FULL PRESCRIBING INFORMATION
WARNING: RISK OF SERIOUS CARDIOVASCULAR AND GASTROINTESTINAL EVENTS
Cardiovascular Thrombotic Events
- Nonsteroidal anti-inflammatory drugs (NSAIDs) cause an increased risk of serious cardiovascular thrombotic events, including myocardial infarction, and stroke, which can be fatal. This risk may occur early in the treatment and may increase with duration of use. [ see Warnings and Precautions (5.1)]
- Celecoxib is contraindicated in the setting of coronary artery bypass graft (CABG) surgery. [ see Contraindications (4) and Warnings and Precautions (5.1)]
Gastrointestinal Bleeding, Ulceration, and Perforation
- NSAIDs cause an increased risk of serious gastrointestinal (GI) adverse events including bleeding, ulceration, and perforation of the stomach or intestines, which can be fatal. These events can occur at any time during use and without warning symptoms. Elderly patients and patients with a prior history of peptic ulcer disease and/or GI bleeding are at greater risk for serious GI events. [ see Warnings and Precautions (5.2)]
1. INDICATIONS AND USAGE
Celecoxib capsules are indicated
1.1 Osteoarthritis (OA)
For the management of the signs and symptoms of OA [ see Clinical Studies (14.1)]
1.2 Rheumatoid Arthritis (RA)
For the management of the signs and symptoms of RA [ see Clinical Studies (14.2)]
1.3 Juvenile Rheumatoid Arthritis (JRA)
For the management of the signs and symptoms of JRA in patients 2 years and older [ see Clinical Studies (14.3)]
1.4 Ankylosing Spondylitis (AS)
For the management of the signs and symptoms of AS [ see Clinical Studies (14.4)]
2. DOSAGE AND ADMINISTRATION
2.1 General Dosing Instructions
Carefully consider the potential benefits and risks of celecoxib capsules and other treatment options before deciding to use celecoxib capsules. Use the lowest effective dosage for the shortest duration consistent with individual patient treatment goals [ see Warnings and Precautions (5)].
These doses can be given without regard to timing of meals.
2.2 Osteoarthritis
For OA, the dosage is 200 mg per day administered as a single dose or as 100 mg twice daily.
2.4 Juvenile Rheumatoid Arthritis
For JRA, the dosage for pediatric patients (age 2 years and older) is based on weight. For patients ≥10 kg to ≤25 kg the recommended dose is 50 mg twice daily. For patients >25 kg the recommended dose is 100 mg twice daily.
For patients who have difficulty swallowing capsules, the contents of a celecoxib capsule can be added to applesauce. The entire capsule contents are carefully emptied onto a level teaspoon of cool or room temperature applesauce and ingested immediately with water. The sprinkled capsule contents on applesauce are stable for up to 6 hours under refrigerated conditions (2 °C to 8 °C/35 °F to 45°F).
2.5 Ankylosing Spondylitis
For AS, the dosage of celecoxib capsules is 200 mg daily in single (once per day) or divided (twice per day) doses. If no effect is observed after 6 weeks, a trial of 400 mg daily may be worthwhile. If no effect is observed after 6 weeks on 400 mg daily, a response is not likely and consideration should be given to alternate treatment options.
2.6 Management of Acute Pain and Treatment of Primary Dysmenorrhea
For management of Acute Pain and Treatment of Primary Dysmenorrhea, the dosage is 400 mg initially, followed by an additional 200 mg dose if needed on the first day. On subsequent days, the recommended dose is 200 mg twice daily as needed.
2.7 Special Populations
Hepatic Impairment
In patients with moderate hepatic impairment (Child-Pugh Class B), reduce the dose by 50%. The use of celecoxib capsules in patients with severe hepatic impairment is not recommended [ see Warnings and Precautions (5.5), Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)].
Poor Metabolizers of CYP2C9 Substrates
In adult patients who are known or suspected to be poor CYP2C9 metabolizers based on genotype or previous history/experience with other CYP2C9 substrates (such as warfarin, phenytoin), initiate treatment with half of the lowest recommended dose.
In patients with JRA who are known or suspected to be poor CYP2C9 metabolizers, consider using alternative treatments. [ see Use in Specific populations (8.8), and Clinical Pharmacology (12.5)] .
3. DOSAGE FORMS AND STRENGTHS
Celecoxib capsules:
50 mg are 4# white capsules, with reverse printed white on red band of body and cap with markings of 0006 on the cap and 50 mg on the body.
100 mg are 2# white capsules, with reverse printed white on blue band of body and cap with markings of 0007 on the cap and 100 mg on the body.
200 mg are 2# white capsules, with reverse printed white on gold band of body and cap with markings of 0008 on the cap and 200 mg on the body.
400 mg are 0# white capsules, with reverse printed white on green band of body and cap with markings of 0009 on the cap and 400 mg on the body.
4. CONTRAINDICATIONS
Celecoxib capsules are contraindicated in the following patients:
- Known hypersensitivity (e.g., anaphylactic reactions and serious skin reactions) to celecoxib, any components of the drug product [see Warnings and Precautions (5.7, 5.9)].
- History of asthma, urticaria, or other allergic-type reactions after taking aspirin or other NSAIDs. Severe, sometimes fatal, anaphylactic reactions to NSAIDs, have been reported in such patients [ see Warnings and Precautions (5.7, 5.8)].
- In the setting of CABG surgery [ see Warnings and Precautions (5.1)].
- In patients who have demonstrated allergic-type reactions to sulfonamides.
5. WARNINGS AND PRECAUTIONS
5.1 Cardiovascular Thrombotic Events
Clinical trials of several cyclooxygenase-2 (COX-2) selective and nonselective NSAIDs of up to three years duration have shown an increased risk of serious cardiovascular (CV) thrombotic events, including myocardial infarction (MI) and stroke, which can be fatal. Based on available data, it is unclear that the risk for CV thrombotic events is similar for all NSAIDs. The relative increase in serious CV thrombotic events over baseline conferred by NSAID use appears to be similar in those with and without known CV disease or risk factors for CV disease. However, patients with known CV disease or risk factors had a higher absolute incidence of excess serious CV thrombotic events, due to their increased baseline rate. Some observational studies found that this increased risk of serious CV thrombotic events began as early as the first weeks of treatment. The increase in CV thrombotic risk has been observed most consistently at higher doses.
In the APC (Adenoma Prevention with Celecoxib) trial, there was about a threefold increased risk of the composite endpoint of cardiovascular death, MI, or stroke for the celecoxib 400 mg twice daily and celecoxib 200 mg twice daily treatment arms compared to placebo. The increases in both celecoxib dose groups versus placebo-treated patients were mainly due to an increased incidence of myocardial infarction [ see Clinical Studies (14.7)].
A randomized controlled trial entitled the Prospective Randomized Evaluation of Celecoxib Integrated Safety vs. Ibuprofen Or Naproxen (PRECISION) was conducted to assess the relative cardiovascular thrombotic risk of a COX-2 inhibitor, celecoxib, compared to the non-selective NSAIDs naproxen and ibuprofen. Celecoxib 100 mg twice daily was non-inferior to naproxen 375 to 500 mg twice daily and ibuprofen 600 to 800 mg three times daily for the composite endpoint of the Antiplatelet Trialists’ Collaboration (APTC), which consists of cardiovascular death (including hemorrhagic death), non-fatal myocardial infarction, and non-fatal stroke [ See Clinical Studies (14.6)].
To minimize the potential risk for an adverse CV event in NSAID-treated patients, use the lowest effective dose for the shortest duration possible. Physicians and patients should remain alert for the development of such events, throughout the entire treatment course, even in the absence of previous CV symptoms. Patients should be informed about the symptoms of serious CV events and the steps to take if they occur.
There is no consistent evidence that concurrent use of aspirin mitigates the increased risk of serious CV thrombotic events associated with NSAID use. The concurrent use of aspirin and an NSAID, such as celecoxib, increases the risk of serious gastrointestinal (GI) events [ see Warnings and Precautions (5.2)].
Status Post Coronary Artery Bypass Graft (CABG) Surgery
Two large, controlled clinical trials of a COX-2 selective NSAID for the treatment of pain in the first 10 to 14 days following CABG surgery found an increased incidence of myocardial infarction and stroke. NSAIDs are contraindicated in the setting of CABG [ see Contraindications (4)].
Post-MI Patients
Observational studies conducted in the Danish National Registry have demonstrated that patients treated with NSAIDs in the post-MI period were at increased risk of reinfarction, CV-related death, and all-cause mortality beginning in the first week of treatment. In this same cohort, the incidence of death in the first year post-MI was 20 per 100 person years in NSAID-treated patients compared to 12 per 100 person years in non-NSAID exposed patients. Although the absolute rate of death declined somewhat after the first year post-MI, the increased relative risk of death in NSAID users persisted over at least the next four years of follow-up.
Avoid the use of celecoxib in patients with a recent MI unless the benefits are expected to outweigh the risk of recurrent CV thrombotic events. If celecoxib is used in patients with a recent MI, monitor patients for signs of cardiac ischemia.
5.2 Gastrointestinal Bleeding, Ulceration, and Perforation
NSAIDs, including celecoxib cause serious gastrointestinal (GI) adverse events including inflammation, bleeding, ulceration, and perforation of the esophagus, stomach, small intestine, or large intestine, which can be fatal. These serious adverse events can occur at any time, with or without warning symptoms, in patients treated with celecoxib. Only one in five patients who develop a serious upper GI adverse event on NSAID therapy is symptomatic. Upper GI ulcers, gross bleeding, or perforation caused by NSAIDs occurred in approximately 1% of patients treated for 3 to 6 months, and in about 2% to 4% of patients treated for one year. However, even short-term NSAID therapy is not without risk.
Risk Factors for GI Bleeding, Ulceration, and Perforation
Patients with a prior history of peptic ulcer disease and/or GI bleeding who used NSAIDs had a greater than 10-fold increased risk for developing a GI bleed compared to patients without these risk factors. Other factors that increase the risk of GI bleeding in patients treated with NSAIDs include longer duration of NSAID therapy; concomitant use of oral corticosteroids, antiplatelet drugs (such as aspirin), anticoagulants; or selective serotonin reuptake inhibitors (SSRIs); smoking; use of alcohol; older age; and poor general health status. Most postmarketing reports of fatal GI events occurred in elderly or debilitated patients. Additionally, patients with advanced liver disease and/or coagulopathy are at increased risk for GI bleeding.
Complicated and symptomatic ulcer rates were 0.78% at nine months for all patients in the CLASS trial, and 2.19% for the subgroup on low-dose ASA. Patients 65 years of age and older had an incidence of 1.40% at nine months, 3.06% when also taking ASA [ see Clinical Studies (14.7)].
Strategies to Minimize the GI Risks in NSAID-treated patients:
- Use the lowest effective dosage for the shortest possible duration.
- Avoid administration of more than one NSAID at a time.
- Avoid use in patients at higher risk unless benefits are expected to outweigh the increased risk of bleeding. For such patients, as well as those with active GI bleeding, consider alternate therapies other than NSAIDs.
- Remain alert for signs and symptoms of GI ulceration and bleeding during NSAID therapy.
- If a serious GI adverse event is suspected, promptly initiate evaluation and treatment, and discontinue celecoxib until a serious GI adverse event is ruled out.
- In the setting of concomitant use of low-dose aspirin for cardiac prophylaxis, monitor patients more closely for evidence of GI bleeding [ see Drug Interactions (7)].
5.3 Hepatotoxicity
Elevations of ALT or AST (three or more times the upper limit of normal [ULN]) have been reported in approximately 1% of NSAID-treated patients in clinical trials. In addition, rare, sometimes fatal, cases of severe hepatic injury, including fulminant hepatitis, liver necrosis, and hepatic failure have been reported.
Elevations of ALT or AST (less than three times ULN) may occur in up to 15% of patients treated with NSAIDs including celecoxib.
In controlled clinical trials of celecoxib, the incidence of borderline elevations (greater than or equal to 1.2 times and less than 3 times the upper limit of normal) of liver associated enzymes was 6% for celecoxib and 5% for placebo, and approximately 0.2% of patients taking celecoxib and 0.3% of patients taking placebo had notable elevations of ALT and AST.
Inform patients of the warning signs and symptoms of hepatotoxicity (e.g., nausea, fatigue, lethargy, diarrhea, pruritus, jaundice, right upper quadrant tenderness, and "flu-like" symptoms). If clinical signs and symptoms consistent with liver disease develop, or if systemic manifestations occur (e.g., eosinophilia, rash), discontinue celecoxib immediately, and perform a clinical evaluation of the patient.
5.4 Hypertension
NSAIDs, including celecoxib capsules can lead to new onset of hypertension or worsening of preexisting hypertension, either of which may contribute to the increased incidence of CV events. Patients taking angiotensin converting enzyme (ACE) inhibitors, thiazide diuretics or loop diuretics may have impaired response to these therapies when taking NSAIDs [ see Drug Interactions (7)].
See Clinical Studies ( 14.6, 14.7) for additional blood pressure data for celecoxib.
Monitor blood pressure (BP) during the initiation of NSAID treatment and throughout the course of therapy.
5.5 Heart Failure and Edema
The Coxib and traditional NSAID Trialists' Collaboration meta-analysis of randomized controlled trials demonstrated an approximately two-fold increase in hospitalizations for heart failure in COX-2 selective-treated patients and nonselective NSAID-treated patients compared to placebo-treated patients. In a Danish National Registry study of patients with heart failure, NSAID use increased the risk of MI, hospitalization for heart failure, and death.
Additionally, fluid retention and edema have been observed in some patients treated with NSAIDs. Use of elecoxib may blunt the CV effects of several therapeutic agents used to treat these medical conditions (e.g., diuretics, ACE inhibitors, or angiotensin receptor blockers [ARBs]) [ see Drug Interactions (7)].
In the CLASS study [ see Clinical Studies (14.7)], the Kaplan-Meier cumulative rates at 9 months of peripheral edema in patients on celecoxib 400 mg twice daily (4-fold and 2-fold the recommended OA and RA doses, respectively), ibuprofen 800 mg three times daily and diclofenac 75 mg twice daily were 4.5%, 6.9% and 4.7%, respectively.
Avoid the use of celecoxib in patients with severe heart failure unless the benefits are expected to outweigh the risk of worsening heart failure. If celecoxib is used in patients with severe heart failure, monitor patients for signs of worsening heart failure.
5.6 Renal Toxicity and Hyperkalemia
Renal Toxicity
Long-term administration of NSAIDs has resulted in renal papillary necrosis and other renal injury.
Renal toxicity has also been seen in patients in whom renal prostaglandins have a compensatory role in the maintenance of renal perfusion. In these patients, administration of an NSAID may cause a dose-dependent reduction in prostaglandin formation and, secondarily, in renal blood flow, which may precipitate overt renal decompensation. Patients at greatest risk of this reaction are those with impaired renal function, dehydration, hypovolemia , heart failure, liver dysfunction, those taking diuretics, ACE inhibitors or the ARBs, and the elderly. Discontinuation of NSAID therapy is usually followed by recovery to the pretreatment state.
No information is available from controlled clinical studies regarding the use of celecoxib in patients with advanced renal disease. The renal effects of celecoxib may hasten the progression of renal dysfunction in patients with preexisting renal disease.
Correct volume status in dehydrated or hypovolemic patients prior to initiating celecoxib. Monitor renal function in patients with renal or hepatic impairment, heart failure, dehydration, or hypovolemia during use of celecoxib [ see Drug Interactions (7)] . Avoid the use of celecoxib in patients with advanced renal disease unless the benefits are expected to outweigh the risk of worsening renal function. If celecoxib is used in patients with advanced renal disease, monitor patients for signs of worsening renal function.
5.7 Anaphylactic Reactions
Celecoxib has been associated with anaphylactic reactions in patients with and without known hypersensitivity to celecoxib and in patients with aspirin sensitive asthma. Celecoxib is a sulfonamide and both NSAIDs and sulfonamides may cause allergic type reactions including anaphylactic symptoms and life-threatening or less severe asthmatic episodes in certain susceptible people [see Contraindications (4) and Warnings and Precautions (5.8)].
Seek emergency help if any anaphylactic reaction occurs.
5.8 Exacerbation of Asthma Related to Aspirin Sensitivity
A subpopulation of patients with asthma may have aspirin-sensitive asthma which may include chronic rhinosinusitis complicated by nasal polyps; severe, potentially fatal bronchospasm; and/or intolerance to aspirin and other NSAIDs. Because cross-reactivity between aspirin and other NSAIDs has been reported in such aspirin-sensitive patients, celecoxib is contraindicated in patients with this form of aspirin sensitivity [ see Contraindications (4)]. When celecoxib is used in patients with preexisting asthma (without known aspirin sensitivity), monitor patients for changes in the signs and symptoms of asthma.
5.9 Serious Skin Reactions
Serious skin reactions have occurred following treatment with celecoxib, including erythema multiforme, exfoliative dermatitis, Stevens-Johnson Syndrome (SJS), toxic epidermal necrolysis (TEN), drug reaction with eosinophilia and systemic symptoms (DRESS), and acute generalized exanthematous pustulosis (AGEP). These serious events may occur without warning and can be fatal.
Inform patients about the signs and symptoms of serious skin reactions, and to discontinue the use of celecoxib at the first appearance of skin rash or any other sign of hypersensitivity. Celecoxib is contraindicated in patients with previous serious skin reactions to NSAIDs [ see Contraindications (4)].
5.10 Premature Closure of Fetal Ductus Arteriosus
Celecoxib may cause premature closure of the ductus arteriosus. Avoid use of NSAIDs, including celecoxib, in pregnant women starting at 30 weeks of gestation (third trimester) [ see Use in Specific Populations (8.1)].
5.11 Hematologic Toxicity
Anemia has occurred in NSAID-treated patients. This may be due to occult or gross blood loss, fluid retention, or an incompletely described effect on erythropoiesis. If a patient treated with celecoxib has any signs or symptoms of anemia, monitor hemoglobin or hematocrit.
In controlled clinical trials the incidence of anemia was 0.6% with celecoxib and 0.4% with placebo. Patients on long-term treatment with celecoxib should have their hemoglobin or hematocrit checked if they exhibit any signs or symptoms of anemia or blood loss.
NSAIDs, including celecoxib, may increase the risk of bleeding events. Co-morbid conditions such as coagulation disorders or concomitant use of warfarin, other anticoagulants, antiplatelet agents (e.g., aspirin), SSRIs and serotonin norepinephrine reuptake inhibitors (SNRIs) may increase this risk. Monitor these patients for signs of bleeding [ see Drug Interactions (7)].
5.12 Masking of Inflammation and Fever
The pharmacological activity of celecoxib in reducing inflammation, and possibly fever, may diminish the utility of diagnostic signs in detecting infections.
5.13 Laboratory Monitoring
Because serious GI bleeding, hepatotoxicity, and renal injury can occur without warning symptoms or signs, consider monitoring patients on long-term NSAID treatment with a CBC and a chemistry profile periodically [ see Warnings and Precautions (5.2, 5.3, 5.6)].
In controlled clinical trials, elevated BUN occurred more frequently in patients receiving celecoxib compared with patients on placebo. This laboratory abnormality was also seen in patients who received comparator NSAIDs in these studies. The clinical significance of this abnormality has not been established.
5.14 Disseminated Intravascular Coagulation (DIC)
Because of the risk of disseminated intravascular coagulation with use of celecoxib in pediatric patients with systemic onset JRA, monitor patients for signs and symptoms of abnormal clotting or bleeding, and inform patients and their caregivers to report symptoms as soon as possible.
6. ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the labeling:
- Cardiovascular Thrombotic Events [ see Warnings and Precautions (5.1)]
- GI Bleeding, Ulceration and Perforation [ see Warnings and Precautions (5.2)]
- Hepatotoxicity [ see Warnings and Precautions (5.3)]
- Hypertension [ see Warnings and Precautions (5.4)]
- Heart Failure and Edema [ see Warnings and Precautions (5.5)]
- Renal Toxicity and Hyperkalemia [ see Warnings and Precautions (5.6)]
- Anaphylactic Reactions [ see Warnings and Precautions (5.7)]
- Serious Skin Reactions [ see Warnings and Precautions (5.9)]
- Hematologic Toxicity [ see Warnings and Precautions (5.11)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice. The adverse reaction information from clinical trials does, however, provide a basis for identifying the adverse events that appear to be related to drug use and for approximating rates.
Of the celecoxib-treated patients in the pre-marketing controlled clinical trials, approximately 4,250 were patients with OA, approximately 2,100 were patients with RA, and approximately 1,050 were patients with post-surgical pain. More than 8,500 patients received a total daily dose of celecoxib of 200 mg (100 mg twice daily or 200 mg once daily) or more, including more than 400 treated at 800 mg (400 mg twice daily). Approximately 3,900 patients received celecoxib at these doses for 6 months or more; approximately 2,300 of these have received it for 1 year or more and 124 of these have received it for 2 years or more.
Pre-marketing Controlled Arthritis Trials
Table 1 lists all adverse events, regardless of causality, occurring in ≥2% of patients receiving celecoxib from 12 controlled studies conducted in patients with OA or RA that included a placebo and/or a positive control group. Since these 12 trials were of different durations, and patients in the trials may not have been exposed for the same duration of time, these percentages do not capture cumulative rates of occurrence.
| CBX
N=4146 | Placebo
N=1864 | NAP
N=1366 | DCF
N=387 | IBU
N=345 |
|
| CBX = Celecoxib 100 mg to 200 mg twice daily or 200 mg once daily; | |||||
| NAP = Naproxen 500 mg twice daily; | |||||
| DCF = Diclofenac 75 mg twice daily; | |||||
| IBU = Ibuprofen 800 mg three times daily. | |||||
| Gastrointestinal | |||||
| Abdominal Pain | 4.1% | 2.8% | 7.7% | 9.0% | 9.0% |
| Diarrhea | 5.6% | 3.8% | 5.3% | 9.3% | 5.8% |
| Dyspepsia | 8.8% | 6.2% | 12.2% | 10.9% | 12.8% |
| Flatulence | 2.2% | 1.0% | 3.6% | 4.1% | 3.5% |
| Nausea | 3.5% | 4.2% | 6.0% | 3.4% | 6.7% |
| Body as a whole | |||||
| Back Pain | 2.8% | 3.6% | 2.2% | 2.6% | 0.9% |
| Peripheral Edema | 2.1% | 1.1% | 2.1% | 1.0% | 3.5% |
| Injury-Accidental | 2.9% | 2.3% | 3.0% | 2.6% | 3.2% |
| Central, Peripheral Nervous system | |||||
| Dizziness | 2.0% | 1.7% | 2.6% | 1.3% | 2.3% |
| Headache | 15.8% | 20.2% | 14.5% | 15.5% | 15.4% |
| Psychiatric | |||||
| Insomnia | 2.3% | 2.3% | 2.9% | 1.3% | 1.4% |
| Respiratory | |||||
| Pharyngitis | 2.3% | 1.1% | 1.7% | 1.6% | 2.6% |
| Rhinitis | 2.0% | 1.3% | 2.4% | 2.3% | 0.6% |
| Sinusitis | 5.0% | 4.3% | 4.0% | 5.4% | 5.8% |
| Upper Respiratory Infection | 8.1% | 6.7% | 9.9% | 9.8% | 9.9% |
| Skin | |||||
| Rash | 2.2% | 2.1% | 2.1% | 1.3% | 1.2% |
In placebo- or active-controlled clinical trials, the discontinuation rate due to adverse events was 7.1% for patients receiving celecoxib and 6.1% for patients receiving placebo. Among the most common reasons for discontinuation due to adverse events in the celecoxib treatment groups were dyspepsia and abdominal pain (cited as reasons for discontinuation in 0.8% and 0.7% of celecoxib patients, respectively). Among patients receiving placebo, 0.6% discontinued due to dyspepsia and 0.6% withdrew due to abdominal pain.
The following adverse reactions occurred in 0.1% to 1.9% of patients treated with celecoxib (100 mg to 200 mg twice daily or 200 mg once daily):
Gastrointestinal: Constipation, diverticulitis, dysphagia, eructation, esophagitis, gastritis, gastroenteritis, gastroesophageal reflux, hemorrhoids, hiatal hernia, melena, dry mouth, stomatitis, tenesmus, vomiting
Cardiovascular: Aggravated hypertension, angina pectoris, coronary artery disorder, myocardial infarction
General: Hypersensitivity, allergic reaction, chest pain, cyst NOS, edema generalized, face edema, fatigue, fever, hot flushes, influenza-like symptoms, pain, peripheral pain
Central, peripheral nervous system: Leg cramps, hypertonia, hypoesthesia, migraine, paresthesia, vertigo
Hearing and vestibular: Deafness, tinnitus
Heart rate and rhythm: Palpitation, tachycardia
Liver and biliary: Hepatic enzyme increased (including SGOT increased, SGPT increased)
Metabolic and nutritional: blood urea nitrogen (BUN) increased, creatine phosphokinase (CPK) increased, hypercholesterolemia, hyperglycemia, hypokalemia, NPN increased, creatinine increased, alkaline phosphatase increased, weight increased
Musculoskeletal: Arthralgia, arthrosis, myalgia, synovitis, tendinitis
Platelets (bleeding or clotting): Ecchymosis, epistaxis, thrombocythemia,
Psychiatric: Anorexia, anxiety, appetite increased, depression, nervousness, somnolence
Hemic: Anemia
Respiratory: Bronchitis, bronchospasm, bronchospasm aggravated, cough, dyspnea, laryngitis, pneumonia
Skin and appendages: Alopecia, dermatitis, photosensitivity reaction, pruritus, rash erythematous, rash maculopapular, skin disorder, skin dry, sweating increased, urticaria
Application site disorders: Cellulitis, dermatitis contact
Urinary: Albuminuria, cystitis, dysuria, hematuria, micturition frequency, renal calculus
The following serious adverse events (causality not evaluated) occurred in < 0.1% of patients:
Cardiovascular: Syncope, congestive heart failure, ventricular fibrillation, pulmonary embolism, cerebrovascular accident, peripheral gangrene, thrombophlebitis
Gastrointestinal: Intestinal obstruction, intestinal per foration, gastrointestinal bleeding, colitis with bleeding, esophageal perforation, pancreatitis, ileus
General: Sepsis, sudden death
Liver and biliary: Cholelithiasis
Hemic and lymphatic: Thrombocytopenia
Nervous: Ataxia, suicide [see Drug Interactions (7.1)]
Renal: Acute renal failure
The Celecoxib Long-Term Arthritis Safety Study [see Special Studies(14.7)]
Hematological Events: The incidence of clinically significant decreases in hemoglobin (>2 g/dL) was lower in patients on celecoxib 400 mg twice daily (0.5%) compared to patients on either diclofenac 75 mg twice daily (1.3%) or ibuprofen 800 mg three times daily 1.9%. The lower incidence of events with celecoxib was maintained with or without aspirin use [ see Clinical Pharmacology (12.2)].
Withdrawals/Serious Adverse Events: Kaplan-Meier cumulative rates at 9 months for withdrawals due to adverse events for celecoxib, diclofenac and ibuprofen were 24%, 29%, and 26%, respectively. Rates for serious adverse events (i.e., causing hospitalization or felt to be life-threatening or otherwise medically significant), regardless of causality, were not different across treatment groups (8%, 7%, and 8%, respectively).
Juvenile Rheumatoid Arthritis Study
In a 12-week, double-blind, active-controlled study, 242 JRA patients 2 years to 17 years of age were treated with Celecoxib or naproxen; 77 JRA patients were treated with celecoxib 3 mg/kg twice daily, 82 patients were treated with celecoxib 6 mg/kg twice daily, and 83 patients were treated with naproxen 7.5 mg/kg twice daily. The most commonly occurring (≥5%) adverse events in celecoxib treated patients were headache, fever (pyrexia), upper abdominal pain, cough, nasopharyngitis, abdominal pain, nausea, arthralgia, diarrhea and vomiting. The most commonly occurring (≥5%) adverse experiences for naproxen-treated patients were headache, nausea, vomiting, fever, upper abdominal pain, diarrhea, cough, abdominal pain, and dizziness (Table 2). Compared with naproxen, celecoxib at doses of 3 and 6 mg/kg twice daily had no observable deleterious effect on growth and development during the course of the 12-week double-blind study. There was no substantial difference in the number of clinical exacerbations of uveitis or systemic features of JRA among treatment groups.
In a 12-week, open-label extension of the double-blind study described above, 202 JRA patients were treated with celecoxib 6 mg/kg twice daily. The incidence of adverse events was similar to that observed during the double-blind study; no unexpected adverse events of clinical importance emerged.
Table 2: Adverse Events Occurring in ≥5% of JRA Patients in Any Treatment Group, by System Organ Class (% of patients with events)
| All Doses Twice Daily | |||
|---|---|---|---|
|
System Organ Class Preferred Term |
Celecoxib 3 mg/kg N = 77 | Celecoxib
6 mg/kg N = 82 | Naproxen
7.5 mg/kg N = 83 |
| Any Event | 64 | 70 | 72 |
| Eye Disorders | 5 | 5 | 5 |
| Gastrointestinal | 26 | 24 | 36 |
| Abdominal pain NOS | 4 | 7 | 7 |
| Abdominal pain upper | 8 | 6 | 10 |
| Vomiting NOS | 3 | 6 | 11 |
| Diarrhea NOS | 5 | 4 | 8 |
| Nausea | 7 | 4 | 11 |
| General | 13 | 11 | 18 |
| Pyrexia | 8 | 9 | 11 |
| Infections | 25 | 20 | 27 |
| Nasopharyngitis | 5 | 6 | 5 |
| Injury and Poisoning | 4 | 6 | 5 |
| Investigations * | 3 | 11 | 7 |
| Musculoskeletal | 8 | 10 | 17 |
| Arthralgia | 3 | 7 | 4 |
| Nervous System | 17 | 11 | 21 |
| Headache NOS | 13 | 10 | 16 |
| Dizziness (excl vertigo) | 1 | 1 | 7 |
| Respiratory | 8 | 15 | 15 |
| Cough | 7 | 7 | 8 |
| Skin & Subcutaneous | 10 | 7 | 18 |
Other Pre-Approval Studies
Adverse Events from Ankylosing Spondylitis Studies: A total of 378 patients were treated with celecoxib in placebo- and active-controlled AS studies. Doses up to 400 mg once daily were studied. The types of adverse events reported in the AS studies were similar to those reported in the OA/RA studies.
Adverse Events from Analgesia and Dysmenorrhea Studies: Approximately 1,700 patients were treated with celecoxib in analgesia and dysmenorrhea studies. All patients in post-oral surgery pain studies received a single dose of study medication. Doses up to 600 mg/day of celecoxib were studied in primary dysmenorrhea and post-orthopedic surgery pain studies. The types of adverse events in the analgesia and dysmenorrhea studies were similar to those reported in arthritis studies. The only additional adverse event reported was post-dental extraction alveolar osteitis (dry socket) in the post-oral surgery pain studies.
The APC and PreSAP Trials
Adverse reactions from long-term, placebo-controlled polyp prevention studies: Exposure to celecoxib in the APC and PreSAP trials was 400 mg to 800 mg daily for up to 3 years [ see Special Studies Adenomatous Polyp Prevention Studies (14.7)].
Some adverse reactions occurred in higher percentages of patients than in the arthritis pre-marketing trials (treatment durations up to 12 weeks; see Adverse events from celecoxib pre-marketing controlled arthritis trials, above). The adverse reactions for which these differences in patients treated with celecoxib were greater as compared to the arthritis pre-marketing trials were as follows:
|
Celecoxib
(400 to 800 mg daily)
| Placebo
N=1303 |
|
| Diarrhea | 10.5% | 7.0% |
| Gastroesophageal reflux disease | 4.7% | 3.1% |
| Nausea | 6.8% | 5.3% |
| Vomiting | 3.2% | 2.1% |
| Dyspnea | 2.8% | 1.6% |
| Hypertension | 12.5% | 9.8% |
| Nephrolithiasis | 2.1% | 0.8% |
The following additional adverse reactions occurred in ≥ 0.1% and < 1% of patients taking celecoxib, at an incidence greater than placebo in the long-term polyp prevention studies, and were either not reported during the controlled arthritis pre-marketing trials or occurred with greater frequency in the long-term, placebo-controlled polyp prevention studies:
Nervous system disorders: Cerebral infarction
Eye disorders: Vitreous floaters, conjunctival hemorrhage
Ear and labyrinth: Labyrinthitis
Cardiac disorders: Angina unstable, aortic valve incompetence, coronary artery atherosclerosis, sinus bradycardia, ventricular hypertrophy
Vascular disorders: Deep vein thrombosis
Reproductive system and breast disorders: Ovarian cyst
Investigations: Blood potassium increased, blood sodium increased, blood testosterone decreased
Injury, poisoning and procedural complications: Epicondylitis, tendon rupture
6.2 Postmarketing Experience
The following adverse reactions have been identified during post approval use of celecoxib. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure
Cardiovascular: Vasculitis, deep venous thrombosis
General: Anaphylactoid reaction, angioedema
Liver and biliary: Liver necrosis, hepatitis, jaundice, hepatic failure
Hemic and lymphatic: Agranulocytosis, aplastic anemia, pancytopenia, leucopenia
Metabolic: Hypoglycemia, hyponatremia
Nervous: Aseptic meningitis, ageusia, anosmia, fatal intracranial hemorrhage
Renal: Interstitial nephritis
7. DRUG INTERACTIONS
See Table 3 for clinically significant drug interactions with Celecoxib.
| Drugs That Interfere with Hemostasis | |
| Clinical Impact: |
|
| Intervention: | Monitor patients with concomitant use of celecoxib with anticoagulants (e.g., warfarin), antiplatelet drugs (e.g., aspirin), SSRIs, and SNRIs for signs of bleeding [ see Warnings and Precautions (5.11)]. |
| Aspirin | |
| Clinical Impact: | Controlled clinical studies showed that the concomitant use of NSAIDs and analgesic doses of aspirin does not produce any greater therapeutic effect than the use of NSAIDs alone. In a clinical study, the concomitant use of an NSAID and aspirin was associated with a significantly increased incidence of GI adverse reactions as compared to use of the NSAID alone [
see
Warnings and Precautions (5.2)].
In two studies in healthy volunteers, and in patients with osteoarthritis and established heart disease respectively, Celecoxib (200 mg to 400 mg daily) has demonstrated a lack of interference with the cardioprotective antiplatelet effect of aspirin (100 mg to 325 mg). |
| Intervention: | Concomitant use of celecoxib and analgesic doses of aspirin is not generally recommended because of the increased risk of bleeding [
see
Warnings and Precautions (5.11)].
Celecoxib is not a substitute for low dose aspirin for cardiovascular protection. |
| ACE Inhibitors, Angiotensin Receptor Blockers, and Beta-Blockers | |
| Clinical Impact: |
|
| Intervention: |
|
| Diuretics | |
| Clinical Impact: | Clinical studies, as well as post-marketing observations, showed that NSAIDs reduced the natriuretic effect of loop diuretics (e.g., furosemide) and thiazide diuretics in some patients. This effect has been attributed to the NSAID inhibition of renal prostaglandin synthesis. |
| Intervention: | During concomitant use of celecoxib with diuretics, observe patients for signs of worsening renal function, in addition to assuring diuretic efficacy including antihypertensive effects [ see Warnings and Precautions (5.6)]. |
| Digoxin | |
| Clinical Impact: | The concomitant use of Celecoxib with digoxin has been reported to increase the serum concentration and prolong the half-life of digoxin. |
| Intervention: | During concomitant use of celecoxib and digoxin, monitor serum digoxin levels. |
| Lithium | |
| Clinical Impact: | NSAIDs have produced elevations in plasma lithium levels and reductions in renal lithium clearance . The mean minimum lithium concentration increased 15%, and the renal clearance decreased by approximately 20%. This effect has been attributed to NSAID inhibition of renal prostaglandin synthesis. |
| Intervention: | During concomitant use of celecoxib and lithium, monitor patients for signs of lithium toxicity. |
| Methotrexate | |
| Clinical Impact: | Concomitant use of NSAIDs and methotrexate may increase the risk for methotrexate toxicity (e.g., neutropenia, thrombocytopenia, renal dysfunction).
Celecoxib has no effect on methotrexate pharmacokinetics. |
| Intervention: | During concomitant use of celecoxib and methotrexate, monitor patients for methotrexate toxicity. |
| Cyclosporine | |
| Clinical Impact: | Concomitant use of celecoxib and cyclosporine may increase cyclosporine's nephrotoxicity. |
| Intervention: | During concomitant use of celecoxib and cyclosporine, monitor patients for signs of worsening renal function. |
| NSAIDs and Salicylates | |
| Clinical Impact: | Concomitant use of Celecoxib with other NSAIDs or salicylates (e.g., diflunisal, salsalate) increases the risk of GI toxicity, with little or no increase in efficacy [ see Warnings and Precautions (5.2)] . |
| Intervention: | The concomitant use of Celecoxib with other NSAIDs or salicylates is not recommended. |
| Pemetrexed | |
| Clinical Impact: | Concomitant use of celecoxib and pemetrexed may increase the risk of pemetrexed-associated myelosuppression, renal, and GI toxicity (see the pemetrexed prescribing information). |
| Intervention: | During concomitant use of celecoxib and pemetrexed, in patients with renal impairment whose creatinine clearance ranges from 45 to 79 mL/min, monitor for myelosuppression, renal and GI toxicity.
NSAIDs with short elimination half-lives (e.g., diclofenac, indomethacin) should be avoided for a period of two days before, the day of, and two days following administration of pemetrexed. In the absence of data regarding potential interaction between pemetrexed and NSAIDs with longer half-lives (e.g., meloxicam, nabumetone), patients taking these NSAIDs should interrupt dosing for at least five days before, the day of, and two days following pemetrexed administration. |
| CYP2C9 Inhibitors or inducers | |
| Clinical Impact: | Celecoxib metabolism is predominantly mediated via cytochrome P450 (CYP) 2C9 in the liver. Co-administration of celecoxib with drugs that are known to inhibit CYP2C9 (e.g. fluconazole) may enhance the exposure and toxicity of celecoxib whereas co-administration with CYP2C9 inducers (e.g. rifampin) may lead to compromised efficacy of celecoxib. |
| Intervention | Evaluate each patient's medical history when consideration is given to prescribing celecoxib. A dosage adjustment may be warranted when celecoxib is administered with CYP2C9 inhibitors or inducers.
[ see Clinical Pharmacology (12.3)]. |
| CYP2D6 substrates | |
| Clinical Impact: | In vitro studies indicate that celecoxib, although not a substrate, is an inhibitor of CYP2D6. Therefore, there is a potential for an in vivo drug interaction with drugs that are metabolized by CYP2D6 (e.g. atomoxetine), and celecoxib may enhance the exposure and toxicity of these drugs. |
| Intervention | Evaluate each patient's medical history when consideration is given to prescribing celecoxib. A dosage adjustment may be warranted when celecoxib is administered with CYP2D6 substrates.
[ see Clinical Pharmacology (12.3)]. |
| Corticosteroids | |
| Clinical Impact: | Concomitant use of corticosteroids with celecoxib may increase the risk of GI ulceration or bleeding. |
| Intervention | Monitor patients with concomitant use of celecoxib with corticosteroids for signs of bleeding [see Warnings and Precautions (5.2)]. |
8. USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category C. Pregnancy category D from 30 weeks of gestation onward .
Risk Summary
Use of NSAIDs, including celecoxib, during the third trimester of pregnancy increases the risk of premature closure of the fetal ductus arteriosus. Avoid use of NSAIDs, including celecoxib, in pregnant women starting at 30 weeks of gestation.
There are no adequate and well-controlled studies of celecoxib in pregnant women. Data from observational studies regarding potential embryofetal risks of NSAID use in women in the first or second trimesters of pregnancy are inconclusive. In animal reproduction studies, embryo-fetal deaths and an increase in diaphragmatic hernias were observed in rats administered celecoxib daily during the period of organogenesis at oral doses approximately 6 times the maximum recommended human dose (MRHD) of 200 mg twice daily. In addition, structural abnormalities (e.g., septal defects, ribs fused, sternebrae fused and sternebrae misshapen) were observed in rabbits given daily oral doses of celecoxib during the period of organogenesis at approximately 2 times the MRHD [see Data]. Based on animal data, prostaglandins have been shown to have an important role in endometrial vascular permeability, blastocyst implantation, and decidualization. In animal studies, administration of prostaglandin synthesis inhibitors such as celecoxib, resulted in increased pre- and post-implantation loss.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the general U.S. population, all clinically recognized pregnancies, regardless of drug exposure, have a background rate of 2% to 4% for major malformations, and 15% to 20% for pregnancy loss.
Data
Human Data
The available data do not establish the presence or absence of developmental toxicity related to the use of celecoxib.
Animal data
Celecoxib at oral doses ≥150 mg/kg/day (approximately 2 times the human exposure at 200 mg twice daily as measured by AUC 0-24), caused an increased incidence of ventricular septal defects, a rare event, and fetal alterations, such as ribs fused, sternebrae fused and sternebrae misshapen when rabbits were treated throughout organogenesis. A dose-dependent increase in diaphragmatic hernias was observed when rats were given celecoxib at oral doses ≥30 mg/kg/day (approximately 6 times human exposure based on the AUC 0-24 at 200 mg twice daily for RA) throughout organogenesis. In rats, exposure to celecoxib during early embryonic development resulted in pre-implantation and post-implantation losses at oral doses ≥50 mg/kg/day (approximately 6 times human exposure based on the AUC 0-24 at 200 mg twice daily for RA).
Celecoxib produced no evidence of delayed labor or parturition at oral doses up to 100 mg/kg in rats (approximately 7-fold human exposure as measured by the AUC 0-24 at 200 mg twice daily). The effects of celecoxib on labor and delivery in pregnant women are unknown.
8.2 Lactation
Risk Summary
Limited data from 3 published reports that included a total of 12 breastfeeding women showed low levels of celecoxib in breast milk. The calculated average daily infant dose was 10 to 40 mcg/kg/day, less than 1% of the weight-based therapeutic dose for a two-year old-child. A report of two breastfed infants 17 and 22 months of age did not show any adverse events. Caution should be exercised when celecoxib is administered to a nursing woman. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for celecoxib and any potential adverse effects on the breastfed infant from the celecoxib or from the underlying maternal condition.
8.3 Females and Males of Reproductive Potential
Infertility
Females
Based on the mechanism of action, the use of prostaglandin-mediated NSAIDs, including celecoxib, may delay or prevent rupture of ovarian follicles, which has been associated with reversible infertility in some women. Published animal studies have shown that administration of prostaglandin synthesis inhibitors has the potential to disrupt prostaglandin mediated follicular rupture required for ovulation. Small studies in women treated with NSAIDs have also shown a reversible delay in ovulation. Consider withdrawal of NSAIDs, including celecoxib, in women who have difficulties conceiving or who are undergoing investigation of infertility.
8.4 Pediatric Use
Celecoxib is approved for relief of the signs and symptoms of Juvenile Rheumatoid Arthritis in patients 2 years and older. Safety and efficacy have not been studied beyond six months in children. The long-term cardiovascular toxicity in children exposed to celecoxib has not been evaluated and it is unknown if long-term risks may be similar to that seen in adults exposed to celecoxib or other COX-2 selective and non-selective NSAIDs [ (see Boxed Warning, Warnings and Precautions (5.12), and Clinical Studies (14.3)].
The use of celecoxib in patients 2 years to 17 years of age with pauciarticular, polyarticular course JRA or in patients with systemic onset JRA was studied in a 12-week, double-blind, active controlled, pharmacokinetic, safety and efficacy study, with a 12-week open-label extension. Celecoxib has not been studied in patients under the age of 2 years, in patients with body weight less than 10 kg (22 lbs), and in patients with active systemic features. Patients with systemic onset JRA (without active systemic features) appear to be at risk for the development of abnormal coagulation laboratory tests. In some patients with systemic onset JRA, both celecoxib and naproxen were associated with mild prolongation of activated partial thromboplastin time (APTT) but not prothrombin time (PT). When NSAIDs including celecoxib are used in patients with systemic onset JRA, monitor patients for signs and symptoms of abnormal clotting or bleeding, due to the risk of disseminated intravascular coagulation. Patients with systemic onset JRA should be monitored for the development of abnormal coagulation tests [see Dosage and Administration (2.4), Warnings and Precautions (5.12), Adverse Reactions (6.3), Animal Toxicology (13.2), Clinical Studies (14.3)] .
Alternative therapies for treatment of JRA should be considered in pediatric patients identified to be CYP2C9 poor metabolizers [see Poor Metabolizers of CYP2C9 substrates (8.8)].
8.5 Geriatric Use
Elderly patients, compared to younger patients, are at greater risk for NSAID-associated serious cardiovascular, gastrointestinal, and/or renal adverse reactions. If the anticipated benefit for the elderly patient outweighs these potential risks, start dosing at the low end of the dosing range, and monitor patients for adverse effects [ see Warnings and Precautions (5.1, 5.2, 5.3, 5.6, 5.13)].
Of the total number of patients who received celecoxib in pre-approval clinical trials, more than 3,300 were 65 to 74 years of age, while approximately 1,300 additional patients were 75 years and over. No substantial differences in effectiveness were observed between these subjects and younger subjects. In clinical studies comparing renal function as measured by the GFR, BUN and creatinine, and platelet function as measured by bleeding time and platelet aggregation, the results were not different between elderly and young volunteers. However, as with other NSAIDs, including those that selectively inhibit COX-2, there have been more spontaneous post-marketing reports of fatal GI events and acute renal failure in the elderly than in younger patients [ see Warnings and Precautions (5.4, 5.6)].
8.6 Hepatic Impairment
The daily recommended dose of celecoxib capsules in patients with moderate hepatic impairment (Child-Pugh Class B) should be reduced by 50%. The use of celecoxib in patients with severe hepatic impairment is not recommended [ see Dosage and Administration (2.6) and Clinical Pharmacology (12.3)].
8.7 Renal Impairment
Celecoxib is not recommended in patients with severe renal insufficiency [ see Warnings and Precautions (5.6) and Clinical Pharmacology (12.3)].
8.8 Poor Metabolizers of CYP2C9 Substrates
In patients who are known or suspected to be poor CYP2C9 metabolizers (i.e., CYP2C9*3/*3), based on genotype or previous history/experience with other CYP2C9 substrates (such as warfarin, phenytoin) administer celecoxib starting with half the lowest recommended dose. Alternative management should be considered in JRA patients identified to be CYP2C9 poor metabolizers. [ see Dosage and Administration (2.6) and Clinical Pharmacology (12.5)].
10. OVERDOSAGE
Symptoms following acute NSAID overdosages have been typically limited to lethargy, drowsiness, nausea, vomiting, and epigastric pain, which have been generally reversible with supportive care. Gastrointestinal bleeding has occurred. Hypertension, acute renal failure, respiratory depression, and coma have occured, but were rare [ see Warnings and Precautions (5.1, 5.2, 5.4, 5.6)].
No overdoses of celecoxib were reported during clinical trials. Doses up to 2400 mg/day for up to 10 days in 12 patients did not result in serious toxicity. No information is available regarding the removal of celecoxib by hemodialysis, but based on its high degree of plasma protein binding (>97%) dialysis is unlikely to be useful in overdose.
Manage patients with symptomatic and supportive care following an NSAID overdosage. There are no specific antidotes. Consider emesis and/or activated charcoal (60 to 100 grams in adults, 1 to 2 grams per kg of body weight in pediatric patients) and/or osmotic cathartic in symptomatic patients seen within four hours of ingestion or in patients with a large overdosage (5 to 10 times the recommended dosage). Forced diuresis, alkalinization of urine, hemodialysis, or hemoperfusion may not be useful due to high protein binding.
For additional information about overdosage treatment contact a poison control center (1-800-222-1222).
11. DESCRIPTION
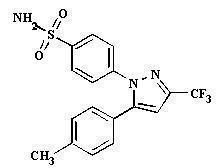
Celecoxib capsule is a nonsteroidal anti-inflammatory drug, available as capsules containing 50 mg, 100 mg, 200 mg and 400 mg celecoxib for oral administration. The chemical name is 4-[5-(4-methylphenyl)- 3-(trifluoromethyl)-1H-pyrazol-1-yl] benzenesulfonamide and is a diaryl-substituted pyrazole. The molecular weight is 381.38. Its molecular formula is C 17H 14F 3N 3O 2S, and it has the following chemical structure:
Celecoxib is a white to off-white powder with a pKa of 11.1 (sulfonamide moiety). Celecoxib is hydrophobic (log P is 3.5) and is practically insoluble in aqueous media at physiological pH range.
The inactive ingredients in celecoxib capsules include: croscarmellose sodium, lactose monohydrate, magnesium stearate, povidone and sodium lauryl sulfate. The empty hard gelatin capsule shells contain gelatin and titanium dioxide. The capsules are imprinted with edible ink containing propylene glycol and shellac. In addition, 50 mg capsules are imprinted with N-butyl alcohol, FD&C yellow No.6, D&C red No.7 and titanium dioxide. 100 mg capsules are imprinted with N-butyl alcohol, strong ammonia solution, titanium dioxide and FD&C blue No.1 aluminum lake. 200 mg capsules are imprinted with iron oxide yellow, N-butyl alcohol, ammonium hydroxide and simethicone. 400 mg capsules are imprinted with N-butyl alcohol, strong ammonia solution, titanium dioxide, FD&C blue No.1 aluminum lake and FD&C yellow No.10 aluminum lake.
12. CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Celecoxib has analgesic, anti-inflammatory, and antipyretic properties.
The mechanism of action of celecoxib is believed to be due to inhibition of prostaglandin synthesis, primarily via inhibition of cyclooxygenase-2 (COX-2).
Celecoxib is a potent inhibitor of prostaglandin synthesis in vitro. Celecoxib concentrations reached during therapy have produced in vivo effects. Prostaglandins sensitize afferent nerves and potentiate the action of bradykinin in inducing pain in animal models. Prostaglandins are mediators of inflammation. Since celecoxib is an inhibitor of prostaglandin synthesis, its mode of action may be due to a decrease of prostaglandins in peripheral tissues.
12.2 Pharmacodynamics
Platelets
In clinical trials using normal volunteers, celecoxib at single doses up to 800 mg and multiple doses of 600 mg twice daily for up to 7 days duration (higher than recommended therapeutic doses) had no effect on reduction of platelet aggregation or increase in bleeding time. Because of its lack of platelet effects, celecoxib is not a substitute for aspirin for cardiovascular prophylaxis. It is not known if there are any effects of celecoxib on platelets that may contribute to the increased risk of serious cardiovascular thrombotic adverse events associated with the use of celecoxib.
Fluid Retention
Inhibition of PGE2 synthesis may lead to sodium and water retention through increased reabsorption in the renal medullary thick ascending loop of Henle and perhaps other segments of the distal nephron. In the collecting ducts, PGE2 appears to inhibit water reabsorption by counteracting the action of antidiuretic hormone.
12.3 Pharmacokinetics
Celecoxib exhibits dose-proportional increase in exposure after oral administration up to 200 mg twice daily and less than proportional increase at hgher doses. It has extensive distribution and high protein binding. It is primarily metabolized by CYP2C9 with a half-life of approximately 11 hours.
Absorption
Peak plasma levels of celecoxib occur approximately 3 hours after an oral dose. Under fasting conditions, both peak plasma levels (C max) and area under the curve (AUC) are roughly dose-proportional up to 200 mg twice daily; at higher doses there are less than proportional increases in C max and AUC [see Food Effects] . Absolute bioavailability studies have not been conducted. With multiple dosing, steady-state conditions are reached on or before Day 5. The pharmacokinetic parameters of celecoxib in a group of healthy subjects are shown in Table 4.
| Mean (%CV) PK Parameter Values | ||||
| Cmax, ng/mL | Tmax, hr | Effective t1/2, hr | V ss/F, L | CL/F, L/hr |
| 705 (38) | 2.8 (37) | 11.2 (31) | 429 (34) | 27.7 (28) |
When celecoxib capsules were taken with a high fat meal, peak plasma levels were delayed for about 1 to 2 hours with an increase in total absorption (AUC) of 10% to 20%. Under fasting conditions, at doses above 200 mg, there is less than a proportional increase in C max and AUC, which is thought to be due to the low solubility of the drug in aqueous media.
Coadministration of celecoxib with an aluminum- and magnesium-containing antacids resulted in a reduction in plasma celecoxib concentrations with a decrease of 37% in C max and 10% in AUC. Celecoxib, at doses up to 200 mg twice daily, can be administered without regard to timing of meals. Higher doses (400 mg twice daily) should be administered with food to improve absorption.
In healthy adult volunteers, the overall systemic exposure (AUC) of celecoxib was equivalent when celecoxib was administered as intact capsule or capsule contents sprinkled on applesauce. There were no significant alterations in C max, T max or t 1/2 after administration of capsule contents on applesauce [ see Dosage and Administration (2)].
Distribution
In healthy subjects, Celecoxib is highly protein bound (~97%) within the clinical dose range. In vitro studies indicate that celecoxib binds primarily to albumin and, to a lesser extent, α 1-acid glycoprotein. The apparent volume of distribution at steady state (V ss/F) is approximately 400 L, suggesting extensive distribution into the tissues. Celecoxib is not preferentially bound to red blood cells.
Elimination
Metabolism
Celecoxib metabolism is primarily mediated via CYP2C9. Three metabolites, a primary alcohol, the corresponding carboxylic acid and its glucuronide conjugate, have been identified in human plasma. These metabolites are inactive as COX-1 or COX-2 inhibitors.
Excretion
Celecoxib is eliminated predominantly by hepatic metabolism with little (<3%) unchanged drug recovered in the urine and feces. Following a single oral dose of radiolabeled drug, approximately 57% of the dose was excreted in the feces and 27% was excreted into the urine. The primary metabolite in both urine and feces was the carboxylic acid metabolite (73% of dose) with low amounts of the glucuronide also appearing in the urine. It appears that the low solubility of the drug prolongs the absorption process making terminal half-life (t 1/2) determinations more variable. The effective half-life is approximately 11 hours under fasted conditions. The apparent plasma clearance (CL/F) is about 500 mL/min.
Specific Populations
Geriatric
At steady state, elderly subjects (over 65 years old) had a 40% higher C max and a 50% higher AUC compared to the young subjects. In elderly females, celecoxib C max and AUC are higher than those for elderly males, but these increases are predominantly due to lower body weight in elderly females. Dose adjustment in the elderly is not generally necessary. However, for patients of less than 50 kg in body weight, initiate therapy at the lowest recommended dose [ see Dosage and Administration (2.7) and Use in Specific Populations (8.5)].
Pediatric
The steady state pharmacokinetics of celecoxib administered as an investigational oral suspension was evaluated in 152 JRA patients 2 years to 17 years of age weighing ≥10 kg with pauciarticular or polyarticular course JRA and in patients with systemic onset JRA. Population pharmacokinetic analysis indicated that the oral clearance (unadjusted for body weight) of celecoxib increases less than proportionally to increasing weight, with 10 kg and 25 kg patients predicted to have 40% and 24% lower clearance, respectively, compared with a 70 kg adult RA patient.
Twice-daily administration of 50 mg capsules to JRA patients weighing ≥12 to ≤25 kg and 100 mg capsules to JRA patients weighing >25 kg should achieve plasma concentrations similar to those observed in a clinical trial that demonstrated the non-inferiority of celecoxib to naproxen 7.5 mg/kg twice daily ( see Dosage and Administration (2.4). Celecoxib has not been studied in JRA patients under the age of 2 years, in patients with body weight less than 10 kg (22 lbs), or beyond 24 weeks.
Race
Meta-analysis of pharmacokinetic studies has suggested an approximately 40% higher AUC of celecoxib in Blacks compared to Caucasians. The cause and clinical significance of this finding is unknown.
Hepatic Impairment
A pharmacokinetic study in subjects with mild (Child-Pugh Class A) and moderate (Child-Pugh Class B) hepatic impairment has shown that steady-state celecoxib AUC is increased about 40% and 180%, respectively, above that seen in healthy control subjects. Therefore, the daily recommended dose of celecoxib capsules should be reduced by approximately 50% in patients with moderate (Child-Pugh Class B) hepatic impairment. Patients with severe hepatic impairment (Child-Pugh Class C) have not been studied. The use of celecoxib in patients with severe hepatic impairment is not recommended [ see Dosage and Administration (2.6) and Use in Specific Populations (8.6)].
Renal Impairment
In a cross-study comparison, celecoxib AUC was approximately 40% lower in patients with chronic renal insufficiency (GFR 35-60 mL/min) than that seen in subjects with normal renal function. No significant relationship was found between GFR and celecoxib clearance. Patients with severe renal insufficiency have not been studied. Similar to other NSAIDs, celecoxib is not recommended in patients with severe renal insufficiency [ see Warnings and Precautions (5.6)].
Drug Interaction Studies
In vitro studies indicate that Celecoxib is not an inhibitor of cytochrome P450 2C9, 2C19 or 3A4.
In vivo studies have shown the following:
Aspirin
When NSAIDs were administered with aspirin, the protein binding of NSAIDs were reduced, although the clearance of free NSAID was not altered. The clinical significance of this interaction is not known. See Table 3 for clinically significant drug interactions of NSAIDs with aspirin [ see Drug Interactions (7)] .
Lithium
In a study conducted in healthy subjects, mean steady-state lithium plasma levels increased approximately 17% in subjects receiving lithium 450 mg twice daily with celecoxib 200 mg twice daily as compared to subjects receiving lithium alone [ see Drug Interactions (7)].
Fluconazole
Concomitant administration of fluconazole at 200 mg once daily resulted in a two-fold increase in celecoxib plasma concentration. This increase is due to the inhibition of celecoxib metabolism via P450 2C9 by fluconazole [ see Drug Interactions (7)].
Other Drugs
The effects of celecoxib on the pharmacokinetics and/or pharmacodynamics of glyburide, ketoconazole, [ see Drug Interactions (7)], phenytoin, and tolbutamide have been studied in vivo and clinically important interactions have not been found.
12.5 Pharmacogenomics
CYP2C9 activity is reduced in individuals with genetic polymorphisms that lead to reduced enzyme activity, such as those homozygous for the CYP2C9*2 and CYP2C9*3 polymorphisms. Limited data from 4 published reports that included a total of 8 subjects with the homozygous CYP2C9*3/*3 genotype showed celecoxib systemic levels that were 3- to 7-fold higher in these subjects compared to subjects with CYP2C9*1/*1 or *I/*3 genotypes. The pharmacokinetics of celecoxib have not been evaluated in subjects with other CYP2C9 polymorphisms, such as *2, *5, *6, *9 and *11. It is estimated that the frequency of the homozygous *3/*3 genotype is 0.3% to 1.0% in various ethnic groups. [ see Dosage and Administration (2.6), Use in Specific Populations (8.8)].
13. NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Celecoxib was not carcinogenic in Sprague-Dawley rats given oral doses up to 200 mg/kg for males and 10 mg/kg for females (approximately 2-to 4-times the human exposure as measured by the AUC 0-24 at 200 mg twice daily) or in mice given oral doses up to 25 mg/kg for males and 50 mg/kg for females (approximately equal to human exposure as measured by the AUC 0-24 at 200 mg twice daily) for two years.
Mutagenesis
Celecoxib was not mutagenic in an Ames test and a mutation assay in Chinese hamster ovary (CHO) cells, nor clastogenic in a chromosome aberration assay in CHO cells and an in vivo micronucleus test in rat bone marrow.
Impairment of Fertility
Celecoxib had no effect on male or female fertility or male reproductive function in rats at oral doses up to 600 mg/kg/day (approximately 11-times human exposure at 200 mg twice daily based on the AUC 0-24). At ≥50 mg/kg/day (approximately 6-times human exposure based on the AUC 0-24 at 200 mg twice daily) there was increased preimplantation loss.
13.2 Animal Toxicology
An increase in the incidence of background findings of spermatocele with or without secondary changes such as epididymal hypospermia as well as minimal to slight dilation of the seminiferous tubules was seen in the juvenile rat. These reproductive findings while apparently treatment-related did not increase in incidence or severity with dose and may indicate an exacerbation of a spontaneous condition. Similar reproductive findings were not observed in studies of juvenile or adult dogs or in adult rats treated with celecoxib. The clinical significance of this observation is unknown.
14. CLINICAL STUDIES
14.1 Osteoarthritis
Celecoxib has demonstrated significant reduction in joint pain compared to placebo. Celecoxib was evaluated for treatment of the signs and the symptoms of OA of the knee and hip in placebo- and active-controlled clinical trials of up to 12 weeks duration. In patients with OA, treatment with celecoxib 100 mg twice daily or 200 mg once daily resulted in improvement in WOMAC (Western Ontario and McMaster Universities) osteoarthritis index, a composite of pain, stiffness, and functional measures in OA. In three 12-week studies of pain accompanying OA flare, celecoxib doses of 100 mg twice daily and 200 mg twice daily provided significant reduction of pain within 24 to 48 hours of initiation of dosing. At doses of 100 mg twice daily or 200 mg twice daily the effectiveness of celecoxib was shown to be similar to that of naproxen 500 mg twice daily. Doses of 200 mg twice daily provided no additional benefit above that seen with 100 mg twice daily. A total daily dose of 200 mg has been shown to be equally effective whether administered as 100 mg twice daily or 200 mg once daily.
14.2 Rheumatoid Arthritis
Celecoxib has demonstrated significant reduction in joint tenderness/pain and joint swelling compared to placebo. Celecoxib was evaluated for treatment of the signs and symptoms of RA in placebo- and active-controlled clinical trials of up to 24 weeks in duration. Celecoxib was shown to be superior to placebo in these studies, using the ACR20 Responder Index, a composite of clinical, laboratory, and functional measures in RA. Celecoxib doses of 100 mg twice daily and 200 mg twice daily were similar in effectiveness and both were comparable to naproxen 500 mg twice daily.
Although celecoxib 100 mg twice daily and 200 mg twice daily provided similar overall effectiveness, some patients derived additional benefit from the 200 mg twice daily dose. Doses of 400 mg twice daily provided no additional benefit above that seen with 100 mg to 200 mg twice daily.
14.3 Juvenile Rheumatoid Arthritis (NCT00652925)
In a 12-week, randomized, double-blind active-controlled, parallel-group, multicenter, non-inferiority study, patients from 2 years to 17 years of age with pauciarticular, polyarticular course JRA or systemic onset JRA (with currently inactive systemic features), received one of the following treatments: celecoxib 3 mg/kg (to a maximum of 150 mg) twice daily; celecoxib 6 mg/kg (to a maximum of 300 mg) twice daily; or naproxen 7.5 mg/kg (to a maximum of 500 mg) twice daily. The response rates were based upon the JRA Definition of Improvement greater than or equal to 30% (JRA DOI 30) criterion, which is a composite of clinical, laboratory, and functional measures of JRA. The JRA DOI 30 response rates at week 12 were 69%, 80% and 67% in the celecoxib 3 mg/kg twice daily, celecoxib 6 mg/kg twice daily, and naproxen 7.5 mg/kg twice daily treatment groups, respectively.
The efficacy and safety of celecoxib for JRA have not been studied beyond six months. The long-term cardiovascular toxicity in children exposed to celecoxib has not been evaluated and it is unknown if the long-term risk may be similar to that seen in adults exposed to celecoxib or other COX-2 selective and non-selective NSAIDs [( see Boxed Warning, Warnings and Precautions (5.12)].
14.4 Ankylosing Spondylitis
Celecoxib was evaluated in AS patients in two placebo- and active-controlled clinical trials of 6 and 12 weeks duration. Celecoxib at doses of 100 mg twice daily, 200 mg once daily and 400 mg once daily was shown to be statistically superior to placebo in these studies for all three co-primary efficacy measures assessing global pain intensity (Visual Analogue Scale), global disease activity (Visual Analogue Scale) and functional impairment (Bath Ankylosing Spondylitis Functional Index). In the 12-week study, there was no difference in the extent of improvement between the 200 mg and 400 mg celecoxib doses in a comparison of mean change from baseline, but there was a greater percentage of patients who responded to celecoxib 400 mg, 53%, than to celecoxib 200 mg, 44%, using the Assessment in Ankylosing Spondylitis response criteria (ASAS 20). The ASAS 20 defines a responder as improvement from baseline of at least 20% and an absolute improvement of at least 10 mm, on a 0 mm to 100 mm scale, in at least three of the four following domains: patient global pain, Bath Ankylosing Spondylitis Functional Index, and inflammation. The responder analysis also demonstrated no change in the responder rates beyond 6 weeks.
14.5 Analgesia, including Primary Dysmenorrhea
In acute analgesic models of post-oral surgery pain, post-orthopedic surgical pain, and primary dysmenorrhea, celecoxib relieved pain that was rated by patients as moderate to severe. Single doses [ see Dosage and Administration (2.6)] of celecoxib provided pain relief within 60 minutes.
14.6 Cardiovascular Outcomes Trial: Prospective Randomized Evaluation of Celecoxib Integrated Safety vs. Ibuprofen Or Naproxen (PRECISION; NCT00346216)
Design
The PRECISION trial was a double-blind randomized controlled trial of cardiovascular safety in OA and RA patients with or at high risk for cardiovascular disease comparing celecoxib with naproxen and ibuprofen. Patients were randomized to a starting dose of 100 mg twice daily of celecoxib, 600 mg three times daily of ibuprofen, or 375 mg twice daily of naproxen, with the option of escalating the dose as needed for pain management. Based on labeled doses, OA patients randomized to celecoxib could not dose escalate.
The primary endpoint, the Antiplatelet Trialists’ Collaboration (APTC) composite, was an independently adjudicated composite of cardiovascular death (including hemorrhagic death), non-fatal myocardial infarction, and non-fatal stroke with 80% power to evaluate non-inferiority. All patients were prescribed open-label esomeprazole (20-40 mg) for gastroprotection. Treatment randomization was stratified by baseline low-dose aspirin use.
Additionally, there was a 4-month substudy assessing the effects of the three drugs on blood pressure as measured by ambulatory monitoring.
Results
Among subjects with OA, only 0.2% (17/7259) escalated celecoxib to the 200 mg twice daily dose, whereas 54.7% (3946/7208) escalated ibuprofen to 800 mg three times daily, and 54.8% (3937/7178) escalated naproxen to the 500 mg twice daily dose. Among subjects with RA, 55.7% (453/813) escalated celecoxib to the 200 mg twice daily dose, 56.5% (470/832) escalated ibuprofen to 800 mg three times daily, and 54.6% (432/791) escalated naproxen to the 500 mg twice daily dose; however, the RA population accounted for only 10% of the trial population.
Because relatively few celecoxib patients overall (5.8% [470/8072]) dose-escalated to 200 mg twice daily, the results of the PRECISION trial are not suitable for determining the relative CV safety of celecoxib at 200 mg twice daily compared to ibuprofen and naproxen at the doses taken.
Primary Endpoint
The trial had two prespecified analysis populations:
- Intent-to-treat population (ITT): Comprised of all randomized subjects followed for a maximum of 30 months
- Modified Intent-to-treat population (mITT): Comprised of all randomized subjects who received at least one dose of study medication and had at least one post-baseline visit followed until the earlier of treatment discontinuation plus 30 days, or 43 months
Celecoxib, at the 100 mg twice daily dose, as compared with either naproxen or ibuprofen at the doses taken, met all four prespecified non-inferiority criteria (p<0.001 for non-inferiority in both comparisons) for the APTC endpoint, a composite of cardiovascular death (including hemorrhagic death), non-fatal myocardial infarction, and non-fatal stroke [ see Table 5]. Non-inferiority was prespecified as a hazard ratio (HR) of ≤1.12 in both ITT and mITT analyses, and upper 95% CI of ≤ 1.33 for ITT analysis and ≤1.40 for mITT analysis.
The primary analysis results for ITT and mITT are described in Table 5.
| Intent-To-Treat Analysis (ITT, through month 30) | |||
| Celecoxib | Ibuprofen | Naproxen | |
| N | 8,072 | 8,040 | 7,969 |
| Subjects with Events | 188 (2.3%) | 218 (2.7%) | 201 (2.5%) |
| Pairwise Comparison | Celecoxib vs. Naproxen | Celecoxib vs. Ibuprofen | Ibuprofen vs. Naproxen |
| HR (95% CI) | 0.93 (0.76, 1.13) | 0.86 (0.70, 1.04) | 1.08 (0.89, 1.31) |
| Modified Intent-To-Treat Analysis (mITT, on treatment plus 30 days, through month 43) | |||
| Celecoxib | Ibuprofen | Naproxen | |
| N | 8,030 | 7,990 | 7,933 |
| Subjects with Events | 134 (1.7%) | 155 (1.9%) | 144 (1.8%) |
| Pairwise Comparison | Celecoxib vs. Naproxen | Celecoxib vs. Ibuprofen | Ibuprofen vs. Naproxen |
| HR (95% CI) | 0.90 (0.72, 1.14) | 0.81 (0.64, 1.02) | 1.12 (0.89, 1.40) |
| Intent-To-Treat Analysis (ITT, through month 30) | |||
| Celecoxib | Ibuprofen | Naproxen | |
| N | 8,072 | 8,040 | 7,969 |
| CV Death | 68 (0.8%) | 80 (1.0%) | 86 (1.1%) |
| Non-Fatal MI | 76 (0.9%) | 92 (1.1%) | 66 (0.8%) |
| Non-Fatal Stroke | 51 (0.6%) | 53 (0.7%) | 57 (0.7%) |
| Modified Intent-To-Treat Analysis (mITT, on treatment plus 30 days, through month 43) | |||
| N | 8,030 | 7,990 | 7,933 |
| CV Death | 35 (0.4%) | 51 (0.6%) | 49 (0.6%) |
| Non-fatal MI | 58 (0.7%) | 76 (1.0%) | 53 (0.7%) |
| Non-fatal Stroke | 43 (0.5%) | 32 (0.4%) | 45 (0.6%) |
*A patient may have experienced more than one component; therefore, the sum of the components is larger than the number of patients who experienced the composite outcome
In the ITT analysis population through 30 months, all-cause mortality was 1.6% in the celecoxib group, 1.8% in the ibuprofen group, and 2.0% in the naproxen group.
Ambulatory Blood Pressure Monitoring (ABPM) Substudy
In the PRECISION-ABPM substudy, among the total of 444 analyzable patients at Month 4, celecoxib dosed at 100 mg twice daily decreased mean 24-hour systolic blood pressure (SBP) by 0.3 mmHg, whereas ibuprofen and naproxen at the doses taken increased mean 24-hour SBP by 3.7 and 1.6 mmHg, respectively. These changes resulted in a statistically significant and clinically meaningful difference of 3.9 mmHg (p=0.0009) between celecoxib and ibuprofen and a non-statistically significant difference of 1.8 (p=0.119) mmHg between celecoxib and naproxen.
14.7 Special Studies
Adenomatous Polyp Prevention Studies (NCT00005094 and NCT00141193)
Cardiovascular safety was evaluated in two randomized, double-blind, placebo-controlled, three year studies involving patients with Sporadic Adenomatous Polyps treated with celecoxib: the APC trial (Adenoma Prevention with Celecoxib) and the PreSAP trial (Prevention of Spontaneous Adenomatous Polyps). In the APC trial, there was a dose-related increase in the composite endpoint (adjudicated) of cardiovascular death, myocardial infarction, or stroke with celecoxib compared to placebo over 3 years of treatment. The PreSAP trial did not demonstrate a statistically significant increased risk for the same composite endpoint (adjudicated):
- In the APC trial, the hazard ratios compared to placebo for a composite endpoint (adjudicated) of cardiovascular death, myocardial infarction, or stroke were 3.4 (95% CI 1.4-8.5) with celecoxib 400 mg twice daily and 2.8 (95% CI 1.1-7.2) with celecoxib 200 mg twice daily. Cumulative rates for this composite endpoint over 3 years were 3.0% (20/671 subjects) and 2.5% (17/685 subjects), respectively, compared to 0.9% (6/679 subjects) with placebo treatment. The increases in both celecoxib dose groups versus placebo-treated patients were mainly due to an increased incidence of myocardial infarction.
- In the PreSAP trial, the hazard ratio for this same composite endpoint (adjudicated) was 1.2 (95% CI 0.6-2.4) with celecoxib 400 mg once daily compared to placebo. Cumulative rates for this composite endpoint over 3 years were 2.3% (21/933 subjects) and 1.9% (12/628 subjects), respectively.
Clinical trials of other COX-2 selective and non-selective NSAIDs of up to three-years duration have shown an increased risk of serious cardiovascular thrombotic events, myocardial infarction, and stroke, which can be fatal. As a result, all NSAIDs are considered potentially associated with this risk.
Celecoxib Long-Term Arthritis Safety Study (CLASS)
This was a prospective, long-term, safety outcome study conducted post-marketing in approximately 5,800 OA patients and 2,200 RA patients. Patients received celecoxib 400 mg twice daily (4-fold and 2-fold the recommended OA and RA doses, respectively), ibuprofen 800 mg three times daily or diclofenac 75 mg twice daily (common therapeutic doses). Median exposures for celecoxib (n=3,987) and diclofenac (n=1,996) were 9 months while ibuprofen (n=1,985) was 6 months. The primary endpoint of this outcome study was the incidence of complicated ulcers (gastrointestinal bleeding, perforation or obstruction). Patients were allowed to take concomitant low-dose (≤325 mg/day) aspirin (ASA) for cardiovascular prophylaxis (ASA subgroups: celecoxib, n=882; diclofenac, n=445; ibuprofen, n=412). Differences in the incidence of complicated ulcers between celecoxib and the combined group of ibuprofen and diclofenac were not statistically significant.
Patients on celecoxib and concomitant low-dose ASA (N=882) experienced 4-fold higher rates of complicated ulcers compared to those not on ASA (N=3105). The Kaplan-Meier rate for complicated ulcers at 9 months was 1.12% versus 0.32% for those on low-dose ASA and those not on ASA, respectively [ see Warnings and Precautions (5.4)].
The estimated cumulative rates at 9 months of complicated and symptomatic ulcers for patients treated with celecoxib 400 mg twice daily are described in Table 7. Table 7 also displays results for patients less than or greater than 65 years of age. The difference in rates between celecoxib alone and celecoxib with ASA groups may be due to the higher risk for GI events in ASA users.
| All Patients | |
| Celecoxib alone (n=3105) | 0.78 |
| Celecoxib with ASA (n=882) | 2.19 |
| Patients < 65 Years | |
| Celecoxib alone (n=2025) | 0.47 |
| Celecoxib with ASA (n=403) | 1.26 |
| Patients ≥ 65 Years | |
| Celecoxib alone (n=1080) | 1.40 |
| Celecoxib with ASA (n=479) | 3.06 |
In a small number of patients with a history of ulcer disease, the complicated and symptomatic ulcer rates in patients taking celecoxib alone or celecoxib with ASA were, respectively, 2.56% (n=243) and 6.85% (n=91) at 48 weeks. These results are to be expected in patients with a prior history of ulcer disease [ see Warnings and Precautions (5.4) and Adverse Reactions (6.1)].
Cardiovascular safety outcomes were also evaluated in the CLASS trial. Kaplan-Meier cumulative rates for investigator-reported serious cardiovascular thromboembolic adverse events (including MI, pulmonary embolism, deep venous thrombosis, unstable angina, transient ischemic attacks, and ischemic cerebrovascular accidents) demonstrated no differences between the celecoxib, diclofenac, or ibuprofen treatment groups. The cumulative rates in all patients at nine months for celecoxib, diclofenac, and ibuprofen were 1.2%, 1.4%, and 1.1%, respectively. The cumulative rates in non-ASA users at nine months in each of the three treatment groups were less than 1%. The cumulative rates for myocardial infarction in non-ASA users at nine months in each of the three treatment groups were less than 0.2%. There was no placebo group in the CLASS trial, which limits the ability to determine whether the three drugs tested had no increased risk of CV events or if they all increased the risk to a similar degree. In the CLASS study, the Kaplan-Meier cumulative rates at 9 months of peripheral edema in patients on celecoxib 400 mg twice daily (4-fold and 2-fold the recommended OA and RA doses, respectively), ibuprofen 800 mg three times daily and diclofenac 75 mg twice daily were 4.5%, 6.9% and 4.7%, respectively. The rates of hypertension from the CLASS trial in the celecoxib, ibuprofen and diclofenac-treated patients were 2.4%, 4.2% and 2.5%, respectively.
Endoscopic Studies
The correlation between findings of short-term endoscopic studies with celecoxib and the relative incidence of clinically significant serious upper GI events with long-term use has not been established. Serious clinically significant upper GI bleeding has been observed in patients receiving celecoxib in controlled and open-labeled trials [ see Warnings and Precautions (5.4) and Clinical Studies (14.7)]
A randomized, double-blind study in 430 RA patients was conducted in which an endoscopic examination was performed at 6 months. The incidence of endoscopic ulcers in patients taking celecoxib 200 mg twice daily was 4% vs. 15% for patients taking diclofenac SR 75 mg twice daily. However, celecoxib was not statistically different than diclofenac for clinically relevant GI outcomes in the CLASS trial [ see Clinical Studies (14.7)].
The incidence of endoscopic ulcers was studied in two 12-week, placebo-controlled studies in 2157 OA and RA patients in whom baseline endoscopies revealed no ulcers. There was no dose relationship for the incidence of gastroduodenal ulcers and the dose of celecoxib (50 mg to 400 mg twice daily). The incidence for naproxen 500 mg twice daily was 16.2 and 17.6% in the two studies, for placebo was 2.0 and 2.3%, and for all doses of celecoxib the incidence ranged between 2.7%-5.9%. There have been no large, clinical outcome studies to compare clinically relevant GI outcomes with celecoxib and naproxen.
In the endoscopic studies, approximately 11% of patients were taking aspirin (≤325 mg/day). In the celecoxib capsules groups, the endoscopic ulcer rate appeared to be higher in aspirin users than in non-users. However, the increased rate of ulcers in these aspirin users was less than the endoscopic ulcer rates observed in the active comparator groups, with or without aspirin.
16. HOW SUPPLIED/STORAGE AND HANDLING
Celecoxib capsules, 50 mg are white, with reverse printed white on red band of body and cap with markings of 0006 on the cap and 50mg on the body, supplied as:
| NDC Number | Size |
| 70247-006-06 | bottle of 60 |
Celecoxib capsules, 100 mg are white, with reverse printed white on blue band of body and cap with markings of 0007 on the cap and 100mg on the body, supplied as:
| NDC Number | Size |
|---|---|
| 70247-007-10 | bottle of 100 |
| 70247-007-50 | bottle of 500 |
Celecoxib capsules, 200 mg are white, with reverse printed white on gold band of body and cap with markings of 0008 on the cap and 200mg on the body, supplied as:
| NDC Number | Size |
|---|---|
| 70247-008-10 | bottle of 100 |
| 70247-008-50 | bottle of 500 |
Celecoxib capsules, 400 mg are white, with reverse printed white on green band of body and cap with markings of 0009 on the cap and 400mg on the body, supplied as:
| NDC Number | Size |
|---|---|
| 70247-009-06 | bottle of 60 |
17. PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide) that accompanies each prescription dispensed. Inform patients, families, or their caregivers of the following information before initiating therapy with celecoxib and periodically during the course of ongoing therapy.
Cardiovascular Thrombotic Events
Advise patients to be alert for the symptoms of cardiovascular thrombotic events, including chest pain, shortness of breath, weakness, or slurring of speech, and to report any of these symptoms to their health care provider immediately [ see Warnings and Precautions (5.1)].
Gastrointestinal Bleeding, Ulceration, and Perforation
Advise patients to report symptoms of ulcerations and bleeding, including epigastric pain, dyspepsia, melena, and hematemesis to their health care provider. In the setting of concomitant use of low-dose aspirin for cardiac prophylaxis, inform patients of the increased risk for and the signs and symptoms of GI bleeding [ see Warnings and Precautions (5.2)].
Hepatotoxicity
Inform patients of the warning signs and symptoms of hepatotoxicity (e.g., nausea, fatigue, lethargy, pruritus, diarrhea jaundice, right upper quadrant tenderness, and "flu-like" symptoms). If these occur, instruct patients to stop celecoxib and seek immediate medical therapy [ see Warnings and Precautions (5.3), Use in Specific Populations (8.6)].
Heart Failure and Edema
Advise patients to be alert for the symptoms of congestive heart failure including shortness of breath, unexplained weight gain, or edema and to contact their healthcare provider if such symptoms occur [ see Warnings and Precautions (5.5)].
Anaphylactic Reactions
Inform patients of the signs of an anaphylactic reaction (e.g., difficulty breathing, swelling of the face or throat). Instruct patients to seek immediate emergency help if these occur [ see Contraindications (4) and Warnings and Precautions (5.7)].
Serious Skin Reactions
Advise patients to stop celecoxib immediately if they develop any type of rash and to contact their healthcare provider as soon as possible [ see Warnings and Precautions (5.9)].
Female Fertility
Advise females of reproductive potential who desire pregnancy that NSAIDs, including celecoxib, may be associated with a reversible delay in ovulation [ see Use in Specific Populations (8.3)].
Fetal Toxicity
Inform pregnant women to avoid use of celecoxib and other NSAIDs starting at 30 weeks of gestation because of the risk of the premature closing of the fetal ductus arteriosus [ see Warnings and Precautions (5.10) and Use in Specific Populations (8.1)].
Avoid Concomitant Use of NSAIDs
Inform patients that the concomitant use of celecoxib with other NSAIDs or salicylates (e.g., diflunisal, salsalate) is not recommended due to the increased risk of gastrointestinal toxicity, and little or no increase in efficacy [see Warnings and Precautions (5.2) and Drug Interactions (7)]. Alert patients that NSAIDs may be present in "over the counter" medications for treatment of colds, fever, or insomnia.
Use of NSAIDS and Low-Dose Aspirin
Inform patients not to use low-dose aspirin concomitantly with celecoxib until they talk to their healthcare provider [see Drug Interactions (7)].
Manufactured by: Qingdao BAHEAL Pharmaceutical Co., Ltd.
No. 268 Yingcheng Road, Jimo Qingdao, Shandong, China, 266200
LAB-0036-18.1
June 2019
| Medication Guide for Nonsteroidal Anti-inflammatory Drugs (NSAIDs) | |
| What is the most important information I should know about medicines called Nonsteroidal Anti-inflammatory Drugs (NSAIDs)? | |
| NSAIDs can cause serious side effects, including: | |
|
|
| Do not take NSAIDs right before or after a heart surgery called a "coronary artery bypass graft (CABG)." | |
| Avoid taking NSAIDs after a recent heart attack, unless your healthcare provider tells you to. You may have an increased risk of another heart attack if you take NSAIDs after a recent heart attack. | |
|
|
|
|
|
|
|
|
|
|
|
|
| What are NSAIDs?
NSAIDs are used to treat pain and redness, swelling, and heat (inflammation) from medical conditions such as different types of arthritis, menstrual cramps, and other types of short-term pain. |
|
| Who should not take NSAIDs?
Do not take NSAIDs:
|
|
Before taking NSAIDS, tell your healthcare provider about all of your medical conditions, including if you:
|
|
| What are the possible side effects of NSAIDs? | |
| NSAIDs can cause serious side effects, including:
See " What is the most important information I should know about medicines called Nonsteroidal Anti-inflammatory Drugs (NSAIDs)?
|
|
| Get emergency help right away if you get any of the following symptoms: | |
|
|
| Stop taking your NSAID and call your healthcare provider right away if you get any of the following symptoms: | |
|
|
| If you take too much of your NSAID, call your healthcare provider or get medical help right away.
These are not all the possible side effects of NSAIDs. For more information, ask your healthcare provider or pharmacist about NSAIDs. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|
Other information about NSAIDs
|
|
|
General information about the safe and effective use of NSAIDs
|
|
| Manufactured for: Qingdao BAHEAL Pharmaceutical Co., Ltd.
No. 268 Yingcheng Road, Jimo Qingdao, Shandong, China, 266200 |
|
| LAB number: LAB-0792-1.1 | |
This Medication Guide has been approved by the U.S. Food and Drug Administration. Revised June 2019
PRINCIPAL DISPLAY PANEL - 50 mg Bottle Label
NDC 70247-006-06
Celecoxib Capsules
50 mg
PHARMACIST: Dispense the enclosed
Medication Guide to each patient
60 Capsules
Rx only

PRINCIPAL DISPLAY PANEL - 100 mg Bottle Label
NDC 70247-007-10
Celecoxib Capsules
100 mg
PHARMACIST: Dispense the enclosed
Medication Guide to each patient
100 Capsules
Rx only


