ATORVASTATIN CALCIUM- atorvastatin calcium tablet
Denton Pharma, Inc. DBA Northwind Pharmaceuticals
----------
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ATORVASTATIN CALCIUM TABLETS safely and effectively. See full prescribing information for ATORVASTATIN CALCIUM TABLETS.
ATORVASTATIN CALCIUM tablets, for oral administration Initial U.S. Approval: 1996 INDICATIONS AND USAGEAtorvastatin calcium tablet is an HMG-CoA inhibitor reductase indicated as an adjunct therapy to diet to:
Limitations of Use: Atorvastatin calcium tablets have not been studied in Fredrickson Types I and V dyslipidemias ( 1.3). DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHSTablets: 10, 20 and 40 mg of atorvastatin (3). CONTRAINDICATIONSWARNINGS AND PRECAUTIONS
ADVERSE REACTIONSThe most commonly reported adverse reactions (incidence ≥ 2%) in patients treated with atorvastatin calcium in placebo-controlled trials regardless of causality were: nasopharyngitis, arthralgia, diarrhea, pain in extremity, and urinary tract infection (6.1). To report SUSPECTED ADVERSE REACTIONS, contact Dr. Reddy’s Laboratories Inc., at 1-888-375-3784or FDA at 1-800-FDA-1088 orwww.fda.gov/medwatch. DRUG INTERACTIONSDrug Interactions Associated with Increased Risk of Myopathy/Rhabdomyolysis ( 2.6, 5.1, 7, 12.3)
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and PATIENT COUNSELING INFORMATION. Revised: 12/2020 |
FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
Therapy with lipid-altering agents should be only one component of multiple risk factor intervention in individuals at significantly increased risk for atherosclerotic vascular disease due to hypercholesterolemia. Drug therapy is recommended as an adjunct to diet when the response to a diet restricted in saturated fat and cholesterol and other nonpharmacologic measures alone has been inadequate. In patients with CHD or multiple risk factors for CHD, atorvastatin calcium tablets can be started simultaneously with diet.
1.1 Prevention of Cardiovascular Disease in Adults
In adult patients without clinically evident coronary heart disease, but with multiple risk factors for coronary heart disease such as age, smoking, hypertension, low HDL-C, or a family history of early coronary heart disease, atorvastatin calcium tablets are indicated to:
- Reduce the risk of myocardial infarction
- Reduce the risk of stroke
- Reduce the risk for revascularization procedures and angina
In adult patients with type 2 diabetes, and without clinically evident coronary heart disease, but with multiple risk factors for coronary heart disease such as retinopathy, albuminuria, smoking, or hypertension, atorvastatin calcium tablets are indicated to:
- Reduce the risk of myocardial infarction
- Reduce the risk of stroke
In adult patients with clinically evident coronary heart disease, atorvastatin calcium tablets are indicated to:
- Reduce the risk of non-fatal myocardial infarction
- Reduce the risk of fatal and non-fatal stroke
- Reduce the risk for revascularization procedures
- Reduce the risk of hospitalization for CHF
- Reduce the risk of angina
1.2 Hyperlipidemia
Atorvastatin calcium tablets are indicated:
- As an adjunct to diet to reduce elevated total-C, LDL-C, apo B, and TG levels and to increase HDL-C in adult patients with primary hypercholesterolemia (heterozygous familial and nonfamilial) and mixed dyslipidemia ( Fredrickson Types IIa and IIb);
- As an adjunct to diet for the treatment of adult patients with elevated serum TG levels ( Fredrickson Type IV);
- For the treatment of adult patients with primary dysbetalipoproteinemia ( Fredrickson Type III) who do not respond adequately to diet;
- To reduce total-C and LDL-C in patients with homozygous familial hypercholesterolemia (HoFH) as an adjunct to other lipid-lowering treatments (e.g., LDL apheresis) or if such treatments are unavailable;
- As an adjunct to diet to reduce total-C, LDL-C, and apo B levels in pediatric patients, 10 years to 17 years of age, with heterozygous familial hypercholesterolemia (HeFH) if after an adequate trial of diet therapy the following findings are present:
a. LDL-C remains ≥ 190 mg/dL or
b. LDL-C remains ≥ 160 mg/dL and:
- there is a positive family history of premature cardiovascular disease or
- two or more other CVD risk factors are present in the pediatric patient
2 DOSAGE AND ADMINISTRATION
2.1 Hyperlipidemia and Mixed Dyslipidemia
The recommended starting dose of atorvastatin calcium tablets is 10 or 20 mg once daily. Patients who require a large reduction in LDL-C (more than 45%) may be started at 40 mg once daily. The dosage range of atorvastatin calcium tablets is 10 to 80 mg once daily. Atorvastatin calcium tablets can be administered as a single dose at any time of the day, with or without food. The starting dose and maintenance doses of atorvastatin calcium tablets should be individualized according to patient characteristics such as goal of therapy and response. After initiation and/or upon titration of atorvastatin calcium tablets, lipid levels should be analyzed within 2 to 4 weeks and dosage adjusted accordingly.
2.2 Heterozygous Familial Hypercholesterolemia in Pediatric Patients (10 Years to 17 Years of Age)
The recommended starting dose of atorvastatin calcium tablets is 10 mg/day; the usual dose range is 10 to 20 mg orally once daily [see Clinical Studies ( 14.6) ]. Doses should be individualized according to the recommended goal of therapy [see Indications and Usage ( 1.2) and Clinical Pharmacology ( 12) ]. Doses should be individualized according to the recommended goal of therapy [see Indications and Usage ( 1.2) and Clinical Pharmacology ( 12) ]. Adjustments should be made at intervals of 4 weeks or more.
2.3 Homozygous Familial Hypercholesterolemia
The dosage of atorvastatin calcium tablets in patients with HoFH is 10 to 80 mg daily. Atorvastatin calcium tablets should be used as an adjunct to other lipid-lowering treatments (e.g., LDL apheresis) in these patients or if such treatments are unavailable.
2.4 Concomitant Lipid-Lowering Therapy
Atorvastatin calcium tablets may be used with bile acid resins. The combination of HMG-CoA reductase inhibitors (statins) and fibrates should generally be used with caution [see Warnings and Precautions (5.1) Drug Interactions (7) ].
2.5 Dosage in Patients with Renal Impairment
Renal disease does not affect the plasma concentrations nor LDL-C reduction of atorvastatin calcium tablets; thus, dosage adjustment in patients with renal dysfunction is not necessary [see Warnings and Precautions (5.1) and Clinical Pharmacology, Pharmacokinetics (12.3) ].
2.6 Dosage in Patients Taking Cyclosporine, Clarithromycin, Itraconazole, or Certain Protease Inhibitors
In patients taking cyclosporine or the HIV protease inhibitors (tipranavir plus ritonavir) or the hepatitis C protease inhibitor (telaprevir), therapy with atorvastatin calcium tablets should be avoided. In patients with HIV taking lopinavir plus ritonavir, caution should be used when prescribing atorvastatin calcium tablets and the lowest dose necessary employed. In patients taking clarithromycin, itraconazole, or in patients with HIV taking a combination of saquinavir plus ritonavir, darunavir plus ritonavir, fosamprenavir, or fosamprenavir plus ritonavir, therapy with atorvastatin calcium tablets should be limited to 20 mg, and appropriate clinical assessment is recommended to ensure that the lowest dose necessary of atorvastatin calcium is employed. In patients taking the HIV protease inhibitor nelfinavir, or the hepatitis C protease inhibitor boceprevir, therapy with atorvastatin calcium tablets should be limited to 40 mg, and appropriate clinical assessment is recommended to ensure that the lowest dose necessary of atorvastatin calcium tablets is employed [see Warnings and Precautions, (5.1) and Drug Interactions (7) ].
3 DOSAGE FORMS AND STRENGTHS
Atorvastatin calcium tablets of 10 mg are white to off-white capsule shaped, biconvex, film coated tablets debossed ‘RDY’ on one side and ‘121’on other side.
Atorvastatin calcium tablets of 20 mg are white to off-white capsule shaped, biconvex, film coated tablets debossed ‘RDY’ on one side and ‘122’ on other side.
Atorvastatin calcium tablets of 40 mg are white to off-white capsule shaped, biconvex, film coated tablets debossed ‘RDY’ on one side and ‘123’ on other side.
5 WARNINGS AND PRECAUTIONS
5.1 Skeletal Muscle
Rare cases of rhabdomyolysis with acute renal failure secondary to myoglobinuria have been reported with atorvastatin calciumand with other drugs in this class. A history of renal impairment may be a risk factor for the development of rhabdomyolysis. Such patients merit closer monitoring for skeletal muscle effects.
Atorvastatin, like other statins, occasionally causes myopathy, defined as muscle aches or muscle weakness in conjunction with increases in creatine phosphokinase (CPK) values >10 times ULN. The concomitant use of higher doses of atorvastatin with certain drugs such as cyclosporine and strong CYP3A4 inhibitors (e.g., clarithromycin, itraconazole, and HIV protease inhibitors) increases the risk of myopathy/rhabdomyolysis.
There have been rare reports of immune-mediated necrotizing myopathy (IMNM), an autoimmune myopathy, associated with statin use. IMNM is characterized by: proximal muscle weakness and elevated serum creatine kinase, which persist despite discontinuation of statin treatment; muscle biopsy showing necrotizing myopathy without significant inflammation; improvement with immunosuppressive agents.
Myopathy should be considered in any patient with diffuse myalgias, muscle tenderness or weakness, and/or marked elevation of CPK. Patients should be advised to report promptly unexplained muscle pain, tenderness, or weakness, particularly if accompanied by malaise or fever or if muscle signs and symptoms persist after discontinuing atorvastatin. Atorvastatin calcium therapy should be discontinued if markedly elevated CPK levels occur or myopathy is diagnosed or suspected.
The risk of myopathy during treatment with drugs in this class is increased with concurrent administration of cyclosporine, fibric acid derivatives, erythromycin, clarithromycin, the hepatitis C protease inhibitor telaprevir, combinations of HIV protease inhibitors, including saquinavir plus ritonavir, lopinavir plus ritonavir, tipranavir plus ritonavir, darunavir plus ritonavir, fosamprenavir, and fosamprenavir plus ritonavir, niacin, or azole antifungals. Physicians considering combined therapy with atorvastatin calcium and fibric acid derivatives, erythromycin, clarithromycin, a combination of saquinavir plus ritonavir, lopinavir plus ritonavir, darunavir plus ritonavir, fosamprenavir, or fosamprenavir plus ritonavir, azole antifungals, or lipid-modifying doses of niacin should carefully weigh the potential benefits and risks and should carefully monitor patients for any signs or symptoms of muscle pain, tenderness, or weakness, particularly during the initial months of therapy and during any periods of upward dosage titration of either drug. Lower starting and maintenance doses of atorvastatin should be considered when taken concomitantly with the aforementioned drugs [see Drug Interactions (7)]. Periodic creatine phosphokinase (CPK) determinations may be considered in such situations, but there is no assurance that such monitoring will prevent the occurrence of severe myopathy.
Prescribing recommendations for interacting agents are summarized in Table 2 [see also Dosage and Administration (2.6), Drug Interactions (7), Clinical Pharmacology (12.3)].
Table 2. Drug Interactions Associated with Increased Risk of Myopathy/Rhabdomyolysis
| Interacting Agents | Prescribing Recommendations |
| Cyclosporine, HIV protease inhibitors (tipranavir plus ritonavir), hepatitis C protease inhibitor (telaprevir) | Avoid atorvastatin |
| HIV protease inhibitor (lopinavir plus ritonavir) | Use with caution and lowest dose necessary |
| Clarithromycin, itraconazole, HIV protease inhibitors (saquinavir plus ritonavir*, darunavir plus ritonavir, fosamprenavir, fosamprenavir plus ritonavir) | Do not exceed 20 mg atorvastatin daily |
| HIV protease inhibitor (nelfinavir)
Hepatitis C protease inhibitor (boceprevir) | Do not exceed 40 mg atorvastatin daily |
*Use with caution and with the lowest dose necessary (12.3)
Cases of myopathy, including rhabdomyolysis, have been reported with atorvastatin co-administered with colchicine, and caution should be exercised when prescribing atorvastatin with colchicine [see Drug Interactions (7.11)]
Atorvastatin calcium therapy should be temporarily withheld or discontinued in any patient with an acute, serious condition suggestive of a myopathy or having a risk factor predisposing to the development of renal failure secondary to rhabdomyolysis (e.g., severe acute infection, hypotension, major surgery, trauma, severe metabolic, endocrine and electrolyte disorders, and uncontrolled seizures).
5.2 Liver Dysfunction
Statins, like some other lipid-lowering therapies, have been associated with biochemical abnormalities of liver function. Persistent elevations (>3 times the upper limit of normal [ULN] occurring on 2 or more occasions) in serum transaminases occurred in 0.7% of patients who received atorvastatin calcium in clinical trials. The incidence of these abnormalities was 0.2%, 0.2%, 0.6%, and 2.3% for 10, 20, 40, and 80 mg, respectively.
One patient in clinical trials developed jaundice. Increases in liver function tests (LFT) in other patients were not associated with jaundice or other clinical signs or symptoms. Upon dose reduction, drug interruption, or discontinuation, transaminase levels returned to or near pretreatment levels without sequelae. Eighteen of 30 patients with persistent LFT elevations continued treatment with a reduced dose of atorvastatin calcium.
It is recommended that liver enzyme tests be obtained prior to initiating therapy with atorvastatin calcium and repeated as clinically indicated. There have been rare postmarketing reports of fatal and non-fatal hepatic failure in patients taking statins, including atorvastatin. If serious liver injury with clinical symptoms and/or hyperbilirubinemia or jaundice occurs during treatment with atorvastatin calcium, promptly interrupt therapy. If an alternate etiology is not found, do not restart atorvastatin calcium.
Atorvastatin calcium should be used with caution in patients who consume substantial quantities of alcohol and/or have a history of liver disease. Active liver disease or unexplained persistent transaminase elevations are contraindications to the use of atorvastatin calcium [see Contraindications (4)].
5.3 Endocrine Function
Increased in HbA1c and fasting serum glucose levels have been reported with HMG-CoA reductase inhibitors, including atorvastatin calcium.
Statins interfere with cholesterol synthesis and theoretically might blunt adrenal and/or gonadal steroid production. Clinical studies have shown that atorvastatin calcium does not reduce basal plasma cortisol concentration or impair adrenal reserve. The effects of statins on male fertility have not been studied in adequate numbers of patients. The effects, if any, on the pituitary-gonadal axis in premenopausal women are unknown. Caution should be exercised if a statin is administered concomitantly with drugs that may decrease the levels or activity of endogenous steroid hormones, such as ketoconazole, spironolactone, and cimetidine.
5.4 CNS Toxicity
Brain hemorrhage was seen in a female dog treated for 3 months at 120 mg/kg/day. Brain hemorrhage and optic nerve vacuolation were seen in another female dog that was sacrificed in moribund condition after 11 weeks of escalating doses up to 280 mg/kg/day. The 120 mg/kg dose resulted in a systemic exposure approximately 16 times the human plasma area-under-the-curve (AUC, 0-24 hours) based on the maximum human dose of 80 mg/day. A single tonic convulsion was seen in each of 2 male dogs (one treated at 10 mg/kg/day and one at 120 mg/kg/day) in a 2-year study. No CNS lesions have been observed in mice after chronic treatment for up to 2 years at doses up to 400 mg/kg/day or in rats at doses up to 100 mg/kg/day. These doses were 6 to 11 times (mouse) and 8 to 16 times (rat) the human AUC (0-24) based on the maximum recommended human dose of 80 mg/day.
CNS vascular lesions, characterized by perivascular hemorrhages, edema, and mononuclear cell infiltration of perivascular spaces, have been observed in dogs treated with other members of this class. A chemically similar drug in this class produced optic nerve degeneration (Wallerian degeneration of retinogeniculate fibers) in clinically normal dogs in a dose-dependent fashion at a dose that produced plasma drug levels about 30 times higher than the mean drug level in humans taking the highest recommended dose.
5.5 Use in Patients with Recent Stroke or TIA
In a post-hoc analysis of the Stroke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL) study where atorvastatin calcium 80 mg vs. placebo was administered in 4,731 subjects without CHD who had a stroke or TIA within the preceding 6 months, a higher incidence of hemorrhagic stroke was seen in the atorvastatin calcium 80 mg group compared to placebo (55, 2.3% atorvastatin vs. 33, 1.4% placebo; HR: 1.68, 95% CI: 1.09, 2.59; p=0.0168). The incidence of fatal hemorrhagic stroke was similar across treatment groups (17 vs. 18 for the atorvastatin and placebo groups, respectively). The incidence of nonfatal hemorrhagic stroke was significantly higher in the atorvastatin group (38, 1.6%) as compared to the placebo group (16, 0.7%). Some baseline characteristics, including hemorrhagic and lacunar stroke on study entry, were associated with a higher incidence of hemorrhagic stroke in the atorvastatin group [see Adverse Reactions (6.1)].
6 ADVERSE REACTIONS
The following serious adverse reactions are discussed in greater detail in other sections of the label:
Rhabdomyolysis and myopathy [see Warnings and Precautions (5.1)]
Liver enzyme abnormalities [see Warnings and Precautions (5.2) ]
Because clinical trials are conducted under widely varying conditions, the adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
In the atorvastatin calcium placebo-controlled clinical trial database of 16,066 patients (8755 atorvastatin calcium vs. 7311 placebo; age range 10 to 93 years, 39% women, 91% Caucasians, 3% Blacks, 2% Asians, 4% other) with a median treatment duration of 53 weeks, 9.7% of patients on atorvastatin calcium and 9.5% of the patients on placebo discontinued due to adverse reactions regardless of causality. The five most common adverse reactions in patients treated with atorvastatin calcium that led to treatment discontinuation and occurred at a rate greater than placebo were: myalgia (0.7%), diarrhea (0.5%), nausea (0.4%), alanine aminotransferase increase (0.4%), and hepatic enzyme increase (0.4%).
The most commonly reported adverse reactions (incidence ≥ 2% and greater than placebo) regardless of causality, in patients treated with atorvastatin calcium in placebo controlled trials (n=8755) were: nasopharyngitis (8.3%), arthralgia (6.9%), diarrhea (6.8%), pain in extremity (6%), and urinary tract infection (5.7%).
Table 2 summarizes the frequency of clinical adverse reactions, regardless of causality, reported in ≥ 2% and at a rate greater than placebo in patients treated with atorvastatin calcium (n=8755), from seventeen placebo-controlled trials.
| Table 3. Clinical adverse reactions occurring in ≥ 2% in patients treated with any dose of atorvastatin calcium and at an incidence greater than placebo regardless of causality (% of patients). | ||||||
| Adverse Reaction* | Any dose N=8755 | 10 mg N=3908 | 20 mg N=188 | 40 mg N=604 | 80 mg N=4055 | Placebo N=7311 |
| Nasopharyngitis | 8.3 | 12.9 | 5.3 | 7 | 4.2 | 8.2 |
| Arthralgia | 6.9 | 8.9 | 11.7 | 10.6 | 4.3 | 6.5 |
| Diarrhea | 6.8 | 7.3 | 6.4 | 14.1 | 5.2 | 6.3 |
| Pain in extremity | 6 | 8.5 | 3.7 | 9.3 | 3.1 | 5.9 |
| Urinary tractinfection | 5.7 | 6.9 | 6.4 | 8 | 4.1 | 5.6 |
| Dyspepsia | 4.7 | 5.9 | 3.2 | 6 | 3.3 | 4.3 |
| Nausea | 4 | 3.7 | 3.7 | 7.1 | 3.8 | 3.5 |
| Musculoskeletal pain | 3.8 | 5.2 | 3.2 | 5.1 | 2.3 | 3.6 |
| Muscle Spasms | 3.6 | 4.6 | 4.8 | 5.1 | 2.4 | 3 |
| Myalgia | 3.5 | 3.6 | 5.9 | 8.4 | 2.7 | 3.1 |
| Insomnia | 3 | 2.8 | 1.1 | 5.3 | 2.8 | 2.9 |
| Pharyngolaryngeal pain | 2.3 | 3.9 | 1.6 | 2.8 | 0.7 | 2.1 |
| * Adverse Reaction ≥ 2% in any dose greater than placebo | ||||||
Other adverse reactions reported in placebo-controlled studies include: Body as a whole: malaise, pyrexia; Digestive system: abdominal discomfort, eructation, flatulence, hepatitis, cholestasis; Musculoskeletal system: musculoskeletal pain, muscle fatigue, neck pain, joint swelling; Metabolic and nutritional system: transaminases increase, liver function test abnormal, blood alkaline phosphatase increase, creatine phosphokinase increase, hyperglycemia; Nervous system: nightmare; Respiratory system: epistaxis; Skin and appendages: urticaria; Special senses: vision blurred, tinnitus; Urogenital system: white blood cells urine positive.
Anglo-Scandinavian Cardiac Outcomes Trial (ASCOT)
In ASCOT [see Clinical Studies (14.1)] involving 10,305 participants (age range 40 to 80 years, 19% women; 94.6% Caucasians, 2.6% Africans, 1.5% South Asians, 1.3% mixed/other) treated with atorvastatin calcium 10 mg daily (n=5,168) or placebo (n=5,137), the safety and tolerability profile of the group treated with atorvastatin calcium was comparable to that of the group treated with placebo during a median of 3.3 years of follow-up.
Collaborative Atorvastatin Diabetes Study (CARDS)
In CARDS [see Clinical Studies (14.1)] involving 2,838 subjects (age range 39 to 77 years, 32% women; 94.3% Caucasians, 2.4% South Asians, 2.3% Afro-Caribbean, 1% other) with type 2 diabetes treated with atorvastatin calcium 10 mg daily (n=1,428) or placebo (n=1,410), there was no difference in the overall frequency of adverse reactions or serious adverse reactions between the treatment groups during a median follow-up of 3.9 years. No cases of rhabdomyolysis were reported.
Treating to New Targets Study (TNT)
In TNT [see Clinical Studies (14.1)] involving 10,001 subjects (age range 29 to 78 years, 19% women; 94.1% Caucasians, 2.9% Blacks, 1% Asians, 2% other) with clinically evident CHD treated with atorvastatin calcium 10 mg daily (n=5006) or atorvastatin calcium 80 mg daily (n=4995), there were more serious adverse reactions and discontinuations due to adverse reactions in the high-dose atorvastatin group (92, 1.8%; 497, 9.9%, respectively) as compared to the low-dose group (69, 1.4%; 404, 8.1%, respectively) during a median follow-up of 4.9 years. Persistent transaminase elevations (≥3 x ULN twice within 4 to 10 days) occurred in 62 (1.3%) individuals with atorvastatin 80 mg and in nine (0.2%) individuals with atorvastatin 10 mg. Elevations of CK (≥ 10 x ULN) were low overall, but were higher in the high-dose atorvastatin treatment group (13, 0.3%) compared to the low-dose atorvastatin group (6, 0.1%).
Incremental Decrease in Endpoints through Aggressive Lipid Lowering Study (IDEAL)
In IDEAL [see Clinical Studies (14.1)] involving 8,888 subjects (age range 26 to 80 years, 19% women; 99.3% Caucasians, 0.4% Asians, 0.3% Blacks, 0.04% other) treated with atorvastatin calcium 80 mg/day (n=4439) or simvastatin 20 to 40 mg daily (n=4449), there was no difference in the overall frequency of adverse reactions or serious adverse reactions between the treatment groups during a median follow-up of 4.8 years.
Stroke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL)
In SPARCL involving 4731 subjects (age range 21 to 92 years, 40% women; 93.3% Caucasians, 3% Blacks, 0.6% Asians, 3.1% other) without clinically evident CHD but with a stroke or transient ischemic attack (TIA) within the previous 6 months treated with atorvastatin calcium 80 mg (n=2365) or placebo (n=2366) for a median follow-up of 4.9 years, there was a higher incidence of persistent hepatic transaminase elevations (≥ 3 x ULN twice within 4 to 10 days) in the atorvastatin group (0.9%) compared to placebo (0.1%). Elevations of CK (>10 x ULN) were rare, but were higher in the atorvastatin group (0.1%) compared to placebo (0%). Diabetes was reported as an adverse reaction in 144 subjects (6.1%) in the atorvastatin group and 89 subjects (3.8%) in the placebo group [see Warnings and Precautions (5.5)].
In a post-hoc analysis, atorvastatin calcium 80 mg reduced the incidence of ischemic stroke (218/2365, 9.2% vs. 274/2366, 11.6%) and increased the incidence of hemorrhagic stroke (55/2365, 2.3% vs. 33/2366, 1.4%) compared to placebo. The incidence of fatal hemorrhagic stroke was similar between groups (17 atorvastatin calcium vs. 18 placebo). The incidence of non-fatal hemorrhagic strokes was significantly greater in the atorvastatin group (38 non-fatal hemorrhagic strokes) as compared to the placebo group (16 non-fatal hemorrhagic strokes). Subjects who entered the study with a hemorrhagic stroke appeared to be at increased risk for hemorrhagic stroke [7 (16%) atorvastatin calcium vs. 2 (4%) placebo].
There were no significant differences between the treatment groups for all-cause mortality: 216 (9.1%) in the atorvastatin calcium 80 mg/day group vs. 211 (8.9%) in the placebo group. The proportions of subjects who experienced cardiovascular death were numerically smaller in the atorvastatin calcium 80 mg group (3.3%) than in the placebo group (4.1%). The proportions of subjects who experienced non-cardiovascular death were numerically larger in the atorvastatin calcium 80 mg group (5%) than in the placebo group (4%).
Adverse Reactions from Clinical Studies of Atorvastatin Calcium in Pediatric Patients
In a 26-week controlled study in boys and post-menarchal girls with HeFH (ages 10 years to 17 years) (n=140, 31% female; 92% Caucasians, 1.6% Blacks, 1.6% Asians, 4.8% other), the safety and tolerability profile of atorvastatin calcium 10 to 20 mg daily, as an adjunct to diet to reduce total cholesterol, LDL-C, and apo B levels, was generally similar to that of placebo [see Use in Special Populations ( 8.4) and Clinical Studies ( 14.6) ].
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of atorvastatin calcium. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Adverse reactions associated with atorvastatin calcium therapy reported since market introduction, that are not listed above, regardless of causality assessment, include the following: anaphylaxis, angioneurotic edema, bullous rashes (including erythema multiforme, Stevens-Johnson syndrome, and toxic epidermal necrolysis), rhabdomyolysis, myositis, fatigue, tendon rupture, fatal and non-fatal hepatic failure, dizziness, depression, peripheral neuropathy, pancreatitis and interstitial lung disease.
There have been rare reports of immune-mediated necrotizing myopathy associated with statin use [see Warnings and Precautions (5.1)]
There have been rare postmarketing reports of cognitive impairment (e.g., memory loss, forgetfulness, amnesia, memory impairment, confusion) associated with statin use. These cognitive issues have been reported for all statins. The reports are generally nonserious, and reversible upon statin discontinuation, with variable times to symptom onset (1 day to years) and symptom resolution (median of 3 weeks).
7 DRUG INTERACTIONS
The risk of myopathy during treatment with statins is increased with concurrent administration of fibric acid derivatives, lipid-modifying doses of niacin, cyclosporine, or strong CYP 3A4 inhibitors (e.g., clarithromycin, HIV protease inhibitors, and itraconazole) [see Warnings and Precautions (5.1)andClinical Pharmacology (12.3)].
7.1 Strong Inhibitors of CYP3A4
Atorvastatin calcium is metabolized by cytochrome P450 3A4. Concomitant administration of atorvastatin calcium with strong inhibitors of CYP 3A4 can lead to increases in plasma concentrations of atorvastatin. The extent of interaction and potentiation of effects depend on the variability of effect on CYP 3A4.
Clarithromycin
Atorvastatin AUC was significantly increased with concomitant administration of atorvastatin calcium 80 mg with clarithromycin (500 mg twice daily) compared to that of atorvastatin calcium alone [see Clinical Pharmacology (12.3)]. Therefore, in patients taking clarithromycin, caution should be used when the atorvastatin calcium dose exceeds 20 mg [see Dosage and Administration ( 2.6) and Warnings and Precautions ( 5.1) ].
Combination of Protease Inhibitors
Atorvastatin AUC was significantly increased with concomitant administration of atorvastatin calcium with several combinations of HIV protease inhibitors, as well as with the hepatitis C protease inhibitor telaprevir, compared to that of atorvastatin calcium alone [see Clinical Pharmacology (12.3)]. Therefore, in patients taking the HIV protease inhibitor tipranavir plus ritonavir, or the hepatitis C protease inhibitor telaprevir, concomitant use of atorvastatin calcium should be avoided. In patients taking the HIV protease inhibitor lopinavir plus ritonavir, caution should be used when prescribing atorvastatin calcium and the lowest dose necessary should be used. In patients taking the HIV protease inhibitors saquinavir plus ritonavir, darunavir plus ritonavir, fosamprenavir, or fosamprenavir plus ritonavir, the dose of atorvastatin calcium should not exceed 20 mg and should be used with caution [see Dosage and Administration ( 2.6) andWarnings and Precautions ( 5.1) ].In patients taking the HIV protease inhibitor nelfinavir or the hepatitis C protease inhibitor boceprevir, the dose of atorvastatin calcium should not exceed 40 mg andclose clinicalmonitoring is recommended.
Itraconazole
Atorvastatin AUC was significantly increased with concomitant administration of atorvastatin calcium 40 mg and itraconazole 200 mg [see Clinical Pharmacology ( 12.3)]. Therefore, in patients taking itraconazole, caution should be used when the atorvastatin calcium dose exceeds 20 mg [see Dosage and Administration ( 2.6) and Warnings and Precautions ( 5.1) ].
7.2 Grapefruit Juice
Contains one or more components that inhibit CYP 3A4 and can increase plasma concentrations of atorvastatin, especially with excessive grapefruit juice consumption (>1.2 liters per day).
7.3 Cyclosporine
Atorvastatin and atorvastatin-metabolites are substrates of the OATP1B1 transporter. Inhibitors of the OATP1B1 (e.g., cyclosporine) can increase the bioavailability of atorvastatin. Atorvastatin AUC was significantly increased with concomitant administration of atorvastatin calcium 10 mg and cyclosporine 5.2 mg/kg/day compared to that of atorvastatin calcium alone [see Clinical Pharmacology (12.3)]. The co-administration of atorvastatin calcium with cyclosporine should be avoided [see Warnings and Precautions (5.1)].
7.4 Gemfibrozil
Due to an increased risk of myopathy/rhabdomyolysis when HMG-CoA reductase inhibitors are co-administered with gemfibrozil, concomitant administration of atorvastatin calcium with gemfibrozil should be avoided [see Warnings and Precautions (5.1)].
7.5 Other Fibrates
Because it is known that the risk of myopathy during treatment with HMG-CoA reductase inhibitors is increased with concurrent administration of other fibrates, atorvastatin calcium should be administered with caution when used concomitantly with other fibrates [see Warnings and Precautions (5.1)].
7.6 Niacin
The risk of skeletal muscle effects may be enhanced when atorvastatin calcium is used in combination with niacin; a reduction in atorvastatin calcium dosage should be considered in this setting [see Warnings and Precautions (5.1)].
7.7 Rifampin or other Inducers of Cytochrome P450 3A4
Concomitant administration of atorvastatin calcium with inducers of cytochrome P450 3A4 (e.g., efavirenz, rifampin) can lead to variable reductions in plasma concentrations of atorvastatin. Due to the dual interaction mechanism of rifampin, simultaneous co-administration of atorvastatin calcium with rifampin is recommended, as delayed administration of atorvastatin calcium after administration of rifampin has been associated with a significant reduction in atorvastatin plasma concentrations.
7.8 Digoxin
When multiple doses of atorvastatin calcium and digoxin were co-administered, steady state plasma digoxin concentrations increased by approximately 20%. Patients taking digoxin should be monitored appropriately.
7.9 Oral Contraceptives
Co-administration of atorvastatin calcium and an oral contraceptive increased AUC values for norethindrone and ethinyl estradiol [see Clinical Pharmacology (12.3)]. These increases should be considered when selecting an oral contraceptive for a woman taking atorvastatin calcium.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Atorvastatin calcium is contraindicated for use in pregnant women since safety in pregnant women has not been established and there is no apparent benefit of lipid lowering drugs during pregnancy. Because HMG-CoA reductase inhibitors decrease cholesterol synthesis and possibly the synthesis of other biologically active substances derived from cholesterol, atorvastatin calcium may cause fetal harm when administered to a pregnant woman. Atorvastatin calcium should be discontinued as soon as pregnancy is recognized [see Contraindications ( 4) ]. Limited published data on the use of atorvastatin are insufficient to determine a drug-associated risk of major congenital malformations or miscarriage. In animal reproduction studies in rats and rabbits there was no evidence of embryo-fetal toxicity or congenital malformations at doses up to 30 and 20 times, respectively, the human exposure at the maximum recommended human dose (MRHD) of 80 mg, based on body surface area (mg/m 2). In rats administered atorvastatin during gestation and lactation, decreased postnatal growth and development was observed at doses ≥ 6 times the MRHD (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Human Data
Limited published data on atorvastatin calcium from observational studies, meta-analyses and case reports have not shown an increased risk of major congenital malformations or miscarriage. Rare reports of congenital anomalies have been received following intrauterine exposure to other HMG-CoA reductase inhibitors. In a review of approximately 100 prospectively followed pregnancies in women exposed to simvastatin or lovastatin, the incidences of congenital anomalies, spontaneous abortions, and fetal deaths/stillbirths did not exceed what would be expected in the general population. The number of cases is adequate to exclude a ≥3 to 4-fold increase in congenital anomalies over the background incidence. In 89% of the prospectively followed pregnancies, drug treatment was initiated prior to pregnancy and was discontinued at some point in the first trimester when pregnancy was identified.
Animal Data
Atorvastatin crosses the rat placenta and reaches a level in fetal liver equivalent to that of maternal plasma. Atorvastatin was administered to pregnant rats and rabbits during organogenesis at oral doses up to 300 mg/kg/day and 100 mg/kg/day, respectively. Atorvastatin was not teratogenic in rats at doses up to 300 mg/kg/day or in rabbits at doses up to 100 mg/kg/day. These doses resulted in multiples of about 30 times (rat) or 20 times (rabbit) the human exposure at the MRHD based on surface area (mg/m 2). In rats, the maternally toxic dose of 300 mg/kg resulted in increased post-implantation loss and decreased fetal body weight. At the maternally toxic doses of 50 and 100 mg/kg/day in rabbits, there was increased post-implantation loss, and at 100 mg/kg/day fetal body weights were decreased.
In a study in pregnant rats administered 20, 100, or 225 mg/kg/day from gestation day 7 through to lactation day 20 (weaning), there was decreased survival at birth, postnatal day 4, weaning, and post-weaning in pups of mothers dosed with 225 mg/kg/day, a dose at which maternal toxicity was observed. Pup body weight was decreased through postnatal day 21 at 100 mg/kg/day, and through postnatal day 91 at 225 mg/kg/day. Pup development was delayed (rotorod performance at 100 mg/kg/day and acoustic startle at 225 mg/kg/day; pinnae detachment and eye-opening at 225 mg/kg/day). These doses correspond to 6 times (100 mg/kg) and 22 times (225 mg/kg) the human exposure at the MRHD, based on AUC.
8.2 Lactation
Risk Summary
Atorvastatin calcium use is contraindicated during breastfeeding [see Contraindications ( 4) ]. There is no available information on the effects of the drug on the breastfed infant or the effects of the drug on milk production. It is not known whether atorvastatin is present in human milk, but it has been shown that another drug in this class passes into human milk and atorvastatin is present in rat milk. Because of the potential for serious adverse reactions in a breastfed infant, advise women that breastfeeding is not recommended during treatment with atorvastatin calcium.
8.3 Females and Males of Reproductive Potential
Contraception
Atorvastatin calcium may cause fetal harm when administered to a pregnant woman. Advise females of reproductive potential to use effective contraception during treatment with atorvastatin calcium [see Use in Specific Populations ( 8.1) ].
8.4 Pediatric Use
Heterozygous Familial Hypercholesterolemia (HeFH)
The safety and effectiveness of atorvastatin calcium have been established in pediatric patients, 10 years to 17 years of age, with HeFH as an adjunct to diet to reduce total cholesterol, LDL-C, and apo B levels when, after an adequate trial of diet therapy, the following are present:
• LDL-C ≥ 190 mg/dL, or
• LDL-C ≥ 160 mg/dLand o two or more other CVD risk factors are present.
o a positive family history of FH, or premature CVD in a first, or second-degreerelative, or
o two or more other CVD risk factors are present.
Use of atorvastatin calcium for this indication is supported by evidence from [see Dosage and Administration ( 2.2), Adverse Reactions ( 6.1), Clinical Pharmacology ( 12.3), and Clinical Studies ( 14.6) ]:
• A placebo-controlled clinical trial of 6 months duration in 187 boys and postmenarchal girls, 10 years to 17 years of age. Patients treated with 10 mg or 20 mg daily atorvastatin calcium had an adverse reaction profile generally similar to that of patients treated with placebo. In this limited controlled study, there was no significant effect on growth or sexual maturation in boys or on menstrual cycle length in girls.
Advise postmenarchal girls of contraception recommendations, if appropriate for the patient [see Use in Specific Populations ( 8.1), ( 8.3) ].
The long-term efficacy of atorvastatin calcium therapy initiated in childhood to reduce morbidity and mortality in adulthood has not been established.
The safety and efficacy of atorvastatin calcium have not been established in pediatric patients younger than 10 years of age with HeFH.
Additional pediatric use information is approved for Pfizer’s LIPITOR (atorvastatin calcium) tablets. However, due to Pfizer’s marketing exclusivity rights, this drug product is not labeled with that pediatric information.
Homozygous Familial Hypercholesterolemia (HoFH)
Clinical efficacy of atorvastatin calcium with dosages up to 80 mg/day for 1 year was evaluated in an uncontrolled study of patients with HoFH including 8 pediatric patients [see Clinical Studies ( 14.5) ].
8.5 Geriatric Use
Of the 39,828 patients who received atorvastatin calcium in clinical studies, 15,813 (40%) were ≥65 years old and 2,800 (7%) were ≥75 years old. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older adults cannot be ruled out. Since advanced age (≥65 years) is a predisposing factor for myopathy, atorvastatin calcium should be prescribed with caution in the elderly.
10 OVERDOSAGE
There is no specific treatment for atorvastatin calcium overdosage. In the event of an overdose, the patient should be treated symptomatically, and supportive measures instituted as required. Due to extensive drug binding to plasma proteins, hemodialysis is not expected to significantly enhance atorvastatin calcium clearance.
11 DESCRIPTION
Atorvastatin calcium is a synthetic lipid-lowering agent. Atorvastatin is an inhibitor of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase. This enzyme catalyzes the conversion of HMG-CoA to mevalonate, an early and rate-limiting step in cholesterol biosynthesis.
Atorvastatin calcium is [R-(R*, R*)]-2-(4-fluorophenyl)-ß, δ-dihydroxy-5-(1-methylethyl)-3-phenyl-4-[(phenylamino) carbonyl]-1H-pyrrole-1-heptanoic acid, calcium salt (2:1). The molecular formula of atorvastatin calcium is C 66H 68CaF 2N 4O 10and its molecular weight is 1155.36. Its structural formula is:
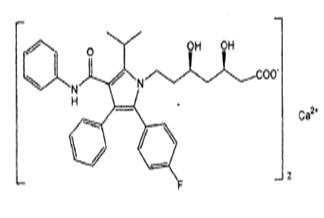
Atorvastatin calcium is a white to off-white colored powder free from visible extraneous matter. Atorvastatin calcium is soluble in dimethyl sulphoxide, slightly soluble in alcohol, very slightly soluble in water, in pH 7.4 phosphate buffer and in acetonitrile and practically insoluble in aqueous solutions of pH 4 and below.
Atorvastatin calcium tablets for oral administration contain 10, 20 and 40 mg atorvastatin and the following inactive ingredients: Basic butylated methacrylate copolymer, crospovidone, hydroxy propyl cellulose, lactose monohydrate, magnesium stearate, methanol, microcrystalline cellulose, sodium bicarbonate and sodium lauryl sulphate. The tablet coating contains isopropyl alcohol, methylene chloride and coloring agent opadry OY-58900 white contains polyethylene glycol, titanium dioxide and hypromellose.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Atorvastatin calcium is a selective, competitive inhibitor of HMG-CoA reductase, the rate-limiting enzyme that converts 3-hydroxy-3methylglutaryl-coenzyme A to mevalonate, a precursor of sterols, including cholesterol. In animal models, LIPITOR lowers plasma cholesterol and lipoprotein levels by inhibiting HMG-CoA reductase and cholesterol synthesis in the liver and by increasing the number of hepatic LDL receptors on the cell surface to enhance uptake and catabolism of LDL; atorvastatin calcium also reduces LDL production and the number of LDL particles.
12.2 Pharmacodynamics
Atorvastatin calcium, as well as some of its metabolites, are pharmacologically active in humans. The liver is the primary site of action and the principal site of cholesterol synthesis and LDL clearance. Drug dosage, rather than systemic drug concentration, correlates better with LDL-C reduction. Individualization of drug dosage should be based on therapeutic response [see Dosage and Administration (2)].
12.3 Pharmacokinetics
Absorption: Atorvastatin calcium is rapidly absorbed after oral administration; maximum plasma concentrations occur within 1 to 2 hours. Extent of absorption increases in proportion to atorvastatin calcium dose. The absolute bioavailability of atorvastatin (parent drug) is approximately 14% and the systemic availability of HMG-CoA reductase inhibitory activity is approximately 30%. The low systemic availability is attributed to presystemic clearance in gastrointestinal mucosa and/or hepatic first-pass metabolism. Although food decreases the rate and extent of drug absorption by approximately 25% and 9%, respectively, as assessed by Cmax and AUC, LDL-C reduction is similar whether atorvastatin calcium is given with or without food. Plasma atorvastatin calcium concentrations are lower (approximately 30% for Cmax and AUC) following evening drug administration compared with morning. However, LDL-C reduction is the same regardless of the time of day of drug administration [see Dosage and Administration (2)].
Distribution: Mean volume of distribution of atorvastatin calcium is approximately 381 liters. Atorvastatin calcium is ≥98% bound to plasma proteins. A blood/plasma ratio of approximately 0.25 indicates poor drug penetration into red blood cells. Based on observations in rats, atorvastatin calcium is likely to be secreted in human milk [see Contraindications, Nursing Mothers (4)and Use in Specific Populations (8.2) ].
Metabolism: Atorvastatin calcium is extensively metabolized to ortho- and parahydroxylated derivatives and various beta-oxidation products. In vitro inhibition of HMG-CoA reductase by ortho- and parahydroxylated metabolites is equivalent to that of atorvastatin calcium. Approximately 70% of circulating inhibitory activity for HMG-CoA reductase is attributed to active metabolites. In vitro studies suggest the importance of atorvastatin calcium metabolism by cytochrome P450 3A4, consistent with increased plasma concentrations of atorvastatin calcium in humans following co-administration with erythromycin, a known inhibitor of this isozyme [see Drug Interactions (7.1)]. In animals, the ortho-hydroxy metabolite undergoes further glucuronidation.
Excretion: Atorvastatin calcium and its metabolites are eliminated primarily in bile following hepatic and/or extra-hepatic metabolism; however, the drug does not appear to undergo enterohepatic recirculation. Mean plasma elimination half-life of atorvastatin calcium in humans is approximately 14 hours, but the half-life of inhibitory activity for HMG-CoA reductase is 20 to 30 hours due to the contribution of active metabolites. Less than 2% of a dose of atorvastatin calcium is recovered in urine following oral administration.
Specific Populations
Geriatric: Plasma concentrations of atorvastatin calcium are higher (approximately 40% for Cmax and 30% for AUC) in healthy elderly subjects (age ≥65 years) than in young adults. Clinical data suggest a greater degree of LDL-lowering at any dose of drug in the elderly patient population compared to younger adults [see Use in Specific Populations (8.5)].
Pediatric: Apparent oral clearance of atorvastatin in pediatric subjects appeared similar to that of adults when scaled allometrically by body weight as the body weight was the only significant covariate in atorvastatin population PK model with data including pediatric HeFH patients (ages 10 years to 17 years of age, n=29) in an open-label, 8-week study.
Gender : Plasma concentrations of atorvastatin calcium in women differ from those in men (approximately 20% higher for Cmax and 10% lower for AUC); however, there is no clinically significant difference in LDL-C reduction with atorvastatin calcium between men and women.
Renal Impairment: Renal disease has no influence on the plasma concentrations or LDL-C reduction of atorvastatin calcium; thus, dose adjustment in patients with renal dysfunction is not necessary [see Dosage and Administration (2.5) and Warnings and Precautions (5.1) ].
Hemodialysis: While studies have not been conducted in patients with end-stage renal disease, hemodialysis is not expected to significantly enhance clearance of atorvastatin calcium since the drug is extensively bound to plasma proteins.
Hepatic Impairment: In patients with chronic alcoholic liver disease, plasma concentrations of atorvastatin calcium are markedly increased. Cmax and AUC are each 4-fold greater in patients with Childs-Pugh A disease. Cmax and AUC are approximately 16-fold and 11-fold increased, respectively, in patients with Childs-Pugh B disease [see Contraindications (4)].
TABLE 4. Effect of Co-administered Drugs on the Pharmacokinetics of Atorvastatin
| Co-administered drug and dosing regimen | Atorvastatin | ||
| Dose (mg) | Change in AUC & | Change in Cmax & | |
| #Cyclosporine 5.2 mg/kg/day, stable dose | 10 mg QD for 28 days | ↑ 8.7 fold | ↑10.7 fold |
| #Tipranavir 500 mg BID/ritonavir 200 mg BID, 7 days | 10 mg, SD | ↑ 9.4 fold | ↑ 8.6 fold |
| #Telaprevir 750 mg q8h, 10 days | 20 mg, SD | ↑ 7.88 fold | ↑ 10.6 fold |
| #, ‡Saquinavir 400 mg BID/ ritonavir 400mg BID,
15 days | 40 mg QD for 4 days | ↑ 3.9 fold | ↑ 4.3 fold |
| #Clarithromycin 500 mg BID, 9 days | 80 mg QD for 8 days | ↑ 4.4 fold | ↑ 5.4 fold |
| #Darunavir 300 mg BID/ritonavir 100 mg BID, 9 days | 10 mg QD for 4 days | ↑3.4 fold | ↑2.25 fold |
| #Itraconazole 200 mg QD, 4 days | 40 mg SD | ↑ 3.3 fold | ↑ 20% |
| #Fosamprenavir 700 mg BID/ritonavir 100 mg BID,
14 days | 10 mg QD for 4 days | ↑ 2.53 fold | ↑2.84 fold |
| #Fosamprenavir 1400 mg BID, 14 days | 10 mg QD for 4 days | ↑ 2.3 fold | ↑ 4.04 fold |
| #Nelfinavir 1250 mg BID, 14 days | 10 mg QD for 28 days | ↑ 74% | ↑ 2.2 fold |
| #Grapefruit Juice, 240 mL QD * | 40 mg, SD | ↑ 37% | ↑ 16% |
| Diltiazem 240 mg QD, 28 days | 40 mg, SD | ↑ 51% | No change |
| Erythromycin 500 mg QID, 7 days | 10 mg, SD | ↑ 33% | ↑ 38% |
| Amlodipine 10 mg, single dose | 80 mg, SD | ↑ 15% | ↓ 12 % |
| Cimetidine 300 mg QID, 2 weeks | 10 mg QD for 2 weeks | ↓ Less than 1% | ↓ 11% |
| Colestipol 10 mg BID, 28 weeks | 40 mg QD for 28 weeks | Not determined | ↓ 26%** |
| Maalox TC® 30 mL QD, 17 days | 10 mg QD for 15 days | ↓ 33% | ↓ 34% |
| Efavirenz 600 mg QD, 14 days | 10 mg for 3 days | ↓ 41% | ↓ 1% |
| #Rifampin 600 mg QD, 7 days (co-administered) † | 40 mg SD | ↑ 30% | ↑ 2.7 fold |
| #Rifampin 600 mg QD, 5 days (doses separated) † | 40 mg SD | ↓ 80% | ↓ 40% |
| #Gemfibrozil 600mg BID, 7 days | 40mg SD | ↑ 35% | ↓ Less than 1% |
| #Fenofibrate 160mg QD, 7 days | 40mg SD | ↑ 3% | ↑ 2% |
| Boceprevir 800 mg TID, 7 days | 40 mg SD | ↑ 2.30 fold | ↑2.66 fold |
& Data given as x-fold change represent a simple ratio between co-administration and atorvastatin alone (i.e., 1-fold = no change). Data given as % change represent % difference relative to atorvastatin alone (i.e., 0% = no change).
# See Sections 5.1 and 7 for clinical significance.
* Greater increases in AUC (up to 2.5 fold) and/or Cmax (up to 71%) have been reported with excessive grapefruit consumption (≥ 750 mL - 1.2 liters per day).
** Single sample taken 8-16 h post dose.
† Due to the dual interaction mechanism of rifampin, simultaneous co-administration of atorvastatin with rifampin is recommended, as delayed administration of atorvastatin after administration of rifampin has been associated with a significant reduction in atorvastatin plasma concentrations.
‡ The dose of saquinavir plus ritonavir in this study is not the clinically used dose. The increase in atorvastatin exposure when used clinically is likely to be higher than what was observed in this study. Therefore, caution should be applied and the lowest dose necessary should be used.
TABLE 5. Effect of Atorvastatin on the Pharmacokinetics of Co-administered Drugs
| Atorvastatin | Co-administered drug and dosing regimen | ||
| Drug/Dose (mg) | Change in AUC | Change in Cmax | |
| 80 mg QD for 15 days | Antipyrine, 600 mg SD | ↑ 3% | ↓ 11% |
| 80 mg QD for 14 days | # Digoxin 0.25 mg QD, 20 days | ↑ 15% | ↑ 20 % |
| 40 mg QD for 22 days | Oral contraceptive QD, 2 months
- norethindrone 1mg - ethinyl estradiol 35μg |
↑ 28% ↑ 19% |
↑ 23% ↑ 30% |
| 10 mg, SD | Tipranavir 500 mg BID/ritonavir 200 mg BID, 7 days | No change | No change |
| 10 mg QD for 4 days | Fosamprenavir 1400 mg BID, 14 days | ↓ 27% | ↓ 18% |
| 10 mg QD for 4 days | Fosamprenavir 700 mgBID/ritonavir 100 mg BID, 14 days | No change | No change |
# See Section 7 for clinical significance.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
In a 2-year carcinogenicity study in rats at dose levels of 10, 30, and 100 mg/kg/day, 2 rare tumors were found in muscle in high-dose females: in one, there was a rhabdomyosarcoma and, in another, there was a fibrosarcoma. This dose represents a plasma AUC (0 to 24) value of approximately 16 times the mean human plasma drug exposure after an 80 mg oral dose.
A 2-year carcinogenicity study in mice given 100, 200, or 400 mg/kg/day resulted in a significant increase in liver adenomas in high-dose males and liver carcinomas in high-dose females. These findings occurred at plasma AUC (0 to 24) values of approximately 6 times the mean human plasma drug exposure after an 80 mg oral dose.
In vitro, atorvastatin was not mutagenic or clastogenic in the following tests with and without metabolic activation: the Ames test with Salmonella typhimurium and Escherichia coli, the HGPRT forward mutation assay in Chinese hamster lung cells, and the chromosomal aberration assay in Chinese hamster lung cells. Atorvastatin was negative in the in vivo mouse micronucleus test.
In female rats, atorvastatin at doses up to 225 mg/kg (56 times the human exposure) did not cause adverse effects on fertility. Studies in male rats performed at doses up to 175 mg/kg (15 times the human exposure) produced no changes in fertility. There was aplasia and aspermia in the epididymis of 2 of 10 rats treated with 100 mg/kg/day of atorvastatin for 3 months (16 times the human AUC at the 80 mg dose); testis weights were significantly lower at 30 and 100 mg/kg and epididymal weight was lower at 100 mg/kg. Male rats given 100 mg/kg/day for 11 weeks prior to mating had decreased spermmotility, spermatid head concentration, and increased abnormal sperm. Atorvastatin caused no adverse effects on semen parameters, or reproductive organ histopathology in dogs given doses of 10, 40, or 120 mg/kg for two years.
14 CLINICAL STUDIES
14.1 Prevention of Cardiovascular Disease
In the Anglo-Scandinavian Cardiac Outcomes Trial (ASCOT), the effect of atorvastatin calcium on fatal and non-fatal coronary heart disease was assessed in 10,305 hypertensive patients 40 to 80 years of age (mean of 63 years), without a previous myocardial infarction and with TC levels ≤251 mg/dL (6.5 mmol/L). Additionally, all patients had at least 3 of the following cardiovascular risk factors: male gender (81.1%), age >55 years (84.5%), smoking (33.2%), diabetes (24.3%), history of CHD in a first-degree relative (26%), TC:HDL >6 (14.3%), peripheral vascular disease (5.1%), left ventricular hypertrophy (14.4%), prior cerebrovascular event (9.8%), specific ECG abnormality (14.3%), proteinuria/albuminuria (62.4%). In this double-blind, placebo-controlled study, patients were treated with anti-hypertensive therapy (Goal BP <140/90 mm Hg for non-diabetic patients; <130/80 mm Hg for diabetic patients) and allocated to either atorvastatin calcium 10 mg daily (n=5168) or placebo (n=5137), using a covariate adaptive method which took into account the distribution of nine baseline characteristics of patients already enrolled and minimized the imbalance of those characteristics across the groups. Patients were followed for a median duration of 3.3 years.
The effect of 10 mg/day of atorvastatin calcium on lipid levels was similar to that seen in previous clinical trials.
Atorvastatin calcium significantly reduced the rate of coronary events [either fatal coronary heart disease (46 events in the placebo group vs. 40 events in the atorvastatin calcium group) or non-fatal MI (108 events in the placebo group vs. 60 events in the atorvastatin calcium group)] with a relative risk reduction of 36% [(based on incidences of 1.9% for atorvastatin calcium vs. 3% for placebo), p=0.0005 (see Figure 1)]. The risk reduction was consistent regardless of age, smoking status, obesity, or presence of renal dysfunction. The effect of atorvastatin calcium was seen regardless of baseline LDL levels. Due to the small number of events, results for women were inconclusive.
Figure 1: Effect of Atorvastatin Calcium 10 mg/day on Cumulative Incidence of Non-Fatal Myocardial Infarction or Coronary Heart Disease Death (in ASCOT-LLA)
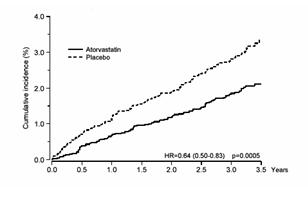
Atorvastatin calcium also significantly decreased the relative risk for revascularization procedures by 42%. Although the reduction of fatal and non-fatal strokes did not reach a pre-defined significance level (p=0.01), a favorable trend was observed with a 26% relative risk reduction (incidences of 1.7% for atorvastatin calcium and 2.3% for placebo). There was no significant difference between the treatment groups for death due to cardiovascular causes (p=0.51) or noncardiovascular causes (p=0.17).
In the Collaborative Atorvastatin Diabetes Study (CARDS), the effect of atorvastatin calcium on cardiovascular disease (CVD) endpoints was assessed in 2838 subjects (94% white, 68% male), ages 40 to 75 with type 2 diabetes based on WHO criteria, without prior history of cardiovascular disease and with LDL ≤ 160 mg/dL and TG ≤ 600 mg/dL. In addition to diabetes, subjects had 1 or more of the following risk factors: current smoking (23%), hypertension (80%), retinopathy (30%), or microalbuminuria (9%) or macroalbuminuria (3%). No subjects on hemodialysis were enrolled in the study. In this multicenter, placebo-controlled, double-blind clinical trial, subjects were randomly allocated to either atorvastatin calcium 10 mg daily (1429) or placebo (1411) in a 1:1 ratio and were followed for a median duration of 3.9 years. The primary endpoint was the occurrence of any of the major cardiovascular events: myocardial infarction, acute CHD death, unstable angina, coronary revascularization, or stroke. The primary analysis was the time to first occurrence of the primary endpoint.
Baseline characteristics of subjects were: mean age of 62 years, mean HbA 1c 7.7%; median LDL-C 120 mg/dL; median TC 207 mg/dL; median TG 151 mg/dL; median HDL-C 52 mg/dL.
The effect of atorvastatin calcium 10 mg/day on lipid levels was similar to that seen in previous clinical trials.
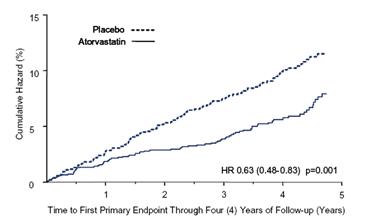
Atorvastatin calcium significantly reduced the rate of major cardiovascular events (primary endpoint events) (83 events in the atorvastatin calcium group vs. 127 events in the placebo group) with a relative risk reduction of 37%, HR 0.63, 95% CI (0.48, 0.83) (p=0.001) (see Figure 2). An effect of atorvastatin calcium was seen regardless of age, sex, or baseline lipid levels.
Atorvastatin calcium significantly reduced the risk of stroke by 48% (21 events in the atorvastatin calcium group vs. 39 events in the placebo group), HR 0.52, 95% CI (0.31, 0.89) (p=0.016) and reduced the risk of MI by 42% (38 events in the atorvastatin calcium group vs. 64 events in the placebo group), HR 0.58, 95.1% CI (0.39, 0.86) (p=0.007). There was no significant difference between the treatment groups for angina, revascularization procedures, and acute CHD death.
There were 61 deaths in the atorvastatin calcium group vs. 82 deaths in the placebo group (HR 0.73, p=0.059).
Figure 2: Effect of Atorvastatin Calcium 10 mg/day on Time to Occurrence of Major Cardiovascular Event (myocardial infarction, acute CHD death, unstable angina, coronary revascularization, or stroke) in CARDS
In the Treating to New Targets Study (TNT), the effect of atorvastatin calcium 80 mg/day vs. atorvastatin calcium 10 mg/day on the reduction in cardiovascular events was assessed in 10,001 subjects (94% white, 81% male, 38% ≥65 years) with clinically evident coronary heart disease who had achieved a target LDL-C level <130 mg/dL after completing an 8-week, open-label, run-in period with atorvastatin calcium 10 mg/day. Subjects were randomly assigned to either 10 mg/day or 80 mg/day of atorvastatin calcium and followed for a median duration of 4.9 years. The primary endpoint was the time-to-first occurrence of any of the following major cardiovascular events (MCVE): death due to CHD, non-fatal myocardial infarction, resuscitated cardiac arrest, and fatal and non-fatal stroke. The mean LDL-C, TC, TG, non-HDL, and HDL cholesterol levels at 12 weeks were 73, 145, 128, 98, and 47 mg/dL during treatment with 80 mg of atorvastatin calcium and 99, 177, 152, 129, and 48 mg/dL during treatment with 10 mg of atorvastatin calcium.
Treatment with atorvastatin calcium 80 mg/day significantly reduced the rate of MCVE (434 events in the 80 mg/day group vs. 548 events in the 10 mg/day group) with a relative risk reduction of 22%, HR 0.78, 95% CI (0.69, 0.89), p=0.0002 (see Figure 3 and Table 6). The overall risk reduction was consistent regardless of age (<65, ≥65) or gender.
Figure 3: Effect of Atorvastatin Calcium 80 mg/day vs. 10 mg/day on Time to Occurrence of Major Cardiovascular Events (TNT)
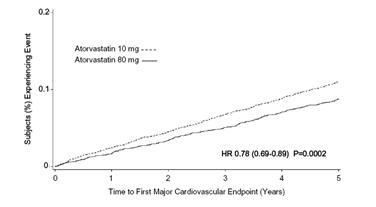
TABLE 6. Overview of Efficacy Results in TNT
| Endpoint | Atorvastatin | Atorvastatin | HR a (95%CI) | ||
| 10 mg | 80 mg | ||||
| (N=5006) | (N=4995) | ||||
| PRIMARY ENDPOINT | n | (%) | n | (%) | |
| First major cardiovascular endpoint | 548 | (10.9) | 434 | (8.7) | 0.78 (0.69, 0.89) |
| Components of the Primary Endpoint | |||||
| CHD death | 127 | (2.5) | 101 | (2) | 0.80 (0.61, 1.03) |
| Non-fatal, non-procedure related MI | 308 | (6.2) | 243 | (4.9) | 0.78 (0.66, 0.93) |
| Resuscitated cardiac arrest | 26 | (0.5) | 25 | (0.5) | 0.96 (0.56, 1.67) |
| Stroke (fatal and non-fatal) | 155 | (3.1) | 117 | (2.3) | 0.75 (0.59, 0.96) |
| SECONDARY ENDPOINTS* | |||||
| First CHF with hospitalization | 164 | (3.3) | 122 | (2.4) | 0.74 (0.59, 0.94) |
| First PVD endpoint | 282 | (5.6) | 275 | (5.5) | 0.97 (0.83, 1.15) |
| First CABG or other coronary revascularization procedure b | 904 | (18.1) | 667 | (13.4) | 0.72 (0.65, 0.80) |
| First documented angina endpoint b | 615 | (12.3) | 545 | (10.9) | 0.88 (0.79, 0.99) |
| All-cause mortality | 282 | (5.6) | 284 | (5.7) | 1.01 (0.85, 1.19) |
| Components of All-Cause Mortality | |||||
| Cardiovascular death | 155 | (3.1) | 126 | (2.5) | 0.81 (0.64, 1.03) |
| Noncardiovascular death | 127 | (2.5) | 158 | (3.2) | 1.25 (0.99, 1.57) |
| Cancer death | 75 | (1.5) | 85 | (1.7) | 1.13 (0.83, 1.55) |
| Other non-CV death | 43 | (0.9) | 58 | (1.2) | 1.35 (0.91, 2) |
| Suicide, homicide, and other traumatic non-CV death | 9 | (0.2) | 15 | (0.3) | 1.67 (0.73, 3.82) |
a Atorvastatin 80 mg: atorvastatin 10 mg
b Component of other secondary endpoints
* Secondary endpoints not included in primary endpoint HR=hazard ratio; CHD=coronary heart disease; CI=confidence interval; MI=myocardial infarction; CHF=congestive heart failure; CV=cardiovascular; PVD=peripheral vascular disease; CABG=coronary artery bypass graft
Confidence intervals for the Secondary Endpoints were not adjusted for multiple comparisons
Of the events that comprised the primary efficacy endpoint, treatment with atorvastatin calcium 80 mg/day significantly reduced the rate of non-fatal, non-procedure related MI and fatal and non-fatal stroke, but not CHD death or resuscitated cardiac arrest (Table 5). Of the predefined secondary endpoints, treatment with atorvastatin calcium 80 mg/day significantly reduced the rate of coronary revascularization, angina, and hospitalization for heart failure, but not peripheral vascular disease. The reduction in the rate of CHF with hospitalization was only observed in the 8% of patients with a prior history of CHF.
There was no significant difference between the treatment groups for all-cause mortality (Table 5). The proportions of subjects who experienced cardiovascular death, including the components of CHD death and fatal stroke, were numerically smaller in the atorvastatin calcium 80 mg group than in the atorvastatin calcium 10 mg treatment group. The proportions of subjects who experienced noncardiovascular death were numerically larger in the atorvastatin calcium 80 mg group than in the atorvastatin calcium 10 mg treatment group.
In the Incremental Decrease in Endpoints Through Aggressive Lipid Lowering Study (IDEAL), treatment with atorvastatin calcium 80 mg/day was compared to treatment with simvastatin 20 to 40 mg/day in 8,888 subjects up to 80 years of age with a history of CHD to assess whether reduction in CV risk could be achieved. Patients were mainly male (81%), white (99%) with an average age of 61.7 years, and an average LDL-C of 121.5 mg/dL at randomization; 76% were on statin therapy. In this prospective, randomized, open-label, blinded endpoint (PROBE) trial with no run-in period, subjects were followed for a median duration of 4.8 years. The mean LDL-C, TC, TG, HDL, and non-HDL cholesterol levels at Week 12 were 78, 145, 115, 45, and 100 mg/dL during treatment with 80 mg of atorvastatin calcium and 105, 179, 142, 47, and 132 mg/dL during treatment with 20 to 40 mg of simvastatin.
There was no significant difference between the treatment groups for the primary endpoint, the rate of first major coronary event (fatal CHD, non-fatal MI, and resuscitated cardiac arrest): 411 (9.3%) in the atorvastatin calcium 80 mg/day group vs. 463 (10.4%) in the simvastatin 20 to 40 mg/day group, HR 0.89, 95% CI ( 0.78, 1.01), p=0.07.
There were no significant differences between the treatment groups for all-cause mortality: 366 (8.2%) in the atorvastatin calcium 80 mg/day group vs. 374 (8.4%) in the simvastatin 20 to 40 mg/day group. The proportions of subjects who experienced CV or non-CV death were similar for the atorvastatin calcium 80 mg group and the simvastatin 20 to 40 mg group.
14.2 Hyperlipidemia and Mixed Dyslipidemia
Atorvastatin calcium reduces total-C, LDL-C, VLDL-C, apo B, and TG, and increases HDL-C in patients with hyperlipidemia (heterozygous familial and nonfamilial) and mixed dyslipidemia (Fredrickson Types IIa and IIb). Therapeutic response is seen within 2 weeks, and maximum response is usually achieved within 4 weeks and maintained during chronic therapy.
Atorvastatin calcium is effective in a wide variety of patient populations with hyperlipidemia, with and without hypertriglyceridemia, in men and women, and in the elderly.
In two multicenter, placebo-controlled, dose-response studies in patients with hyperlipidemia, atorvastatin calcium given as a single dose over 6 weeks, significantly reduced total-C, LDL-C, apo B, and TG. (Pooled results are provided in Table 7.)
TABLE 7. Dose Response in Patients With Primary Hyperlipidemia (Adjusted Mean % Change From Baseline) a
| Dose | N | TC | LDL-C | Apo B | TG | HDL-C | Non-HDLC/ HDL-C |
| Placebo | 21 | 4 | 4 | 3 | 10 | -3 | 7 |
| 10 | 22 | -29 | -39 | -32 | -19 | 6 | -34 |
| 20 | 20 | -33 | -43 | -35 | -26 | 9 | -41 |
| 40 | 21 | -37 | -50 | -42 | -29 | 6 | -45 |
| 80 | 23 | -45 | -60 | -50 | -37 | 5 | -53 |
a Results are pooled from 2 dose-response studies.
In patients with Fredrickson Types IIa and IIb hyperlipoproteinemia pooled from 24 controlled trials, the median (25 th and 75 th percentile) percent changes from baseline in HDL-C for atorvastatin calcium 10, 20, 40, and 80 mg were 6.4 (-1.4, 14), 8.7 (0, 17), 7.8 (0, 16), and 5.1 (-2.7, 15), respectively. Additionally, analysis of the pooled data demonstrated consistent and significant decreases in total-C, LDL-C, TG, total-C/HDL-C, and LDL-C/HDL-C.
In three multicenter, double-blind studies in patients with hyperlipidemia, atorvastatin calcium was compared to other statins. After randomization, patients were treated for 16 weeks with either atorvastatin calcium 10 mg per day or a fixed dose of the comparative agent (Table 8).
TABLE 8. Mean Percentage Change From Baseline at Endpoint (Double-Blind, Randomized, Active-Controlled Trials)
| Treatment
(Daily Dose) | N | Total-C | LDL-C | Apo B | TG | HDL-C | Non-HDL-C/ HDL-C |
| Study 1 | |||||||
| Atorvastatin Calcium 10 mg | 707 | -27 a | -36 a | -28 a | -17 a | +7 | -37 a |
| Lovastatin 20 mg 95% CI for Diff 1 | 191 | -19
-9.2, -6.5 | -27
-10.7, -7.1 | -20
-10, -6.5 | -6
-15.2, -7.1 | +7
-1.7, 2 | -28
-11.1, -7.1 |
| Study 2 Atorvastatin Calcium 10 mg | 222 | -25 b | -35 b | -27 b | -17 b | +6 | -36 b |
| Pravastatin 20 mg 95% CI for Diff 1 | 77 | -17
-10.8, -6.1 | -23
-14.5, -8.2 | -17
-13.4, -7.4 | -9
-14.1, -0.7 | +8
-4.9, 1.6 | -28
-11.5, -4.1 |
| Study 3 | |||||||
| Atorvastatin Calcium 10 mg | 132 | -29 c | -37 c | -34 c | -23 c | +7 | -39 c |
| Simvastatin 10 mg 95% CI for Diff 1 | 45 | -24
-8.7, -2.7 | -30
-10.1, -2.6 | -30
-8, -1.1 | -15
-15.1, -0.7 | +7
-4.3, 3.9 | -33
-9.6, -1.9 |
1 A negative value for the 95% CI for the difference between treatments favors atorvastatin calcium for all except HDL-C, for which a positive value favors atorvastatin calcium. If the range does not include 0, this indicates a statistically significant difference.
a Significantly different from lovastatin, ANCOVA, p ≤0.05
b Significantly different from pravastatin, ANCOVA, p ≤0.05
c Significantly different from simvastatin, ANCOVA, p ≤0.05
The impact on clinical outcomes of the differences in lipid-altering effects between treatments shown in Table 7 is not known. Table 7 does not contain data comparing the effects of atorvastatin calcium 10 mg and higher doses of lovastatin, pravastatin, and simvastatin. The drugs compared in the studies summarized in the table are not necessarily interchangeable.
14.3 Hypertriglyceridemia
The response to atorvastatin calcium in 64 patients with isolated hypertriglyceridemia(Fredrickson Type IV) treated across several clinical trials is shown in the table below (Table 9). For the atorvastatin calcium-treated patients, median (min, max) baseline TG level was 565 (267 to 1502).
TABLE 9. Combined Patients With Isolated Elevated TG: Median (min, max) Percentage Change From Baseline
| Placebo | Atorvastatin Calcium | Atorvastatin Calcium | Atorvastatin Calcium | |
| 10 mg | 20 mg | 80 mg | ||
| (N=12) | (N=37) | (N=13) | (N=14) | |
| Triglycerides | -12.4 (-36.6, 82.7) | -41 (-76.2, 49.4) | -38.7 (-62.7, 29.5) | -51.8 (-82.8, 41.3) |
| Total-C | -2.3 (-15.5, 24.4) | -28.2 (-44.9, -6.8) | -34.9 (-49.6, -15.2) | -44.4 (-63.5, -3.8) |
| LDL-C | 3.6 (-31.3, 31.6) | -26.5 (-57.7, 9.8) | -30.4 (-53.9, 0.3) | -40.5 (-60.6, -13.8) |
| HDL-C | 3.8 (-18.6, 13.4) | 13.8 (-9.7, 61.5) | 11 (-3.2, 25.2) | 7.5 (-10.8, 37.2) |
| VLDL-C | -1 (-31.9, 53.2) | -48.8 (-85.8, 57.3) | -44.6 (-62.2, -10.8) | -62 (-88.2, 37.6) |
| non-HDL-C | -2.8 (-17.6, 30) | -33 (-52.1, -13.3) | -42.7 (-53.7, -17.4) | -51.5 (-72.9, -4.3) |
14.4 Dysbetalipoproteinemia
The results of an open-label crossover study of 16 patients (genotypes: 14 apo E2/E2 and 2 apo E3/E2) with dysbetalipoproteinemia ( Fredrickson Type III) are shown in the table below (Table 10).
TABLE 10. Open-Label Crossover Study of 16 Patients With Dysbetalipoproteinemia (Fredrickson Type III)
| Median % Change (min, max) | |||
| Median (min, max) at | Atorvastatin Calcium | Atorvastatin Calcium | |
| Baseline (mg/dL) | 10 mg | 80 mg | |
| Total-C | 442 (225, 1320) | -37 (-85, 17) | -58 (-90, -31) |
| Triglycerides | 678 (273, 5990) | -39 (-92, -8) | -53 (-95, -30) |
| IDL-C + VLDL-C | 215 (111, 613) | -32 (-76, 9) | -63 (-90, -8) |
| non-HDL-C | 411 (218, 1272) | -43 (-87, -19) | -64 (-92, -36) |
14.5 Homozygous Familial Hypercholesterolemia
In a study without a concurrent control group, 29 patients ages 6 years to 37 years with HoFH received maximum daily doses of 20 to 80 mg of atorvastatin calcium. The mean LDL-C reduction in this study was 18%. Twenty-five patients with a reduction in LDL-C had a mean response of 20% (range of 7% to 53%, median of 24%); the remaining 4 patients had 7% to 24% increases in LDL-C. Five of the 29 patients had absent LDL-receptor function. Of these, 2 patients also had a portacaval shunt and had no significant reduction in LDL-C. The remaining 3 receptor-negative patients had a mean LDL-C reduction of 22%.
14.6 Heterozygous Familial Hypercholesterolemia in Pediatric Patients
In a double-blind, placebo-controlled study followed by an open-label phase, 187 boys and post-menarchal girls 10 years to 17 years of age (mean age 14.1 years) with heterozygous familial hypercholesterolemia (HeFH) or severe hypercholesterolemia, were randomized to atorvastatin calcium (n=140) or placebo (n=47) for 26 weeks and then all received atorvastatin calcium for 26 weeks. Inclusion in the study required 1) a baseline LDL-C level ≥ 190 mg/dL or 2) a baseline LDL-C level ≥ 160 mg/dL and positive family history of FH or documented premature cardiovascular disease in a first or second-degree relative. The mean baseline LDL-C value was 218.6 mg/dL (range: 138.5 to 385 mg/dL) in the atorvastatin calcium group compared to 230 mg/dL (range: 160 to 324.5 mg/dL) in the placebo group. The dosage of atorvastatin calcium (once daily) was 10 mg for the first 4 weeks and uptitrated to 20 mg if the LDL-C level was > 130 mg/dL. The number of atorvastatin calcium-treated patients who required uptitration to 20 mg after Week 4 during the double-blind phase was 78 (55.7%).
Atorvastatin calcium significantly decreased plasma levels of total-C, LDL-C, triglycerides, and apolipoprotein B during the 26-week double-blind phase (see Table 11).
TABLE 11. Lipid-altering Effects of Atorvastatin Calcium in Adolescent Boys and Girls with Heterozygous Familial Hypercholesterolemia or Severe Hypercholesterolemia (Mean Percentage Change From Baseline at Endpoint in Intention-to-Treat Population)
| DOSAGE | N | Total-C | LDL-C | HDL-C | TG | Apolipoprotein B |
| Placebo | 47 | -1.5 | -0.4 | -1.9 | 1 | 0.7 |
| Atorvastatin Calcium | 140 | -31.4 | -39.6 | 2.8 | -12 | -34 |
The mean achieved LDL-C value was 130.7 mg/dL (range: 70 to 242 mg/dL) in the atorvastatin calcium group compared to 228.5 mg/dL (range: 152 to 385 mg/dL) in the placebo group during the 26-week double-blind phase.
The long-term efficacy of atorvastatin calcium therapy in childhood to reduce morbidity and mortality in adulthood has not been established.
Additional pediatric use information is approved for Pfizer’s LIPITOR (atorvastatin calcium) tablets. However, due to Pfizer’s marketing exclusivity rights, this drug product is not labeled with that pediatric information.
16 HOW SUPPLIED/STORAGE AND HANDLING
Atorvastatin calcium tablets of 20 mg are white to off-white, capsule shaped, biconvex, film coated tablets debossed ‘RDY’ on one side and ‘122’ on other side and are supplied in bottles of 90’s.
Bottles of 90 NDC 70934-201-90
Storage
Store atorvastatin calcium tablets at 20° - 25°C (68° - 77°F) [see USP Controlled Room Temperature].
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Patients taking atorvastatin calcium should be advised that cholesterol is a chronic condition and they should adhere to their medication along with their National Cholesterol Education Program (NCEP)-recommended diet, a regular exercise program as appropriate, and periodic testing of a fasting lipid panel to determine goal attainment.
Patients should be advised about substances they should not take concomitantly with atorvastatin [see Warnings and Precautions (5.1)]. Patients should also be advised to inform other healthcare professionals prescribing a new medication that they are taking atorvastatin calcium.
17.1 Muscle Pain
All patients starting therapy with atorvastatin calcium should be advised of the risk of myopathy and told to report promptly any unexplained muscle pain, tenderness, or weakness particularly if accompanied by malaise or fever or if these muscle signs or symptoms persist after discontinuing atorvastatin. The risk of this occurring is increased when taking certain types of medication or consuming larger quantities (>1 liter) of grapefruit juice. They should discuss all medication, both prescription and over the counter, with their healthcare professional.
17.2 Liver Enzymes
It is recommended that liver enzyme tests be performed before the initiation of atorvastatin calcium and if signs or symptoms of liver injury occur. All patients treated with atorvastatin calcium should be advised to report promptly any symptoms that may indicate liver injury, including fatigue, anorexia, right upper abdominal discomfort, dark urine, or jaundice.
PATIENT INFORMATION
Atorvastatin Calcium Tablets
(a tor'' va stat' in kal' see um)
Read the Patient Information that comes with atorvastatin calcium tablets before you start taking it and each time you get a refill. There may be new information. This leaflet does not take the place of talking with your doctor about your condition or treatment.
If you have any questions about atorvastatin calcium tablets, ask your doctor or pharmacist.
What is atorvastatin calcium tablets?
Atorvastatin calcium is a prescription medicine that lowers cholesterol in your blood. It lowers the LDL-C ("bad" cholesterol) and triglycerides in your blood. It can raise your HDL-C ("good" cholesterol) as well. Atorvastatin calcium tablets are for adults and children over 10 whose cholesterol does not come down enough with exercise and a low-fat diet alone.
Atorvastatin calcium tablets can lower the risk for heart attack, stroke, certain types of heart surgery, and chest pain in patients who have heart disease or risk factors for heart disease such as:
- age, smoking, high blood pressure, low HDL-C, heart disease in the family.
Atorvastatin calcium tablets can lower the risk for heart attack or stroke in patients with diabetes and risk factors such as:
- eye problems, kidney problems, smoking, or high blood pressure.
Atorvastatin calcium tablets starts to work in about 2 weeks.
What is cholesterol?
Cholesterol and triglycerides are fats that are made in your body. They are also found in foods. You need some cholesterol for good health, but too much is not good for you. Cholesterol and triglycerides can clog your blood vessels. It is especially important to lower your cholesterol if you have heart disease, smoke, have diabetes or high blood pressure, are older, or if heart disease starts early in your family.
Who should not take atorvastatin calcium tablets?
Do not take atorvastatin calcium tablets if you:
- are pregnant or think you may be pregnant, or are planning to become pregnant. Atorvastatin calcium tablets may harm your unborn baby. If you get pregnant, stop taking atorvastatin calcium tablets and call your doctor right away.
- are breast feeding . Atorvastatin calcium tablets can pass into your breast milk and may harm your baby.
- have liver problems.
- are allergic to atorvastatin calcium tablets or any of its ingredients. The active ingredient is atorvastatin. See the end of this leaflet for a complete list of ingredients in atorvastatin calcium tablets.
Atorvastatin calcium tablets dosing has not been established in children under 10 years of age.
Before you start atorvastatin calcium tablets
Tell your doctor if you:
- have muscle aches or weakness
- drink more than 2 glasses of alcohol daily
- have diabetes
- have a thyroid problem
- have kidney problems
Some medicines should not be taken with atorvastatin calcium tablets. Tell your doctor about all the medicines you take, including prescription and non-prescription medicines, vitamins, and herbal supplements. Atorvastatin calcium tablets and certain other medicines can interact causing serious side effects. Especially tell your doctor if you take medicines for:
- your immune system
- cholesterol
- infections
- birth control
- heart failure
- HIV or AIDS
Know all the medicines you take.Keep a list of them with you to show your doctor and pharmacist.
How should I take atorvastatin calcium tablets?
- Take atorvastatin calcium tablets exactly as prescribed by your doctor. Do not change your dose or stop atorvastatin calcium tablets without talking to your doctor. Your doctor may do blood tests to check your cholesterol levels during your treatment with atorvastatin calcium tablets. Your dose of atorvastatin calcium tablets may be changed based on these blood test results.
- Take atorvastatin calcium tablets each day at any time of day at about the same time each day. atorvastatin calcium tablets can be taken with or without food. Don't break atorvastatin calcium tablets before taking.
- Your doctor should start you on a low-fat diet before giving you atorvastatin calcium tablets. Stay on this low-fat diet when you take atorvastatin calcium tablets.
- If you miss a dose of atorvastatin calcium tablets, take it as soon as you remember. Do not take atorvastatin calcium tablets if it has been more than 12 hours since you missed your last dose. Wait and take the next dose at your regular time. Do not take 2 doses of atorvastatin calcium tablets at the same time.
- If you take too much atorvastatin calcium tablets or overdose, call your doctor or Poison Control Center right away. Or go to the nearest emergency room.
What should I avoid while taking atorvastatin calcium tablets?
- Talk to your doctor before you start any new medicines. This includes prescription and non-prescription medicines, vitamins, and herbal supplements. Atorvastatin calcium tablets and certain other medicines can interact causing serious side effects.
- Do not get pregnant. If you get pregnant, stop taking atorvastatin calcium right away and call your doctor.
What are the possible side effects of atorvastatin calcium tablets?
Atorvastatin calcium tablets can cause serious side effects. These side effects have happened only to a small number of people. Your doctor can monitor you for them. These side effects usually go away if your dose is lowered or atorvastatin calcium tablets are stopped. These serious side effects include:
- Muscle problems. Atorvastatin calcium tablets can cause serious muscle problems that can lead to kidney problems, including kidney failure. You have a higher chance for muscle problems if you are taking certain other medicines with atorvastatin calcium tablets.
-
Liver problems. Your doctor should do blood tests to check your liver before you start taking atorvastatin calcium tablets and if you have symptoms of liver problems while you take atorvastatin calcium tablets. Call your doctor right away if you have the following symptoms of liver problems:
- feel tired or week
- loss of appetite
- upper belly pain
- dark amber colored urine
- yellowing of your skin or the whites of your eyes
Call your doctor right away if you have:
- muscle problems like weakness, tenderness, or pain that happen without a good reason, especially if you also have a fever or feel more tired than usual. This may be an early sign of a rare muscle problem.
- muscle problems that do not go away even after your doctor has advised you to stop taking atorvastatin tablets. Your doctor may to further tests to diagnose the cause of your muscle problems.
- allergic reactions including swelling of the face, lips, tongue, and/or throat that may cause difficulty in breathing or swallowing which may require treatment right away.
- nausea and vomiting.
- passing brown or dark-colored urine.
- you feel more tired than usual
- your skin and whites of your eyes get yellow.
- stomach pain.
- allergic skin reactions.
In clinical studies, patients reported the following common side effects while taking atorvastatin calcium tablets: diarrhea, upset stomach, muscle and joint pain, and alterations in some laboratory blood tests.
The following additional side effects have been reported with atorvastatin calcium tablets: tiredness, tendon problems, memory loss, and confusion.
Talk to your doctor or pharmacist if you have side effects that bother you or that will not go away.
These are not all the side effects of atorvastatin calcium tablets. Ask your doctor or pharmacist for a complete list.
How do I store atorvastatin calcium tablets
- Store atorvastatin calcium tablets at 20°C - 25°C (68°F - 77°F) .
- Do not keep medicine that is out of date or that you no longer need.
- Keep atorvastatin calcium tablets and all medicines out of the reach of children. Be sure that if you throw medicine away, it is out of the reach of children.
General information about atorvastatin calciumtablets
Medicines are sometimes prescribed for conditions that are not mentioned in patient information leaflets. Do not use atorvastatin calcium tablets for a condition for which it was not prescribed. Do not give atorvastatin calcium tablets to other people, even if they have the same problem you have. It may harm them.
This leaflet summarizes the most important information about atorvastatin calcium tablets. If you would like more information, talk with your doctor. You can ask your doctor or pharmacist for information about atorvastatin calcium tablets that is written for health professionals.
What are the Ingredients in atorvastatin calcium tablets?
Active Ingredient:atorvastatin calcium
Inactive Ingredients: Basic butylated methacrylate copolymer, crospovidone, hydroxy propyl cellulose, lactose monohydrate, magnesium stearate, methanol, microcrystalline cellulose, sodium bicarbonate and sodium lauryl sulphate. The tablet coating contains isopropyl alcohol, methylene chloride and coloring agent opadry OY-58900 white contains polyethylene glycol, titanium dioxide and hypromellose.
Rx Only
Manufactured by:
Dr. Reddy’s Laboratories Limited
Bachupally – 500 090 INDIA
Revised: 0218
or
Manufactured by
Dr. Reddy’s Laboratories Limited
Srikakulam - 532 409 INDIA
Revised: 0218
Dispense with Patient Information Sheet available at:
www.drreddys.com/pi/atorvastatintabs.pdf

| ATORVASTATIN CALCIUM
atorvastatin calcium tablet |
||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||
| Labeler - Denton Pharma, Inc. DBA Northwind Pharmaceuticals (080355546) |
| Registrant - Denton Pharma, Inc. DBA Northwind Pharmaceuticals (080355546) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Denton Pharma, Inc. DBA Northwind Pharmaceuticals | 080355546 | repack(70934-201) | |