FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
CERDELGA is indicated for the long-term treatment of adult patients with Gaucher disease type 1 (GD1) who are CYP2D6 extensive metabolizers (EMs), intermediate metabolizers (IMs), or poor metabolizers (PMs) as detected by an FDA-cleared test [see Dosage and Administration (2.1)].
Limitations of Use:
- Patients who are CYP2D6 ultra-rapid metabolizers (URMs) may not achieve adequate concentrations of CERDELGA to achieve a therapeutic effect [see Clinical Studies (14)].
- A specific dosage cannot be recommended for those patients whose CYP2D6 genotype cannot be determined (indeterminate metabolizers) [see Clinical Studies (14)].
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
Select patients with Gaucher disease type 1 based on their CYP2D6 metabolizer status. It is recommended patient genotypes be established using an FDA-cleared test for determining CYP2D6 genotype [see Indications and Usage (1)].
2.2 Recommended Adult Dosage
The recommended dosage of CERDELGA in adults is based on the patient's CYP2D6 metabolizer status.
| CYP2D6 Metabolizer Status | CERDELGA Dosage |
|---|---|
| EMs | 84 mg twice daily |
| IMs | |
| PMs | 84 mg once daily |
2.3 Dosage Adjustment in EMs and IMs With or Without Hepatic Impairment and Concomitant Use of CYP2D6 or CYP3A Inhibitors
Reduce dosage frequency of CERDELGA 84 mg to once daily in CYP2D6 EMs and IMs with or without hepatic impairment taking CYP2D6 or CYP3A inhibitors, as shown in Table 2 [see Warnings and Precautions (5.1), Drug Interactions (7.1), Use in Specific Populations (8.7)].
| CYP2D6 Metabolizer Status | Hepatic Impairment Status | Concomitant CYP Inhibitor |
|---|---|---|
| EMs | Without Hepatic Impairment |
|
| Mild (Child-Pugh Class A) Hepatic Impairment |
|
|
| IMs | Without Hepatic Impairment |
|
2.4 Important Administration Instructions
- Swallow capsules whole, preferably with water, and do not crush, dissolve, or open the capsules.
- CERDELGA can be taken with or without food.
- Avoid the consumption of grapefruit or grapefruit juice (strong CYP3A inhibitors) with CERDELGA [see Drug Interactions (7.1)].
- If a dose of CERDELGA is missed, take the prescribed dose at the next scheduled time; do not double the next dose.
- For patients currently treated with imiglucerase, velaglucerase alfa, or taliglucerase alfa, CERDELGA may be administered 24 hours after the last dose of the previous enzyme replacement therapy (ERT).
3 DOSAGE FORMS AND STRENGTHS
Capsules: 84 mg of eliglustat is in a capsule with a pearl blue-green opaque cap and pearl white opaque body imprinted with "GZ02" in black.
4 CONTRAINDICATIONS
CERDELGA is contraindicated in the following patients based on CYP2D6 metabolizer status due to the risk of cardiac arrhythmias from prolongation of the PR, QTc, and/or QRS cardiac intervals.
EMs
- Taking a strong or moderate CYP2D6 inhibitor concomitantly with a strong or moderate CYP3A inhibitor [see Drug Interactions (7.1)]
- Moderate or severe hepatic impairment [see Use in Specific Populations (8.7)]
- Mild hepatic impairment and taking a strong or moderate CYP2D6 inhibitor [see Use in Specific Populations (8.7)]
IMs
- Taking a strong or moderate CYP2D6 inhibitor concomitantly with a strong or moderate CYP3A inhibitor [see Drug Interactions (7.1)]
- Taking a strong CYP3A inhibitor [see Drug Interactions (7.1)]
- Any degree of hepatic impairment [see Use in Specific Populations (8.7)]
PMs
- Taking a strong CYP3A inhibitor [see Drug Interactions (7.1)]
- Any degree of hepatic impairment [see Use in Specific Populations (8.7)]
5 WARNINGS AND PRECAUTIONS
5.1 ECG Changes and Potential for Cardiac Arrhythmias
CERDELGA is predicted to cause increases in ECG intervals (PR, QTc, and QRS) at substantially elevated eliglustat plasma concentrations and may increase the risk of cardiac arrhythmias.
- Use of CERDELGA is contraindicated, to be avoided, or requires dosage adjustment in patients taking CYP2D6 or CYP3A inhibitors, depending on CYP2D6 metabolizer status, type of inhibitor, or degree of hepatic impairment [see Dosage and Administration (2.3), Contraindications (4), Drug Interactions (7.1)].
Use of CERDELGA in patients with pre-existing cardiac conditions has not been studied during clinical trials. Avoid use of CERDELGA in patients with:
- pre-existing cardiac disease (congestive heart failure, recent acute myocardial infarction, bradycardia, heart block, ventricular arrhythmia)
- long QT syndrome
- in combination with Class IA (e.g., quinidine, procainamide) or Class III (e.g., amiodarone, sotalol) antiarrhythmic medications [see Clinical Pharmacology (12.2)]
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The most common adverse reactions to CERDELGA (occurring in ≥10% of the 126 GD1 patients treated with CERDELGA across Trials 1 and 2) were fatigue, headache, nausea, diarrhea, back pain, pain in extremities, and upper abdominal pain.
The adverse reaction profile of CERDELGA is based on two controlled studies, Trials 1 and 2 [see Clinical Studies (14.1, 14.2)]. Table 3 presents the profile from the 9-month double-blind, randomized, placebo-controlled trial of 40 treatment-naive patients (Trial 1). Patients were between the ages of 16 and 63 on the date of the first dose of study drug, and included 20 males and 20 females.
| CERDELGA (N=20) | Placebo (N=20) |
|
|---|---|---|
| Adverse Reaction | Patients n (%) | Patients n (%) |
| Arthralgia | 9 (45) | 2 (10) |
| Headache | 8 (40) | 6 (30) |
| Migraine | 2 (10) | 0 (0) |
| Flatulence | 2 (10) | 1 (5) |
| Nausea | 2 (10) | 1 (5) |
| Oropharyngeal pain | 2 (10) | 1 (5) |
Table 4 presents the profile from the 12-month open-label, randomized, imiglucerase-controlled trial of 159 treated patients switching from enzyme replacement therapy (ERT) (Trial 2). Patients were between the ages of 18 and 69 on the date of the first dose of CERDELGA, and included 87 females and 72 males.
| CERDELGA (N=106) | Imiglucerase (N=53) |
|
|---|---|---|
| Adverse Reaction | Patients n (%) | Patients n (%) |
|
||
| Fatigue | 15 (14) | 1 (2) |
| Headache | 14 (13) | 1 (2) |
| Nausea | 13 (12) | 0 (0) |
| Diarrhea | 13 (12) | 2 (4) |
| Back pain | 13 (12) | 3 (6) |
| Pain in extremity | 12 (11) | 1 (2) |
| Upper abdominal pain | 11 (10) | 0 (0) |
| Dizziness | 9 (8) | 0 (0) |
| Asthenia | 9 (8) | 0 (0) |
| Cough | 7 (7) | 2 (4) |
| Dyspepsia | 7 (7) | 1 (2) |
| Gastroesophageal reflux disease | 7 (7) | 0 (0) |
| Constipation | 5 (5) | 0 (0) |
| Palpitations | 5 (5) | 0 (0) |
| Rash | 5 (5) | 0 (0) |
In a separate uncontrolled study, with up to 4 years of treatment in 26 naive GD1 patients, the types and incidences of adverse reactions were similar to Trials 1 and 2.
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on CERDELGA
Coadministration of CERDELGA with:
- CYP2D6 or CYP3A inhibitors may increase eliglustat concentrations which may increase the risk of cardiac arrhythmias from prolongation of the PR, QTc, and/or QRS cardiac interval [see Warnings and Precautions (5.1), Clinical Pharmacology (12.3)].
- strong CYP3A inducers decrease eliglustat concentrations which may reduce CERDELGA efficacy [see Clinical Pharmacology (12.3)].
See Table 5 for prevention and management of interactions with drugs affecting CERDELGA. Use of CERDELGA is contraindicated, to be avoided, or may require dosage adjustment depending on the concomitant drug and CYP2D6 metabolizer status [see Dosage and Administration (2.2, 2.3), Contraindications (4), Drug Interactions (7.1)].
| Concomitant Drug(s) | CYP2D6 Metabolizer Status | ||
|---|---|---|---|
| EMs | IMs | PMs | |
|
|||
| CYP2D6 Inhibitor | |||
| Strong | Reduce frequency of CERDELGA 84 mg to once daily | Continue CERDELGA 84 mg once daily* | |
| Moderate | |||
| Weak | Continue CERDELGA 84 mg twice daily | ||
| CYP3A Inhibitor | |||
| Strong | Reduce frequency of CERDELGA 84 mg to once daily | Contraindicated | |
| Moderate | Avoid coadministration | ||
| Weak | Continue CERDELGA 84 mg twice daily | Avoid coadministration | |
| CYP2D6 Inhibitor Concomitantly with a strong CYP3A Inhibitor | |||
| Strong | Contraindicated | ||
| Moderate | |||
| CYP2D6 Inhibitor Concomitantly with a moderate CYP3A Inhibitor | |||
| Strong | Contraindicated | Avoid coadministration* | |
| Moderate | |||
| CYP3A Inducer | |||
| Strong | Avoid coadministration | ||
7.2 Effect of CERDELGA on Other Drugs
See Table 6 for clinically relevant interactions affecting P-gp or CYP2D6 substrates when coadministered with CERDELGA.
| Substrates for P-gp or CYP2D6 | ||
|---|---|---|
| Clinical Impact | Coadministration of CERDELGA may increase concentrations of drugs that are substrates for P-gp or CYP2D6 [see Clinical Pharmacology (12.3)] and may increase the risk of toxicity of these drugs. | |
| Prevention or Management | Digoxin | Monitor serum digoxin concentrations before initiating CERDELGA. Reduce digoxin dose by 30% and continue monitoring. |
| Other P-gp substrates or CYP2D6 substrates | Monitor therapeutic drug concentrations, as indicated, or consider reducing the dosage of the concomitant drug and titrate to clinical effect. | |
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Available data on CERDELGA use in pregnant women includes 20 pregnancies that occurred during the clinical development program and a small number of post-marketing case reports. These data are not sufficient to assess drug-associated risks major birth defects, miscarriage, or adverse maternal or fetal outcomes. In animal reproduction studies in pregnant rats administered oral eliglustat during organogenesis, a spectrum of various developmental abnormalities were observed at doses 6 times the recommended human dose. No adverse developmental outcomes were observed with oral administration of eliglustat to pregnant rabbits at dose levels 10 times the recommended human dose [see Data].
The estimated background risk of major birth defects and miscarriage in the indicated population is unknown. All pregnancies have a background risk of birth defect, loss or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Clinical Considerations
Disease-associated maternal and/or embryo/fetal risk
Women with Gaucher disease type 1 have an increased risk of spontaneous abortion, especially if disease symptoms are not treated and controlled pre-conception and during a pregnancy. Pregnancy may exacerbate existing Gaucher disease type 1 symptoms or result in new disease manifestations. Gaucher disease type 1 manifestations may lead to adverse pregnancy outcomes including, hepatosplenomegaly which can interfere with the normal growth of a pregnancy and thrombocytopenia which can lead to increased bleeding and possible hemorrhage.
Data
Animal data
Reproduction studies have been performed in pregnant rats at oral doses up to 120 mg/kg/day (about 6 times the recommended human dose based on body surface area) and in pregnant rabbits at oral doses up to 100 mg/kg/day (about 10 times the recommended human dose based on body surface area) following administration of eliglustat during the period of organogenesis (gestation days 6 to 17 in the rat and 6 to 18 in the rabbit).
In rats, at 120 mg/kg/day (about 6 times the recommended human dose based on body surface area), eliglustat increased the number of late resorptions, dead fetuses and post implantation loss, reduced fetal body weight, and caused fetal cerebral variations (dilated cerebral ventricles), fetal skeletal variations (poor bone ossification) and fetal skeletal malformations (abnormal number of ribs or lumbar vertebra). Eliglustat-related effects on fetal rats were observed in association with signs of maternal toxicity.
Eliglustat did not cause fetal harm in rabbits at oral doses up to 100 mg/kg/day (about 10 times the recommended human dose based on body surface area). Mild maternal toxicity was observed at the 100 mg/kg/day dose.
In a pre and postnatal development study in rats (dosed daily from gestation day 6 to postpartum day 21), eliglustat did not show any significant adverse effects on pre and postnatal development at doses up to 100 mg/kg/day (about 5 times the recommended human dose based on body surface area).
8.2 Lactation
Risk Summary
There are no human data available on the presence of eliglustat in human milk, the effects on the breastfed infant, or the effects on milk production. Eliglustat and its metabolites were present in the milk of lactating rats [see Data]. When a drug is present in animal milk, it is likely that the drug will be present in human milk. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for CERDELGA and any potential adverse effects on the breastfed child from CERDELGA or from the underlying maternal condition.
Data
In a milk excretion study in the rat, a single oral dose of 30 mg/kg [14C]-labeled eliglustat was administered to lactating female rats at day 11 postpartum. Approximately 0.23% of the administered radioactivity was excreted into the milk within 24 hours of dose administration. The concentration in the milk at 24 hours post dose was 16.3-fold higher than the plasma concentration.
8.5 Geriatric Use
Clinical studies of CERDELGA did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Clinical experience has not identified differences in responses between the elderly and younger patients.
8.6 Renal Impairment
Use CERDELGA in patients with renal impairment based on the patient's CYP2D6 metabolizer status [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
Use CERDELGA in patients with hepatic impairment based on CYP2D6 metabolizer status and concomitant use of CYP2D6 or CYP3A inhibitors [see Clinical Pharmacology (12.3)].
EMs
- CERDELGA is contraindicated in patients with [see Contraindications (4)]:
- severe (Child-Pugh Class C) hepatic impairment
- moderate (Child-Pugh Class B) hepatic impairment
- mild (Child-Pugh Class A) hepatic impairment taking a strong or moderate CYP2D6 inhibitor
- Reduce dosage frequency of CERDELGA 84 mg to once daily [see Dosage and Administration (2.3)] in patients with mild hepatic impairment taking:
- a weak CYP2D6 inhibitor
- a strong, moderate, or weak CYP3A inhibitor
- No dosage adjustment is recommended in patients with mild hepatic impairment, unless otherwise specified above.
IMs and PMs
- CERDELGA is contraindicated in patients with any degree of hepatic impairment [see Contraindications (4)].
10 OVERDOSAGE
The highest eliglustat plasma concentration experienced to date occurred in a single-dose, dose escalation study in healthy subjects, in a subject taking a dose equivalent to approximately 21 times the recommended dose for GD1 patients. At the time of the highest plasma concentration (59-fold higher than normal therapeutic conditions), the subject experienced dizziness marked by disequilibrium, hypotension, bradycardia, nausea, and vomiting.
In the event of acute overdose, the patient should be carefully observed and given symptomatic and supportive treatment.
Hemodialysis is unlikely to be beneficial given that eliglustat has a large volume of distribution [see Clinical Pharmacology (12.3)].
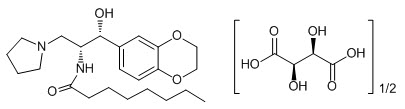
11 DESCRIPTION
CERDELGA (eliglustat) capsules contain eliglustat tartrate, which is a small molecule inhibitor of glucosylceramide synthase that resembles the ceramide substrate for the enzyme, with the chemical name N-((1R,2R)-1-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-1-hydroxy-3-(pyrrolidin-1-yl)propan-2-yl)octanamide (2R,3R)-2,3-dihydroxysuccinate. Its molecular weight is 479.59, and the empirical formula is C23H36N2O4+½(C4H6O6) with the following chemical structure:
Each capsule of CERDELGA for oral use contains 84 mg of eliglustat (equivalent to 100 mg of eliglustat tartrate). The inactive ingredients are candurin silver fine, FD&C blue 2, gelatin, glyceryl behenate, hypromellose, lactose monohydrate, microcrystalline cellulose, and yellow iron oxide.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Gaucher disease is caused by a deficiency of the lysosomal enzyme acid β-glucosidase. Acid β-glucosidase catalyzes the conversion of the sphingolipid glucocerebroside into glucose and ceramide. The enzymatic deficiency causes an accumulation of glucosylceramide (GL-1) primarily in the lysosomal compartment of macrophages, giving rise to foam cells or "Gaucher cells."
The clinical features of this lysosomal storage disorder (LSD) are reflective of the accumulation of Gaucher cells in the reticuloendothelial system (liver, spleen, bone marrow, and other organs). The accumulation of Gaucher cells in the liver, spleen, and bone marrow leads to organomegaly and skeletal disease. Presence of Gaucher cells in the bone marrow and spleen leads to clinically significant anemia and thrombocytopenia.
CERDELGA is a specific inhibitor of glucosylceramide synthase (IC50=10 ng/mL) and acts as a substrate reduction therapy for GD1 by reducing the production of GL-1. By reducing GL-1 production, CERDELGA alleviates the accumulation of GL-1 in the target organs.
12.2 Pharmacodynamics
Effects on spleen and liver volume, hemoglobin, and platelets increased with increasing steady-state average trough concentrations of eliglustat ranging up to 14 ng/mL in treatment naive patients in Trial 1. In patients previously treated with enzyme-replacement therapy in Trial 2 [see Clinical Studies (14.2)], no clinically relevant exposure-response relationship was observed.
Cardiac Electrophysiology
Concentration-related increases were observed for the placebo-corrected change from baseline in the PR, QRS, and QTc intervals. At the mean peak concentration of 237 ng/mL at a dose of 800 mg eliglustat tartrate (8 times the recommended dose), CERDELGA did not prolong the QT/QTc interval to any clinically relevant extent. However, pharmacokinetic/pharmacodynamic modeling predicts mean (upper bound of the 95% one-sided confidence interval) increases in the PR, QRS, and QTcF intervals of 22 (26), 7 (10), and 13 (19) msec, respectively, at eliglustat plasma concentration of 500 ng/mL [see Warnings and Precautions (5.1)].
12.3 Pharmacokinetics
Absorption
The oral bioavailability of eliglustat was less than 5% in CYP2D6 EMs following a single 84 mg dose of CERDELGA.
In CYP2D6 EMs, the eliglustat pharmacokinetics is time-dependent and the systemic exposure increases in a more than dose-proportional manner over the dose range of 42 to 294 mg (0.5 to 3.5 times the recommended dosage). In addition, after multiple oral doses of 84 mg twice daily in EMs, eliglustat systemic exposure (AUC0–12) increased up to about 2-fold at steady state compared to after the first dose (AUC0–∞). The pharmacokinetics of eliglustat in CYP2D6 PMs is expected to be linear and time-independent. Compared to EMs, the systemic exposure following 84 mg twice daily at steady state is 7-fold to 9-fold higher in PMs.
Dosing of CERDELGA 84 mg once daily has not been studied in PMs. The predicted Cmax and AUC0–24hr in PMs using a physiologically based pharmacokinetic (PBPK) model with 84 mg once daily were 75 ng/mL and 956 hr∙ng/mL, respectively.
Table 7 describes the pharmacokinetic parameters for eliglustat in healthy subjects following multiple doses of 84 mg CERDELGA twice daily.
| Parameter | CYP2D6 Metabolizer Status | ||
|---|---|---|---|
| EMs (n=96) | IMs (n=1) | PMs*
(n=9) |
|
|
|||
| Cmax (ng/mL)† | 12.1 (42%) to 25.0 (141%) | 44.6 | 113 (32%) to 137 (40%) |
| AUCtau (ng∙hr/mL)† | 76.3 (37%) to 143 (160%) | 306 | 922 (33%) to 1057 (38%) |
| Median Tmax (hr) [min to max]‡ | 1.5 [0.5 to 3.0] to 2 [1.5 to 2.1] | 2 | 3 [2 to 4] |
Administration of CERDELGA with a high fat meal (approximately 1000 calories with 50% calories from fat) resulted in a 15% decrease in Cmax (not clinically significant) but no change in AUC.
Distribution
Following intravenous administration, the volume of distribution of eliglustat was 835 L in EMs. Plasma protein binding of eliglustat ranges from 76% to 83%.
Elimination
Eliglustat terminal elimination half-life was approximately 6.5 hours in CYP2D6 EMs, and 8.9 hours in PMs. Following intravenous administration of 42 mg (0.5 times the recommended oral dose) in healthy subjects, the mean (range) of eliglustat total body clearance was 88 L/h (80 to 105 L/h) in EMs.
Specific Populations
No clinically significant differences in the pharmacokinetics of eliglustat were observed based on age (18 to 71 years), sex, race (mostly were Caucasian, including those of Ashkenazi Jewish descent; however, it included the following populations: African American, American Indians, Hispanics, and Asians), or body weight (41 to 136 kg).
Patients with renal impairment
Eliglustat pharmacokinetics was similar in CYP2D6 EMs with severe renal impairment and healthy CYP2D6 EMs. Eliglustat pharmacokinetics in EMs with ESRD and in IMs or PMs with any degree of renal impairment is unknown [see Use in Specific Populations (8.6)].
Patients with hepatic impairment
Table 8 describes the effect of mild and moderate hepatic impairment on the pharmacokinetics of eliglustat in CYP2D6 EMs compared to EMs with normal hepatic function following a single 84 mg dose. The effect of hepatic impairment is highly variable with the coefficients of variation (CVs%) of 135% and 110% for Cmax and 171% and 121% for AUC in CYP2D6 EMs with mild and moderate hepatic impairment, respectively.
| Mild Hepatic Impairment (n=6) | Moderate Hepatic Impairment (n=7) | |
|---|---|---|
| Cmax | ↑ 1.2-fold | ↑ 2.8-fold |
| AUC | ↑ 1.2-fold | ↑ 5.2-fold |
Steady-state pharmacokinetics of eliglustat in CYP2D6 IMs and PMs with mild and moderate hepatic impairment is unknown. The effect of severe hepatic impairment in subjects with any CYP2D6 phenotype is unknown [see Use in Specific Populations (8.7)].
Drug Interaction Studies
Effect of other drugs on CERDELGA
Table 9 describes the effect of drug interactions on the pharmacokinetics of eliglustat [see Drug Interactions (7.1)].
| Concomitant Drug(s) | CYP2D6 Metabolizer Status | |||||
|---|---|---|---|---|---|---|
| EMs | IMs | PMs | ||||
| Cmax | AUCtau | Cmax | AUCtau | Cmax | AUCtau | |
| ↑ = Increased; ↓ = Decreased | ||||||
| CYP2D6 Inhibitor | ||||||
| Paroxetine (strong) | ↑ 7.0-fold | ↑ 8.4-fold | ↑ 2.1-fold* | ↑ 2.3-fold* | No increase expected† | |
| Terbinafine (moderate) | ↑ 3.8-fold* | ↑ 4.5-fold* | ↑ 1.6-fold* | |||
| CYP3A Inhibitor | ||||||
| Ketoconazole (strong) | ↑ 4.0-fold | ↑ 4.4-fold | ↑ 4.4-fold* | ↑ 5.4-fold* | ↑ 4.3- fold*,‡ | ↑ 6.2- fold*,‡ |
| Fluconazole (moderate) | ↑ 2.8-fold* | ↑ 3.2-fold* | ↑ 2.5-fold* | ↑ 2.9-fold* | ↑ 2.4- fold*,‡ | ↑ 3.0- fold*,‡ |
| CYP2D6 Inhibitors Concomitantly with CYP3A Inhibitors | ||||||
| Paroxetine with ketoconazole | ↑ 16.7-fold* | ↑ 24.2-fold* | ↑ 7.5-fold* | ↑ 9.8-fold* | Expected similar increase as with CYP3A inhibitors alone† | |
| Terbinafine with fluconazole | ↑ 10.2-fold* | ↑ 13.6-fold* | ↑ 4.2-fold* | ↑ 5.0-fold* | ||
| CYP3A Inducers | ||||||
| Rifampin (strong) | ↓ 90%§ | ↓ 95% | ||||
No clinically significant pharmacokinetic changes were observed for eliglustat when coadministered with intravenous rifampin (an OATP inhibitor), or gastric pH modifying drugs (e.g., aluminum hydroxide, magnesium hydroxide, calcium carbonate, pantoprazole).
In vitro, eliglustat is a substrate of P-glycoprotein (P-gp). The effect of P-gp inhibitors on eliglustat pharmacokinetics is unknown.
Effect of CERDELGA on other drugs
CYP2D6 substrates
Following multiple doses of CERDELGA 127 mg twice daily (1.5 times the recommended dosage), metoprolol (a CYP2D6 substrate) mean Cmax and AUC increased by 1.7-fold and 2.3-fold in CYP2D6 EMs, respectively, and by 1.2-fold and 1.6-fold in IMs, respectively [see Drug Interactions (7.2)].
P-gp substrates
Following multiple doses of CERDELGA 127 mg twice daily (1.5 times the recommended dosage) in CYP2D6 EMs and IMs, or 84 mg twice daily in PMs, digoxin (a P-gp substrate) mean Cmax increased by 1.7-fold and AUC increased by 1.5-fold [see Drug Interactions (7.2)].
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Carcinogenic potential of CERDELGA was assessed in 2-year carcinogenicity studies in rats and mice. In Sprague-Dawley rats, eliglustat was administered by oral gavage at doses up to 75 mg/kg/day in males (about 3.6 times the recommended human daily dose of 84 mg twice daily, based on body surface area) and 50 mg/kg/day in females (about 2.4 times the recommended human daily dose based on body surface area). In CD-1 mice, eliglustat was administered to males and females at up to 75 mg/kg/day (about 1.8 times the recommended human daily dose based on body surface area) via dietary admixture. Eliglustat did not produce any treatment-related neoplasms in rats or mice.
Mutagenesis
Eliglustat was negative in the Ames test, chromosome aberration test in human peripheral blood lymphocytes, mouse lymphoma gene mutation assay and in vivo oral mouse micronucleus test.
Impairment of Fertility
In a fertility and early embryonic development study in rats, eliglustat increased pre-implantation loss at 30 (about 1.5 times the recommended human oral dose based on body surface area) and 100 mg/kg/day (about 5 times the recommended human oral dose based on body surface area).
In mature male rats, eliglustat showed reversible adverse effects on sperm morphology, testes (germ cell necrosis), and sloughed cells in the epididymis at 200 mg/kg/day (about 10 times the recommended human oral dose based on body surface area). Similar effects on sperm were not seen in mature cynomolgus monkeys at 72 mg/kg/day (about 7 times the recommended human oral dose based on body surface area).
14 CLINICAL STUDIES
14.1 CERDELGA in Treatment-Naive GD1 Patients – Trial 1
Trial 1 (NCT00891202) was a randomized, double-blind, placebo-controlled, multicenter clinical study evaluating the efficacy and safety of CERDELGA in 40 treatment- naive GD1 patients 16 years of age or older (median age 30.4 years) with pre-existing splenomegaly and hematological abnormalities. Patients were required to have received no treatment with substrate reduction therapy within 6 months or ERT within 9 months prior to randomization; all but 5 patients in the study had no prior therapy. Patients were stratified according to baseline spleen volume (≤20 or >20 multiples of normal [MN]) and randomized in a 1:1 ratio to receive CERDELGA or placebo for the duration of the 9-month blinded primary analysis period. The CERDELGA treatment group was comprised of IM (5%), EM (90%) and URM (5%) patients. Patients randomized to CERDELGA treatment received a starting dose of 42 mg twice daily, with a dose increase to 84 mg twice daily possible at Week 4 based on the plasma trough concentration at Week 2. The majority of patients (17 [85%]) received a dose escalation to 84 mg twice daily at Week 4, and 3 (15%) continued to receive 42 mg twice daily for the duration of the 9-month blinded primary analysis period.
The primary endpoint was the percentage change in spleen volume (in MN) from baseline to 9 months as compared to placebo. Secondary endpoints were absolute change in hemoglobin level, percentage change in liver volume (in MN), and percentage change in platelet count from baseline to 9 months compared to placebo.
At baseline, mean spleen volumes were 12.5 and 13.9 MN in the placebo and CERDELGA groups, respectively, and mean liver volumes were 1.4 MN for both groups. Mean hemoglobin levels were 12.8 and 12.1 g/dL, and platelet counts were 78.5 and 75.1 × 109/L, respectively.
During the 9-month primary analysis period, CERDELGA demonstrated statistically significant improvements in all primary and secondary endpoints compared to placebo, as shown in Table 10.
| Placebo (n=20) | CERDELGA (n=20) | Difference (CERDELGA – Placebo) [95% CI] | p value* | |
|---|---|---|---|---|
| MN = Multiples of Normal, CI = confidence interval, NA = Not applicable | ||||
|
||||
| Percentage Change in Spleen Volume MN (%) | 2.3 | -27.8 | -30.0 [-36.8, -23.2] | <0.0001 |
| Absolute Change in Spleen Volume (MN) | 0.3 | -3.7 | -4.1 [-5.3, -2.9] | NA |
| Absolute Change in Hemoglobin Level (g/dL) | -0.5 | 0.7 | 1.2 [0.6, 1.9] | 0.0006 |
| Percentage Change in Liver Volume MN (%) | 1.4 | -5.2 | -6.6 [-11.4, -1.9] | 0.0072 |
| Absolute Change in Liver Volume (MN) | 0.0 | -0.1 | -0.1 [-0.2, 0.0] | NA |
| Percentage Change in Platelet Count (%) | -9.1 | 32.0 | 41.1 [24.0, 58.2] | <0.0001 |
| Absolute Change in Platelet Count (× 109/L) | -7.2 | 24.1 | 31.3 [18.8, 43.8] | NA |
In the open-label extension phase of Trial 1 in naive GD1 patients, 38 of 40 patients who continued treatment with CERDELGA for 2 years demonstrated the following changes in clinical parameters from baseline to 2 years: mean (SD) percent change in spleen volume (MN) -51.1% (10.7); mean (SD) percent change in liver volume (MN) -16.1% (11.3); mean (SD) absolute change in hemoglobin level (g/dL) 1.3 (1.2), and mean (SD) percent change in platelet count (mm3) 65.3% (40.9).
In a separate uncontrolled study (NCT00358150) of treatment-naive GD1 patients, improvements in spleen and liver volume, hemoglobin level, and platelet count continued through the 4-year treatment period.
14.2 Patients Switching from Enzyme Replacement Therapy to CERDELGA – Trial 2
Trial 2 (NCT00943111) was a randomized, open-label, active-controlled, non- inferiority, multicenter clinical study evaluating the efficacy and safety of CERDELGA compared with imiglucerase in 159 treated GD1 patients (median age 37.4 years) previously treated with enzyme replacement therapy (≥3 years of enzyme replacement therapy, dosed at 30–130 U/kg/month in at least 6 of the prior 9 months) who met pre-specified therapeutic goals at baseline. Pre-specified baseline therapeutic goals included: no bone crisis and free of symptomatic bone disease within the last year; mean hemoglobin level of ≥11 g/dL in females and ≥12 g/dL in males; mean platelet count ≥100,000/mm3; spleen volume <10 times normal and liver volume <1.5 times normal.
Patients were randomized 2:1 to receive CERDELGA or imiglucerase for the duration of the 12-month primary analysis period. Seventy-five percent of patients randomized to CERDELGA were previously treated with imiglucerase; 21% with velaglucerase alfa and 4% were unreported. Patients randomized to CERDELGA treatment received a starting dose of 42 mg twice daily, with dose increases to 84 mg twice daily and 127 mg twice daily possible at Weeks 4 and 8 based on plasma trough concentrations of CERDELGA at Weeks 2 and 6, respectively. The percentage of patients receiving the 3 possible CERDELGA doses was: 42 mg twice daily (20%), 84 mg twice daily (32%) and 127 mg twice daily (48%). The CERDELGA treatment group was comprised of PM (4%), IM (10%), EM (80%) and URM (4%) patients.
At baseline, mean spleen volumes were 2.6 and 3.2 MN in the imiglucerase and CERDELGA groups, respectively, and liver volumes were 0.9 MN in both groups. Mean hemoglobin levels were 13.8 and 13.6 g/dL, and platelet counts were 192 and 207 × 109/L, respectively.
The primary composite endpoint required stability in all four component domains (hemoglobin level, platelet count, liver volume, and spleen volume) based on changes between baseline and 12 months. Stability was defined by the following pre-specified thresholds of change: hemoglobin level <1.5 g/dL decrease, platelet count <25% decrease, liver volume <20% increase and spleen volume <25% increase. The percentages of patients meeting the criteria for stability in the individual components of the composite endpoint were assessed as secondary efficacy endpoints.
CERDELGA met the criteria to be declared non-inferior to imiglucerase in maintaining patient stability. After 12 months of treatment, the percentage of patients meeting the primary composite endpoint was 84.8% for the CERDELGA group compared to 93.6% for the imiglucerase group. The lower bound of the 95% CI of the 8.8% difference, -17.6%, was within the pre-specified non-inferiority margin of -25%. At Month 12, the percentages of CERDELGA and imiglucerase patients respectively, who met stability criteria for the individual components of the composite endpoint were: hemoglobin level, 94.9% and 100%; platelet count, 92.9% and 100%; spleen volume, 95.8% and 100%; and liver volume, 96.0% and 93.6%. Of the patients who did not meet stability criteria for the individual components, 12 of 15 CERDELGA patients and 3 of 3 imiglucerase patients remained within therapeutic goals for GD1.
Mean changes from baseline in the hematological and visceral parameters through 12 months of treatment are shown in Table 11. There were no clinically meaningful differences between groups for any of the four parameters.
| Imiglucerase (N=47) Mean [95% CI] | CERDELGA (N=99) Mean [95% CI] |
|
|---|---|---|
| MN = Multiples of Normal, CI = confidence interval | ||
|
||
| Percentage Change in Spleen Volume MN (%)* | -3.0 [-6.4, 0.4] | -6.2 [-9.5, -2.8] |
| Absolute Change in Spleen Volume (MN)* | -0.1 [-0.2, 0.0] | -0.2 [-0.3, -0.1] |
| Absolute Change in Hemoglobin Level (g/dL) | 0.0 [-0.2, 0.2] | -0.2 [-0.4, -0.1] |
| Percentage Change in Liver Volume MN (%) | 3.6 [0.6, 6.6] | 1.8 [-0.2, 3.7] |
| Absolute Change in Liver Volume (MN) | 0.0 [0.0, 0.1] | 0.0 [0.0, 0.0] |
| Percentage Change in Platelet Count (%) | 2.9 [-0.6, 6.4] | 3.8 [0.0, 7.6] |
| Absolute Change in Platelet Count (× 109/L) | 6.0 [-0.9, 13.0] | 9.5 [1.4, 17.6] |
| Patients Stable for 52 Weeks, n (%) (Composite Primary Endpoint) | 44 (93.6) | 84 (84.8) |
In the open-label extension phase of Trial 2, 141 of 146 patients (42 patients previously treated with enzyme treatment therapy and 99 who continued treatment with CERDELGA) were evaluated for stability, as defined in the initial 12 months of the trial, in clinical parameters (composite of spleen and liver volume, hemoglobin level, and platelet count). Stability was shown in 120/141 (85%) patients at one year and 111/129 (86%) patients at 2 years of CERDELGA exposure.
16 HOW SUPPLIED/STORAGE AND HANDLING
CERDELGA is supplied as 84 mg eliglustat in a capsule with a pearl blue-green opaque cap and pearl white opaque body imprinted with "GZ02" in black.
CERDELGA 84 mg capsules are supplied as:
NDC 58468-0220-1 – Carton containing 4 packs of capsules (56 capsules total). Each pack is composed of 1 blister card of 14 capsules and a cardboard wallet.
NDC 58468-0220-2 – Carton containing 1 pack of capsules (14 capsules total). Each pack is comprised of 1 blister card of 14 capsules and a cardboard wallet.
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Drug Interactions
Advise patients to discuss all the medications they are taking, including any herbal supplements or vitamins with their healthcare provider [see Contraindications (4), Warnings and Precautions (5), Drug Interactions (7)].
ECG Changes and Potential for Cardiac Arrhythmias
Advise patients to inform their healthcare provider of the following: history of congestive heart failure; recent acute myocardial infarction; bradycardia; heart block; ventricular arrhythmia; and long QT syndrome [see Warnings and Precautions (5.1)].
Advise patients to inform their healthcare provider if they develop new symptoms such as palpitations, fainting, and dizziness.
Administration Instructions
[See Dosage and Administration (2.4).]
Advise patients:
- Swallow capsules whole, preferably with water, and do not crush, dissolve, or open the capsules.
- CERDELGA can be taken with or without food.
- If a dose of CERDELGA is missed, take the prescribed dose at the next scheduled time; do not double the next dose.
- Avoid consumption of grapefruit or its juice.
- For patients currently treated with imiglucerase, velaglucerase alfa, or taliglucerase alfa, CERDELGA may be administered 24 hours after the last dose of the previous enzyme replacement therapy (ERT).
Genzyme Corporation
450 Water Street
Cambridge, MA 02141
A SANOFI COMPANY
For patent information: https://www.sanofi.us/en/products-and-resources/patents
CERDELGA is a registered trademark of Genzyme Corporation.
| MEDICATION GUIDE CERDELGA® (sir-DEL-guh) (eliglustat) capsules |
|
|---|---|
| This Medication Guide has been approved by the U.S. Food and Drug Administration. | Issued: January 2024 |
| What is the most important information I should know about CERDELGA?
CERDELGA can affect the way other medicines work and other medicines can affect how CERDELGA works. Using CERDELGA with other medicines or herbal supplements may cause an increased risk of side effects. Especially tell your doctor if you take:
|
|
| What is CERDELGA?
CERDELGA is a prescription medicine used for the long-term treatment of Gaucher disease type 1 (GD1) in adults. CERDELGA is not used in certain people with Gaucher disease type 1. Your doctor will perform a test to make sure that CERDELGA is right for you. It is not known if CERDELGA is safe and effective in children. |
|
| What should I tell my doctor before taking CERDELGA? Before taking CERDELGA, tell your doctor about all of your medical conditions, including if you:
|
|
How should I take CERDELGA?
|
|
| What should I avoid while taking CERDELGA?
Avoid eating or drinking grapefruit products while taking CERDELGA. Grapefruit products can increase the amount of CERDELGA in your body. |
|
| What are the possible side effects of CERDELGA?
See "What is the most important information I should know about CERDELGA?"
Tell your doctor if you have any side effect that bothers you or that does not go away. These are not all the possible side effects of CERDELGA. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|
How should I store CERDELGA?
|
|
| General information about the safe and effective use of CERDELGA.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use CERDELGA for a condition for which it was not prescribed. Do not give CERDELGA to other people, even if they have the same symptoms you have. It may harm them. If you would like more information, talk with your doctor. You can ask your doctor or pharmacist for information about CERDELGA that is written for health professionals. For more information, go to www.cerdelga.com or call 1-800-745-4447. |
|
| What are the ingredients in CERDELGA? Active ingredient: eliglustat Inactive ingredients: microcrystalline cellulose, lactose monohydrate, hypromellose, glyceryl behenate, gelatin, candurin silver fine, yellow iron oxide, and FD&C blue 2. Genzyme Corporation, 450 Water Street, Cambridge, MA 02141 A SANOFI COMPANY CERDELGA is a registered trademark of Genzyme Corporation. ©2024 Genzyme Corporation. All rights reserved. |
|