of aspirin, desipramine, diazepam, flurazepam, ibuprofen, propranolol, thioridazine, and tolbutamide had only a limited effect on the extent of binding of buspirone to plasma proteins (see CLINICAL PHARMACOLOGY).
Drug/Laboratory Test Interactions
Buspirone hydrochloride may interfere with the urinary metanephrine/catecholamine assay. It has been mistakenly read as metanephrine during routine assay testing for pheochromocytoma, resulting in a false positive laboratory result. Buspirone hydrochloride should therefore be discontinued for at least 48 hours prior to undergoing a urine collection for catecholamines.
Carcinogenesis, Mutagenesis, Impairment of Fertility
No evidence of carcinogenic potential was observed in rats during a 24-month study at approximately 133 times the maximum recommended human oral dose; or in mice, during an 18-month study at approximately 167 times the maximum recommended human oral dose.
With or without metabolic activation, buspirone did not induce point mutations in five strains of Salmonella typhimurium (Ames Test) or mouse lymphoma L5178YTK+ cell cultures, nor was DNA damage observed with buspirone in Wi-38 human cells. Chromosomal aberrations or abnormalities did not occur in bone marrow cells of mice given one or five daily doses of buspirone.
Pregnancy
Teratogenic Effects. Pregnancy Category B
No fertility impairment or fetal damage was observed in reproduction studies performed in rats and rabbits at buspirone doses of approximately 30 times the maximum recommended human dose. In humans, however, adequate and well controlled studies during pregnancy have not been performed. Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed.
Labor and Delivery
The effect of buspirone on labor and delivery in women is unknown. No adverse effects were noted in reproduction studies in rats.
Nursing Mothers
The extent of the excretion in human milk of buspirone or its metabolites is not known. In rats, however, buspirone and its metabolites are excreted in milk. Buspirone administration to nursing women should be avoided if clinically possible.
Pediatric Use
The safety and effectiveness of buspirone were evaluated in two placebo-controlled 6-week trials involving a total of 559 pediatric patients (ranging from 6 to 17 years of age) with GAD. Doses studied were 7.5 mg to 30 mg b.i.d. (15 to 60 mg/day). There were no significant differences between buspirone and placebo with regard to the symptoms of GAD following doses recommended for the treatment of GAD in adults. Pharmacokinetic studies have shown that, for identical doses, plasma exposure to buspirone and its active metabolite, 1-PP, are equal to or higher in pediatric patients than adults. No unexpected safety findings were associated with buspirone in these trials. There are no long-term safety or efficacy data in this population.
Geriatric Use
In one study of 6,632 patients who received buspirone for the treatment of anxiety, 605 patients were ≥ 65 years old and 41 were ≥ 75 years old; the safety and efficacy profiles for these 605 elderly patients (mean age = 70.8 years) were similar to those in the younger population (mean age = 43.3 years). Review of spontaneously reported adverse clinical events has not identified differences between elderly and younger patients, but greater sensitivity of some older patients cannot be ruled out.
There were no effects of age on the pharmacokinetics of buspirone (see CLINICAL PHARMACOLOGY: Special Populations).
Use in Patients with Impaired Hepatic or Renal Function
Buspirone is metabolized by the liver and excreted by the kidneys. A pharmacokinetic study in patients with impaired hepatic or renal function demonstrated increased plasma levels and a lengthened half-life of buspirone. Therefore, the administration of buspirone to patients with severe hepatic or renal impairment cannot be recommended (see CLINICAL PHARMACOLOGY).
ADVERSE REACTIONS
(See also PRECAUTIONS)
Commonly Observed
The more commonly observed untoward events associated with the use of buspirone not seen at an equivalent incidence among placebo-treated patients include dizziness, nausea, headache, nervousness, lightheadedness, and excitement.
Associated with Discontinuation of Treatment
One guide to the relative clinical importance of adverse events associated with buspirone is provided by the frequency with which they caused drug discontinuation during clinical testing. Approximately 10% of the 2,200 anxious patients who participated in the buspirone premarketing clinical efficacy trials in anxiety disorders lasting 3 to 4 weeks discontinued treatment due to an adverse event. The more common events causing discontinuation included: central nervous system disturbances (3.4%), primarily dizziness, insomnia, nervousness, drowsiness, and lightheaded feeling; gastrointestinal disturbances (1.2%), primarily nausea; and miscellaneous disturbances (1.1%), primarily headache and fatigue. In addition, 3.4% of patients had multiple complaints, none of which could be characterized as primary.
Incidence in Controlled Clinical Trials
The table that follows enumerates adverse events that occurred at a frequency of 1% or more among buspirone patients who participated in 4-week, controlled trials comparing buspirone with placebo. The frequencies were obtained from pooled data for 17 trials. The prescriber should be aware that these figures cannot be used to predict the incidence of side effects in the course of usual medical practice where patient characteristics and other factors differ from those which prevailed in the clinical trials. Similarly, the cited frequencies cannot be compared with figures obtained from other clinical investigations involving different treatments, uses, and investigators. Comparison of the cited figures, however, does provide the prescribing physician with some basis for estimating the relative contribution of drug and nondrug factors to the side effect incidence rate in the population studied.
| Adverse Experience | Buspirone (n=477) | Placebo (n=464) |
| — Incidence less than 1%. | ||
|
||
| Cardiovascular | ||
| Tachycardia/Palpitations | 1 | 1 |
| CNS | ||
| Dizziness | 12 | 3 |
| Drowsiness | 10 | 9 |
| Nervousness | 5 | 1 |
| Insomnia | 3 | 3 |
| Lightheadedness | 3 | — |
| Decreased Concentration | 2 | 2 |
| Excitement | 2 | — |
| Anger/Hostility | 2 | — |
| Confusion | 2 | — |
| Depression | 2 | 2 |
| EENT | ||
| Blurred Vision | 2 | — |
| Gastrointestinal | ||
| Nausea | 8 | 5 |
| Dry Mouth | 3 | 4 |
| Abdominal/Gastric Distress | 2 | 2 |
| Diarrhea | 2 | — |
| Constipation | 1 | 2 |
| Vomiting | 1 | 2 |
| Musculoskeletal | ||
| Musculoskeletal Aches/Pains | 1 | — |
| Neurological | ||
| Numbness | 2 | — |
| Parasthesia | 1 | — |
| Incoordination | 1 | — |
| Tremor | 1 | — |
| Skin | ||
| Skin Rash | 1 | — |
| Miscellaneous | ||
| Headache | 6 | 3 |
| Fatigue | 4 | 4 |
| Weakness | 2 | — |
| Sweating/Clamminess | 1 | — |
Other Events Observed During the Entire Premarketing Evaluation of Buspirone
During its premarketing assessment, buspirone was evaluated in over 3,500 subjects. This section reports event frequencies for adverse events occurring in approximately 3,000 subjects from this group who took multiple doses of buspirone in the dose range for which buspirone hydrochloride is being recommended (i.e., the modal daily dose of buspirone hydrochloride fell between 10 mg and 30 mg for 70% of the patients studied) and for whom safety data were systematically collected. The conditions and duration of exposure to buspirone varied greatly, involving well controlled studies as well as experience in open and uncontrolled clinical settings. As part of the total experience gained in clinical studies, various adverse events were reported. In the absence of appropriate controls in some of the studies, a causal relationship to buspirone treatment cannot be determined. The list includes all undesirable events reasonably associated with the use of the drug.
The following enumeration by organ system describes events in terms of their relative frequency of reporting in this data base. Events of major clinical importance are also described in the PRECAUTIONS section.
The following definitions of frequency are used: Frequent adverse events are defined as those occurring in at least 1/100 patients. Infrequent adverse events are those occurring in 1/100 to 1/1,000 patients, while rare events are those occurring in less than 1/1,000 patients.
Cardiovascular: Frequent was nonspecific chest pain; infrequent were syncope, hypotension, and hypertension; rare were cerebrovascular accident, congestive heart failure, myocardial infarction, cardiomyopathy, and bradycardia.
Central Nervous System:Frequent were dream disturbances; infrequent were depersonalization, dysphoria, noise intolerance, euphoria, akathisia, fearfulness, loss of interest, dissociative reaction, hallucinations, involuntary movements, slowed reaction time, suicidal ideation, and seizures; rare were feelings of claustrophobia, cold intolerance, stupor, and slurred speech and psychosis.
EENT: Frequent were tinnitus, sore throat, and nasal congestion; infrequent were redness and itching of the eyes, altered taste, altered smell, and conjunctivitis; rare were inner ear abnormality, eye pain, photophobia, and pressure on eyes.
Endocrine: Rare were galactorrhea and thyroid abnormality.
Gastrointestinal: Infrequent were flatulence, anorexia, increased appetite, salivation, irritable colon, and rectal bleeding; rare was burning of the tongue.
Genitourinary: Infrequent were urinary frequency, urinary hesitancy, menstrual irregularity and spotting, and dysuria; rare were amenorrhea, pelvic inflammatory disease, enuresis, and nocturia.
Musculoskeletal:Infrequent were muscle cramps, muscle spasms, rigid/stiff muscles, and arthralgias; rare was muscle weakness.
Respiratory: Infrequent were hyperventilation, shortness of breath, and chest congestion; rare was epistaxis.
Sexual Function: Infrequent were decreased or increased libido; rare were delayed ejaculation and impotence.
Skin: Infrequent were edema, pruritus, flushing, easy bruising, hair loss, dry skin, facial edema, and blisters; rare were acne and thinning of nails.
Clinical Laboratory: Infrequent were increases in hepatic aminotransferases (SGOT, SGPT); rare were eosinophilia, leukopenia, and thrombocytopenia.
Miscellaneous: Infrequent were weight gain, fever, roaring sensation in the head, weight loss, and malaise; rare were alcohol abuse, bleeding disturbance, loss of voice, and hiccoughs.
Post-Marketing Experience
Post-marketing experience has shown an adverse experience profile similar to that given above. Voluntary reports since introduction have included rare occurrences of allergic reactions (including urticaria), angioedema, cogwheel rigidity, dizziness (rarely reported as vertigo), dystonic reactions (including dystonia), ataxias, extrapyramidal symptoms, dyskinesias (acute and tardive), ecchymosis, emotional lability, serotonin syndrome, transient difficulty with recall, urinary retention, visual changes (including tunnel vision), parkinsonism, akathisia, restless leg syndrome, and restlessness. Because of the uncontrolled nature of these spontaneous reports, a causal relationship to buspirone treatment has not been determined.
DRUG ABUSE AND DEPENDENCE
Physical and Psychological Dependence
In human and animal studies, buspirone has shown no potential for abuse or diversion and there is no evidence that it causes tolerance, or either physical or psychological dependence. Human volunteers with a history of recreational drug or alcohol usage were studied in two double-blind clinical investigations. None of the subjects were able to distinguish between buspirone and placebo. By contrast, subjects showed a statistically significant preference for methaqualone and diazepam. Studies in monkeys, mice, and rats have indicated that buspirone lacks potential for abuse.
Following chronic administration in the rat, abrupt withdrawal of buspirone did not result in the loss of body weight commonly observed with substances that cause physical dependency.
Although there is no direct evidence that buspirone causes physical dependence or drug seeking behavior, it is difficult to predict from experiments the extent to which a CNS active drug will be misused, diverted, and/or abused once marketed. Consequently, physicians should carefully evaluate patients for a history of drug abuse and follow such patients closely, observing them for signs of buspirone misuse or abuse (e.g., development of tolerance, incrementation of dose, drug seeking behavior).
OVERDOSAGE
Signs and Symptoms
In clinical pharmacology trials, doses as high as 375 mg/day were administered to healthy male volunteers. As this dose was approached, the following symptoms were observed: nausea, vomiting, dizziness, drowsiness, miosis, and gastric distress. A few cases of overdosage have been reported, with complete recovery as the usual outcome. No deaths have been reported following overdosage with buspirone alone. Rare cases of intentional overdosage with a fatal outcome were invariably associated with ingestion of multiple drugs and/or alcohol, and a causal relationship to buspirone could not be determined. Toxicology studies of buspirone yielded the following LD50 values: mice, 655 mg/kg; rats, 196 mg/kg; dogs, 586 mg/kg; and monkeys, 356 mg/kg. These dosages are 160 to 550 times the recommended human daily dose.
Recommended Overdose Treatment
General symptomatic and supportive measures should be used along with immediate gastric lavage. Respiration, pulse, and blood pressure should be monitored as in all cases of drug overdosage. No specific antidote is known to buspirone, and dialyzability of buspirone has not been determined.
DOSAGE AND ADMINISTRATION
The recommended initial dose is 15 mg daily (7.5 mg b.i.d.). To achieve an optimal therapeutic response, at intervals of 2 to 3 days the dosage may be increased 5 mg per day, as needed. The maximum daily dosage should not exceed 60 mg per day. In clinical trials allowing dose titration, divided doses of 20 mg to 30 mg per day were commonly employed.
The bioavailability of buspirone is increased when given with food as compared to the fasted state (see CLINICAL PHARMACOLOGY). Consequently, patients should take buspirone in a consistent manner with regard to the timing of dosing; either always with or always without food.
When buspirone is to be given with a potent inhibitor of CYP3A4 the dosage recommendations described in the PRECAUTIONS: Drug Interactions section should be followed.
HOW SUPPLIED
Buspirone Hydrochloride Tablets, USP are available containing 30 mg of buspirone hydrochloride, USP.
The 30 mg tablet is a white, capsule-shaped tablet debossed with M to the left of the score and B4 to the right of the score on one side of the tablet and 10 on each trisect section on the other side. They are available as follows:Bottles of 30 - NDC # 16590-375-30
Bottles of 60 - NDC # 16590-375-60
Bottles of 90 - NDC # 16590-375-90
Dispense in a tight, light-resistant container as defined in the USP using a child-resistant closure.
PHARMACIST: Patient Instruction Sheet attached to prescribing information. Additional copies are provided separately.
Relabeling and Repackaging by:
STAT Rx USA LLC
Gainesville, GA 30501
REFERENCE
- American Psychiatric Association, Ed.: Diagnostic and Statistical Manual of Mental Disorders - III, American Psychiatric Association, May 1980.
Mylan Pharmaceuticals Inc.
Morgantown, WV 26505 U.S.A.
REVISED SEPTEMBER 2011
BUSPAS:R12ppt
BUSPIRONE HYDROCHLORIDE
TABLETS, USP
Patient Instruction Sheet
HOW TO USE:
BUSPIRONE HYDROCHLORIDE
TABLETS, USP
15 mg and 30 mg
In convenient multi-scored tablet form
Response to buspirone varies among individuals. Your physician may find it necessary to adjust your dosage to obtain the proper response.
This multi-scored tablet design makes dosage adjustments easy. Each tablet is scored and can be broken accurately to provide any of the following dosages.
If your doctor prescribed the
15 mg tablet:

15 mg (the entire tablet)

10 mg (two-thirds of a tablet)
5 mg (one-third of a tablet)

7.5 mg (one-half of a tablet)
If your doctor prescribed the 30 mg tablet:
30 mg (the entire tablet)

20 mg (two-thirds of a tablet)
10 mg (one-third of a tablet)

15 mg (one-half of a tablet)
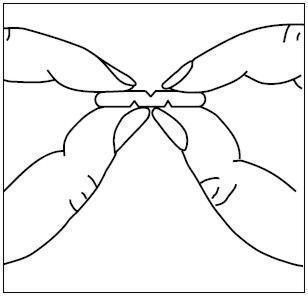
To break a multi-scored tablet accurately and easily, hold the tablet between your thumbs and index fingers close to the appropriate tablet score (groove) as shown in the picture. Then, with the tablet score facing you, apply pressure and snap the tablet segments apart (segments breaking incorrectly should not be used).
Mylan Pharmaceuticals Inc.
Morgantown, WV 26505 U.S.A.
REVISED DECEMBER 2010
PL: BUSPAS:R4
