FULL PRESCRIBING INFORMATION
WARNING: SERIOUS CARDIOPULMONARY REACTIONS
Serious cardiopulmonary reactions, including fatalities, have occurred uncommonly during or following perflutren-containing microsphere administration. Most serious reactions occur within 30 minutes of administration.
- Assess all patients for the presence of any condition that precludes DEFINITY administration [see Contraindications (4)].
- Always have resuscitation equipment and trained personnel readily available [see Warnings and Precautions (5.1)] .
1 INDICATIONS AND USAGE
DEFINITY is indicated, after activation, for use in adult and pediatric patients with suboptimal echocardiograms to opacify the left ventricular chamber and to improve the delineation of the left ventricular endocardial border.
2 DOSAGE AND ADMINISTRATION
2.1 Important Preparation and Administration Instructions
- There are two formulations, DEFINITY and DEFINITY RT, that have differences in preparation and storage. Ensure of the correct product when following the directions for preparation and storage.
- DEFINITY must be activated using the VIALMIX or VIALMIX RFID device before administration according to the instructions outlined below [see Dosage and Administration (2.5)].
- DEFINITY is for intravenous use only and must not be administered by intra-arterial injection [see Warnings and Precautions (5.3)].
- For adult patients, DEFINITY can be administered by either an intravenous bolus or infusion. The maximum dose is either two bolus doses or one single infusion. Do not administer the bolus and infusion dosing in combination or in sequence [see Dosage and Administration (2.2)].
- For pediatric patients, DEFINITY is administered by an intravenous bolus injection only. The maximum dose is two bolus doses [see Dosage and Administration (2.3)].
2.2 Recommended Dosage in Adult Patients
Bolus Dosing
The recommended bolus dose in adult patients is 10 microL/kg of activated DEFINITY administered intravenously over 30 to 60 seconds followed by a 10 mL flush of 0.9% Sodium Chloride Injection, USP. If necessary, administer a second 10 microL/kg dose followed by a second 10 mL flush of 0.9% Sodium Chloride Injection, USP 30 minutes after the first injection to prolong contrast enhancement.
Infusion Dosing
The recommended infusion dose in adult patients is 1.3 mL of activated DEFINITY added to 50 mL of preservative-free 0.9% Sodium Chloride Injection, USP administered intravenously. Initiate infusion at 4 mL/minute, titrating the infusion rate as necessary to achieve optimal image enhancement, not to exceed 10 mL/minute.
2.3 Recommended Dosage in Pediatric Patients
Bolus Dosing
The recommended bolus dose in pediatric patients is 3 microL/kg of activated DEFINITY administered intravenously over 30 seconds to 60 seconds followed by a 5 mL flush of 0.9% Sodium Chloride Injection, USP. If necessary, administer a second bolus at a dose of 3 microL/kg to 5 microL/kg, followed by a second 5 mL flush of 0.9% Sodium Chloride Injection, USP 30 minutes after the first injection to prolong contrast enhancement.
2.4 Imaging Instructions
Adult Patients
After baseline non-contrast echocardiography is completed, set the mechanical index for the ultrasound device at 0.8 or below [see Warnings and Precautions (5.4)]. Then inject activated DEFINITY (as described above) and begin ultrasound imaging immediately [see Clinical Pharmacology(12.2). Evaluate the DEFINITY echocardiogram images in combination with the non-contrast echocardiogram images.
Pediatric Patients
After baseline non-contrast echocardiography is completed, set the mechanical index for the ultrasound transducer at 0.3 or below [see Warnings and Precautions (5.4)]. Then inject activated DEFINITY (as described above) and begin ultrasound imaging immediately [see Clinical Pharmacology(12.2). Evaluate the DEFINITY echocardiogram images in combination with the non-contrast echocardiogram images.
2.5 Instructions for Activation, Preparation, and Handling of DEFINITY
- Adhere to strict aseptic procedures during preparation.
- Allow the DEFINITY vial to warm to room temperature before starting the activation procedure.
- Activate DEFINITY by shaking the vial for 45 seconds using a VIALMIX device or VIALMIX RFID device.
Note: Use only the drug activated in a properly functioning VIALMIX or VIALMIX RFID for a full 45 second activation cycle. Do not reactivate the vial if VIALMIX or VIALMIX RFID did not properly activate the vial. Never reactivate a successfully activated DEFINITY vial (see step 4). Refer to the VIALMIX or VIALMIX RFID User's Guide for illustrations of the activation procedure and a properly functioning VIALMIX or VIALMIX RFID. -
Activated DEFINITY appears as a homogeneous milky white suspension with a presence of foam/bubbles. Use immediately after activation. If the product is not used within 5 minutes of activation, resuspend the microspheres by 10 seconds of hand agitation by inverting the vial before the product is withdrawn in a syringe.
- The activated DEFINITY may be used for up to 12 hours from the time of activation, but only after the microspheres are resuspended by hand agitation for 10 seconds. Store the activated DEFINITY at room temperature 20° to 25°C (68° to 77°F) in the original product vial.
- Invert the vial and withdraw the activated homogeneous milky white suspension using the Intellipin (Dispensing Pin), the PINSYNC (Vented Vial Adapter 13mm), or 18 gauge to 20 gauge syringe needle from the middle of the liquid. Do not inject air into the DEFINITY Vial.
- Use the product immediately after its withdrawal from the vial; do not allow the product to stand in the syringe.
- For infusion dosing in adult patients, dilute 1.3 mL of activated DEFINITY in 50 mL of preservative-free 0.9% Sodium Chloride Injection, USP.
- Parenteral drug products should be inspected visually for foreign particulate matter and discoloration prior to administration, whenever solution and container permit.
2.6 Instructions for RFID-Tagged Vials
The radio frequency identification (RFID) tag integrated on the back of the vial label when used with VIALMIX RFID allows for the exchange of product information such as activation time and activation rate. If the RFID tag is damaged or otherwise non-functional, the vial with the non-functional RFID tag cannot be activated with VIALMIX RFID. Discard the non-functional RFID-tagged vial. Do not activate RFID-tagged DEFINITY vials in the VIALMIX RFID within 6 inches (15 cm) of a pacemaker and/or defibrillator (see the VIALMIX RFID User's Guide).
3 DOSAGE FORMS AND STRENGTHS
DEFINITY is supplied in a single-patient use vial containing 6.52 mg/mL perflutren in the headspace and 0.75mg/mL of a lipid blend as a clear colorless liquid with 1.5 mL volume. After activation, each mL contains a maximum of 1.2 × 1010 perflutren lipid microspheres as a homogeneous milky white injectable suspension, and about 150 microL/mL (1.1 mg/mL) perflutren.
4 CONTRAINDICATIONS
DEFINITY is contraindicated in patients with known or suspected hypersensitivity to perflutren lipid microsphere or its components, such as polyethylene glycol (PEG) [see Warnings and Precautions (5.2) and Description (11)].
5 WARNINGS AND PRECAUTIONS
5.1 Serious Cardiopulmonary Reactions
Serious cardiopulmonary reactions including fatalities have occurred uncommonly during or shortly following perflutren-containing microsphere administration, typically within 30 minutes of administration. The risk for these reactions may be increased among patients with unstable cardiopulmonary conditions (acute myocardial infarction, acute coronary artery syndromes, worsening or unstable congestive heart failure, or serious ventricular arrhythmias). Always have cardiopulmonary resuscitation personnel and equipment readily available prior to DEFINITY administration and monitor all patients for acute reactions.
The reported reactions include: fatal cardiac or respiratory arrest, shock, syncope, symptomatic arrhythmias (atrial fibrillation, tachycardia, bradycardia, supraventricular tachycardia, ventricular fibrillation, ventricular tachycardia), hypertension, hypotension, dyspnea, hypoxia, chest pain, respiratory distress, stridor, wheezing, loss of consciousness, and convulsions [see Adverse Reactions (6.2)].
5.2 Hypersensitivity Reactions
In postmarketing use, serious hypersensitivity reactions were observed during or shortly following perflutren-containing microsphere administration including:
Anaphylaxis, with manifestations that may include death, shock, bronchospasm, throat tightness, angioedema, edema (pharyngeal, palatal, mouth, peripheral, localized), swelling (face, eye, lip, tongue, upper airway), facial hypoesthesia, rash, urticaria, pruritus, flushing, and erythema.
These reactions have occurred in patients with no prior exposure to perflutren-containing microsphere products. DEFINITY contains PEG. There may be increased risk of serious reactions including death in patients with prior hypersensitivity reaction(s) to PEG [see Adverse Reactions (6.2) and Description (11)]. Clinically assess patients for prior hypersensitivity reactions to products containing PEG, such as certain colonoscopy bowel preparations and laxatives. DEFINITY is contraindicated in patients with known or suspected hypersensitivity to perflutren lipid microsphere or its components such as PEG [see Contraindications (4)]. Always have cardiopulmonary resuscitation personnel and equipment readily available prior to DEFINITY administration and monitor all patients for hypersensitivity reactions.
5.3 Systemic Embolization
When administering DEFINITY to patients with a cardiac shunt, the microspheres can bypass filtering by the lung and enter the arterial circulation. Assess patients with shunts for embolic phenomena following DEFINITY administration. DEFINITY is only for intravenous administration and must not be administered by intra-arterial injection [see Dosage and Administration (2.1,2.2,2.3)].
5.4 Ventricular Arrhythmia Related to High Mechanical Index
High ultrasound mechanical index values may cause microsphere cavitation or rupture and lead to ventricular arrhythmias. Additionally, end-systolic triggering with high mechanical indices has been reported to cause ventricular arrhythmias. The maximum recommended mechanical index for use with DEFINITY is 0.8 in adult patients and 0.3 in pediatric patients [see Dosage and Administration (2.4)].
5.5 Pain Episodes in Patients with Sickle Cell Disease
In postmarketing reports, acute pain episodes shortly following DEFINITY administration have been reported in patients with sickle cell disease (SCD). The pain episodes included moderate to severe back pain and vaso-occlusive crisis [see Adverse Reactions (6.2)]. If a patient with sickle cell disease experiences new or worsening pain, discontinue DEFINITY.
6 ADVERSE REACTIONS
The following serious adverse reactions are described elsewhere in the labeling:
- Serious Cardiopulmonary Reactions [see Warnings and Precautions (5.1)]
- Hypersensitivity Reactions [see Warnings and Precautions(5.2)]
- Pain Episodes in Patients with Sickle Cell Disease [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adverse Reactions in Adults
Safety of DEFINITY was evaluated in a total of 1,716 adult subjects in pre-market clinical trials. In this group, 1,063 (62%) were male and 653 (38%) were female, 1,328 (77%) were White, 258 (15%) were Black or African American, 74 (4%) were Hispanic, and 56 (3%) were classified as other racial or ethnic groups. The mean age was 56 years (range 18 to 93).
Among the 1,716 subjects, 19 (1.1%) suffered serious cardiopulmonary adverse reactions.
Adverse reactions that led to discontinuation in a total of 15 (0.9%) subjects receiving DEFINITY in the clinical trials included urticaria, pruritus, dizziness, chest pain, dyspnea, and back pain.
Table 1 summarizes the most common adverse reactions occurring at ≥0.5%.
| DEFINITY (N=1716) |
||
|---|---|---|
| Total Number of Adverse Reactions | 269 | |
| Total Number of Subjects with an Adverse Reaction | 144 | (8.4%) |
| Body system | ||
| Preferred term | n | (%) |
| N=Sample size 1716 subjects who received activated DEFINITY | ||
| n=Number of subjects reporting at least one Adverse Reaction | ||
| Application Site Disorders | 11 | (0.6) |
| Injection Site Reactions | 11 | (0.6) |
| Body as a Whole | 41 | (2.4) |
| Back/renal pain | 20 | (1.2) |
| Chest pain | 13 | (0.8) |
| Central and peripheral nervous system disorder | 54 | (3.1) |
| Headache | 40 | (2.3) |
| Dizziness | 11 | (0.6) |
| Gastrointestinal system | 31 | (1.8) |
| Nausea | 17 | (1.0) |
| Vascular (extracardiac) disorders | 19 | (1.1) |
| Flushing | 19 | (1.1) |
Other adverse reactions that occurred in ≤0.5% of the activated DEFINITY-dosed subjects were:
Body as a Whole: Fatigue, fever, hot flushes, pain, rigors, and syncope
Cardiovascular: Abnormal ECGs, bradycardia, tachycardia, palpitation, hypertension and hypotension
Digestive: Dyspepsia, dry mouth, tongue disorder, toothache, abdominal pain, diarrhea and vomiting
Hematology: Granulocytosis, leukocytosis, leukopenia, and eosinophilia
Musculoskeletal: Arthralgia
Nervous System: Leg cramps, hypertonia, vertigo and paresthesia
Platelet, Bleeding, and Clotting: Hematoma
Respiratory: Coughing, hypoxia, pharyngitis, rhinitis and dyspnea
Special Senses: Decreased hearing, conjunctivitis, abnormal vision and taste perversion
Skin: Pruritus, rash, erythematous rash, urticaria, increased sweating, and dry skin
Urinary: Albuminuria
Adverse Reactions in Pediatric Patients
In a study of DEFINITY in 40 pediatric patients 1 month of age and older, no new safety signals were observed [see Use in Specific Populations (8.4) and Clinical Pharmacology (12.2)].
In published studies of DEFINITY in 149 patients 5 years to 24 years of age (mean age 16.8 years; 56% male) clinically indicated for echocardiography, no additional safety signals were observed.
6.2 Postmarketing Experience
Adverse Reactions from Observational Studies
In a prospective, multicenter, open-label registry of 1,053 patients receiving DEFINITY in routine clinical practice, heart rate, respiratory rate, and pulse oximetry were monitored for 30 minutes after DEFINITY administration. No deaths or serious adverse reactions were reported.
Adverse Reactions from Postmarketing Spontaneous Reports
The following adverse reactions have been identified during the post-marketing use of perflutren and PEG-containing microsphere products. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Cardiopulmonary
Fatal cardiac or respiratory arrest, shock, syncope, symptomatic arrhythmias (atrial fibrillation, tachycardia, bradycardia, supraventricular tachycardia, ventricular fibrillation, ventricular tachycardia), hypertension, hypotension, dyspnea, hypoxia, chest pain, respiratory distress, stridor, wheezing.
Immune System
Anaphylaxis, with manifestations that may include death, shock, bronchospasm, throat tightness, angioedema, edema (pharyngeal, palatal, mouth, peripheral, localized), swelling (face, eye, lip, tongue, upper airway), facial hypoesthesia, rash, urticaria, pruritus, flushing, and erythema.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Available data from case reports with DEFINITY use in pregnant women have not identified a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. DEFINITY has a very short half-life; therefore, administration of DEFINITY to a pregnant woman is not expected to result in clinically relevant fetal exposure. No adverse developmental outcomes were observed in animal reproduction studies with administration of activated DEFINITY in pregnant rats and rabbits during organogenesis at doses up to 8 and 16 times, respectively, the maximum human dose based on body surface area (see Data).
All pregnancies have a background risk of birth defects, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
DEFINITY was administered intravenously to rats at doses of 0.1 mL/kg, 0.3 mL/kg, and 1 mL/kg (approximately 0.8, 2.4, and 8 times the recommended maximum human dose based on body surface area); DEFINITY doses were administered daily from day 6 to day 17 of gestation. DEFINITY was administered intravenously to rabbits at doses of 0.1mL/kg, 0.3 mL/kg, and 1 mL/kg (approximately, 1.6, 4.8, and 16 times the recommended maximum human dose based on body surface area); DEFINITY doses were administered daily from day 7 to day 19 of gestation. No significant findings on the fetus were observed.
8.2 Lactation
Risk Summary
There are no data on the presence of perflutren lipid microspheres in human milk, the effects on the breastfed infant, or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for DEFINITY and any potential adverse effects on the breastfed infant from DEFINITY or from the underlying maternal condition.
8.4 Pediatric Use
The safety and effectiveness of DEFINITY have been established for use in pediatric patients with suboptimal echocardiograms to opacify the left ventricular chamber and to improve delineation of the left endocardial border. Use of DEFINITY for this indication is supported by evidence from adequate and well-controlled studies in adults [see Clinical Studies (14)], a pharmacodynamic and safety study in 40 pediatric patients 1 month of age and older [see Adverse Reactions (6.1) and Clinical Pharmacology (12.2)], and published studies in 149 patients 5 years to 24 years of age [see Adverse Reactions (6.1)].
8.5 Geriatric Use
Of the total number of subjects in clinical trials of DEFINITY, 144 (33%) were 65 and over. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences between the elderly and younger patients.
11 DESCRIPTION
DEFINITY (perflutren lipid microsphere) injectable suspension is, after activation an ultrasound contrast agent for intravenous use.
The perflutren lipid microspheres are composed of perflutren encapsulated in an outer lipid shell consisting of (R) – hexadecanoic acid, 1-[(phosphonoxy)methyl]-1,2-ethanediyl ester, monosodium salt (abbreviated DPPA); (R) - 4-hydroxy-N,N,N-trimethyl-10-oxo-7-[(1-oxohexadecyl)oxy]-3,4,9-trioxa-4-phosphapentacosan-1-aminium, 4-oxide, inner salt (abbreviated DPPC); and (R)-∝-[6-hydroxy-6-oxido-9-[(1-oxohexadecyl)oxy]-5,7,11-trioxa-2-aza-6-phosphahexacos-1-yl]- ω-methoxypoly(ox-1,2-ethanediyl), monosodium salt; commonly called N-(methoxypolyethylene glycol 5000 carbamoyl)-1,2-dipalmitoyl-sn-glycero-3- phosphatidylethanolamine, monosodium salt (abbreviated MPEG5000 DPPE).
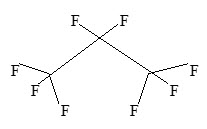
Perflutren is chemically characterized as 1,1,1,2,2,3,3,3-octafluoropropane. It has a molecular weight of 188, empirical formula of C3F8 and has the following structural formula:
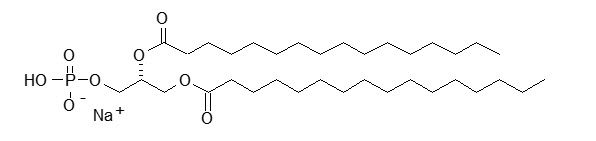
DPPA has a molecular weight of 670, empirical formula of C35H68O8PNa, and following structural formula:
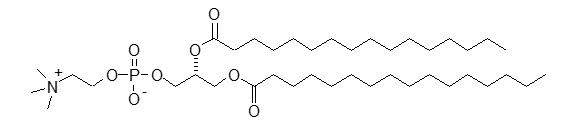
DPPC has a molecular weight of 734, empirical formula of C40H80NO8P, and following structural formula:
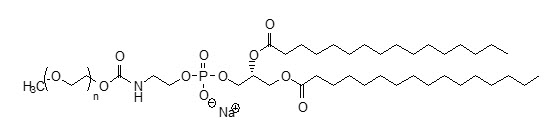
MPEG5000 DPPE has an approximate molecular weight of 5750 represented by empirical formula C265H527NO123PNa, contains <100ppm Ca2+ and Mg2+ and the following structural formula:
Prior to activation, perflutren is in the headspace of the vial with a concentration of 6.52 mg/mL which is confirmed by positive IR spectroscopic testing in every vial. The lipid blend is in the clear, colorless, hypertonic, sterile liquid. Each mL of the liquid contains 0.75 mg lipid blend (consisting of 0.045 mg DPPA, 0.401 mg DPPC, and 0.304 mg MPEG5000 DPPE) and the following inactive ingredients: 103.5 mg propylene glycol, 126.2 mg glycerin, 2.34 mg sodium phosphate monobasic monohydrate, 2.16 mg sodium phosphate dibasic heptahydrate, and 4.87 mg sodium chloride in water for injection. The pH is 6.2 to 6.8. DEFINITY does not contain bacterial preservative.
After activation with the aid of the VIALMIX or VIALMIX RFID, each mL of the activated DEFINITY as homogenous milky white suspension contains a maximum of 1.2 × 1010 perflutren lipid microspheres, and about 150 microL/mL (1.1 mg/mL) perflutren. The microsphere particle size parameters are listed in Table 2 below:
| Microsphere particle size parameters | |
| Mean diameter range | 1.1 µm – 3.3 µm |
| Percent less than 10 µm | 98% |
| Maximum diameter | 20 µm |
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Perflutren lipid microspheres exhibit lower acoustic impedance than blood and enhance the intrinsic backscatter of blood. These physical acoustic properties of activated DEFINITY provide contrast enhancement of the left ventricular chamber and aid delineation of the left ventricular endocardial border during echocardiography.
In animal models the acoustic properties of activated DEFINITY were established at or below a mechanical index of 0.7 (1.8 MHz frequency). In clinical trials, the majority of the patients were imaged at or below a mechanical index of 0.8.
12.2 Pharmacodynamics
Contrast Enhancement Duration in Adults
In a crossover trial of 64 adult subjects randomized to both bolus and infusion, the duration of clinically useful contrast enhancement for fundamental imaging was approximately 3.4 minutes after a 10 microL/kg bolus and was approximately 7.1 minutes during the continuous infusion of 1.3 mL activated DEFINITY in 50 mL saline at a rate of 4 mL/min.
Dose-Ranging and Contrast Enhancement Duration in Pediatric Patients
In a pharmacodynamic and safety study, DEFINITY was administered to 40 pediatric patients 1 month of age and older clinically indicated for echocardiography (60% male; 10 patients were 1 month to younger than 1 year of age; 9 patients were 1 to 5 years of age; 10 patients were 6 to 10 years of age; 10 patients were 11 to 16 years of age; and 1 patient was 17 years of age). Patients underwent echocardiography with a mechanical index of ≤ 0.3 and received DEFINITY as a series of sequential bolus injections (3 microL/kg and 5 microL/kg) with each injection separated by approximately 7 minutes. Patients had acceptable cardiac imaging at baseline. Left ventricular opacification with DEFINITY was graded as adequate or full in 80% to 95% of patients (depending on view) at the 3 microL/kg dose and 85% to 88% of patients at the 5 microL/kg dose [see Dosage and Administration (2.3)]. The mean duration of clinically useful contrast enhancement was approximately 2.7 minutes after a bolus dose of 3 microL/kg and 3.9 minutes after a bolus dose of 5 microL/kg.
Pulmonary Hemodynamic Effects
The impact of DEFINITY on pulmonary hemodynamics was explored in a prospective, open-label study of patients with normal (≤ 35 mmHg, 16 patients) and elevated (> 35 mmHg, ≤ 75 mmHg, 16 patients) pulmonary artery systolic pressure undergoing right heart catheterization. Patients with pulmonary artery systolic pressure greater than 75 mmHg were excluded from this study. Systemic hemodynamic parameters and ECGs were also evaluated. No clinically important pulmonary hemodynamic, systemic hemodynamic, or ECG changes were observed.
12.3 Pharmacokinetics
Human pharmacokinetics information is not available for the intact or degassed lipid microspheres. The pharmacokinetics of perflutren gas was evaluated in healthy subjects (n=8) after the intravenous administration of activated DEFINITY at 50 microL/kg (5 times the recommended dose).
Distribution
Perflutren gas binding to plasma proteins or partitioning into blood cells has not been studied. However, perflutren protein binding is expected to be minimal due to its low partition coefficient into whole blood.
Elimination
Specific Populations
Patients with Obstructive Pulmonary Disease
The pharmacokinetics of perflutren gas was evaluated in subjects (n=11) with chronic obstructive pulmonary disease (COPD). The mean half-life of perflutren in blood was 1.9 minutes. The total lung clearance of perflutren was similar to that in healthy subjects.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, and Impairment of Fertility
Studies with activated DEFINITY have not been performed to evaluate carcinogenic potential. Evidence of genotoxicity was not found in the following studies with activated DEFINITY: 1) bacterial mutagenesis assay (Ames assay), 2) in vitro mammalian mutagenesis assay, 3) in vitro human lymphocyte chromosome aberration assay, and 4) in vivo rat micronucleus assay.
Impairment of male or female fertility was not observed in rats and rabbits treated with activated DEFINITY at doses up to 24 and 15 times the human dose based on body surface area (in rats and rabbits respectively).
14 CLINICAL STUDIES
Effectiveness and safety of DEFINITY were evaluated in four controlled clinical trials (two open-label baseline controlled, unpaired blinded image evaluation studies and two identical placebo-controlled, unpaired blinded image evaluation studies) in 249 subjects who had two or more (of six) non-evaluable segments in either the apical 2- or 4-chamber view in non-contrast fundamental echocardiography.
In this group, 154 (62%) were male and 95 (38%) were female; 183 (74%) were White, 38 (15%) were Black or African American, 21 (8%) were Hispanic, and 7 (3%) were classified as other racial or ethnic groups. The mean age was 54 years (range 18 to 87).
In the two open-label baseline controlled studies, a total of 126 (67 in study A and 59 in study B) subjects received a bolus dose of 10 microL/kg activated DEFINITY. The outcome measures in these studies included the blinded assessment of ejection fraction (EF), endocardial border length (EBL) obtained by direct measurement, and qualitative assessment of wall motion.
In the two placebo-controlled studies a total of 123 subjects were randomized in 1:2 ratio to receive two intravenous bolus doses of either 0.9% Sodium Chloride Injection (placebo) or activated DEFINITY 10 microL/kg (17 placebo vs. 33 DEFINITY subjects and 24 placebo vs. 49 DEFINITY subjects, respectively). The outcome measure for assessing the effectiveness of DEFINITY was the blinded assessment of improvement in ventricular chamber enhancement (measured by videodensitometry at end-diastole and end-systole).
Endocardial Border Length
As shown in Table 3, compared to baseline, a single bolus dose of 10 microL/kg of activated DEFINITY increased the length of endocardial border that could be measured at both end-systole and end-diastole. The mean change in border length from baseline at end-diastole was statistically significant for all readers in the apical 4-chamber view and for 3 out of 4 readers for the apical 2-chamber view. The mean change in border length from baseline at end-systole was statistically significant for 3 out of 4 readers for the apical 4-chamber view and for 2 out of 4 readers for the apical 2-chamber view.
Ventricular Chamber Enhancement
Left ventricular chamber enhancement after an activated DEFINITY dose of 10 microL/kg was significantly increased from baseline compared to placebo in both views at the mid-ventricular and apical levels at end-diastole. Similar results were noted at end-systole, with the exception of the 4-chamber view.
Wall Motion
In a retrospective analysis, in a subset of subjects (n=12 to 47, depending on reader) having at least 2 adjacent segments non-evaluable on non-contrast imaging, DEFINITY converted a baseline non-evaluable image to an evaluable image in 58 to 91% of the subjects, depending on the reader. In the converted images, the accuracy of wall motion (i.e., normal versus abnormal) improved in 42 to 71% of the subjects, depending on the reader, however, improvement in the specific diagnostic accuracy (e.g., hypokinetic, akinetic etc.) was not established. Also, in 13 to 37% of the subjects, depending on the reader, DEFINITY was found to obscure the wall motion rendering the image non-evaluable.
Ejection Fraction
In the 2 baseline controlled studies, ejection fraction results were evaluated in comparison to MRI. The results were evaluated by 3 blinded, independent radiologists. In these studies, although there was a statistically significant increase in ventricular chamber enhancement, DEFINITY did not significantly improve the assessment of ejection fraction compared to the baseline images.
| Study/View | Endocardial Border Length – Blinded Read | |||
|---|---|---|---|---|
| Mean(SD) at End-Diastole | Mean(SD) at End-Systole | |||
| Reader 1 | Reader 2 | Reader 1 | Reader 2 | |
| Activated DEFINITY Bolus Dose = 10 µL/kg | ||||
|
||||
| Study A: (N = 67) | ||||
| Apical 2-chamber | ||||
| Baseline | 8.0(3.4) | 4.7(2.8) | 7.1(3.3) | 4.3(2.6) |
| Post-DEFINITY | 12.8(5.2)* | 5.8(2.6)* | 10.6(5.0)* | 4.4(2.3) |
| Apical 4-chamber | ||||
| Baseline | 8.1(3.3) | 4.5(2.6) | 7.6(3.2) | 4.5(2.7) |
| Post-DEFINITY | 13.5(5.2)* | 6.8(3.3)* | 11.5(4.4)* | 5.3(3.1) |
| Study B: (N = 59) | ||||
| Apical 2-chamber | ||||
| Baseline | 4.3(2.6) | 7.8(5.3) | 4.1(2.4) | 6.5(5.1) |
| Post-DEFINITY | 5.7(4.7)* | 8.2(6.5) | 5.5(4.4)* | 6.9(6.3) |
| Apical 4-chamber | ||||
| Baseline | 4.0(2.7) | 9.2(5.9) | 3.8(2.6) | 7.3(5.6) |
| Post-DEFINITY | 7.1(5.5)* | 11.5(7.5)* | 5.9(5.3)* | 8.7(6.3)* |
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
DEFINITY (perflutren lipid microsphere) injectable suspension is supplied in a single- patient use clear glass vial or a single patient use clear glass Radio Frequency Identification (RFID)-tagged vial containing 6.52 mg/mL perflutren in the headspace of the vial and 0.75 mg/mL of a lipid blend in a clear, colorless liquid phase with 1.5 mL volume. DEFINITY is available in the following packages:
- One (1) vial or RFID-tagged vial per carton NDC (11994-011-01)
- Four (4) vials or RFID-tagged vials per carton - NDC (11994-011-04)
- Sixteen (16) vials or RFID-tagged vials per carton - NDC (11994-011-16)
Order VIALMIX or VIALMIX RFID from Lantheus Medical Imaging.
17 PATIENT COUNSELING INFORMATION
Hypersensitivity Reactions
Advise patients to inform their healthcare provider if they develop any symptoms of hypersensitivity after DEFINITY administration, including rash, wheezing, or shortness of breath [see Warnings and Precautions (5.2)].
Distributed By
Lantheus Medical Imaging
331 Treble Cove Road
N. Billerica, Massachusetts 01862 USA
For ordering, call toll free: 800-299-3431
All Other Business: 800-362-2668
(For Massachusetts and International, call 978-667-9531)
Patent: http://www.lantheus.com/patents/index.html
515987-0324
PRINCIPAL DISPLAY PANEL - 2 mL Vial Carton
DEFINITY®
VIAL
FOR
(Perflutren Lipid Microsphere)
INJECTABLE SUSPENSION
NDC 11994-011-16
Sterile
CAUTION: Rx Only
16x2 mL Single-Dose Containers
Non-Pyrogenic
For Intravenous Use Only, After Activation
Store refrigerated, 2° to 8° C (36° to 46° F)
For Single Use Only, Discard Unused Portion
Use within 12 hours of activation (see Insert)
IMPORTANT: Read enclosed Package Insert for full information on preparation, use and indications.
CONTAINS NO BACTERIOSTATIC PRESERVATIVE
Lantheus
Medical Imaging
