FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
IRESSA is indicated for the first-line treatment of patients with metastatic non-small cell lung cancer (NSCLC) whose tumors have epidermal growth factor receptor (EGFR) exon 19 deletions or exon 21 (L858R) substitution mutations as detected by an FDA-approved test [see Clinical Studies (14)].
Limitation of Use: Safety and efficacy of IRESSA have not been established in patients with metastatic NSCLC whose tumors have EGFR mutations other than exon 19 deletions or exon 21 (L858R) substitution mutations [see Clinical Studies (14)].
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
Select patients for the first-line treatment of metastatic NSCLC with IRESSA based on the presence of EGFR exon 19 deletions or exon 21 L858R mutations in their tumor or plasma specimens [see Indications and Usage (1), Clinical Studies (14)]. If these mutations are not detected in a plasma specimen, test tumor tissue if feasible.
Information on FDA-approved tests for the detection of EGFR mutations in NSCLC is available at: http://www.fda.gov/CompanionDiagnostics.
2.2 Recommended Dose
The recommended dose of IRESSA is 250 mg orally once daily with or without food until disease progression or unacceptable toxicity.
Do not take a missed dose within 12 hours of the next dose.
2.3 Administration to Patients Who Have Difficulty Swallowing Solids
Immerse IRESSA tablets in 4 to 8 ounces of water by dropping the tablet in water, and stir for approximately 15 minutes. Immediately drink the liquid or administer through a naso-gastric tube. Rinse the container with 4 to 8 ounces of water and immediately drink or administer through the naso-gastric tube.
2.4 Dose Modification
Dose Modifications for Adverse Drug Reactions
Withhold IRESSA (for up to 14 days) for any of the following:
- •
- Acute onset or worsening of pulmonary symptoms (dyspnea, cough, fever) [see Warnings and Precautions (5.1)]
- •
- NCI CTCAE Grade 2 or higher in ALT and/or AST elevations [see Warnings and Precautions (5.2)]
- •
- NCI CTCAE Grade 3 or higher diarrhea [see Warnings and Precautions (5.4)]
- •
- Signs and symptoms of severe or worsening ocular disorders including keratitis [see Warnings and Precautions (5.5)]
- •
- NCI CTCAE Grade 3 or higher skin reactions [see Warnings and Precautions (5.6)]
Resume treatment with IRESSA when the adverse reaction fully resolves or improves to NCI CTCAE Grade 1.
Permanently discontinue IRESSA for:
- •
- Confirmed interstitial lung disease (ILD) [see Warnings and Precautions (5.1)]
- •
- Severe hepatic impairment [see Warnings and Precautions (5.2)]
- •
- Gastrointestinal perforation [see Warnings and Precautions (5.3)]
- •
- Persistent ulcerative keratitis [see Warnings and Precautions (5.5)]
Dose Modifications for Drug Interactions
Strong CYP3A4 Inducers
Increase IRESSA to 500 mg daily in the absence of severe adverse drug reaction, and resume IRESSA at 250 mg seven days after discontinuation of the strong CYP3A4 inducer [see Drug Interactions (7), Clinical Pharmacology (12.3)].
3 DOSAGE FORMS AND STRENGTHS
250 mg tablets: round, biconvex, brown film-coated, debossed with "IRESSA 250" on one side and plain on the other side.
5 WARNINGS AND PRECAUTIONS
5.1 Interstitial Lung Disease (ILD)
ILD or ILD-like adverse drug reactions (e.g., lung infiltration, pneumonitis, acute respiratory distress syndrome, or pulmonary fibrosis) occurred in 1.3% of the 2462 patients who received IRESSA across clinical trials; of these, 0.7% were Grade 3 or higher and 3 cases were fatal.
Withhold IRESSA and promptly investigate for ILD in any patient who presents with worsening of respiratory symptoms such as dyspnea, cough and fever. Permanently discontinue IRESSA if ILD is confirmed [see Dosage and Administration (2.4), Adverse Reactions (6.1)].
5.2 Hepatotoxicity
In patients who received IRESSA across clinical trials, 11.4% of patients had increased alanine aminotransferase (ALT), 7.9% of patients had increased aspartate aminotransferase (AST), and 2.7% of patients had increased bilirubin. Grade 3 or higher liver test abnormalities occurred in 5.1% (ALT), 3.0% (AST), and 0.7% (bilirubin) of patients. The incidence of fatal hepatotoxicity was 0.04%.
Obtain periodic liver function testing. Withhold IRESSA in patients with worsening liver function and discontinue in patients with severe hepatic impairment [see Dosage and Administration (2.4), Adverse Reactions (6.1), Use in Specific Populations (8.7)].
5.3 Gastrointestinal Perforation
Gastrointestinal perforation occurred in three (0.1%) of the 2462 IRESSA-treated patients across clinical trials [see Adverse Reactions (6.1)]. Permanently discontinue IRESSA in patients who develop gastrointestinal perforation [see Dosage and Administration (2.4)].
5.4 Severe or Persistent Diarrhea
Grade 3 or 4 diarrhea occurred in 3% of 2462 IRESSA-treated patients across clinical trials. Withhold IRESSA for severe or persistent (up to 14 days) diarrhea [see Dosage and Administration (2.4), Adverse Reactions (6.1)].
5.5 Ocular Disorders including Keratitis
Ocular disorders [keratitis (0.1%), corneal erosion and aberrant eyelash growth (0.2%), conjunctivitis, blepharitis and dry eye (6.7%)] occurred in the 2462 IRESSA-treated patients across clinical trials. The incidence of Grade 3 ocular disorders was 0.1% [see Adverse Reactions (6.1)]. Interrupt or discontinue IRESSA for severe, or worsening ocular disorders [see Dosage and Administration (2.4)].
5.6 Bullous and Exfoliative Skin Disorders
Bullous conditions including toxic epidermal necrolysis, Stevens Johnson syndrome and erythema multiforme have been reported from treatment with IRESSA. Erythema multiforme and dermatitis bullous have been reported in two patients (0.08%) across NSCLC trials (Study 2, Study 3 and Study 4). IRESSA treatment should be interrupted or discontinued if the patient develops severe bullous, blistering or exfoliating conditions.
5.7 Embryo-fetal Toxicity
Based on its mechanism of action and data from animal reproduction studies IRESSA can cause fetal harm when administered to a pregnant woman. In animal reproductive studies, oral administration of gefitinib from organogenesis through weaning resulted in fetotoxicity and neonatal death at doses below the recommended human dose. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with IRESSA and for at least two weeks following completion of therapy [see Use in Specific Populations (8.1, 8.3)].
6 ADVERSE REACTIONS
The following adverse drug reactions are discussed in more detail in other sections of the labeling:
- •
- Interstitial Lung Disease [see Warnings and Precautions (5.1)]
- •
- Hepatotoxicity [see Warnings and Precautions (5.2)]
- •
- Gastrointestinal Perforation [see Warnings and Precautions (5.3)]
- •
- Severe or Persistent Diarrhea [see Warnings and Precautions (5.4)]
- •
- Ocular Disorders including Keratitis [see Warnings and Precautions (5.5)]
- •
- Bullous and Exfoliative Skin Disorders [see Warning and Precautions (5.6)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of IRESSA is based on the data from 2462 patients with NSCLC who received IRESSA 250 mg daily monotherapy in three randomized clinical studies (Study 2, Study 3 and Study 4). Patients with a history of interstitial lung disease, drug-induced interstitial disease, radiation pneumonitis that required steroid treatment or any evidence of clinically active interstitial lung disease were excluded from these studies.
Controlled Studies:
Study 2 was a randomized, multicenter, open-label trial in which 1217 patients were randomized to receive first-line treatment for metastatic NSCLC; 607 patients received IRESSA 250 mg daily and 589 patients received carboplatin/paclitaxel. The median duration of treatment with IRESSA was 5.9 months. The study population characteristics were: median age 57 years, age less than 65 years (73%), female (79%), Asian (100%), NSCLC adenocarcinoma histology (100%), never smoker (94%), light ex-smoker (6%), ECOG PS 0 or 1 (90%).
Study 3 was a randomized, multicenter, double-blind, placebo-controlled trial in which 1692 patients were randomized to receive second- or third-line treatment for metastatic NSCLC; of which 1126 patients received IRESSA 250 mg daily and 562 patients received placebo. The median duration of treatment with IRESSA was 2.9 months. The study population characteristics were: median age 62 years, age less than 65 years (60%), female (33%), Caucasian (75%), Asian (21%), NSCLC adenocarcinoma histology (48%), never smoker (22%), ECOG PS 0 or 1 (65%), PS 2 (29%), PS 3 (5%) and two or more prior therapies (51%).
Study 4 was a randomized, multicenter, open-label trial in which 1466 patients were randomized to receive second-line treatment for metastatic NSCLC; 729 patients received IRESSA 250 mg daily and 715 patients received docetaxel. The median duration of treatment with IRESSA was 2.4 months. The study population characteristics were: median age 61 years, age less than 65 years (61%), female (36%), Caucasian (79%), Asian (21%), NSCLC adenocarcinoma histology (54%), never smoker (20%), ECOG PS 0 or 1 (88%) and two or more prior therapies (16%).
The pooled safety database from the three randomized trials was used to evaluate for serious and uncommon adverse drug reactions. Common adverse reactions were evaluated in Study 3. The most frequent adverse reactions in Study 3 (incidence of >20% and greater than placebo) reported in IRESSA-treated patients were skin reactions (47%) and diarrhea (29%). The most frequent fatal adverse reactions in IRESSA-treated patients were respiratory failure (0.9%), pneumonia (0.8%), and pulmonary embolism (0.5%).
Approximately 5% of IRESSA-treated patients and 2.3% of placebo-treated patients discontinued treatment due to an adverse event. The most frequent adverse reactions that led to discontinuation in patients treated with IRESSA were nausea (0.5%), vomiting (0.5%) and diarrhea (0.4%).
|
||||
|
Adverse Reaction |
Percentage (%) of patients |
|||
|
IRESSA (N=1126) |
Placebo (N=562) |
|||
|
All Grades |
Grade 3 and 4 |
All Grades |
Grade 3 and 4 |
|
|
Skin and subcutaneous tissue disorders |
||||
|
Skin reactions* |
47% |
2% |
17% |
0.4% |
|
Nail disorders† |
5% |
0.1% |
0.7% |
0% |
|
Gastrointestinal disorders |
||||
|
Diarrhea‡ |
29% |
3% |
10% |
1% |
|
Vomiting |
14% |
1.2% |
10% |
0.4% |
|
Stomatitis§ |
7% |
0.3% |
4% |
0.2% |
|
Metabolism and nutrition disorders |
||||
|
Decreased appetite |
17% |
2.3% |
14% |
2.0% |
|
Eye disorders |
||||
|
Conjunctivitis/blepharitis/dry eye¶ |
6% |
0% |
3.2% |
0% |
|
||||
|
Adverse Reaction |
IRESSA |
Placebo |
||
|
All Grades % |
Grade 3 and 4 % |
All Grades % |
Grade 3 and 4 % |
|
|
Alanine aminotransferase increased* |
38%† |
2.4% |
23%† |
1.4%‡ |
|
Aspartate aminotransferase increased* |
40%§ |
2.0% |
25%§ |
1.3%¶ |
|
Proteinuria |
35% |
4.7% |
31% |
3.3% |
The following adverse reactions have been reported with IRESSA across NSCLC trials (Study 2, Study 3 and Study 4) and are not listed elsewhere in Section 6: nausea (18%), asthenia (17%), pyrexia (9%), alopecia (4.7%), hemorrhage (including epistaxis and hematuria) (4.3%), dry mouth (2%), dehydration (1.8%), elevations in blood creatinine (1.5%), allergic reactions including angioedema and urticaria (1.1%), palmar-plantar erythrodysesthesia syndrome (0.2%) and pancreatitis (0.1%).
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of IRESSA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Renal and urinary disorders: cystitis, hemorrhagic cystitis
Skin and subcutaneous tissue disorders: cutaneous vasculitis
7 DRUG INTERACTIONS
7.1 Drugs Affecting Gefitinib Exposure
CYP3A4 Inducer
Drugs that are strong inducers of CYP3A4 increase the metabolism of gefitinib and decrease gefitinib plasma concentrations. Increase IRESSA to 500 mg daily in patients receiving a strong CYP3A4 inducer (e.g., rifampicin, phenytoin, or tricyclic antidepressant) and resume IRESSA at 250 mg 7 days after discontinuation of the strong inducer [see Dosage and Administration (2.4), Clinical Pharmacology (12.3)].
CYP3A4 Inhibitor
Drugs that are strong inhibitors of CYP3A4 (e.g., ketoconazole and itraconazole) decrease gefitinib metabolism and increase gefitinib plasma concentrations. Monitor adverse reactions when administering strong CYP3A4 inhibitors with IRESSA.
Drugs Affecting Gastric pH
Drugs that elevate gastric pH (e.g., proton pump inhibitors, histamine H2-receptor antagonists, and antacids) may reduce plasma concentrations of gefitinib. Avoid concomitant use of IRESSA with proton pump inhibitors, if possible. If treatment with a proton-pump inhibitor is required, take IRESSA 12 hours after the last dose or 12 hours before the next dose of the proton-pump inhibitor. Take IRESSA 6 hours after or 6 hours before an H2-receptor antagonist or an antacid [see Clinical Pharmacology (12.3)].
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on its mechanism of action and animal data, IRESSA can cause fetal harm when administered to a pregnant woman. In animal reproductive studies, oral administration of gefitinib from organogenesis through weaning resulted in fetotoxicity and neonatal death at doses below the recommended human dose (see Animal Data). Advise pregnant women of the potential hazard to a fetus or potential risk for loss of the pregnancy.
The background risk of major birth defects and miscarriage for the indicated population is unknown; however, the background risk in the U.S. general population of major birth defects is 2-4% and miscarriage is 15-20% of clinically recognized pregnancies.
Data
Animal Data
A single dose study in rats showed that gefitinib crosses the placenta after an oral dose of 5 mg/kg (30 mg/m2, about 0.2 times the recommended human dose on a mg/m2 basis). When pregnant rats were treated with 5 mg/kg from the beginning of organogenesis to the end of weaning there was a reduction in the number of offspring born alive. This effect was more severe at 20 mg/kg (approximate the human clinical dose on a mg/m2 basis) and was accompanied by high neonatal mortality soon after parturition. In rabbits, a dose of 20 mg/kg/day (240 mg/m2, about twice the recommended dose in humans on a mg/m2 basis) caused reduced fetal weight.
8.2 Lactation
Risk Summary
It is not known whether IRESSA is excreted in human milk. Animal studies indicate the gefitinib and its metabolites are present in rat milk at a concentration higher than those in maternal plasma. Because of the potential for serious adverse reactions in nursing infants from IRESSA, advise women to discontinue breast-feeding during treatment with IRESSA.
Data
Animal Data
Levels of gefitinib and its metabolites were 11-to-19-fold higher in milk than in blood, after oral exposure of lactating rats to a dose of 5 mg/kg.
8.3 Females and Males of Reproductive Potential
Contraception
Based on its mechanism of action and animal data, IRESSA can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception during treatment with IRESSA and for at least two weeks following completion of therapy.
Infertility
IRESSA may result in reduced fertility in females of reproductive potential [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of IRESSA in pediatric patients have not been established.
8.5 Geriatric Use
Of the 823 patients enrolled in two randomized, active-controlled clinical trials 374 patients (45%) were 65 years and older, and 93 patients (11%) were 75 years and older. No overall differences in safety were observed between patients 65 years and older and those younger than 65 years. There is insufficient information to assess for differences in efficacy between older and younger patients.
8.6 Renal Impairment
Less than four percent (<4%) of gefitinib and its metabolites are excreted via the kidney. No clinical studies were conducted with IRESSA in patients with severe renal impairment.
8.7 Hepatic Impairment
The systemic exposure of gefitinib was compared in patients with mild, moderate, or severe hepatic impairment due to cirrhosis (according to Child-Pugh classification) and healthy subjects with normal hepatic function (N=10/group). The mean systemic exposure (AUC0-∞) was increased by 40% in patients with mild impairment, 263% in patients with moderate impairment, and 166% in patients with severe hepatic impairment. Monitor adverse reactions when IRESSA is administered to patients with moderate and severe hepatic impairment.
In a study comparing 13 patients with liver metastases and moderate hepatic impairment (addition of CTC grade of baseline AST/SGOT, ALP, and bilirubin equals 3 to 5) to 14 patients with liver metastases and normal hepatic function, the systemic exposure of gefitinib was similar [see Warnings and Precautions (5.2)].
10 OVERDOSAGE
Twenty three patients were treated weekly with doses from 1500 mg to 3500 mg, and IRESSA exposure did not increase with increasing dose. Adverse events were mostly mild to moderate in severity, and were consistent with the known safety profile of IRESSA. In the event of suspected overdose, interrupt IRESSA, institute supportive care, and observe until clinical stabilization. There are no specific measures/treatments that should be taken following IRESSA overdosing.
11 DESCRIPTION
Gefitinib is a kinase inhibitor.
The chemical name of gefitinib is 4-Quinazolinamine N-(3-chloro-4-fluorophenyl)-7-methoxy-6-[3-(4-morpholinyl) propoxy] and the following structural formula:
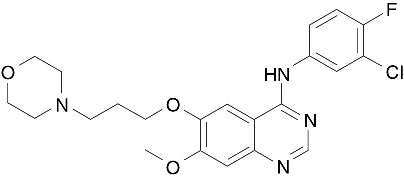
Gefitinib has the molecular formula C22H24ClFN4O3, a relative molecular mass of 446.9 daltons and is a white-colored powder. Gefitinib is a free base. The molecule has pKas of 5.4 and 7.2. Gefitinib can be defined as sparingly soluble at pH 1, but is practically insoluble above pH 7, with the solubility decreasing sharply between pH 4 and pH 6. In non-aqueous solvents, gefitinib is freely soluble in glacial acetic acid and dimethyl sulfoxide, soluble in pyridine, sparingly soluble in tetrahydrofuran, and slightly soluble in methanol, ethanol (99.5%), ethyl acetate, propan-2-ol and acetonitrile.
IRESSA® (gefitinib) tablets are available as brown film-coated tablets, containing 250 mg of gefitinib, for oral administration. The inactive ingredients of the tablet core of IRESSA tablets are lactose monohydrate, microcrystalline cellulose, croscarmellose sodium, povidone, sodium lauryl sulfate and magnesium stearate. The tablet coating is composed of hypromellose, polyethylene glycol 300, titanium dioxide, red ferric oxide and yellow ferric oxide.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The epidermal growth factor receptor (EGFR) is expressed on the cell surface of both normal and cancer cells and plays a role in the processes of cell growth and proliferation. Some EGFR activating mutations (exon 19 deletion or exon 21 point mutation L858R) within NSCLC cells have been identified as contributing to the promotion of tumor cell growth, blocking of apoptosis, increasing the production of angiogenic factors and facilitating the processes of metastasis.
Gefitinib reversibly inhibits the kinase activity of wild-type and certain activating mutations of EGFR, preventing autophosphorylation of tyrosine residues associated with the receptor, thereby inhibiting further downstream signaling and blocking EGFR-dependent proliferation.
Gefitinib binding affinity for EGFR exon 19 deletion or exon 21 point mutation L858R mutations is higher than its affinity for the wild-type EGFR. Gefitinib also inhibits IGF and PDGF-mediated signaling at clinically relevant concentrations; inhibition of other tyrosine kinase receptors has not been fully characterized.
12.3 Pharmacokinetics
Absorption and Distribution
The mean oral bioavailability of gefitinib is 60%, with peak plasma levels occurring 3-7 hours after dosing. Food does not alter gefitinib bioavailability to a clinically meaningful extent. IRESSA can be administered with or without food. Gefitinib is extensively distributed throughout the body with a mean steady state volume of distribution of 1400 L following intravenous administration. In vitro binding of gefitinib to human plasma proteins (serum albumin and α1-acid glycoprotein) is 90%, independent of drug concentrations. Gefitinib is a substrate for the membrane transport P-glycoprotein (P-gp), but it is unlikely to influence gefitinib absorption as P-gp is saturated at higher concentrations.
Metabolism and Elimination
Gefitinib undergoes extensive hepatic metabolism in humans, predominantly by CYP3A4. Three sites of biotransformation have been identified: metabolism of the N-propoxymorpholino-group, demethylation of the methoxy-substituent on the quinazoline, and oxidative defluorination of the halogenated phenyl group. Five metabolites have been fully identified in fecal extracts and the major active component was O-desmethyl gefitinib produced by CYP2D6 metabolism and accounted for 14% of the dose.
Eight metabolites were identified in human plasma. Only O-desmethyl gefitinib has exposure comparable to gefitinib. Although this metabolite has similar EGFR-TK activity to gefitinib in the isolated enzyme assay, it had only 1/14 of the potency of gefitinib in one of the cell-based assays.
Gefitinib is cleared primarily by the liver, with total plasma clearance and elimination half-life of 48 hours after intravenous administration. The inter-subject variability (coefficient of variation) for AUC in healthy subjects was 67%. Daily oral administration of gefitinib to cancer patients resulted in a two-fold accumulation compared to single dose administration. Steady state plasma concentrations are achieved within 10 days after daily dosing. Excretion of gefitinib and its metabolites is predominantly via the feces (86%), with renal elimination accounting for less than 4% of the administered dose.
Specific Populations
Age, gender, body weight, ethnicity or renal function: Population pharmacokinetic analyses suggest that patient age, body weight, ethnicity (populations included) or creatinine clearance (above 20 mL/min) has no clinically meaningful effect on predicted steady state trough concentration of gefitinib. Population pharmacokinetic analyses of Study 1 showed that women had 27% higher exposure than men; however, this difference was not identified in the analyses of other gefitinib clinical studies. No dose adjustment based on patient gender is recommended.
Hepatic Impairment: The systemic exposure of gefitinib was compared between patients with mild, moderate, or severe hepatic impairment due to cirrhosis (according to Child-Pugh classification) and healthy subjects with normal hepatic function (N=10/group). The mean systemic exposure (AUC0-∞) was increased by 40% in patients with mild impairment, 263% in patients with moderate impairment, and 166% in patients with severe hepatic impairment. In a study comparing 13 patients with liver metastases and moderate hepatic impairment to 14 patients with liver metastases and normal hepatic function, the systemic exposure of gefitinib was similar [see Warnings and Precautions (5.2), Use in Specific Populations (8.7)].
CYP2D6 Poor metabolizer: CYP2D6 metabolizes gefitinib to O-desmethyl gefitinib in vitro. In healthy CYP2D6 poor metabolizers, O-desmethyl gefitinib concentration was not measurable and the mean exposure to gefitinib was 2-fold higher as compared to the extensive metabolizers. This increase in exposure in CYP2D6 poor metabolizers may be clinically important because some adverse drug reactions are related to higher exposure of gefitinib. No dose adjustment is recommended in patients with a known CYP2D6 poor metabolizer genotype, but these patients should be closely monitored for adverse reactions. The impact of CYP2D6 inhibiting drugs on gefitinib pharmacokinetics has not been evaluated. However, similar precautions should be used when administering CYP2D6 inhibitors with IRESSA because of the possibility of increased exposure in these patients.
An exploratory exposure response analysis showed an increase in the incidence of interstitial lung disease (ILD) with a greater than 2-fold increase in the gefitinib exposure [see Warnings and Precautions (5.1)].
Drug-Drug Interactions
Strong CYP3A4 Inducer:
Concomitant administration of rifampicin (600 mg QD for 16 days), a strong inducer of CYP3A4, with gefitinib (500 mg single dose on Day 10 of gefitinib administration) reduced mean AUC of gefitinib by 83% [see Dosage and Administration (2.4), Drug Interactions (7)].
CYP3A4 Inhibitor:
Concomitant administration of itraconazole (200 mg QD for 12 days), an inhibitor of CYP3A4, with gefitinib (250 mg single dose on Day 4 of itraconazole administration) to healthy male subjects, increased mean gefitinib AUC by 80% [see Drug Interactions (7)].
Drugs Affecting Gastric pH:
Co-administration of high doses of ranitidine with sodium bicarbonate (to maintain the gastric pH above pH 5.0) to healthy subjects decreased mean gefitinib AUC by 47% [see Drug Interactions (7)].
In human liver microsome studies, gefitinib had no inhibitory effect on CYP1A2, CYP2C9, and CYP3A4 activities at concentrations ranging from 2-5000 ng/mL. At the highest concentration studied (5000 ng/mL), gefitinib inhibited CYP2C19 by 24% and CYP2D6 by 43%.
Exposure to metoprolol, a substrate of CYP2D6, was increased by 30% when it was given on Day 15 of gefitinib dosing (500 mg daily for 28 days) in patients with solid tumors.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Gefitinib has been tested for genotoxicity in a series of in vitro (bacterial mutation, mouse lymphoma, and human lymphocyte) assays and an in vivo rat micronucleus test. Under the conditions of these assays, gefitinib did not cause genetic damage.
In a two-year carcinogenicity study in mice, administration of gefitinib at a dose of 270 mg/m2/day (approximately twice the recommended daily dose of 250 mg on a mg/m2 basis; dose reduced from 375 mg/m2/day from week 22) caused hepatocellular adenomas in females. In a two-year carcinogenicity study in rats, administration of gefitinib at 60 mg/m2/day (approximately 0.4 times the recommended daily clinical dose on a mg/m2 basis) caused hepatocellular adenomas and hemangiomas/hemagiosarcomas of the mesenteric lymph nodes in female rats. The clinical relevance of these findings is unknown.
In a dedicated fertility study in rats at doses ≥120 mg/m2 (approximately equal to the recommended human dose of gefitinib on a mg/m2 basis), animals presented with an increased incidence of irregular estrous, decreased corpora lutea, and decreases in uterine implants and live embryos per litter.
14 CLINICAL STUDIES
Non-Small Cell Lung Cancer (NSCLC)
Study 1
The efficacy and safety of IRESSA for the first-line treatment of patients with metastatic NSCLC containing EGFR exon 19 deletions or L858R substitution mutations was demonstrated in a multicenter, single-arm, open-label clinical study (Study 1). A total of 106 treatment-naive patients with metastatic EGFR mutation positive NSCLC received IRESSA at a dose of 250 mg once daily until disease progression or intolerable toxicity. The major efficacy outcome measure was objective response rate (ORR) according to RECIST v1.1 as evaluated by both a Blinded Independent Central Review (BICR) and investigators. Duration of response (DOR) was an additional outcome measure. Eligible patients were required to have a deletion in EGFR exon 19 or L858R, L861Q, or G719X substitution mutation and no T790M or S768I mutation or exon 20 insertion in tumor specimens as prospectively determined by a clinical trial assay. Tumor samples from 87 patients were tested retrospectively using the therascreen® EGFR RGQ PCR Kit.
The study population characteristics were: median age 65 years, age 75 years or older (25%), age less than 65 years (49%), white (100%), female (71%), never smokers (64%), WHO PS 0 (45%), WHO PS 1 (48%), WHO PS 2 (7%), and adenocarcinoma histology (97%). Sixty patients had exon 19 deletions (65%), 29 patients had L858R substitution (31%), while two patients each had tumors harboring L861Q or G719X substitution mutation.
The median duration of treatment was 8.0 months. Efficacy results from Study 1 are summarized below.
|
Efficacy Parameter |
BICR* Assessment (n=106)† |
Investigator Assessment
|
|
Objective Response Rate‡ (95% CI) |
50% (41, 59) |
70% (61, 78) |
|
Complete Response Rate |
0.9% |
1.9% |
|
Partial Response Rate |
49% |
68% |
|
Median Duration of Response (months) (95% CI) |
6.0 (5.6, 11.1) |
8.3 (7.6, 11.3) |
The response rates were similar in patients whose tumors had EGFR exon 19 deletions and exon 21 L858R substitution mutations. Two partial responses were observed in both patients whose tumors had G719X substitution mutation with duration of response of at least 2.8 months and 5.6 months, respectively. One of two patients whose tumors had L861Q substitution mutation also achieved a partial response with duration of response of at least 2.8 months.
Study 2
The results of Study 1 were supported by an exploratory analysis of a subset of a randomized, multicenter, open-label trial (Study 2) conducted in patients with metastatic adenocarcinoma histology NSCLC receiving first-line treatment. Patients were randomized (1:1) to receive IRESSA 250 mg orally once daily or up to 6 cycles of carboplatin/paclitaxel. The efficacy outcomes included progression-free survival (PFS) and objective response rate (ORR) as assessed by BICR.
The subset population consisted of 186 of 1217 patients (15%) determined to be EGFR positive by the same clinical trial assay as used in Study 1 and had radiographic scans available for a retrospective assessment by BICR. In this subset, there were 88 IRESSA-treated patients and 98 carboplatin/paclitaxel-treated patients.
Demographic and baseline characteristics of this subset were a median age of 59 years, age 75 years or older (7%), age less than 65 (70%), Asian (100%), female (83%), never smokers (96%), adenocarcinoma histology (100%), and PS 0-1 (94%).
The median duration of treatment for IRESSA-treated patients was 9.8 months. The hazard ratio for PFS favored the IRESSA-treated patients [HR of 0.54 (95% CI: 0.38, 0.79)] with a median PFS of 10.9 months for the IRESSA-treated patients and 7.4 months for the carboplatin/paclitaxel-treated patients as assessed by BICR. In addition, the objective response rate was 67% (95% CI: 56, 77) for IRESSA-treated patients and 41% (95% CI: 31, 51) for carboplatin/paclitaxel-treated patients based on BICR assessment. The median duration of response was 9.6 months for IRESSA-treated patients and 5.5 months for carboplatin/paclitaxel-treated patients.
16 HOW SUPPLIED/STORAGE AND HANDLING
IRESSA® (gefitinib) is available as 250 mg tablets.
IRESSA 250 mg tablets are round, biconvex, brown film-coated, debossed with "IRESSA 250" on one side and plain on the other side.
IRESSA® (gefitinib) tablets are supplied as:
Bottles of 30 Tablets (NDC 0310-0482-30)
Store at controlled room temperature 20°C-25°C (68°F-77°F) [see USP Controlled Room Temperature].
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labelling (Patient Information).
Interstitial Lung Disease: Advise patients to immediately contact their healthcare provider for new onset or worsening of pulmonary symptoms such as dyspnea, cough and fever [see Warnings and Precautions (5.1)].
Hepatotoxicity: Inform patients that they will need to undergo lab tests to monitor for liver function. Advise patients to contact their healthcare provider to report any new symptoms indicating hepatic toxicity [see Warnings and Precautions (5.2)].
Gastrointestinal Perforation: Advise patients that IRESSA can increase the risk of gastrointestinal perforation and to seek immediate medical attention for severe abdominal pain [see Warnings and Precautions (5.3)].
Severe or Persistent Diarrhea: Advise patients to contact their healthcare provider for severe or persistent diarrhea [see Warnings and Precautions (5.4)].
Ocular Disorders including Keratitis: Advise patients promptly to contact their healthcare provider if they develop eye symptoms, lacrimation, light sensitivity, blurred vision, eye pain, red eye or changes in vision [see Warnings and Precautions (5.5)].
Bullous and Exfoliative Skin Disorders: Advise patients that IRESSA can increase the risk of bullous and exfoliative skin disorders and to seek immediately medical attention for severe skin reactions [see Warnings and Precautions (5.6)].
Embryo-fetal Toxicity: Advise pregnant women of the potential risk to a fetus or potential risk for loss of the pregnancy [see Warnings and Precautions (5.7), Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception during treatment with IRESSA and for at least two weeks following completion of therapy [see Use in Specific Populations (8.3)].
Lactation: Advise women to discontinue breast-feeding during treatment with IRESSA [see Use in Specific Populations (8.2)].
IRESSA is a trademark of the AstraZeneca group of companies.
©AstraZeneca 2023
Distributed by:
AstraZeneca Pharmaceuticals LP
Wilmington, DE 19850
|
Patient Information IRESSA (eye-RES-sah) (gefitinib) tablets |
|
|
|
What is IRESSA? IRESSA is a prescription medicine used to treat people with non-small cell lung cancer (NSCLC) that has spread to other parts of the body and:
Your healthcare provider will perform a test to make sure that IRESSA is right for you. It is not known if IRESSA is safe and effective in people with NSCLC that have other types of EGFR genes. It is not known if IRESSA is safe and effective in children.
|
|
Before taking IRESSA, tell your healthcare provider about all of your medical conditions, including if you:
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, or herbal supplements. If you take a proton pump inhibitor (PPI), H2 blocker, or an antacid medicine, talk to your healthcare provider about the best time to take it during treatment with IRESSA. If you take a blood thinner called warfarin, your healthcare provider should do blood tests regularly to check how fast your blood clots, during treatment with IRESSA.
|
|
How should I take IRESSA?
|
|
What are the possible side effects of IRESSA? IRESSA may cause serious side effects, including:
Your healthcare provider will do blood tests to check your liver function during your treatment with IRESSA.
IRESSA may cause fertility problems in females. Talk to your healthcare provider if you plan to become pregnant. Tell your healthcare provider if you have any side effect that bothers you or that does not go away. These are not all the possible side effects of IRESSA. For more information, ask your healthcare provider or pharmacist. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
|
|
How should I store IRESSA? Store IRESSA at room temperature between 68˚F to 77˚F (20˚C to 25˚C) Keep IRESSA and all medicines out of the reach of children. General information about the safe and effective use of IRESSA. Medicines are sometimes prescribed for purposes other than those listed in Patient Information leaflet. Do not use IRESSA for a condition for which it was not prescribed. Do not give IRESSA to other people, even if they have the same symptoms you have. It may harm them. You can ask your healthcare provider or pharmacist for information about IRESSA that is written for health professionals.
|
|
What are the ingredients in IRESSA? Active ingredient: gefitinib Inactive ingredients: lactose monohydrate, microcrystalline cellulose, croscarmellose sodium, povidone, sodium lauryl sulfate, magnesium stearate Tablet coating contains: hypromellose, polyethylene glycol 300, titanium dioxide, yellow iron oxide and red iron oxide.
|
|
IRESSA is a trademark of the AstraZeneca group of companies. ©AstraZeneca 2021 Distributed by: AstraZeneca Pharmaceuticals LP Wilmington, DE 19850 For more information, go to www.iressa.com or call 1-800-236-9933 |
This Patient Information has been approved by the U.S. Food and Drug Administration. Issued: May 2021
