FULL PRESCRIBING INFORMATION
WARNING: CARDIOPULMONARY ARREST and HYPOMAGNESEMIA
- Cardiopulmonary arrest and/or sudden death occurred in 3.0% of patients treated with PORTRAZZA in combination with gemcitabine and cisplatin. Closely monitor serum electrolytes, including serum magnesium, potassium, and calcium, with aggressive replacement when warranted during and after PORTRAZZA administration [see Warnings and Precautions (5.1, 5.2)].
- Hypomagnesemia occurred in 83% of patients receiving PORTRAZZA in combination with gemcitabine and cisplatin, and was severe in 20% of patients. Monitor patients for hypomagnesemia, hypocalcemia, and hypokalemia prior to each dose of PORTRAZZA during treatment and for at least 8 weeks following completion of PORTRAZZA. Withhold PORTRAZZA for Grade 3 or 4 electrolyte abnormalities. Replete electrolytes as medically appropriate [see Warnings and Precautions (5.2)].
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dose and Schedule
The recommended dose of PORTRAZZA is 800 mg administered as an intravenous infusion over 60 minutes on Days 1 and 8 of each 3-week cycle prior to gemcitabine and cisplatin infusion. Continue PORTRAZZA until disease progression or unacceptable toxicity.
2.2 Premedication
- For patients who have experienced a previous Grade 1 or 2 infusion-related reaction (IRR), pre-medicate with diphenhydramine hydrochloride (or equivalent) prior to all subsequent PORTRAZZA infusions [see Dosage and Administration (2.3)].
- For patients who have experienced a second Grade 1 or 2 occurrence of IRR, pre-medicate for all subsequent infusions, with diphenhydramine hydrochloride (or equivalent), acetaminophen (or equivalent), and dexamethasone (or equivalent) prior to each PORTRAZZA infusion [see Dosage and Administration (2.3)].
2.3 Dose Modifications
Infusion-Related Reactions (IRR)
- Reduce the infusion rate of PORTRAZZA by 50% for Grade 1 IRR [see Dosage and Administration (2.2) and Warnings and Precautions (5.5)].
- Stop the infusion for Grade 2 IRR until signs and symptoms have resolved to Grade 0 or 1; resume PORTRAZZA at 50% reduced rate for all subsequent infusions [see Dosage and Administration (2.2) and Warnings and Precautions (5.5)].
- Permanently discontinue PORTRAZZA for Grade 3 or 4 IRR [see Dosage and Administration (2.2) and Warnings and Precautions (5.5)].
Dermatologic Toxicity
- Withhold PORTRAZZA for Grade 3 rash or acneiform rash until symptoms resolve to Grade ≤2, then resume PORTRAZZA at reduced dose of 400 mg for at least 1 treatment cycle. If symptoms do not worsen, may increase dose to 600 mg and 800 mg in subsequent cycles.
- Permanently discontinue PORTRAZZA if:
- -
- Grade 3 rash or acneiform rash do not resolve to Grade ≤2 within 6 weeks,
- -
- Reactions worsen or become intolerable at a dose of 400 mg
- -
- Patient experiences Grade 3 skin induration/fibrosis [see Warnings and Precautions (5.4) and Adverse Reactions (6.1)] or
- -
- Grade 4 dermatologic toxicity [see Warnings and Precautions (5.4) and Adverse Reactions (6.1)].
2.4 Preparation for Administration
Inspect vial contents for particulate matter and discoloration prior to dilution [see Description (11)]. Discard the vial if particulate matter or discoloration is identified. Store vials in a refrigerator at 2° to 8°C (36˚ to 46˚F) until time of use. Keep the vial in the outer carton in order to protect from light [see How Supplied/Storage and Handling (16.2)].
- Dilute the required volume of PORTRAZZA with 0.9% Sodium Chloride Injection, USP in an intravenous infusion container to a final volume of 250 mL. Do not use solutions containing dextrose.
- Gently invert the container to ensure adequate mixing.
- DO NOT FREEZE OR SHAKE the infusion solution. DO NOT dilute with other solutions or co-infuse with other electrolytes or medication.
- Store diluted infusion solution for no more than 24 hours at 2° to 8°C (36° to 46°F), or no more than 4 hours at room temperature (up to 25°C [77°F]).
- Discard vial with any unused portion of PORTRAZZA.
2.5 Administration
Visually inspect the diluted solution for particulate matter and discoloration prior to administration. If particulate matter or discoloration is identified, discard the solution.
Administer diluted PORTRAZZA infusion via infusion pump over 60 minutes through a separate infusion line. Flush the line with 0.9% Sodium Chloride Injection, USP at the end of the infusion.
5 WARNINGS AND PRECAUTIONS
5.1 Cardiopulmonary Arrest
Cardiopulmonary arrest or sudden death occurred in 15 (3%) of 538 patients treated with PORTRAZZA plus gemcitabine and cisplatin as compared to 3 (0.6%) of 541 patients treated with gemcitabine and cisplatin alone in Study 1. Twelve of the fifteen patients died within 30 days of the last dose of PORTRAZZA and had comorbid conditions including history of coronary artery disease (n=3), hypomagnesemia (n=4), chronic obstructive pulmonary disease (n=7), and hypertension (n=5). Eleven of the 12 patients had an unwitnessed death. Patients with significant coronary artery disease, myocardial infarction within 6 months, uncontrolled hypertension, and uncontrolled congestive heart failure were not enrolled in Study 1. The incremental risk of cardiopulmonary arrest or sudden death in patients with a history of coronary artery disease, congestive heart failure, or arrhythmias as compared to those without these comorbid conditions is not known.
Closely monitor serum electrolytes, including serum magnesium, potassium, and calcium prior to each infusion of PORTRAZZA during treatment and after PORTRAZZA administration for at least 8 weeks after the last dose. Withhold PORTRAZZA for Grade 3 or 4 electrolyte abnormalities; subsequent cycles of PORTRAZZA may be administered in these patients once electrolyte abnormalities have improved to Grade ≤2. Replete electrolytes as medically appropriate [see Boxed Warning and Warnings and Precautions (5.2)].
5.2 Hypomagnesemia
Hypomagnesemia occurred in 83% of 461/538 patients with available laboratory results treated with PORTRAZZA as compared to 70% of 457/541 patients with available laboratory results treated with gemcitabine and cisplatin alone in Study 1. Hypomagnesemia was severe (Grade 3 or 4) in 20% of the patients treated with PORTRAZZA compared to 7% of the patients treated with gemcitabine and cisplatin alone. The median time to development of hypomagnesemia and accompanying electrolyte abnormalities was 6 weeks (25th percentile 4 weeks; 75th percentile 9 weeks) after initiation of PORTRAZZA. Monitor patients for hypomagnesemia, hypocalcemia, and hypokalemia prior to each infusion of PORTRAZZA during treatment and for at least 8 weeks following the completion of PORTRAZZA. Withhold PORTRAZZA for Grade 3 or 4 electrolyte abnormalities; subsequent cycles of PORTRAZZA may be administered in these patients once hypomagnesemia and related electrolyte abnormalities have improved to Grade ≤2. Replete electrolytes as medically appropriate [see Boxed Warning, Warnings and Precautions (5.1), and Adverse Reactions (6.1)].
5.3 Venous and Arterial Thromboembolic Events
Venous and arterial thromboembolic events (VTE and ATE), some fatal, were observed with PORTRAZZA in combination with gemcitabine and cisplatin. In Study 1, the incidence of VTE was 9% in patients receiving PORTRAZZA plus gemcitabine and cisplatin versus 5% in patients receiving gemcitabine and cisplatin alone and the incidence of Grade 3 or higher VTE was 5% versus 3%, respectively. The incidence of fatal VTEs was similar between arms (0.2% versus 0.2%). The most common VTEs were pulmonary embolism (5%) and deep-vein thrombosis (2%).
The incidence of ATEs of any grade was 5% versus 4% and the incidence of Grade 3 or higher ATE was 4% versus 2% in the PORTRAZZA containing and gemcitabine and cisplatin arms, respectively, in Study 1. The most common ATEs were cerebral stroke and ischemia (2%) and myocardial infarction (1%).
In an exploratory analysis of Study 1, the relative risk of VTE or ATE was approximately 3-fold higher in patients with a reported history of VTE or ATE than in patients with no reported history of VTE or ATE.
Discontinue PORTRAZZA for patients with serious or life threatening VTE or ATE.
5.4 Dermatologic Toxicities
Dermatologic toxicities, including rash, dermatitis acneiform, acne, dry skin, pruritus, generalized rash, skin fissures, maculo-papular rash and erythema, occurred in 79% of patients receiving PORTRAZZA in Study 1. Skin toxicity was severe in 8% of patients. Skin toxicity usually developed within the first 2 weeks of therapy and resolved within 17 weeks after onset. For Grade 3 skin reactions, modify the dose of PORTRAZZA [see Dosage and Administration (2.3) and Adverse Reactions (6.1)]. Limit sun exposure [see Patient Counseling Information (17)].
Discontinue PORTRAZZA for severe (Grade 4) skin reactions, or for Grade 3 skin induration/fibrosis.
5.5 Infusion-Related Reactions
In Study 1, 1.5% of PORTRAZZA treated patients experienced IRRs of any severity with 0.4% Grade 3 IRR. No patients received premedication for IRR for the first dose of PORTRAZZA in Study 1. Most IRRs occurred after the first or second administration of PORTRAZZA. Monitor patients during and following PORTRAZZA infusion for signs and symptoms of IRR. Discontinue PORTRAZZA for serious or life-threatening IRR [see Dosage and Administration (2.3) and Adverse Reactions (6.1)].
5.6 Non-Squamous NSCLC - Increased Toxicity and Increased Mortality
PORTRAZZA is not indicated for the treatment of patients with non-squamous NSCLC. In a study of PORTRAZZA plus pemetrexed and cisplatin (PC) versus PC alone (Study 2), patients treated with PORTRAZZA and PC experienced more serious (51% versus 41%) and fatal toxicities (16% versus 10%) and cardiopulmonary arrest/sudden death within 30 days of the last study drug (3.3% versus 1.3%) compared to patients who received PC alone [see Clinical Studies (14.2)].
5.7 Embryo-Fetal Toxicity
Based on animal data and its mechanism of action, PORTRAZZA can cause fetal harm when administered to a pregnant woman. Disruption or depletion of EGFR in animal models results in impairment of embryofetal development including effects on placental, lung, cardiac, skin, and neural development. The absence of EGFR signaling has resulted in embryolethality as well as post-natal death in animals. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with PORTRAZZA and for three months following the final dose [see Use in Specific Populations (8.1, 8.3) and Clinical Pharmacology (12.1)].
6 ADVERSE REACTIONS
The following adverse drug reactions are discussed in greater detail in other sections of the label:
- Cardiopulmonary Arrest [see Boxed Warning and Warnings and Precautions (5.1)].
- Hypomagnesemia [see Boxed Warning and Warnings and Precautions (5.2)].
- Venous and Arterial Thromboembolic Events [see Warnings and Precautions (5.3)].
- Dermatologic Toxicities [see Dosage and Administration (2.3) and Warnings and Precautions (5.4)].
- Infusion-Related Reactions [see Dosage and Administration (2.2, 2.3) and Warnings and Precautions (5.5)].
- Non-Squamous NSCLC - Increased Toxicity and Increased Mortality [see Warnings and Precautions (5.6) and Clinical Studies (14.2)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of PORTRAZZA was evaluated in two randomized, open-label trials comparing PORTRAZZA plus gemcitabine and cisplatin to gemcitabine and cisplatin alone in patients with squamous NSCLC (Study 1), and PORTRAZZA plus pemetrexed and cisplatin to pemetrexed and cisplatin alone in patients with non-squamous NSCLC (Study 2). Since the data in Study 2 demonstrated similar incidence of adverse reactions over control as observed in Study 1, the safety data from Study 1 alone is described below.
For patients who received at least 1 dose of treatment in Study 1, the median age was 62 years (range 32 to 84), 83% were male; 84% were Caucasian; and 92% were smokers. Baseline ECOG performance status was 0 or 1 for 91%, and 2 for 9% of patients; 90% had metastatic disease in 2 or more sites. Patients received PORTRAZZA 800 mg intravenously on days 1 and 8 of each 21 day cycle in combination with up to six cycles of gemcitabine (1250 mg/m2 on days 1 and 8) and cisplatin (75 mg/m2 on day 1). Patients received PORTRAZZA until progressive disease or unacceptable toxicity.
Patients in the gemcitabine and cisplatin alone arm received a maximum of 6 cycles, while patients in the PORTRAZZA plus gemcitabine and cisplatin arm demonstrating at least stable disease were permitted to continue to receive additional cycles of PORTRAZZA until disease progression or unacceptable toxicity. The median duration of exposure to PORTRAZZA in 538 patients who received at least 1 dose of treatment in Study 1 was 4.6 months (range 0.5 months to 34 months), including 182 patients exposed for at least 6 months and 41 patients exposed for greater than 1 year. Patients were monitored for safety until 30 days after treatment discontinuation and resolution of treatment-emergent adverse events.
The most common adverse reactions (all grades) observed in PORTRAZZA-treated patients at a rate of ≥15% and ≥2% higher than gemcitabine and cisplatin alone were rash (44%), vomiting (29%), diarrhea (16%), and dermatitis acneiform (15%). The most common severe (Grade 3 or higher) adverse events that occurred at a ≥2% higher rate in PORTRAZZA-treated patients compared to patients treated with gemcitabine and cisplatin alone were venous thromboembolic events (5%; including pulmonary embolism), rash (4%), and vomiting (3%).
Table 1 contains selected adverse drug reactions observed in Study 1 at an incidence of ≥5% in the PORTRAZZA arm and at ≥2% higher incidence than the control arm.
|
a Pulmonary embolism is also included in the composite term venous thromboembolic events under system organ class vascular disorders. |
||||
|
b VTE is a composite term which includes: pulmonary embolism, deep vein thrombosis, thrombosis, mesenteric veins thrombosis, pulmonary artery thrombosis, pulmonary venous thrombosis, venous thrombosis limb, axillary vein thrombosis, thrombophlebitis, thrombosis in device, vena cava thrombosis, venous thrombosis, subclavian vein thrombosis, superior vena cava syndrome, and thrombophlebitis superficial. |
||||
|
c Conjunctivitis is a composite term that includes conjunctivitis, eye irritation, vision blurred, conjunctivitis bacterial, dry eye, visual acuity reduced, blepharitis, allergic blepharitis, conjunctiva hemorrhage, eye infection, eye pain, lacrimation increased, ocular hyperemia, Sjogren's syndrome, visual impairment, and eye pruritus. |
||||
| Adverse Reactions (MedDRA)
System Organ Class | PORTRAZZA PLUS GEMCITABINE AND CISPLATIN
N=538 (%) | GEMCITABINE AND CISPLATIN
N=541 (%) |
||
| All Grades
(Frequency %) | Grade 3-4
(Frequency %) | All Grades
(Frequency %) | Grade 3-4
(Frequency %) |
|
| Skin and Subcutaneous Tissue Disorders | ||||
| Rash | 44 | 4 | 6 | 0.2 |
| Dermatitis Acneiform | 15 | 1 | 0.6 | 0 |
| Acne | 9 | 0.4 | 0.6 | 0 |
| Pruritus | 7 | 0.2 | 0.9 | 0.2 |
| Dry Skin | 7 | 0 | 1 | 0 |
| Skin fissures | 5 | 0.4 | 0 | 0 |
| Gastrointestinal Disorders | ||||
| Vomiting | 29 | 3 | 25 | 0.9 |
| Diarrhea | 16 | 2 | 11 | 1 |
| Stomatitis | 11 | 1 | 6 | 0.6 |
| Investigations | ||||
| Weight decreased | 13 | 0.7 | 6 | 0.6 |
| Respiratory, Thoracic and Mediastinal Disorders | ||||
| Hemoptysis | 10 | 1 | 5 | 0.9 |
| Pulmonary embolisma | 5 | 4 | 2 | 2 |
| Nervous System Disorders | ||||
| Headache | 11 | 0 | 6 | 0.4 |
| Vascular Disorders | ||||
| Venous Thromboembolic Events (VTE)b | 9 | 5 | 5 | 3 |
| Infections and Infestations | ||||
| Paronychia | 7 | 0.4 | 0.2 | 0 |
| Eye Disorders | ||||
| Conjunctivitisc | 7 | 0.4 | 2 | 0 |
Clinically relevant adverse reactions (all grades) reported in ≥1% and <5% of patients treated with PORTRAZZA were: dysphagia (3%), oropharyngeal pain (1%), muscle spasms (2%), phlebitis (2%), and hypersensitivity/IRR (1.5%).
In Study 1, 12% of the patients on the PORTRAZZA arm discontinued study treatment due to an adverse reaction. The most common PORTRAZZA related toxicity leading to PORTRAZZA discontinuation was skin rash (1%).
Table 2 contains selected electrolyte abnormalities observed in Study 1 according to laboratory assessment at an incidence of >10% in the PORTRAZZA arm and at >2% higher incidence than the control arm.
The median time to onset of hypomagnesemia was 6 weeks (25th percentile 4 weeks; 75th percentile 9 weeks). Hypomagnesemia was reported as resolved in 43% of the patients who received PORTRAZZA. In Study 1, 32% of the patients in the PORTRAZZA arm and 16% of the patients who received gemcitabine and cisplatin alone received magnesium replacement.
|
a Only patients with baseline and at least one post-baseline result are included. |
||||||
| LABORATORY PARAMETER | PORTRAZZA PLUS GEMCITABINE AND CISPLATIN
N=538 | GEMCITABINE AND CISPLATIN
N=541 |
||||
| Na
| All Grades
(Frequency %) | Grade 3 or 4
(Frequency %) | Na
| All Grades
(Frequency %) | Grade 3 or 4
(Frequency %) |
|
| Hypomagnesemia | 461 | 83 | 20 | 457 | 70 | 7 |
| Hypokalemia | 505 | 28 | 5 | 505 | 18 | 3 |
| Hypocalcemia | 502 | 45 | 6 | 499 | 30 | 2 |
| Hypocalcemia (albumin corrected) | 477 | 36 | 4 | 480 | 23 | 2 |
| Hypophosphatemia | 462 | 31 | 8 | 454 | 23 | 6 |
6.2 Immunogenicity
As with all therapeutic proteins, there is the potential for immunogenicity. In clinical trials, treatment-emergent anti-necitumumab antibodies (ADA) were detected in 4.1% (33/814) of patients using an enzyme-linked immunosorbent assay (ELISA). Neutralizing antibodies were detected in 1.4% (11/814) of patients post exposure to PORTRAZZA. No relationship was found between the presence of ADA and incidence of infusion-related reactions. The impact of ADA on efficacy (overall survival) could not be assessed due to the limited number of patients with treatment-emergent ADA. In Study 1, the exposure to necitumumab was lower in patients with ADA post-treatment than in patients without detectable ADA [see Clinical Pharmacology (12.3)].
The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of incidence of antibodies to PORTRAZZA with the incidences of antibodies to other products may be misleading.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on animal data and its mechanism of action, PORTRAZZA can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. Disruption or depletion of EGFR in animal models results in impairment of embryo-fetal development including effects on placental, lung, cardiac, skin, and neural development. The absence of EGFR signaling has resulted in embryolethality as well as post-natal death in animals (see Data). No animal reproduction studies have been conducted with necitumumab. There are no available data for PORTRAZZA exposure in pregnant women. Advise pregnant women of the potential risk to a fetus, and the risk to postnatal development.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Animal Data
No animal studies have been conducted to evaluate the effect of necitumumab on reproduction and fetal development; however, based on its mechanism of action, PORTRAZZA can cause fetal harm or developmental anomalies. In mice, EGFR is critically important in reproductive and developmental processes including blastocyst implantation, placental development, and embryo-fetal/postnatal survival and development. Reduction or elimination of embryo-fetal or maternal EGFR signaling can prevent implantation, can cause embryo-fetal loss during various stages of gestation (through effects on placental development) and can cause developmental anomalies and early death in surviving fetuses. Adverse developmental outcomes were observed in multiple organs in embryos/neonates of mice with disrupted EGFR signaling. Human IgG1 is known to cross the placenta; therefore, necitumumab has the potential to be transmitted from the mother to the developing fetus.
In monkeys, administration of a chimeric anti-EGFR antibody that binds to an epitope overlapping that of necitumumab during the period of organogenesis resulted in detectable exposure of the antibody in the amniotic fluid and in the serum of embryos from treated dams. While no fetal malformations or other clear teratogenic effects occurred in offspring, there was an increased incidence of embryolethality and abortions.
8.2 Lactation
Risk Summary
There is no information regarding the presence of necitumumab in human milk, the effects on the breastfed infant, or the effects on milk production. Because of the potential for serious adverse reactions in breastfed infants from PORTRAZZA, advise a nursing woman not to breastfeed during treatment with PORTRAZZA and for three months following the final dose.
8.3 Females and Males of Reproductive Potential
Females
Based on its mechanism of action, PORTRAZZA can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception during treatment with PORTRAZZA and for three months following the final dose.
8.4 Pediatric Use
The safety and effectiveness of PORTRAZZA have not been established in pediatric patients.
8.5 Geriatric Use
Of the 545 patients in the PORTRAZZA plus gemcitabine and cisplatin arm in Study 1, 213 (39%) were 65 years and over, while 108 (20%) were 70 years and over. In an exploratory subgroup analysis of Study 1, the hazard ratio for overall survival in patients 70 years or older was 1.03 (95% CI: 0.75, 1.42). Of the adverse reactions listed in Table 1 [see Adverse Reactions (6.1)], there was a higher incidence (≥3%) of venous thromboembolic events including pulmonary embolism in patients age 70 and over compared to those who were younger than age 70.
8.6 Renal Impairment
No formal studies have been conducted to evaluate the effect of renal impairment on the exposure to necitumumab. Renal function has no influence on the exposure to necitumumab based on the population pharmacokinetic analysis of data from clinical trials [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
No formal studies have been conducted to evaluate the effect of hepatic impairment on the exposure to necitumumab. Mild or moderate hepatic impairment has no influence on the exposure to necitumumab based on the population pharmacokinetic analysis. No patients with severe hepatic impairment were enrolled in the clinical trials with PORTRAZZA [see Clinical Pharmacology (12.3)].
10 OVERDOSAGE
There has been limited experience with PORTRAZZA overdose in human clinical trials.
The highest dose of PORTRAZZA studied clinically in a human dose-escalation Phase 1 study was 1000 mg once a week and once every other week. Two out of 9 patients in the every other week cohort experienced dose-limiting toxicities (e.g., a combination of Grade 3 headache, vomiting, and nausea). There is no known antidote for PORTRAZZA overdose.
11 DESCRIPTION
Necitumumab is an anti-EGFR recombinant human monoclonal antibody of the IgG1 kappa isotype that specifically binds to the ligand binding site of the human EGFR. Necitumumab has an approximate molecular weight of 144.8 kDa. Necitumumab is produced in genetically engineered mammalian NS0 cells.
PORTRAZZA is a sterile, preservative free, clear to slightly opalescent and colorless to slightly yellow solution. PORTRAZZA is available in single-dose vials for intravenous infusion following dilution. Each vial contains 800 mg PORTRAZZA in 50 mL (16 mg/mL).
Each mL contains necitumumab (16 mg), citric acid anhydrous (0.256 mg), glycine (9.984 mg), mannitol (9.109 mg), polysorbate 80 (0.1 mg), sodium chloride (2.338 mg), sodium citrate dihydrate (2.55 mg), and water for injection, pH 6.0.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Necitumumab is a recombinant human lgG1 monoclonal antibody that binds to the human epidermal growth factor receptor (EGFR) and blocks the binding of EGFR to its ligands. Expression and activation of EGFR has been correlated with malignant progression, induction of angiogenesis, and inhibition of apoptosis. Binding of necitumumab induces EGFR internalization and degradation in vitro. In vitro, binding of necitumumab also led to antibody-dependent cellular cytotoxicity (ADCC) in EGFR-expressing cells.
In in vivo studies using xenograft models of human cancer, including non-small cell lung carcinoma, administration of necitumumab to implanted mice resulted in increased antitumor activity in combination with gemcitabine and cisplatin as compared to mice receiving gemcitabine and cisplatin alone.
12.3 Pharmacokinetics
Based on population pharmacokinetic (popPK) analysis of serum concentration data from patients in clinical studies with PORTRAZZA, necitumumab exhibits dose-dependent kinetics. Following the administration of PORTRAZZA 800 mg on Days 1 and 8 of each 21 day cycle, the estimated mean total systemic clearance (CLtot) at steady state is 14.1 mL/h (CV=39%), the steady state volume of distribution (Vss) is 7.0 L (CV=31%) and the elimination half-life is approximately 14 days. The predicted time to reach steady state is approximately 100 days.
Effect of Age, Body Weight, Sex and Race:
Based on the popPK analysis with data obtained in 807 patients, age (range 19-84 years), sex (75% males), and race (85% Whites) have no effect on the systemic exposure of necitumumab.
Body weight is identified as a covariate in the popPK analysis; however, weight-based dosing is not expected to significantly decrease the variability in exposure. No dose adjustment based on body weight is necessary.
Renal Impairment
Patients with Renal Impairment — PopPK analysis did not identify a correlation between necitumumab exposure and renal function as assessed by estimated creatinine clearance ranging from 11-250 mL/min.
Hepatic Impairment
Patients with Hepatic Impairment — PopPK analysis did not identify a correlation between the exposure of necitumumab and hepatic function as assessed by alanine aminotransferase (ranging from 2-615 U/L), aspartate transaminase (ranging from 1.2-619 U/L) and total bilirubin (ranging from 0.1-106 μmol/L).
Effect of Necitumumab on Gemcitabine and Cisplatin
In 12 patients with advanced solid tumors who received gemcitabine and cisplatin in combination with PORTRAZZA, the geometric mean dose-normalized AUC of gemcitabine was increased by 22% and Cmax increased by 63% compared to administration of gemcitabine and cisplatin alone while exposure to cisplatin was unchanged.
14 CLINICAL STUDIES
14.1 Squamous Non-Small Cell Lung Cancer
Study 1 was a randomized, multi-center open-label, controlled trial conducted in 1093 patients receiving gemcitabine and cisplatin first-line chemotherapy for metastatic squamous NSCLC. Patients were randomized (1:1) to receive PORTRAZZA plus gemcitabine and cisplatin or gemcitabine and cisplatin alone. Stratification factors were ECOG performance status (0, 1 versus 2) and geographic region (North America, Europe, and Australia versus South America, South Africa, and India versus Eastern Asia). Gemcitabine (1250 mg/m2, Days 1 and 8) plus cisplatin (75 mg/m2, Day 1) were administered every 3 weeks (1 cycle) for a maximum of 6 cycles in the absence of disease progression or unacceptable toxicity. PORTRAZZA (800 mg by intravenous infusion on Days 1 and 8 of each 3-week cycle) was administered prior to gemcitabine and cisplatin. Patients demonstrating at least stable disease on PORTRAZZA plus gemcitabine and cisplatin were to continue PORTRAZZA as a single agent in the absence of disease progression or unacceptable toxicity after completion of 6 planned courses of chemotherapy or if chemotherapy was discontinued for toxicity.
Of the 1093 randomized patients, the median age was 62 years (range 32 to 86), 83% were male; 84% were Caucasian; and 91% were smokers. The majority of the patients (87%) were enrolled in North America, Europe and Australia, 36 patients (3%) were enrolled at clinical sites in the U.S., 6% of the patients were enrolled in South America, South Africa and India and 8% enrolled at clinical sites in Eastern Asia. Baseline ECOG performance status was 0 or 1 for 91%, and 2 for 9% of patients; 91% had metastatic disease in 2 or more sites. In the PORTRAZZA plus gemcitabine and cisplatin arm, 51% of patients continued PORTRAZZA after completion or discontinuation of chemotherapy. Use of post-study systemic therapy was 47% in the PORTRAZZA plus gemcitabine and cisplatin arm, and 45% in the gemcitabine and cisplatin arm.
The main outcome measure was overall survival (OS). Investigator-assessed progression-free survival (PFS) and overall response rate (ORR) were also assessed. Overall survival and PFS were statistically significantly improved in patients randomized to receive PORTRAZZA plus gemcitabine and cisplatin compared to gemcitabine and cisplatin alone. There was no difference in ORR between arms, with an ORR of 31% (95% CI 27, 35) for PORTRAZZA plus gemcitabine and cisplatin arm and an ORR of 29% (95% CI 25, 33) for gemcitabine and cisplatin arm, p-value 0.40.
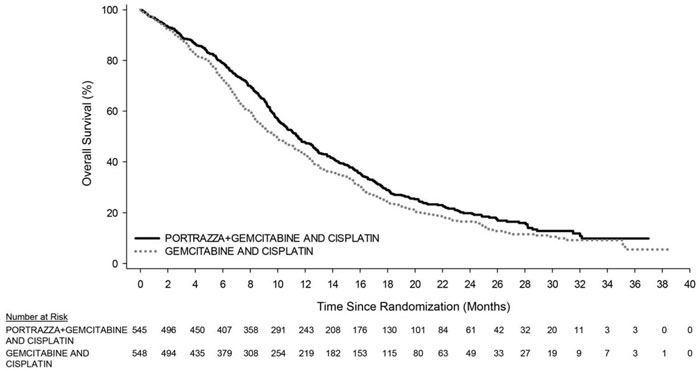
Efficacy results are shown in Table 3 and Figure 1.
|
a Abbreviations: CI = confidence interval |
||
|
b Investigator assessed |
||
| PORTRAZZA PLUS GEMCITABINE AND CISPLATIN
N=545 | GEMCITABINE AND CISPLATIN
N=548 |
|
| Overall Survival | ||
| Number of deaths (%) | 418 (77%) | 442 (81%) |
| Median – months (95% CI)a | 11.5 (10.4, 12.6) | 9.9 (8.9, 11.1) |
| Stratified Hazard Ratio (95% CI) | 0.84 (0.74, 0.96) | |
| Stratified Log-rank p-value | 0.01 | |
| Progression-Free Survivalb | ||
| Number of events (%) | 431 (79%) | 417 (76%) |
| Median – months (95% CI) | 5.7 (5.6, 6.0) | 5.5 (4.8, 5.6) |
| Stratified Hazard Ratio (95% CI) | 0.85 (0.74, 0.98) | |
| Stratified Log-rank p-value | 0.02 | |
Figure 1: Kaplan-Meier Curves of Overall Survival in Patients with Metastatic Squamous Non-Small Cell Lung Cancer
14.2 Non-Squamous NSCLC - Lack of Efficacy
Lack of efficacy of PORTRAZZA in combination with pemetrexed and cisplatin for the treatment of patients with metastatic non-squamous non-small cell lung cancer was determined in one randomized, open-label, multicenter trial (Study 2). The study was closed prematurely after 633 patients were enrolled due to increased incidence of death due to any cause and of thromboembolic events in the PORTRAZZA arm. Patients with no prior chemotherapy for metastatic disease were randomized (1:1) to receive PORTRAZZA plus pemetrexed and cisplatin or pemetrexed and cisplatin alone. Stratification factors were smoking status (non-smokers versus light smokers versus smokers), ECOG performance status (0 - 1 versus 2), histology (adenocarcinoma/large cell versus others), and geographic region. PORTRAZZA (800 mg, Days 1 and 8 of each 3-week cycle) was administered prior to pemetrexed and cisplatin. Patients demonstrating at least stable disease on PORTRAZZA plus pemetrexed and cisplatin were to continue PORTRAZZA as a single agent in the absence of disease progression or unacceptable toxicity after completion of 6 planned courses of chemotherapy.
Of the 633 patients, 315 were randomized to PORTRAZZA plus pemetrexed and cisplatin arm and 318 in the pemetrexed and cisplatin arm. The median age was 61 years, 67 % were male, 93% were Caucasian and 94% had ECOG PS 0 or 1. More than 75% were smokers and 89% had adenocarcinoma histology.
The main efficacy outcome was OS. Progression-free survival and ORR were also assessed. Addition of PORTRAZZA to pemetrexed and cisplatin did not improve OS [HR=1.01; 95%CI (0.84, 1.21); p-value = 0.96)]; PFS [HR=0.96; 95% CI (0.8, 1.16)] or ORR (31% in the PORTRAZZA plus pemetrexed and cisplatin arm and 32% in the pemetrexed and cisplatin alone arm).
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
Hypomagnesemia
Advise patients of risk of decreased blood levels of magnesium, potassium and calcium. Take medicines to replace the electrolytes exactly as advised by the physician. [see Boxed Warning and Warnings and Precautions (5.2)]
Venous and Arterial Thromboembolic Events
Advise patients of increased risk of venous and arterial thromboembolic events [see Warnings and Precautions (5.3)].
Skin reactions
Advise patients to minimize sun exposure with protective clothing and use of sunscreen while receiving PORTRAZZA [see Dosage and Administration (2.3) and Warnings and Precautions (5.4)].
Infusion-Related Reactions
Advise patients to report signs and symptoms of infusion reactions such as fever, chills, or breathing problems [see Warnings and Precautions (5.5)].
Embryo-Fetal Toxicity
Advise pregnant women of the potential risk to a fetus [see Use in Specific Populations (8.1)].
Advise females of reproductive potential to use effective contraception during treatment with PORTRAZZA and for three months following final dose [see Use in Specific Populations (8.3)].
Lactation
Advise women not to breastfeed during treatment with PORTRAZZA and for three months following the final dose [see Use in Specific Populations (8.2)].
Literature issued November 2015
Manufactured by: Eli Lilly and Company, Indianapolis, IN 46285
US License No. 1891
Marketed by: Lilly USA, LLC, Indianapolis, IN 46285, USA
Copyright © 2015, Eli Lilly and Company. All rights reserved.
POR-0001-USPI-20151124
