FULL PRESCRIBING INFORMATION
WARNING: INFUSION REACTIONS and CARDIOPULMONARY ARREST
Infusion Reactions: ERBITUX can cause serious and fatal infusion reactions [see Warnings and Precautions (5.1), Adverse Reactions (6)]. Immediately interrupt and permanently discontinue ERBITUX for serious infusion reactions [see Dosage and Administration (2.5)].
Cardiopulmonary Arrest: Cardiopulmonary arrest or sudden death occurred in patients with squamous cell carcinoma of the head and neck receiving ERBITUX with radiation therapy or a cetuximab product with platinum-based therapy and fluorouracil. Monitor serum electrolytes, including serum magnesium, potassium, and calcium, during and after ERBITUX administration [see Warnings and Precautions (5.2, 5.6)].
1 INDICATIONS AND USAGE
1.1 Squamous Cell Carcinoma of the Head and Neck (SCCHN)
ERBITUX® is indicated:
- in combination with radiation therapy for the initial treatment of locally or regionally advanced squamous cell carcinoma of the head and neck (SCCHN).
- in combination with platinum-based therapy with fluorouracil for the first-line treatment of patients with recurrent locoregional disease or metastatic SCCHN.
- as a single-agent for the treatment of patients with recurrent or metastatic SCCHN for whom prior platinum-based therapy has failed.
1.2 K-Ras Wild-type, EGFR-expressing Colorectal Cancer (CRC)
ERBITUX is indicated for the treatment of K-Ras wild-type, epidermal growth factor receptor (EGFR)-expressing, metastatic colorectal cancer (mCRC) as determined by an FDA-approved test [see Dosage and Administration (2.2)]:
- in combination with FOLFIRI (irinotecan, fluorouracil, leucovorin) for first-line treatment,
- in combination with irinotecan in patients who are refractory to irinotecan-based chemotherapy,
- as a single-agent in patients who have failed oxaliplatin- and irinotecan-based chemotherapy or who are intolerant to irinotecan.
Limitations of Use: ERBITUX is not indicated for treatment of Ras-mutant colorectal cancer or when the results of the Ras mutation tests are unknown [see Warnings and Precautions (5.7)].
1.3 BRAF V600E Mutation-Positive Metastatic Colorectal Cancer (CRC)
ERBITUX is indicated, in combination with encorafenib, for the treatment of adult patients with metastatic colorectal cancer (CRC) with a BRAF V600E mutation, as detected by an FDA-approved test, after prior therapy [see Dosage and Administration (2.3)].
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
Select patients with metastatic colorectal cancer (CRC) for treatment with ERBITUX based on the presence of:
- Ras wild-type, EGFR-expressing CRC [see Clinical Studies (14.2)], or
- BRAF V600E mutation-positive metastatic CRC [see Clinical Studies (14.3)]
Information on FDA-approved tests for the detection of K-Ras or BRAF V600E mutations in CRC in patients with metastatic CRC is available at: http://www.fda.gov/CompanionDiagnostics.
2.2 Recommended Dosage for Squamous Cell Carcinoma of the Head and Neck (SCCHN)
In combination with radiation therapy
- Initial dose: 400 mg/m2 administered as a 120-minute intravenous infusion one week prior to initiating a course of radiation therapy.
- Subsequent doses: 250 mg/m2 administered as a 60-minute infusion every week for the duration of radiation therapy (6–7 weeks).
- Complete ERBITUX administration 1 hour prior to radiation therapy.
As a single-agent or in combination with platinum-based therapy and fluorouracil
Administer Erbitux as a single-agent or in combination with platinum-based therapy and fluorouracil on a weekly or biweekly schedule.
2.3 Recommended Dosage for Colorectal Cancer (CRC)
As a single-agent or in combination with irinotecan or FOLFIRI (irinotecan, fluorouracil, leucovorin)
Administer Erbitux as a single-agent or in combination with irinotecan or FOLFIRI (irinotecan, fluorouracil, leucovorin) on a weekly or biweekly schedule.
Weekly Dosage
- Initial dose: 400 mg/m2 administered as a 120-minute intravenous infusion
- Subsequent doses: 250 mg/m2 administered as a 60-minute infusion every week
Biweekly Dosage
- Initial and subsequent doses: 500 mg/m2 administered as a 120-minute intravenous infusion every 2 weeks
Complete ERBITUX administration 1 hour prior to irinotecan or FOLFIRI. Continue treatment until disease progression or unacceptable toxicity.
In combination with encorafenib
- The recommended initial dose is 400 mg/m2 administered as a 120-minute intravenous infusion in combination with encorafenib.
- The recommended subsequent dosage is 250 mg/m2 weekly as a 60-minute infusion in combination with encorafenib until disease progression or unacceptable toxicity.
Refer to the encorafenib prescribing information for recommended encorafenib dosage information.
2.4 Premedication
Premedicate with a histamine-1 (H1) receptor antagonist intravenously 30–60 minutes prior to the first dose or subsequent doses as deemed necessary [see Warnings and Precautions (5.1)].
2.5 Dosage Modifications for Adverse Reactions
Reduce, delay, or discontinue ERBITUX to manage adverse reactions as described in Table 1.
| Adverse Reaction | Severitya | Dosage Modification |
|---|---|---|
|
a National Cancer Institute (NCI) Common Toxicity Criteria (CTC), version 2.0. |
||
| Infusion reactions [see Warnings and Precautions (5.1)] | Grade 1 or 2 | Reduce the infusion rate by 50%. |
| Grade 3 or 4 | Immediately and permanently, discontinue ERBITUX. | |
| Dermatologic toxicities and infectious sequelae (e.g., acneiform rash, mucocutaneous disease) [see Warnings and Precautions (5.4)] | 1st occurrence; Grade 3 or 4 | Delay infusion 1 to 2 weeks; if condition improves, continue at 250 mg/m2. If no improvement, discontinue ERBITUX. |
| 2nd occurrence; Grade 3 or 4 | Delay infusion 1 to 2 weeks; if condition improves, continue at 200 mg/m2. If no improvement, discontinue ERBITUX. |
|
| 3rd occurrence; Grade 3 or 4 | Delay infusion 1 to 2 weeks; if condition improves, continue at 150 mg/m2. If no improvement, discontinue ERBITUX. |
|
| 4th occurrence; Grade 3 or 4 | Discontinue ERBITUX. | |
| Pulmonary toxicity [see Warnings and Precautions (5.3)] | Acute onset or worsening pulmonary symptoms | Delay infusion 1 to 2 weeks; if condition improves, continue at the dose that was being administered at the time of occurrence. If no improvement in 2 weeks or interstitial lung disease (ILD) is confirmed, discontinue ERBITUX. |
2.6 Preparation for Administration
- The solution should be clear and colorless and may contain a small amount of easily visible, white, amorphous, cetuximab particulates. Do not shake or dilute.
- Visually inspect for foreign particulate matter and discoloration prior to administration, whenever solution and container permit. Do not use if solution is discolored, cloudy, or contains foreign particulate matter.
- Do not administer ERBITUX as an intravenous push or bolus.
- Administer via infusion pump or syringe pump. Do not exceed an infusion rate of 10 mg/min.
- Administer through a low protein binding 0.22-micrometer in-line filter.
3 DOSAGE FORMS AND STRENGTHS
Injection: 100 mg/50 mL (2 mg/mL) or 200 mg/100 mL (2 mg/mL) as a clear, colorless solution in a single-dose vial.
5 WARNINGS AND PRECAUTIONS
5.1 Infusion Reactions
ERBITUX can cause serious and fatal infusion reactions. Infusion reactions of any grade occurred in 8.4% of 1373 patients who received ERBITUX across clinical trials. Severe (Grades 3 and 4) infusion reactions occurred in 2.2% of patients [see Adverse Reactions (6.1)]. Signs and symptoms included rapid onset of airway obstruction (bronchospasm, stridor, hoarseness), hypotension, shock, loss of consciousness, myocardial infarction, and/or cardiac arrest.
The risk of anaphylactic reactions may be increased in patients with a history of tick bites, red meat allergy, or in the presence of IgE antibodies directed against galactose-α-1,3-galactose (alpha-gal). Consider testing patients for alpha-gal IgE antibodies using FDA-cleared methods prior to initiating ERBITUX. Negative results for alpha-gal antibodies do not rule out the risk of severe infusion reactions.
Approximately 90% of severe infusion reactions occurred with the first infusion despite premedication with antihistamines. Infusion reactions may occur during or several hours following completion of the infusion.
Premedicate with a histamine-1(H1) receptor antagonist as recommended [see Dosage and Administration (2.4)]. Monitor patients for at least 1 hour following each ERBITUX infusion, in a setting with resuscitation equipment and other agents necessary to treat anaphylaxis. In patients requiring treatment for infusion reactions, monitor for more than 1 hour to confirm resolution of the reaction. Interrupt the infusion and upon recovery, resume the infusion at a slower rate or permanently discontinue ERBITUX based on severity [see Dosage and Administration (2.5)].
5.2 Cardiopulmonary Arrest
ERBITUX can cause cardiopulmonary arrest. Cardiopulmonary arrest or sudden death occurred in 2% of 208 patients treated with radiation therapy and ERBITUX in BONNER. Three patients with prior history of coronary artery disease died at home, with myocardial infarction as the presumed cause of death. One of these patients had arrhythmia and one had congestive heart failure. Death occurred 27, 32, and 43 days after the last dose of ERBITUX. One patient with no prior history of coronary artery disease died one day after the last dose of ERBITUX.
In EXTREME, fatal cardiac disorders and/or sudden death occurred in 3% of 219 patients treated with a cetuximab product in combination with platinum-based therapy and fluorouracil.
Carefully consider use of ERBITUX with radiation therapy or platinum-based therapy with fluorouracil in patients with SCCHN with a history of coronary artery disease, congestive heart failure, or arrhythmias. Monitor serum electrolytes, including serum magnesium, potassium, and calcium, during and after ERBITUX [see Warnings and Precautions (5.6)].
5.3 Pulmonary Toxicity
ERBITUX can cause interstitial lung disease (ILD). ILD, including 1 fatality, occurred in <0.5% of 1570 patients receiving ERBITUX in clinical trials.
Monitor patients for signs and symptoms of pulmonary toxicity. Interrupt or permanently discontinue ERBITUX for acute onset or worsening of pulmonary symptoms. Permanently discontinue ERBITUX for confirmed ILD [see Dosage and Administration (2.5)].
5.4 Dermatologic Toxicity
ERBITUX can cause dermatologic toxicities, including acneiform rash, skin drying and fissuring, paronychial inflammation, infectious sequelae (for example, S. aureus sepsis, abscess formation, cellulitis, blepharitis, conjunctivitis, keratitis/ulcerative keratitis with decreased visual acuity, cheilitis), and hypertrichosis.
Acneiform rash occurred in 82% of the 1373 patients who received ERBITUX across clinical trials. Severe (Grades 3 or 4) acneiform rash occurred in 10% of patients [see Adverse Reactions (6.1)]. Acneiform rash usually developed within the first two weeks of therapy; the rash lasted more than 28 days after stopping ERBITUX in most patients.
Life-threatening and fatal bullous mucocutaneous disease with blisters, erosions, and skin sloughing, has been observed in patients who received ERBITUX. It could not be determined whether these mucocutaneous adverse reactions were directly related to EGFR inhibition or to idiosyncratic immune-related effects (e.g., Stevens-Johnson syndrome or toxic epidermal necrolysis).
Monitor patients receiving ERBITUX for dermatologic toxicities and infectious sequelae. Instruct patients to limit sun exposure during ERBITUX therapy. Withhold, reduce dose or permanently discontinue ERBITUX based on severity of acneiform rash or mucocutaneous disease [see Dosage and Administration (2.5)].
5.5 Risks Associated with Use in Combination with Radiation and Cisplatin
In a controlled study, 940 patients with locally advanced SCCHN were randomized 1:1 to receive either ERBITUX in combination with radiation therapy and cisplatin or radiation therapy and cisplatin alone. The addition of ERBITUX resulted in an increase in the incidence of Grade 3 and 4 mucositis, radiation recall syndrome, acneiform rash, cardiac events, and electrolyte disturbances compared to radiation and cisplatin alone. Adverse reactions with fatal outcome were reported in 4% of patients in the ERBITUX combination arm and 3% in the control arm. In the ERBITUX arm, 2% experienced myocardial ischemia compared to 0.9% in the control arm. The main efficacy outcome of the study was progression-free survival (PFS). The addition of ERBITUX to radiation and cisplatin did not improve PFS. ERBITUX is not indicated for the treatment of SCCHN in combination with radiation and cisplatin.
5.6 Hypomagnesemia and Accompanying Electrolyte Abnormalities
ERBITUX can cause hypomagnesemia. Hypomagnesemia occurred in 55% of 365 patients receiving ERBITUX in Study CA225-025 and two other clinical trials in patients with colorectal cancer (CRC) or head and neck cancer, including Grades 3 and 4 in 6% to 17%.
In EXTREME, where a cetuximab product was administered in combination with platinum-based therapy, the addition of cetuximab to cisplatin and fluorouracil resulted in an increased incidence of hypomagnesemia of any grade (14%) and of Grade 3 or 4 hypomagnesemia (7%). Hypomagnesemia of any grade occurred in 4% of patients who received cetuximab, carboplatin, and fluorouracil. No patient experienced Grade 3 or 4 hypomagnesemia [see Adverse Reactions (6.1)].
Hypomagnesemia and accompanying electrolyte abnormalities can occur days to months after initiating ERBITUX. Monitor patients weekly during treatment for hypomagnesemia, hypocalcemia, and hypokalemia, and for at least 8 weeks following the completion of ERBITUX. Replete electrolytes as necessary.
5.7 Increased Tumor Progression, Increased Mortality, or Lack of Benefit in Patients with Ras-Mutant mCRC
ERBITUX is not indicated for the treatment of patients with CRC that harbor somatic mutations in exon 2 (codons 12 and 13), exon 3 (codons 59 and 61), and exon 4 (codons 117 and 146) of either K-Ras or N-Ras and hereafter is referred to as “Ras” or when the Ras status is unknown.
Retrospective subset analyses of Ras-mutant and wild-type populations across several randomized clinical trials, including CRYSTAL, were conducted to investigate the role of Ras mutations on the clinical effects of anti-EGFR-directed monoclonal antibodies. Use of cetuximab in patients with Ras mutations resulted in no clinical benefit with treatment related toxicity. Confirm Ras mutation status in tumor specimens prior to initiating ERBITUX [see Indications and Usage (1.2)].
5.8 Embryo-Fetal Toxicity
Based on animal data and its mechanism of action, ERBITUX can cause fetal harm when administered to a pregnant woman. There are no available data for ERBITUX exposure in pregnant women. In an animal reproduction study, intravenous administration of cetuximab once weekly to pregnant cynomolgus monkeys during the period of organogenesis resulted in an increased incidence of embryolethality and abortion. Disruption or depletion of EGFR in animal models results in impairment of embryo-fetal development including effects on placental, lung, cardiac, skin, and neural development. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with ERBITUX and for 2 months after the last dose of ERBITUX [see Use in Specific Populations (8.1, 8.3)].
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the label:
- Infusion reactions [see Warnings and Precautions (5.1)].
- Cardiopulmonary arrest [see Warnings and Precautions (5.2)].
- Pulmonary toxicity [see Warnings and Precautions (5.3)].
- Dermatologic toxicity [see Warnings and Precautions (5.4)].
- Hypomagnesemia and Electrolyte Abnormalities [see Warnings and Precautions (5.6)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described in Warnings and Precautions reflect exposure to ERBITUX in 1373 patients with SCCHN or CRC enrolled in clinical trials and treated at the recommended dosage for a median of 7 to 14 weeks [see Clinical Studies (14.1, 14.2)]. The most common adverse reactions in clinical trials with ERBITUX as a single-agent or in combination with radiotherapy or chemotherapy [FOLFIRI, irinotecan and 5-fluorouracil/platinum] (incidence ≥25%) include cutaneous adverse reactions (including rash, pruritus, and nail changes), headache, diarrhea, and infection.
In Combination with Radiation Therapy
The safety of ERBITUX in combination with radiation therapy compared to radiation therapy alone was evaluated in BONNER. The data described below reflect exposure to ERBITUX in 420 patients with locally or regionally advanced SCCHN. ERBITUX was administered at the recommended dosage (400 mg/m2 initial dose, followed by 250 mg/m2 weekly). Patients received a median of 8 infusions (range 1 to 11) [see Clinical Studies (14.1)].
Table 2 provides the frequency and severity of adverse reactions in BONNER.
| Adverse Reaction | ERBITUX with Radiation (n=208) | Radiation Therapy Alone (n=212) |
||
|---|---|---|---|---|
| Grades 1–4b | Grades 3 and 4 | Grades 1–4 | Grades 3 and 4 |
|
|
a Adverse reactions occurring in ≥10% of patients in the ERBITUX combination arm and at a higher incidence (≥5%) compared to the radiation alone arm. |
||||
|
b Adverse reactions were graded using the NCI CTC, version 2.0. |
||||
|
c Includes cases also reported as infusion reaction. |
||||
|
d Infusion reaction defined as any event described at any time during the clinical study as “allergic reaction” or “anaphylactoid reaction”, or any event occurring on the first day of dosing described as “allergic reaction”, “anaphylactoid reaction”, “fever”, “chills”, “chills and fever”, or “dyspnea”. |
||||
|
e Based on laboratory measurements, not on reported adverse reactions, the number of subjects with tested samples varied from 205–206 for ERBITUX with Radiation arm; 209–210 for Radiation alone. |
||||
|
f Acneiform rash defined as any event described as “acne”, “rash”, “maculopapular rash”, “pustular rash”, “dry skin”, or “exfoliative dermatitis”. |
||||
| General | ||||
| Asthenia | 56 | 4 | 49 | 5 |
| Feverc | 29 | 1 | 13 | 1 |
| Headache | 19 | <1 | 8 | <1 |
| Chillsc | 16 | 0 | 5 | 0 |
| Infusion Reactiond | 15 | 3 | 2 | 0 |
| Infection | 13 | 1 | 9 | 1 |
| Gastrointestinal | ||||
| Nausea | 49 | 2 | 37 | 2 |
| Emesis | 29 | 2 | 23 | 4 |
| Diarrhea | 19 | 2 | 13 | 1 |
| Dyspepsia | 14 | 0 | 9 | 1 |
| Metabolism and Nutrition | ||||
| Weight Loss | 84 | 11 | 72 | 7 |
| Dehydration | 25 | 6 | 19 | 8 |
| Increased Alanine Transaminasee | 43 | 2 | 21 | 1 |
| Increased Aspartate Transaminasee | 38 | 1 | 24 | 1 |
| Increased Alkaline Phosphatasee | 33 | <1 | 24 | 0 |
| Respiratory | ||||
| Pharyngitis | 26 | 3 | 19 | 4 |
| Dermatologic | ||||
| Acneiform Rashf | 87 | 17 | 10 | 1 |
| Radiation Dermatitis | 86 | 23 | 90 | 18 |
| Application Site Reaction | 18 | 0 | 12 | 1 |
| Pruritus | 16 | 0 | 4 | 0 |
The overall incidence of late radiation toxicities (any grade) was higher for patients receiving ERBITUX in combination with radiation therapy compared with radiation therapy alone. The following sites were affected: salivary glands (65% versus 56%), larynx (52% versus 36%), subcutaneous tissue (49% versus 45%), mucous membrane (48% versus 39%), esophagus (44% versus 35%), skin (42% versus 33%). The incidence of Grade 3 or 4 late radiation toxicities was similar between the radiation therapy alone and the ERBITUX with radiation treatment groups.
In Combination with Platinum-based Therapy and Fluorouracil
The safety of a cetuximab product in combination with platinum-based therapy and fluorouracil or platinum-based therapy and fluorouracil alone was evaluated in EXTREME. The data described below reflect exposure to a cetuximab product in 434 patients with recurrent locoregional disease or metastatic SCCHN. Because ERBITUX provides approximately 22% higher exposure relative to the cetuximab product, the data provided below may underestimate the incidence and severity of adverse reactions anticipated with ERBITUX for this indication; however, the tolerability of the recommended dose is supported by safety data from additional studies of ERBITUX [see Clinical Pharmacology (12.3)]. Cetuximab was administered intravenously at a dosage of 400 mg/m2 for the initial dose, followed by 250 mg/m2 weekly. Patients received a median of 17 infusions (range 1 to 89) [see Clinical Studies (14.1)].
Table 3 provides the frequency and severity of adverse reactions in EXTREME.
| Adverse Reaction | Cetuximab with Platinum-based Therapy and fluorouracil (n=219) | Platinum-based Therapy and fluorouracil Alone (n=215) |
||
|---|---|---|---|---|
| Grades 1–4b | Grades 3 and 4 | Grades 1–4 | Grades 3 and 4 |
|
|
a Adverse reactions occurring in ≥10% of patients in the cetuximab combination arm and at a higher incidence (≥5%) compared to the platinum-based therapy and fluorouracil alone arm. |
||||
|
b Adverse reactions were graded using the NCI CTC, version 2.0. |
||||
|
c Infusion reaction defined as “anaphylactic reaction”, “hypersensitivity”, “fever and/or chills”, “dyspnea”, or “pyrexia” on the first day of dosing. |
||||
|
d Infection excludes sepsis-related events which are presented separately. |
||||
|
e Acneiform rash defined as “acne”, “dermatitis acneiform”, “dry skin”, “exfoliative rash”, “rash”, “rash erythematous”, “rash macular”, “rash papular”, or “rash pustular”. |
||||
|
Chemotherapy = cisplatin and fluorouracil or carboplatin and fluorouracil |
||||
| Eye | ||||
| Conjunctivitis | 10 | 0 | 0 | 0 |
| Gastrointestinal | ||||
| Nausea | 54 | 4 | 47 | 4 |
| Diarrhea | 26 | 5 | 16 | 1 |
| General and Administration Site | ||||
| Pyrexia | 22 | 0 | 13 | 1 |
| Infusion Reactionc | 10 | 2 | <1 | 0 |
| Infections | ||||
| Infectiond | 44 | 11 | 27 | 8 |
| Metabolism and Nutrition | ||||
| Anorexia | 25 | 5 | 14 | 1 |
| Hypocalcemia | 12 | 4 | 5 | 1 |
| Hypokalemia | 12 | 7 | 7 | 5 |
| Hypomagnesemia | 11 | 5 | 5 | 1 |
| Dermatologic | ||||
| Acneiform Rashe | 70 | 9 | 2 | 0 |
| Rash | 28 | 5 | 2 | 0 |
| Acne | 22 | 2 | 0 | 0 |
| Dermatitis Acneiform | 15 | 2 | 0 | 0 |
| Dry Skin | 14 | 0 | <1 | 0 |
| Alopecia | 12 | 0 | 7 | 0 |
For cardiac disorders, approximately 9% of patients in both treatment arms in EXTREME experienced a cardiac event. The majority of these events occurred in patients who received cisplatin and fluorouracil with or without cetuximab. Cardiac disorders were observed in 11% and 12% of patients who received cisplatin and fluorouracil with or without cetuximab, respectively, and 6% and 4% in patients who received carboplatin and fluorouracil with or without cetuximab, respectively. In both arms, the incidence of cardiovascular events was higher in the cisplatin and fluorouracil containing subgroup. Death attributed to cardiovascular events or sudden death was reported in 3% of the patients in the cetuximab with platinum-based therapy and fluorouracil arm and in 2% of the patients in the platinum-based therapy and fluorouracil alone arm.
K-Ras Wild-type, EGFR-expressing, Metastatic Colorectal Cancer (mCRC)
In Combination with FOLFIRI
The safety of a cetuximab product in combination with FOLFIRI or FOLFIRI alone was evaluated in CRYSTAL. The data described below reflect exposure to a cetuximab product in 667 patients with K-Ras wild-type, EGFR-expressing, mCRC. ERBITUX provides approximately 22% higher exposure compared to this product; however, the safety data from CRYSTAL is consistent in incidence and severity of adverse reactions with those seen for ERBITUX in this indication. Cetuximab was administered intravenously at a dosage of 400 mg/m2 initial dose, followed by 250 mg/m2 weekly. Patients received a median of 24 infusions (range 1 to 224) [see Clinical Studies (14.2)].
Serious adverse reactions included pulmonary embolism, which was reported in 4.4% of patients treated with cetuximab with FOLFIRI as compared to 3.4% of patients treated with FOLFIRI alone.
Table 4 provides the frequency and severity of adverse reactions in CRYSTAL.
| Adverse Reaction | Cetuximab with FOLFIRI (n=317) | FOLFIRI Alone (n=350) |
||
|---|---|---|---|---|
| Grades 1–4b | Grades 3 and 4 | Grades 1–4 | Grades 3 and 4 |
|
|
a Adverse reactions occurring in ≥10% of patients in the cetuximab combination arm and at a higher incidence (≥5%) compared to the FOLFIRI alone arm. |
||||
|
b Adverse reactions were graded using the NCI CTC, version 2.0. |
||||
|
c Infusion reaction defined as any event meeting the medical concepts of allergy/anaphylaxis at any time during the clinical study or any event occurring on the first day of dosing and meeting the medical concepts of dyspnea and fever or by the following events: “acute myocardial infarction”, “angina pectoris”, “angioedema”, “autonomic seizure”, “blood pressure abnormal”, “blood pressure decreased”, “blood pressure increased”, “cardiac failure”, “cardiopulmonary failure”, “cardiovascular insufficiency”, “clonus”, “convulsion”, “coronary no-reflow phenomenon”, “epilepsy”, “hypertension”, “hypertensive crisis”, “hypertensive emergency”, “hypotension”, “infusion related reaction”, “loss of consciousness”, “myocardial infarction”, “myocardial ischemia”, “prinzmetal angina”, “shock”, “sudden death”, “syncope”, or “systolic hypertension”. |
||||
|
d Acne-like rash defined by the following events: “acne”, “acne pustular”, “butterfly rash”, “dermatitis acneiform”, “drug rash with eosinophilia and systemic symptoms”, “dry skin”, “erythema”, “exfoliative rash”, “folliculitis”, “genital rash”, “mucocutaneous rash”, “pruritus”, “rash”, “rash erythematous”, “rash follicular”, “rash generalized”, “rash macular”, “rash maculopapular”, “rash maculovesicular”, “rash morbilliform”, “rash papular”, “rash papulosquamous”, “rash pruritic”, “rash pustular”, “rash rubelliform”, “rash scarlatiniform”, “rash vesicular”, “skin exfoliation”, “skin hyperpigmentation”, “skin plaque”, “telangiectasia”, or “xerosis”. |
||||
| Hematologic | ||||
| Neutropenia | 49 | 31 | 42 | 24 |
| Eye | ||||
| Conjunctivitis | 18 | <1 | 3 | 0 |
| Gastrointestinal | ||||
| Diarrhea | 66 | 16 | 60 | 10 |
| Stomatitis | 31 | 3 | 19 | 1 |
| Dyspepsia | 16 | 0 | 9 | 0 |
| General and Administration Site | ||||
| Pyrexia | 26 | 1 | 14 | 1 |
| Weight Decreased | 15 | 1 | 9 | 1 |
| Infusion Reactionc | 14 | 2 | <1 | 0 |
| Infections | ||||
| Paronychia | 20 | 4 | <1 | 0 |
| Metabolism and Nutrition | ||||
| Anorexia | 30 | 3 | 23 | 2 |
| Dermatologic | ||||
| Acne-like Rashd | 86 | 18 | 13 | <1 |
| Rash | 44 | 9 | 4 | 0 |
| Dermatitis Acneiform | 26 | 5 | <1 | 0 |
| Dry Skin | 22 | 0 | 4 | 0 |
| Acne | 14 | 2 | 0 | 0 |
| Pruritus | 14 | 0 | 3 | 0 |
| Palmar-plantar Erythrodysesthesia Syndrome | 19 | 4 | 4 | <1 |
| Skin Fissures | 19 | 2 | 1 | 0 |
As Single-Agent
The safety of ERBITUX with best supportive care (BSC) or BSC alone was evaluated in Study CA225-025. The data described below reflect exposure to ERBITUX in 242 patients with K-Ras wild-type, EGFR-expressing, metastatic colorectal cancer (mCRC) [see Warnings and Precautions (5.8)]. ERBITUX was administered intravenously at the recommended dosage (400 mg/m2 initial dose, followed by 250 mg/m2 weekly). Patients received a median of 17 infusions (range 1 to 51) [see Clinical Studies (14.2)].
Table 5 provides the frequency and severity of adverse reactions in Study CA225-025.
| Adverse Reaction | ERBITUX with BSC (n=118) | BSC alone (n=124) |
||
|---|---|---|---|---|
| Grades 1–4b | Grades 3 and 4 | Grades 1–4 | Grades 3 and 4 |
|
|
a Adverse reactions occurring in ≥10% of patients in the ERBITUX with BSC arm and at a higher incidence (≥5%) compared to the BSC alone arm. |
||||
|
b Adverse reactions were graded using the NCI CTC, version 2.0. |
||||
|
c Infusion reaction defined as any event (chills, rigors, dyspnea, tachycardia, bronchospasm, chest tightness, swelling, urticaria, hypotension, flushing, rash, hypertension, nausea, angioedema, pain, sweating, tremors, shaking, drug fever, or other hypersensitivity reaction) recorded by the investigator as infusion-related. |
||||
| Dermatologic | ||||
| Rash/Desquamation | 95 | 16 | 21 | 1 |
| Dry Skin | 57 | 0 | 15 | 0 |
| Pruritus | 47 | 2 | 11 | 0 |
| Other-Dermatology | 35 | 0 | 7 | 2 |
| Nail Changes | 31 | 0 | 4 | 0 |
| General | ||||
| Fatigue | 91 | 31 | 79 | 29 |
| Fever | 25 | 3 | 16 | 0 |
| Infusion Reactionsc | 18 | 3 | 0 | 0 |
| Rigors, Chills | 16 | 1 | 3 | 0 |
| Pain | ||||
| Pain-Other | 59 | 18 | 37 | 10 |
| Headache | 38 | 2 | 11 | 0 |
| Bone Pain | 15 | 4 | 8 | 2 |
| Pulmonary | ||||
| Dyspnea | 49 | 16 | 44 | 13 |
| Cough | 30 | 2 | 19 | 2 |
| Gastrointestinal | ||||
| Nausea | 64 | 6 | 50 | 6 |
| Constipation | 53 | 3 | 38 | 3 |
| Diarrhea | 42 | 2 | 23 | 2 |
| Vomiting | 40 | 5 | 26 | 5 |
| Stomatitis | 32 | 1 | 10 | 0 |
| Other | 22 | 12 | 16 | 5 |
| Dehydration | 13 | 5 | 3 | 0 |
| Mouth Dryness | 12 | 0 | 6 | 0 |
| Taste Disturbance | 10 | 0 | 5 | 0 |
| Infection | ||||
| Infection without neutropenia | 38 | 11 | 19 | 5 |
| Musculoskeletal | ||||
| Arthralgia | 14 | 3 | 6 | 0 |
| Neurological | ||||
| Neuropathy-sensory | 45 | 1 | 38 | 2 |
| Insomnia | 27 | 0 | 13 | 0 |
| Confusion | 18 | 6 | 10 | 2 |
| Anxiety | 14 | 1 | 5 | 1 |
| Depression | 14 | 0 | 5 | 0 |
In Combination with Irinotecan
ERBITUX at the recommended dosage was administered in combination with irinotecan in 354 patients with EGFR-expressing recurrent mCRC in Study CP02-9923 and BOND.
The most common adverse reactions were acneiform rash (88%), asthenia/malaise (73%), diarrhea (72%), and nausea (55%). The most common Grades 3–4 adverse reactions included diarrhea (22%), leukopenia (17%), asthenia/malaise (16%), and acneiform rash (14%).
BRAF V600E Mutation-Positive Metastatic Colorectal Cancer (CRC) in Combination with Encorafenib
The safety of ERBITUX (400 mg/m2 initial dose, followed by 250 mg/m2 weekly) in combination with encorafenib (300 mg once daily) was evaluated in 216 patients with BRAF V600E mutation-positive metastatic CRC in a randomized, open-label, active-controlled trial (BEACON CRC). The BEACON CRC trial [see Clinical Studies (14.3)] excluded patients with a history of Gilbert’s syndrome, abnormal left ventricular ejection fraction, prolonged QTc (>480 ms), uncontrolled hypertension, and history or current evidence of retinal vein occlusion. The median duration of exposure was 4.4 months for patients treated with ERBITUX in combination with encorafenib and 1.6 months for patients treated with either irinotecan or infusional 5-fluorouracil (5-FU)/folinic acid (FA)/irinotecan (FOLFIRI) in combination with ERBITUX.
The most common (≥ 25%) adverse reactions in patients receiving ERBITUX in combination with encorafenib were fatigue, nausea, diarrhea, dermatitis acneiform, abdominal pain, decreased appetite, arthralgia, and rash.
Table 6 and Table 7 present adverse drug reactions and laboratory abnormalities, respectively, identified in BEACON CRC.
| Adverse Reaction | ERBITUX with encorafenib N=216 | ERBITUX with irinotecan or ERBITUX with FOLFIRI N=193 |
||
|---|---|---|---|---|
| All Grades (%) | ≥ Grade 3b (%) | All Grades (%) | ≥ Grade 3 (%) | |
|
a Grades per National Cancer Institute CTCAE v4.03. |
||||
|
b Grade 4-5 adverse reactions in the ERBITUX with encorafenib arm were limited to Grade 5 hemorrhage (n=1). |
||||
|
c Represents a composite of multiple, related preferred terms. |
||||
| General Disorders and Administration Site Conditions | ||||
| Fatiguec | 51 | 7 | 50 | 8 |
| Pyrexiac | 17 | 1 | 15 | 1 |
| Gastrointestinal Disorders | ||||
| Nausea | 34 | 1 | 41 | 1 |
| Diarrheac | 33 | 2 | 48 | 10 |
| Abdominal painc | 30 | 4 | 32 | 5 |
| Vomiting | 21 | 1 | 29 | 3 |
| Constipation | 15 | 0 | 18 | 1 |
| Metabolism and Nutrition Disorders | ||||
| Decreased appetite | 27 | 2 | 27 | 3 |
| Musculoskeletal and Connective Tissue Disorders | ||||
| Arthralgiac | 27 | 4 | 3 | 0 |
| Myopathyc | 15 | 1 | 4 | 0 |
| Pain in extremity | 10 | 0 | 1 | 0 |
| Skin and Subcutaneous Tissue Disorders | ||||
| Dermatitis acneiformc | 32 | 1 | 43 | 3 |
| Rashc | 26 | 0 | 26 | 2 |
| Pruritusc | 14 | 0 | 6 | 0 |
| Melanocytic nevus | 14 | 0 | 0 | 0 |
| Dry skinc | 13 | 0 | 12 | 1 |
| Nervous System Disorders | ||||
| Headachec | 20 | 0 | 3 | 0 |
| Peripheral neuropathyc | 12 | 1 | 6 | 0 |
| Vascular Disorders | ||||
| Hemorrhagec | 19 | 2 | 9 | 0 |
| Psychiatric Disorders | ||||
| Insomniac | 13 | 0 | 6 | 0 |
Other clinically important adverse reactions occurring in <10% of patients who received ERBITUX in combination with encorafenib were:
Gastrointestinal disorders: Pancreatitis
| Laboratory Abnormalityb | ERBITUX with encorafenib | ERBITUX with irinotecan or ERBITUX with FOLFIRI | ||
|---|---|---|---|---|
| All Grades (%)bb | Grades 3 and 4 (%) | All Grades (%) | Grades 3 and 4 (%) | |
|
a Grades per National Cancer Institute CTCAE v4.03. |
||||
|
b Based on the number of patients with available baseline and at least one on-treatment laboratory test. |
||||
| Hematology | ||||
| Anemia | 34 | 4 | 48 | 5 |
| Lymphopenia | 24 | 7 | 35 | 5 |
| Increased Activated Partial Thromboplastin Time | 13 | 1 | 7 | 1 |
| Chemistry | ||||
| Hypomagnesemia | 19 | 0 | 22 | 1 |
| Increased Alkaline Phosphatase | 18 | 4 | 30 | 7 |
| Increased ALT | 17 | 0 | 29 | 3 |
| Increased AST | 15 | 1 | 22 | 2 |
| Hypokalemia | 12 | 3 | 32 | 5 |
| Hyponatremia | 11 | 2 | 13 | 2 |
6.2 Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to cetuximab in the studies below with the incidence of antibodies to cetuximab in other studies or to other products may be misleading.
An ELISA methodology was used to characterize the incidence of anti-cetuximab antibodies. The incidence of anti-cetuximab binding antibodies in 105 patients (from studies I4E-MC-JXBA, I4E-MC-JXBB, and I4E-MC-JXBD) with at least one post-baseline blood sample (≥4 weeks post first ERBITUX administration) was <5%.
6.3 Postmarketing Experience
The following adverse reactions have been identified during post approval use of ERBITUX. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Neurologic: Aseptic meningitis
- Gastrointestinal: Mucosal inflammation
- Dermatologic: Stevens-Johnson syndrome, toxic epidermal necrolysis, life-threatening and fatal bullous mucocutaneous disease
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal studies and its mechanism of action [see Clinical Pharmacology (12.1)], ERBITUX can cause fetal harm when administered to a pregnant woman. There are no available data for ERBITUX exposure in pregnant women. In an animal reproduction study, intravenous administration of cetuximab once weekly to pregnant cynomolgus monkeys during the period of organogenesis resulted in an increased incidence of embryolethality and abortion. Disruption or depletion of EGFR in animal models results in impairment of embryo-fetal development including effects on placental, lung, cardiac, skin, and neural development (see Data). Human IgG is known to cross the placental barrier; therefore, cetuximab may be transmitted from the mother to the developing fetus. Advise pregnant women of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20% respectively.
Data
Animal Data
Pregnant cynomolgus monkeys were administered cetuximab intravenously once weekly during the period of organogenesis (gestation day [GD] 20-48) at dose levels 0.4 to 4 times the recommended dose of ERBITUX based on body surface area (BSA). Cetuximab was detected in the amniotic fluid and in the serum of embryos from treated dams on GD 49. While no fetal malformations occurred in offspring, there was an increased incidence of embryolethality and abortions at doses approximately 1 to 4 times the recommended dose of ERBITUX based on BSA.
In mice, EGFR is critically important in reproductive and developmental processes including blastocyst implantation, placental development, and embryo-fetal/postnatal survival and development. Reduction or elimination of embryo-fetal or maternal EGFR signaling can prevent implantation, can cause embryo-fetal loss during various stages of gestation (through effects on placental development), and can cause developmental anomalies and early death in surviving fetuses. Adverse developmental outcomes were observed in multiple organs in embryos/neonates of mice with disrupted EGFR signaling.
8.2 Lactation
Risk Summary
There is no information regarding the presence of ERBITUX in human milk, the effects on the breastfed infant, or the effects on milk production. Human IgG antibodies can be excreted in human milk. Due to the potential for serious adverse reactions in breastfed infants from ERBITUX, advise women not to breastfeed during treatment with ERBITUX and for 2 months after the last dose of ERBITUX.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Verify pregnancy status in females of reproductive potential prior to initiating ERBITUX [see Use in Specific Population (8.1)].
Contraception
Based on its mechanism of action, ERBITUX can cause harm to the fetus when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Infertility
Females
Based on animal studies, ERBITUX may impair fertility in females of reproductive potential [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of ERBITUX in pediatric patients have not been established. The pharmacokinetics of cetuximab, in combination with irinotecan, were evaluated in pediatric patients with refractory solid tumors in an open-label, single-arm, dose-finding study. ERBITUX was administered once-weekly, at doses up to 250 mg/m2, to 27 patients ranging from 1 to 12 years old; and in 19 patients ranging from 13 to 18 years old. No new safety signals were identified in pediatric patients. The pharmacokinetics of cetuximab between the two age groups were similar following a single dose of 75 mg/m2 and 150 mg/m2. The volume of the distribution appears to be independent of dose and approximates the vascular space of 2 L/m2 to 3 L/m2. Following a single dose of 250 mg/m2, the mean AUC0-inf (CV%) was 17.7 mg*h/mL (34%) in the younger age group (1–12 years, n=9) and 13.4 mg*h/mL (38%) in the adolescent group (13–18 years, n=6). The mean half-life of cetuximab was 110 hours (69 to 188 hours) in the younger group and 82 hours (55 to 117 hours) in the adolescent group.
8.5 Geriatric Use
Of the 1662 patients with advanced colorectal cancer who received ERBITUX with irinotecan, with FOLFIRI or as single-agent in six studies (BOND, IMCL-CP02-9923, IMCL-CP02-0141, IMCL-CP02-0144, CA225-025 and CRYSTAL), 35% of patients were 65 years of age or older. No overall differences in safety or efficacy were observed between these patients and younger patients.
Clinical studies of ERBITUX conducted in patients with head and neck cancer did not include sufficient number of subjects aged 65 and over to determine whether they respond differently from younger subjects.
Of the 216 patients with BRAF V600E mutation positive metastatic CRC who received ERBITUX in combination with encorafenib 300 mg, once daily, 29% were 65 years of age to up to 75 years of age, while 20 (9%) were 75 years of age and over [see Clinical Studies (14.3)].
No overall differences in the safety or effectiveness of ERBITUX plus encorafenib were observed in elderly patients as compared to younger patients.
11 DESCRIPTION
Cetuximab is an epidermal growth factor receptor (EGFR) antagonist. It is a recombinant, human/mouse chimeric monoclonal antibody that binds specifically to the extracellular domain of the human epidermal growth factor receptor (EGFR). Cetuximab is composed of the Fv regions of a murine anti-EGFR antibody with human IgG1 heavy and kappa light chain constant regions and has an approximate molecular weight of 152 kDa. Cetuximab is produced in mammalian (murine myeloma) cell culture.
ERBITUX (cetuximab) injection for intravenous use, is a sterile, preservative-free, clear, colorless solution, which may contain a small amount of visible, white, amorphous cetuximab particulates in a single-dose vial. Each 1 mL of solution contains 2 mg of cetuximab, sodium chloride (8.48 mg), sodium phosphate dibasic heptahydrate (1.88 mg), sodium phosphate monobasic monohydrate (0.41 mg), and Water for Injection, USP at pH of 7.0 to 7.4.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The epidermal growth factor receptor (EGFR, HER1, c-ErbB-1) is a transmembrane glycoprotein that is a member of a subfamily of type I receptor tyrosine kinases including EGFR, HER2, HER3, and HER4. The EGFR is constitutively expressed in many normal epithelial tissues, including the skin and hair follicle. Expression of EGFR is also detected in many human cancers including those of the head and neck, colon, and rectum.
Cetuximab binds specifically to the EGFR on both normal and tumor cells, and competitively inhibits the binding of epidermal growth factor (EGF) and other ligands, such as transforming growth factor-alpha. In vitro assays and in vivo animal studies have shown that binding of cetuximab to the EGFR blocks phosphorylation and activation of receptor-associated kinases, resulting in inhibition of cell growth, induction of apoptosis, and decreased matrix metalloproteinase and vascular endothelial growth factor production. Signal transduction through the EGFR results in activation of wild-type Ras proteins, but in cells with activating Ras somatic mutations, the resulting mutant Ras proteins are continuously active regardless of EGFR regulation.
In vitro, cetuximab can mediate antibody-dependent cellular cytotoxicity (ADCC) against certain human tumor types. In vitro assays and in vivo animal studies have shown that cetuximab inhibits the growth and survival of tumor cells that express the EGFR. No anti-tumor effects of cetuximab were observed in human tumor xenografts lacking EGFR expression. The addition of cetuximab to radiation therapy or irinotecan in human tumor xenograft models in mice resulted in an increase in anti-tumor effects compared to radiation therapy or chemotherapy alone.
In the setting of BRAF-mutant CRC, induction of EGFR-mediated MAPK pathway activation has been identified as a mechanism of resistance to BRAF inhibitors. Combinations of a BRAF inhibitor and agents targeting EGFR have been shown to overcome this resistance mechanism in nonclinical models. Coadministration of cetuximab and encorafenib had an anti-tumor effect greater than either drug alone, in a mouse model of colorectal cancer with mutated BRAF V600E.
12.3 Pharmacokinetics
ERBITUX administered as a single-agent or in combination with concomitant chemotherapy or radiation therapy exhibits nonlinear pharmacokinetics. The area under the concentration time curve (AUC) increased in a greater than dose proportional manner while clearance of cetuximab decreased from 0.08 L/h/m2 to 0.02 L/h/m2 as the dose increased from 20 mg/m2 to 200 mg/m2 and plateaued at doses >200 mg/m2.
The systemic exposure of cetuximab after ERBITUX administration was 22% (90% CI: 6%, 38%) higher than that of another cetuximab product used in EXTREME and CRYSTAL.
Distribution
The volume of the distribution for cetuximab appeared to be independent of dose and approximated the vascular space of 2–3 L/m2.
Elimination
Following the recommended dosage (400 mg/m2 initial dose; 250 mg/m2 weekly dose), concentrations of cetuximab reached steady-state levels by the third weekly infusion with mean peak and trough concentrations across studies ranging from 168 μg/mL to 235 μg/mL and 41 μg/mL to 85 μg/mL, respectively. The mean half-life of cetuximab was approximately 112 hours (63 to 230 hours).
Specific Population
Age, sex, race, hepatic and renal function had no clinically significant effect on the pharmacokinetics of cetuximab. Clearance of cetuximab increased 1.8-fold as body surface area increased from 1.3 m2 to 2.3 m2, which is consistent with the recommended dosing of cetuximab on mg/m2 basis.
Drug Interaction Studies
No pharmacokinetic interaction was observed between cetuximab and irinotecan, cetuximab and cisplatin, and cetuximab and carboplatin.
No clinically significant differences in the pharmacokinetics of cetuximab or encorafenib were observed when the recommended ERBITUX initial dose of 400 mg/m2 was co-administered with encorafenib.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term animal studies have not been performed to test cetuximab for carcinogenic potential, and no mutagenic or clastogenic potential of cetuximab was observed in the Salmonella-Escherichia coli (Ames) assay or in the in vivo rat micronucleus test. Menstrual cyclicity was impaired in female cynomolgus monkeys receiving weekly doses of 0.4 to 4 times the recommended dose of ERBITUX (based on total body surface area). Cetuximab-treated animals exhibited increased incidences of irregular or absent cycles, as compared to control animals. These effects were initially noted beginning on week 25 and continued through the 6-week recovery period. No effects of cetuximab were observed on measured male fertility parameters (i.e., serum testosterone levels and analysis of sperm counts, viability, and motility) as compared to control male monkeys.
14 CLINICAL STUDIES
14.1 Squamous Cell Carcinoma of the Head and Neck (SCCHN)
In Combination with Radiation Therapy
BONNER (NCT00004227) was a randomized, multicenter, controlled trial of 424 patients with locally or regionally advanced SCCHN. Patients with Stage III/IV SCCHN of the oropharynx, hypopharynx, or larynx with no prior therapy were randomized (1:1) to receive either ERBITUX in combination with radiation therapy or radiation therapy alone. Stratification factors were Karnofsky performance status (60–80 versus 90–100), nodal stage (N0 versus N+), tumor stage (T1–3 versus T4 using American Joint Committee on Cancer 1998 staging criteria), and radiation therapy fractionation (concomitant boost versus once-daily versus twice-daily). Radiation therapy was administered for 6–7 weeks as once-daily, twice-daily, or concomitant boost. ERBITUX was administered intravenously as a 400 mg/m2 initial dose beginning one week prior to initiation of radiation therapy, followed by 250 mg/m2 weekly administered 1 hour prior to radiation therapy for the duration of radiation therapy (6–7 weeks). The main efficacy outcome measure was duration of locoregional control. Another outcome measure was overall survival (OS).
Of the 424 randomized patients, the median age was 57 years, 80% were male, 83% were White, and 90% had baseline Karnofsky performance status ≥80. There were 258 patients enrolled in U.S. sites (61%). Sixty percent of patients had oropharyngeal, 25% laryngeal, and 15% hypopharyngeal primary tumors; 28% had AJCC T4 tumor stage. Fifty-six percent of the patients received radiation therapy with concomitant boost, 26% received once-daily regimen, and 18% twice-daily regimen.
Efficacy results are presented in Table 8.
| ERBITUX plus Radiation (n=211) | Radiation Alone (n=213) | Hazard Ratio (95% CIa) | Stratified Log-rank p-value |
|
|---|---|---|---|---|
|
a CI = confidence interval. |
||||
| Locoregional Control | ||||
| Median duration (months) | 24.4 | 14.9 | 0.68 (0.52–0.89) | 0.005 |
| Overall Survival | ||||
| Median duration (months) | 49.0 | 29.3 | 0.74 (0.57–0.97) | 0.03 |
In Combination with Platinum-based Therapy with Fluorouracil
EXTREME (NCT00122460) was an open-label, randomized, multicenter, controlled trial of 442 patients with recurrent locoregional disease or metastatic SCCHN. Patients with no prior therapy for recurrent locoregional disease or metastatic SCCHN were randomized (1:1) to receive a cetuximab product in combination with platinum-based therapy and fluorouracil or platinum-based therapy and fluorouracil alone. Choice of cisplatin or carboplatin was at the discretion of the investigator. Stratification factors were Karnofsky performance status (<80 versus ≥80) and previous chemotherapy. Cisplatin (100 mg/m2 intravenously on Day 1) or carboplatin (AUC 5 mg/mL*min intravenously on Day 1) and fluorouracil (1000 mg/m2/day intravenously on Days 1–4) were administered every 3 weeks (1 cycle) for a maximum of 6 cycles in the absence of disease progression or unacceptable toxicity. Cetuximab was administered intravenously at a 400 mg/m2 initial dose, followed by a 250 mg/m2 weekly dose. In the absence of disease progression or unacceptable toxicity after completion of 6 planned courses of platinum-based therapy, weekly cetuximab as a single-agent could be continued until disease progression or unacceptable toxicity. If chemotherapy was delayed because of adverse reactions, weekly cetuximab was continued. If chemotherapy was discontinued for adverse reactions, weekly cetuximab as a single-agent could be continued until disease progression or unacceptable toxicity. The main efficacy outcome measure was OS. Other outcome measures were PFS and objective response rate (ORR).
Of the 442 randomized patients, the median age was 57 years, 90% were male, 98% were White, and 88% had baseline Karnofsky performance status ≥80. Thirty-four percent of patients had oropharyngeal, 25% laryngeal, 20% oral cavity, and 14% hypopharyngeal primary tumors. Fifty-three percent of patients had recurrent locoregional disease only and 47% had metastatic disease. Fifty-eight percent had AJCC Stage IV disease and 21% had Stage III disease. Sixty-four percent of patients received cisplatin therapy and 34% received carboplatin as initial therapy. Approximately fifteen percent of the patients in the cisplatin alone arm switched to carboplatin during the treatment period.
Efficacy results are presented in Table 9 and Figure 1.
| Cetuximab with Platinum-based Therapy and Fluorouracil (n=222) | Platinum-based Therapy and Fluorouracil (n=220) |
|
|---|---|---|
|
a CI = confidence interval. |
||
|
b CMH = Cochran-Mantel-Haenszel. |
||
| Overall Survival | ||
| Median Duration (months) | 10.1 | 7.4 |
| Hazard Ratio (95% CIa) | 0.80 (0.64, 0.98) | |
| Stratified Log-rank p-value | 0.034 | |
| Progression-free Survival | ||
| Median Duration (months) | 5.5 | 3.3 |
| Hazard Ratio (95% CIa) | 0.57 (0.46, 0.72) | |
| Stratified Log-rank p-value | <0.0001 | |
| Objective Response Rate | 35.6% | 19.5% |
| Odds Ratio (95% CIa) | 2.33 (1.50, 3.60) | |
| CMHb Test p-value | 0.0001 | |
Figure 1: Kaplan-Meier Curves for Overall Survival in Patients with Recurrent Locoregional Disease or Metastatic SCCHN in EXTREME
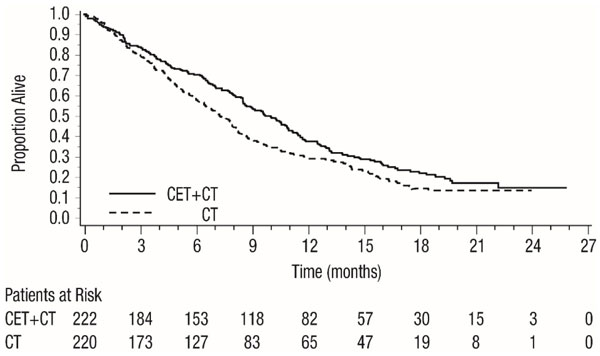
CT = Platinum-based therapy with fluorouracil
CET = another cetuximab product
In exploratory subgroup analyses by initial platinum therapy (cisplatin or carboplatin), for patients (N=284) receiving cetuximab in combination with cisplatin and fluorouracil compared to cisplatin and fluorouracil alone, the difference in median OS was 3.3 months (10.6 versus 7.3 months; HR 0.71; 95% CI 0.54, 0.93). The difference in median PFS was 2.1 months (5.6 versus 3.5 months; HR 0.55; 95% CI 0.41, 0.73). The ORR was 39% and 23%, respectively (OR 2.18; 95% CI 1.29, 3.69).
For patients (N=149) receiving cetuximab in combination with carboplatin and fluorouracil compared to carboplatin and fluorouracil alone, the difference in median OS was 1.4 months (9.7 versus 8.3 months; HR 0.99; 95% CI 0.69, 1.43). The difference in median PFS was 1.7 months (4.8 versus 3.1 months, respectively; HR 0.61; 95% CI 0.42, 0.89). The ORR was 30% and 15%, respectively (OR 2.45; 95% CI 1.10, 5.46).
As Single-Agent
EMR 62202-016 was a single-arm, multicenter clinical trial in 103 patients with recurrent or metastatic SCCHN. All patients had documented disease progression within 30 days of a platinum-based chemotherapy regimen. Patients were administered intravenously a 20-mg test dose of ERBITUX on Day 1, followed by a 400 mg/m2 initial dose, and 250 mg/m2 weekly until disease progression or unacceptable toxicity.
The median age was 57 years, 82% were male, 100% White, and 62% had a Karnofsky performance status of ≥80.
The ORR was 13% (95% CI 7%, 21%). Median duration of response (DoR) was 5.8 months (range 1.2 to 5.8 months).
14.2 K-Ras Wild-type, EGFR-expressing, Metastatic Colorectal Cancer (CRC)
In Combination with FOLFIRI
CRYSTAL (NCT00154102) was a randomized, open-label, multicenter, study of 1217 patients with EGFR-expressing, mCRC. Patients were randomized (1:1) to receive either a cetuximab product in combination with FOLFIRI or FOLFIRI alone as first-line treatment. Stratification factors were Eastern Cooperative Oncology Group (ECOG) performance status (0 and 1 versus 2) and region (Western Europe versus Eastern Europe versus other).
FOLFIRI regimen included 14-day cycles of irinotecan (180 mg/m2 intravenously on Day 1), folinic acid (400 mg/m2 [racemic] or 200 mg/m2 [L-form] intravenously on Day 1), and fluorouracil (400 mg/m2 bolus on Day 1 followed by 2400 mg/m2 as a 46-hour continuous infusion). Cetuximab was administered intravenously as a 400 mg/m2 initial dose , followed by 250 mg/m2 weekly administered 1 hour prior to chemotherapy. Study treatment continued until disease progression or unacceptable toxicity. The main efficacy outcome measure was PFS assessed by an independent review committee (IRC). Other outcome measures were OS and ORR.
Of the 1217 randomized patients, the median age was 61 years, 60% were male, 86% were White, and 96% had a baseline ECOG performance status 0–1, 60% had primary tumor localized in colon, 84% had 1–2 metastatic sites and 20% had received prior adjuvant and/or neoadjuvant chemotherapy. Demographics and baseline characteristics were similar between study arms.
K-Ras mutation status was available for 89% of the patients: 63% had K-Ras wild-type tumors and 37% had K-Ras mutant tumors where testing assessed for the following somatic mutations in codons 12 and 13 (exon 2): G12A, G12D, G12R, G12C, G12S, G12V, G13D. Baseline characteristics and demographics in the K-Ras wild-type subset were similar to that seen in the overall population.
A statistically significant improvement in PFS was observed for the cetuximab with FOLFIRI arm compared with the FOLFIRI arm (median PFS 8.9 vs. 8.1 months, HR 0.85 [95% CI 0.74, 0.99], p-value=0.036). OS was not significantly different at the planned, final analysis based on 838 events (HR=0.93, 95% CI [0.8, 1.1], p-value 0.327).
Results of the planned PFS and ORR analysis in all randomized patients and post-hoc PFS and ORR analysis in subgroups of patients defined by K-Ras mutation status, and post-hoc analysis of updated OS based on additional follow-up (1000 events) in all randomized patients and in subgroups of patients defined by K-Ras mutation status are presented in Table 10 and Figure 2. The treatment effect in the all-randomized population for PFS was driven by treatment effects limited to patients who have K-Ras wild-type tumors. There is no evidence of effectiveness in the subgroup of patients with K-Ras mutant tumors.
| All Randomized | K-Ras Wild-type | K-Ras Mutant | ||||
|---|---|---|---|---|---|---|
| Cetuximab with FOLFIRI (n=608) | FOLFIRI (n=609) | Cetuximab with FOLFIRI (n=320) | FOLFIRI (n=356) | Cetuximab with FOLFIRI (n=216) | FOLFIRI (n=187) |
|
|
a Based on the Stratified Log-rank test. |
||||||
|
b Post-hoc updated OS analysis, results based on an additional 162 events. |
||||||
| Progression-Free Survival | ||||||
| Number of Events (%) | 343 (56) | 371 (61) | 165 (52) | 214 (60) | 138 (64) | 112 (60) |
| Median (months) (95% CI) | 8.9 (8.0, 9.4) | 8.1 (7.6, 8.8) | 9.5 (8.9, 11.1) | 8.1 (7.4, 9.2) | 7.5 (6.7, 8.7) | 8.2 (7.4, 9.2) |
| HR (95% CI) | 0.85 (0.74, 0.99) | 0.70 (0.57, 0.86) | 1.13 (0.88, 1.46) | |||
| p-valuea | 0.0358 | |||||
| Overall Survivalb | ||||||
| Number of Events (%) | 491 (81) | 509 (84) | 244 (76) | 292 (82) | 189 (88) | 159 (85) |
| Median (months) (95% CI) | 19.6 (18, 21) | 18.5 (17, 20) | 23.5 (21, 26) | 19.5 (17, 21) | 16.0 (15, 18) | 16.7 (15, 19) |
| HR (95% CI) | 0.88 (0.78, 1.0) | 0.80 (0.67, 0.94) | 1.04 (0.84, 1.29) | |||
| Objective Response Rate | ||||||
| ORR (95% CI) | 46% (42, 50) | 38% (34, 42) | 57% (51, 62) | 39% (34, 44) | 31% (25, 38) | 35% (28, 43) |
Figure 2: Kaplan-Meier Curves for Overall Survival in the K-Ras Wild-type Population in CRYSTAL
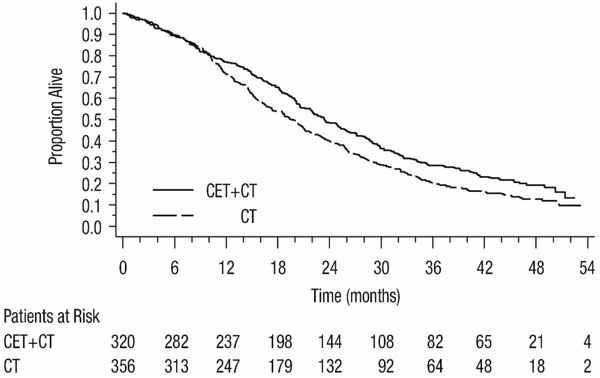
As Single-Agent
Study CA225-025 (NCT00079066) was a multicenter, open-label, randomized, clinical trial conducted in 572 patients with EGFR-expressing, previously treated, recurrent mCRC. Patients were randomized (1:1) to receive either ERBITUX with best supportive care (BSC) or BSC alone. ERBITUX was administered intravenously as a 400 mg/m2 initial dose, followed by 250 mg/m2 weekly until disease progression or unacceptable toxicity. The main efficacy outcome measure was OS.
Of the 572 randomized patients, the median age was 63 years, 64% were male, 89% were White, and 77% had baseline ECOG performance status of 0–1. Demographics and baseline characteristics were similar between study arms. All patients were to have received and progressed on prior therapy including an irinotecan-containing regimen and an oxaliplatin-containing regimen.
K-Ras status was available for 79% of the patients: 54% had K-Ras wild-type tumors and 46% had K-Ras mutant tumors where testing assessed for the following somatic mutations in codons 12 and 13 (exon 2): G12A, G12D, G12R, G12C, G12S, G12V, G13D.
Efficacy results are presented in Table 11 and Figure 3.
|
a Based on the Stratified Log-rank test. |
||||||
| All Randomized | K-Ras Wild-type | K-Ras Mutant | ||||
| ERBITUX with BSC (N=287) | BSC (N=285) | ERBITUX with BSC (N=117) | BSC (N=128) | ERBITUX with BSC (N=108) | BSC (N=100) |
|
| Median (months) (95% CI) | 6.1 (5.4, 6.7) | 4.6 (4.2, 4.9) | 8.6 (7.0, 10.3) | 5.0 (4.3, 5.7) | 4.8 (3.9, 5.6) | 4.6 (3.6, 4.9) |
| HR (95% CI) | 0.77 (0.64, 0.92) | 0.63 (0.47, 0.84) | 0.91 (0.67, 1.24) |
|||
| p-valuea | 0.0046 | |||||
Figure 3: Kaplan-Meier Curves for Overall Survival in Patients with K-Ras Wild-type Metastatic Colorectal Cancer in Study CA225-025
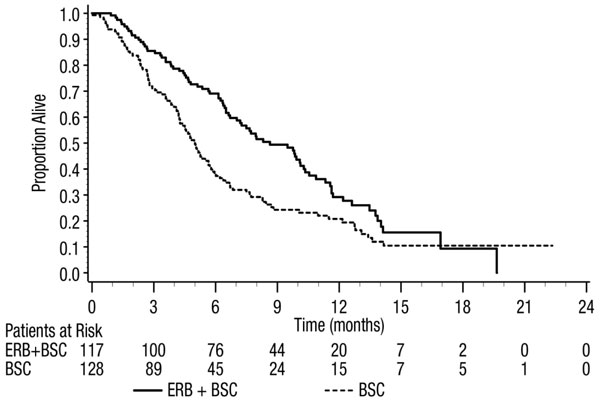
In Combination with Irinotecan
BOND was a multicenter, clinical trial conducted in 329 patients with EGFR-expressing recurrent mCRC. Tumor specimens were not available for testing for K-Ras mutation status. Patients were randomized (2:1) to receive either ERBITUX in combination with irinotecan (218 patients) or ERBITUX single-agent (111 patients). ERBITUX was administered intravenously as a 400 mg/m2 initial dose, followed by 250 mg/m2 weekly until disease progression or unacceptable toxicity. In the ERBITUX with irinotecan arm, irinotecan was added to ERBITUX using the same dosage for irinotecan as the patient had previously failed. Acceptable irinotecan schedules were 350 mg/m2 every 3 weeks, 180 mg/m2 every 2 weeks, or 125 mg/m2 weekly times four doses every 6 weeks. The efficacy of ERBITUX with irinotecan or ERBITUX single-agent, based on durable objective responses, was evaluated in all randomized patients and in two pre-specified subpopulations: irinotecan refractory and irinotecan and oxaliplatin failures.
Of the 329 patients, the median age was 59 years, 63% were male, 98% were White, and 88% had baseline Karnofsky performance status ≥80. Approximately two-thirds had previously failed oxaliplatin treatment.
In patients receiving ERBITUX with irinotecan, the ORR was 23% (95% CI 18%, 29%), median DoR was 5.7 months, and median time to progression was 4.1 months. In patients receiving ERBITUX as a single-agent, the ORR was 11% (95% CI 6%, 18%), median DoR was 4.2 months, and median time to progression was 1.5 months. Similar response rates were observed in the pre-defined subsets in both the combination arm and single-agent arm.
14.3 BRAF V600E Mutation-Positive Metastatic Colorectal Cancer (CRC)
ERBITUX in combination with encorafenib was evaluated in a randomized, active-controlled, open label, multicenter trial (BEACON CRC; NCT02928224). Eligible patients were required to have BRAF V600E mutation-positive metastatic CRC, as detected using the Qiagen therascreen BRAF V600E RGQ polymerase chain reaction (PCR) Kit, with disease progression after 1 or 2 prior regimens. Other key eligibility criteria included absence of prior treatment with a RAF, MEK, or EGFR inhibitor, eligibility to receive cetuximab per local labeling with respect to tumor RAS status, and ECOG performance status (PS) 0–1. Randomization was stratified by ECOG performance status (0 versus 1), prior use of irinotecan (yes versus no), and cetuximab product used (US-licensed versus EU-approved).
Patients were randomized 1:1:1 to one of the following treatment arms:
- ERBITUX in combination with encorafenib 300 mg orally once daily (ERBITUX/encorafenib arm)
- ERBITUX in combination with binimetinib and encorafenib 300 mg orally once daily
- ERBITUX with irinotecan or ERBITUX with FOLFIRI (control arm)
The dosage of cetuximab in all patients was 400 mg/m2 intravenously for the first dose followed by 250 mg/m2 weekly.
Patients in the control arm received ERBITUX with either irinotecan 180 mg/m2 intravenously on Days 1 and 15 of each 28-day cycle or FOLFIRI intravenously (irinotecan 180 mg/m2 on Days 1 and 15; folinic acid 400 mg/m2 on Days 1 and 15; then fluorouracil 400 mg/m2 bolus on Days 1 and 15 followed by fluorouracil 2400 mg/m2 /day by continuous infusion over 2 days).
Treatment continued until disease progression or unacceptable toxicity. Only the results of the approved regimen (ERBITUX in combination with encorafenib) are described below.
The major efficacy outcome measure was OS. Additional efficacy outcome measures included PFS, ORR, and DoR as assessed by blinded independent central review (BICR). OS and PFS were assessed in all randomized patients. ORR and DoR were assessed in the subset of the first 220 patients included in the randomized portion of the ERBITUX/encorafenib and control arm of the study.
A total of 220 patients were randomized to the ERBITUX/encorafenib arm and 221 to the control arm. Of these 441 patients, the median age was 61 years; 53% were female; 80% were White and 15% were Asian. Fifty percent (50%) had baseline ECOG performance status of 0; 66% received 1 prior therapy and 34% received 2; 93% received prior oxaliplatin and 52% received prior irinotecan.
ERBITUX in combination with encorafenib demonstrated a statistically significant improvement in OS, ORR, and PFS compared to the active comparator. Efficacy results are summarized in Table 12 and Figure 4.
| ERBITUX with encorafenib N=220 | ERBITUX with irinotecan or ERBITUX with FOLFIRI N=221 |
|
|---|---|---|
|
CI = Confidence interval; CR = Complete response; DoR = Duration of response; HR = Hazard ratio; NR = Not reached; ORR = Overall response rate; OS = Overall survival; PFS = Progression-free survival; PR = Partial response. |
||
|
a Stratified by ECOG PS, source of cetuximab (US-licensed versus EU-approved) and prior irinotecan use at randomization. |
||
|
b Stratified Cox proportional hazard model. |
||
|
c Stratified log-rank test, tested at alpha level of 0.0084. |
||
|
d ERBITUX/encorafenib arm (n=113) and control arm (n=107). |
||
|
e Cochran-Mantel-Haenszel test; tested at alpha level of 0.05. |
||
|
f Stratified log-rank test, tested at alpha level of 0.0234. |
||
| Overall Survival | ||
| Number of Events (%) | 93 (42) | 114 (52) |
| Median OS, months (95% CI) | 8.4 (7.5, 11.0) | 5.4 (4.8, 6.6) |
| HR (95% CI)a,b | 0.60 (0.45, 0.79) | |
| p-valuea,c | 0.0003 | |
| Overall Response Rate (per BICR) | ||
| ORR (95% CI)d | 20% (13%, 29%) | 2% (0%, 7%) |
| CR | 5% | 0% |
| PR | 15% | 2% |
| p-valuea,e | <0.0001 | |
| Median DoR, months (95% CI) | 6.1 (4.1, 8.3) | NR (2.6, NR) |
| Progression Free Survival (per BICR) | ||
| Number of events (%) | 133 (60) | 128 (58) |
| Progressive disease | 110 (50) | 101 (46) |
| Death | 23 (10) | 27 (12) |
| Median PFS, months (95% CI) | 4.2 (3.7, 5.4) | 1.5 (1.4, 1.7) |
| HR (95% CI)a,b | 0.40 (0.31, 0.52) | |
| p-valuea,f | <0.0001 | |
Figure 4: Kaplan-Meier Curves for Overall Survival in Patients with BRAF V600E Mutation-Positive Metastatic Colorectal Cancer in BEACON
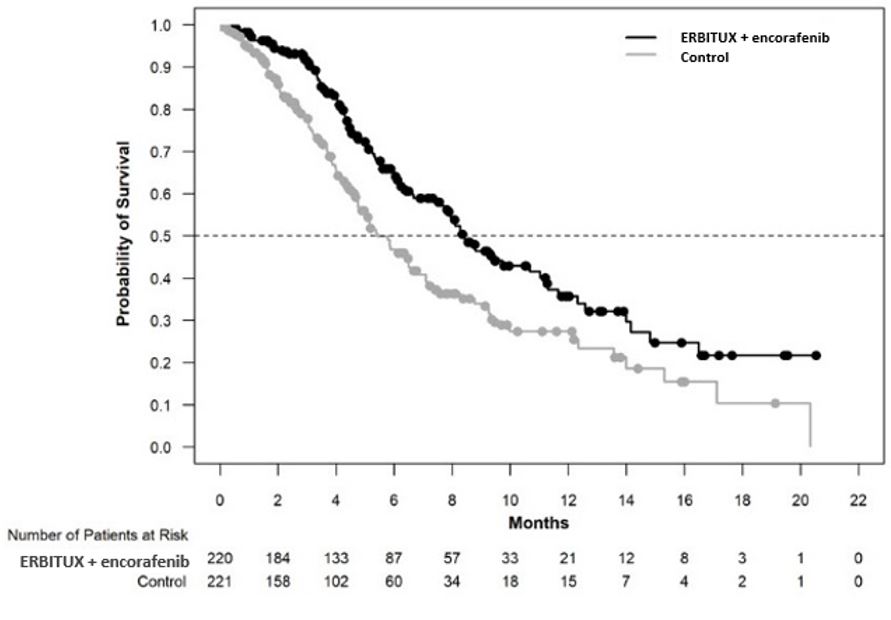
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
ERBITUX® (cetuximab) injection is a sterile, preservative-free, clear, colorless solution in a 2 mg/mL single-dose vial supplied as follows:
- 100 mg/50 mL individually packaged in a carton (NDC 66733-948-23)
- 200 mg/100 mL individually packaged in a carton (NDC 66733-958-23)
Storage and Handling
- Store vials under refrigeration at 2° C to 8° C (36° F to 46° F).
- Do not freeze or shake.
- Increased particulate formation may occur at temperatures at or below 0° C (32° F).
- Discard any remaining solution in the infusion container after 8 hours at controlled room temperature or after 12 hours at 2° C to 8° C.
- Discard any unused portion of the vial.
17 PATIENT COUNSELING INFORMATION
Infusion Reactions
Advise patients that the risk of serious infusion reactions may be increased in patients who have had a tick bite or red meat allergy. Advise patients to contact their healthcare provider and to report signs and symptoms of infusion reactions, including late onset infusion reactions, such as fever, chills, or breathing problems [see Warnings and Precautions (5.1)].
Cardiopulmonary Arrest
Advise patients of the risk of cardiopulmonary arrest or sudden death and to report any history of coronary artery disease, congestive heart failure, or arrhythmias [see Warnings and Precautions (5.2)].
Pulmonary Toxicity
Advise patients to contact their healthcare provider immediately for new or worsening cough, chest pain, or shortness of breath [see Warnings and Precautions (5.3)].
Dermatologic Toxicities
Advise patients to limit sun exposure during ERBITUX treatment and for 2 months after the last dose of ERBITUX. Advise patients to notify their healthcare provider of any sign of acne-like rash, (which can include itchy, dry, scaly, or cracking skin and inflammation, infection or swelling at the base of the nails or loss of the nails), conjunctivitis, blepharitis, or decreased vision [see Warnings and Precautions (5.4)].
Embryo-Fetal Toxicity
Advise female patients of reproductive potential of the potential risk to a fetus and to use effective contraception during ERBITUX treatment and for 2 months after the last dose of ERBITUX. Advise patients to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.8), Use in Specific Populations (8.3)].
Lactation
Advise patients not to breastfeed during ERBITUX treatment and for 2 months after the last dose of ERBITUX [see Use in Specific Populations (8.2)].
ERBITUX® is a registered trademark of ImClone LLC a wholly-owned subsidiary of Eli Lilly and Company.
Manufactured by ImClone LLC a wholly-owned subsidiary of Eli Lilly and Company, Branchburg, NJ 08876 USA
Eli Lilly and Company, Indianapolis, IN 46285, USA
US License No. 1827
Copyright © 2004, 2021, ImClone LLC a wholly-owned subsidiary of Eli Lilly and Company. All rights reserved.
ERB-0009-USPI-20210927

