FULL PRESCRIBING INFORMATION
WARNING: THROMBOSIS, RENAL DYSFUNCTION AND ACUTE RENAL FAILURE
Thrombosis may occur with immune globulin products1-3, including PRIVIGEN. Risk factors may include: advanced age, prolonged immobilization, hypercoagulable conditions, history of venous or arterial thrombosis, use of estrogens, indwelling central vascular catheters, hyperviscosity, and cardiovascular risk factors. Thrombosis may occur in the absence of known risk factors [see Warnings and Precautions (5.3), Patient Counseling Information (17)].
Renal dysfunction, acute renal failure, osmotic nephrosis, and death may occur with immune globulin intravenous (IGIV) products in predisposed patients. Patients predisposed to renal dysfunction include those with any degree of pre-existing renal insufficiency, diabetes mellitus, age greater than 65, volume depletion, sepsis, paraproteinemia, or patients receiving known nephrotoxic drugs.
Renal dysfunction and acute renal failure occur more commonly in patients receiving IGIV products containing sucrose.4 PRIVIGEN does not contain sucrose.
For patients at risk of thrombosis, renal dysfunction or failure, administer PRIVIGEN at the minimum dose and infusion rate practicable. Ensure adequate hydration in patients before administration. Monitor for signs and symptoms of thrombosis and assess blood viscosity in patients at risk for hyperviscosity [see Dosage and Administration (2.5), Warnings and Precautions (5.2, 5.3)].
1 INDICATIONS AND USAGE
PRIVIGEN is an Immune Globulin Intravenous (Human), 10% Liquid indicated for the treatment of the following conditions.
1.1 Primary Humoral Immunodeficiency
PRIVIGEN is indicated as replacement therapy for primary humoral immunodeficiency (PI). This includes, but is not limited to, the humoral immune defect in congenital agammaglobulinemia, common variable immunodeficiency (CVID), X-linked agammaglobulinemia, Wiskott-Aldrich syndrome, and severe combined immunodeficiencies.
1.2 Chronic Immune Thrombocytopenic Purpura
PRIVIGEN is indicated for the treatment of patients age 15 years and older with chronic immune thrombocytopenic purpura (ITP) to raise platelet counts.
1.3 Chronic Inflammatory Demyelinating Polyneuropathy
PRIVIGEN is indicated for the treatment of adults with chronic inflammatory demyelinating polyneuropathy (CIDP) to improve neuromuscular disability and impairment.
Limitation of Use:
PRIVIGEN maintenance therapy in CIDP has not been studied for periods longer than 6 months. After responding during an initial treatment period, not all patients require indefinite maintenance therapy with PRIVIGEN in order to remain free of CIDP symptoms. Individualize the duration of any treatment beyond 6 months based upon the patient's response and demonstrated need for continued therapy.
2 DOSAGE AND ADMINISTRATION
| Indication | Dose | Initial infusion rate | Maintenance infusion rate (as tolerated) |
|---|---|---|---|
| Primary Immunodeficiency | 200-800 mg/kg (2-8 mL/kg) every 3-4 weeks | 0.5 mg/kg/min (0.005 mL/kg/min) | Increase to 8 mg/kg/min (0.08 mL/kg/min) |
| Chronic Immune Thrombocytopenic Purpura | 1 g/kg (10 mL/kg) for 2 consecutive days | 0.5 mg/kg/min (0.005 mL/kg/min) | Increase to 4 mg/kg/min (0.04 mL/kg/min) |
| Chronic Inflammatory Demyelinating Polyneuropathy | Loading dose: 2 g/kg (20 mL/kg) in divided doses over 2 to 5 consecutive days Maintenance dose: 1 g/kg (10 mL/kg) administered in 1 to 2 infusions on consecutive days, every 3 weeks | 0.5 mg/kg/min (0.005 mL/kg/min) | Increase to 8 mg/kg/min (0.08 mL/kg/min) |
2.1 Dosage for Primary Humoral Immunodeficiency (PI)
As there are significant differences in the half-life of IgG among patients with PI, the frequency and amount of immunoglobulin therapy may vary from patient to patient. The proper amount can be determined by monitoring clinical response.
The recommended dose of PRIVIGEN for patients with PI is 200 to 800 mg/kg (2 to 8 mL/kg), administered every 3 to 4 weeks. If a patient misses a dose, administer the missed dose as soon as possible, and then resume scheduled treatments every 3 or 4 weeks, as applicable.
Adjust the dosage over time to achieve the desired serum IgG trough levels and clinical responses. No randomized, controlled trial data are available to determine an optimal trough level in patients receiving immune globulin therapy.
Measles Exposure
If a patient has been exposed to measles, it may be prudent to administer an extra dose of Immune Globulin Intravenous as soon as possible and within 6 days of exposure. A dose of 400 mg/kg should provide a serum level > 240 mIU/mL of measles antibodies for at least two weeks.
If a patient is at risk of future measles exposure and receives a dose of less than 530 mg/kg every 3-4 weeks, the dose should be increased to at least 530 mg/kg. This should provide a serum level of 240 mIU/mL of measles antibodies for at least 22 days after infusion.
2.2 Dosage for Chronic Immune Thrombocytopenic Purpura (ITP)
The recommended dose of PRIVIGEN for patients with chronic ITP is 1 g/kg (10 mL/kg) administered daily for 2 consecutive days, resulting in a total dosage of 2 g/kg.
Carefully consider the relative risks and benefits before prescribing the high dose regimen (e.g., 1 g/kg/day for 2 days) in patients at increased risk of thrombosis, hemolysis, acute kidney injury, or volume overload [see Warnings and Precautions (5.9)].
2.3 Dosage for Chronic Inflammatory Demyelinating Polyneuropathy (CIDP)
PRIVIGEN may be initially administered as a total loading dose of 2 g/kg (20 mL/kg) given in divided doses over two to five consecutive days. PRIVIGEN may be administered as a maintenance infusion of 1 g/kg (10 mL/kg) administered in a single infusion given in one day or divided into two doses given on two consecutive days, every 3 weeks. Maintenance therapy beyond 6 months has not been studied.
The recommended initial infusion rate is 0.5 mg/kg/min (0.005 mL/kg/min). If the infusion is well tolerated, the rate may be gradually increased to a maximum of 8 mg/kg/min (0.08 mL/kg/min). For patients judged to be at risk for thrombosis, renal dysfunction, or volume overload, administer PRIVIGEN at the minimum infusion rate practicable [see Warnings and Precautions (5.2, 5.3)].
2.4 Preparation and Handling
- PRIVIGEN is a clear or slightly opalescent, colorless to pale yellow solution. Inspect parenteral drug products visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Do not use if the solution is cloudy, turbid, or if it contains particulate matter.
- DO NOT SHAKE.
- Do not freeze. Do not use if PRIVIGEN has been frozen.
- PRIVIGEN should be at room temperature (up to 25ºC [77ºF]) at the time of administration.
- Do not use PRIVIGEN beyond the expiration date on the product label.
- The PRIVIGEN vial is for single-use only. Promptly use any vial that has been entered. PRIVIGEN contains no preservative. Discard partially used vials or unused product in accordance with local requirements.
- Infuse PRIVIGEN using a separate infusion line. Prior to use, the infusion line may be flushed with Dextrose Injection, USP (D5W) or 0.9% Sodium Chloride for Injection, USP.
- Do not mix PRIVIGEN with other IGIV products or other intravenous medications. However, PRIVIGEN may be diluted with Dextrose Injection, USP (D5W).
- An infusion pump may be used to control the rate of administration.
- If large doses of PRIVIGEN are to be administered, several vials may be pooled using aseptic technique. Begin infusion within 8 hours of pooling.
2.5 Administration
PRIVIGEN is for intravenous administration only.
Monitor the patient's vital signs throughout the infusion. Slow or stop the infusion if adverse reactions occur. If symptoms subside promptly, the infusion may be resumed at a lower rate that is comfortable for the patient.
Ensure that patients with pre-existing renal insufficiency are not volume depleted. For patients judged to be at risk for renal dysfunction or thrombosis, administer PRIVIGEN at the minimum dose and infusion rate practicable, and discontinue PRIVIGEN administration if renal function deteriorates [see Boxed Warning, Warnings and Precautions (5.2, 5.3)].
The following patients may be at risk of developing systemic reactions (mimicking symptoms of an inflammatory response or infection) on rapid infusion of PRIVIGEN (greater than 4 mg/kg/min [0.04 mL/kg/min]): 1) those who have never received PRIVIGEN or another IgG product or who have not received it within the past 8 weeks, and 2) those who are switching from another IgG product. These patients should be started at a slow rate of infusion (e.g., 0.5 mg/kg/min [0.005 mL/kg/min] or less) and gradually increase as tolerated.
3 DOSAGE FORMS AND STRENGTHS
PRIVIGEN is a liquid solution containing 10% IgG (0.1 g/mL) for intravenous infusion.
4 CONTRAINDICATIONS
- PRIVIGEN is contraindicated in patients who have a history of anaphylactic or severe systemic reaction to the administration of human immune globulin.
- PRIVIGEN is contraindicated in patients with hyperprolinemia because it contains the stabilizer L-proline [see Description (11)].
- PRIVIGEN is contraindicated in IgA-deficient patients with antibodies to IgA and a history of hypersensitivity [see Warnings and Precautions (5.1)].
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity
Severe hypersensitivity reactions may occur [see Contraindications (4)]. In case of hypersensitivity, discontinue the PRIVIGEN infusion immediately and institute appropriate treatment. Medications such as epinephrine should be available for immediate treatment of acute hypersensitivity reactions.
PRIVIGEN contains trace amounts of IgA (≤25 mcg/mL) [see Description (11)]. Individuals with IgA deficiency can develop anti-IgA antibodies and anaphylactic reactions (including anaphylaxis and shock) after administration of blood components containing IgA. Patients with known antibodies to IgA may have a greater risk of developing potentially severe hypersensitivity and anaphylactic reactions with administration of PRIVIGEN. PRIVIGEN is contraindicated in patients with antibodies against IgA and a history of hypersensitivity.
5.2 Renal Dysfunction and Acute Renal Failure
Renal dysfunction, acute renal failure, osmotic nephrosis, and death may occur with immune globulin intravenous (IGIV) products in predisposed patients. Renal dysfunction and acute renal failure occur more commonly in patients receiving IGIV products containing sucrose. 4 PRIVIGEN does not contain sucrose. Acute renal failure may also occur as a result of PRIVIGEN-induced hemolysis. Ensure that patients are not volume depleted and assess renal function, including measurement of blood urea nitrogen (BUN) and serum creatinine, before the initial infusion of PRIVIGEN and at appropriate intervals thereafter.
Periodic monitoring of renal function and urine output is particularly important in patients judged to be at increased risk of developing acute renal failure.4 If renal function deteriorates, consider discontinuing PRIVIGEN. For patients judged to be at risk of developing renal dysfunction because of pre-existing renal insufficiency, or predisposition to acute renal failure (such as those with diabetes mellitus or hypovolemia, those who are obese, those who use concomitant nephrotoxic medicinal products, or those who are over 65 years of age), administer PRIVIGEN at the minimum rate of infusion practicable [see Boxed Warning, Dosage and Administration (2.5)].
5.3 Thrombosis
Thrombosis may occur following treatment with immune globulin products1-3, including PRIVIGEN. Risk factors may include: advanced age, prolonged immobilization, hypercoagulable conditions, history of venous or arterial thrombosis, use of estrogens, indwelling central vascular catheters, hyperviscosity, and cardiovascular risk factors. Thrombosis may occur in the absence of known risk factors.
Consider baseline assessment of blood viscosity in patients at risk for hyperviscosity, including those with cryoglobulins, fasting chylomicronemia/markedly high triacylglycerols (triglycerides), or monoclonal gammopathies. For patients at risk of thrombosis, administer PRIVIGEN at the minimum dose and infusion rate practicable. Ensure adequate hydration in patients before administration. Monitor for signs and symptoms of thrombosis and assess blood viscosity in patients at risk for hyperviscosity [see Boxed Warning, Dosage and Administration (2.5), Patient Counseling Information (17)].
5.4 Hyperproteinemia, Increased Serum Viscosity, and Hyponatremia
Hyperproteinemia, increased serum viscosity, and hyponatremia may occur following treatment with IGIV products, including PRIVIGEN. The hyponatremia is likely to be a pseudohyponatremia, as demonstrated by a decreased calculated serum osmolality or elevated osmolar gap. It is critical to distinguish true hyponatremia from pseudohyponatremia, as treatment aimed at decreasing serum free water in patients with pseudohyponatremia may lead to volume depletion, a further increase in serum viscosity, and a possible predisposition to thromboembolic events.5
5.5 Aseptic Meningitis Syndrome (AMS)
AMS may occur infrequently following treatment with PRIVIGEN [see Adverse Reactions (6)] and other human immune globulin products. Discontinuation of treatment has resulted in remission of AMS within several days without sequelae.6 AMS usually begins within several hours to 2 days following IGIV treatment.
AMS is characterized by the following signs and symptoms: severe headache, nuchal rigidity, drowsiness, fever, photophobia, painful eye movements, nausea, and vomiting. Cerebrospinal fluid (CSF) studies are frequently positive with pleocytosis up to several thousand cells per cubic millimeter, predominantly from the granulocytic series, and with elevated protein levels up to several hundred mg/dL, but negative culture results. Conduct a thorough neurological examination on patients exhibiting such signs and symptoms, including CSF studies, to rule out other causes of meningitis.
AMS may occur more frequently in association with high doses (2 g/kg) and/or rapid infusion of IGIV.
5.6 Hemolysis
PRIVIGEN may contain blood group antibodies that can act as hemolysins and induce in vivo coating of red blood cells (RBCs) with immunoglobulin, causing a positive direct antiglobulin test (DAT) (Coombs' test) result and hemolysis.7-9 Delayed hemolytic anemia can develop subsequent to PRIVIGEN therapy due to enhanced RBC sequestration, and acute hemolysis, consistent with intravascular hemolysis, has been reported.10 Cases of severe hemolysis-related renal dysfunction/failure or disseminated intravascular coagulation have occurred following infusion of PRIVIGEN.
The following risk factors may be associated with the development of hemolysis: high doses (e.g., ≥2 g/kg), given either as a single administration or divided over several days, and non-O blood group.11 Other individual patient factors, such as an underlying inflammatory state (as may be reflected by, for example, elevated C-reactive protein or erythrocyte sedimentation rate), have been hypothesized to increase the risk of hemolysis following administration of IGIV,12 but their role is uncertain. Hemolysis has been reported following administration of IGIV for a variety of indications, including ITP, CIDP, and PI.9
Closely monitor patients for clinical signs and symptoms of hemolysis, particularly patients with risk factors noted above and those with pre-existing anemia and/or cardiovascular or pulmonary compromise. Consider appropriate laboratory testing in higher risk patients, including measurement of hemoglobin or hematocrit prior to infusion and within approximately 36 hours and again 7 to 10 days post infusion. If clinical signs and symptoms of hemolysis or a significant drop in hemoglobin or hematocrit have been observed, perform additional confirmatory laboratory testing. If transfusion is indicated for patients who develop hemolysis with clinically compromising anemia after receiving IGIV, perform adequate cross-matching to avoid exacerbating on-going hemolysis.
5.7 Hypertension
Elevations of systolic blood pressure to ≥180 mm Hg and/or of diastolic blood pressure to >120 mm Hg (hypertensive urgency) have been observed during and/or shortly following infusion of PRIVIGEN. These blood pressure elevations were resolved or significantly improved within hours with either observation alone or changes in oral anti-hypertensive therapy [see Adverse Reactions (6.1)]. Such elevations were reported more often among patients with a history of hypertension. Check patients for a history of hypertension and current antihypertensive medication use. Monitor blood pressure prior to, during, and following PRIVIGEN infusion.
5.8 Transfusion-Related Acute Lung Injury (TRALI)
Noncardiogenic pulmonary edema may occur following treatment with IGIV products, including PRIVIGEN.13 TRALI is characterized by severe respiratory distress, pulmonary edema, hypoxemia, normal left ventricular function, and fever. Symptoms typically appear within 1 to 6 hours following treatment.
Monitor patients for pulmonary adverse reactions. If TRALI is suspected, perform appropriate tests for the presence of anti-neutrophil antibodies and anti-human leukocyte antigen (HLA) antibodies in both the product and the patient's serum.
TRALI may be managed using oxygen therapy with adequate ventilatory support.
5.9 Volume Overload
Carefully consider the relative risks and benefits before prescribing the high dose regimen (for chronic ITP and CIDP) in patients at increased risk of thrombosis, hemolysis, acute kidney injury, or volume overload.
5.10 Transmissible Infectious Agents
Because PRIVIGEN is made from human blood, it may carry a risk of transmitting infectious agents (eg, viruses, the variant Creutzfeldt Jakob disease [vCJD] agent and, theoretically, the Creutzfeldt Jakob disease [CJD] agent). The risk of infectious agent transmission has been reduced by screening plasma donors for prior exposure to certain viruses, testing for the presence of certain current virus infections, and including virus inactivation/removal steps in the manufacturing process for PRIVIGEN.
Report any infection thought to be possibly transmitted by PRIVIGEN to CSL Behring Pharmacovigilance at 1-866-915-6958.
6 ADVERSE REACTIONS
The following important adverse reactions are reported with IGIV: hypersensitivity, renal dysfunction and acute renal failure, thrombosis, hyperproteinemia, increased serum viscosity, hyponatremia, aseptic meningitis syndrome, hemolysis, hypertension, transfusion related acute lung injury, volume overload, and transmissible infectious agents [see Warnings and Precautions (5)] and are described elsewhere in the prescribing information.
Adverse reactions (ARs) [see Adverse Reactions (6.1)] are defined as adverse events at least possibly related or events occurring during or within 72 hours of a PRIVIGEN infusion.
Primary Humoral Immunodeficiency
The most serious adverse reaction observed in clinical study subjects receiving PRIVIGEN for PI was hypersensitivity in one subject [see Warnings and Precautions (5.1)]. The most common adverse reactions observed in >5% of clinical study subjects with PI were headache, fatigue, nausea, chills, vomiting, back pain, pain, elevated body temperature, abdominal pain, diarrhea, cough, stomach discomfort, chest pain, joint swelling/effusion, influenza-like illness, pharyngolaryngeal pain, urticaria, and dizziness.
Chronic Immune Thrombocytopenic Purpura
The most serious adverse reactions observed in the premarketing clinical study subjects receiving PRIVIGEN for chronic ITP were aseptic meningitis syndrome in one subject and hemolysis in two subjects [see Warnings and Precautions (5.5, 5.6)]. A total of 8 subjects (14%) in the premarketing ITP study experienced hemolysis as documented from clinical laboratory data. No serious adverse reactions were observed in the postmarketing chronic ITP study. A total of 12 subjects (21%) in the postmarketing ITP study were adjudicated to have mild hemolysis as documented from clinical laboratory data [see Warnings and Precautions (5.6)]. The most common adverse reactions observed in >5% of subjects in both clinical studies of subjects with chronic ITP were laboratory findings consistent with hemolysis (hemoglobin and hematocrit decrease without blood loss in conjunction with positive direct antiglobulin test (DAT) and elevated blood lactate dehydrogenase (LDH) and/or indirect bilirubin), headache, elevated body temperature, anemia, nausea, and vomiting.
Chronic Inflammatory Demyelinating Polyneuropathy
The most serious adverse reactions observed in clinical study subjects receiving PRIVIGEN for CIDP was hemolysis. The most common adverse reactions observed in >5% of subjects in both clinical studies of subjects with CIDP were headache, asthenia, hypertension, nausea, pain in extremity, hemolysis, influenza like illness, leukopenia, and rash.
6.1 Clinical Trials Experience
Because different clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Treatment of Primary Humoral Immunodeficiency
In a prospective, open-label, single-arm, multicenter clinical study, 80 subjects with PI (with a diagnosis of XLA or CVID) received PRIVIGEN every 3 or 4 weeks for up to 12 months [see Clinical Studies (14.1)]. All subjects had been on regular IGIV replacement therapy for at least 6 months prior to participating in the study. Subjects ranged in age from 3 to 69; 46 (57.5%) were male and 34 (42.5%) were female.
The safety analysis included all 80 subjects, 16 (20%) on the 3-week schedule and 64 (80%) on the 4-week schedule. The median dose of PRIVIGEN administered was 428 mg/kg (3-week schedule) or 441 mg/kg (4-week schedule) and ranged from 200 to 888 mg/kg. A total of 1038 infusions of PRIVIGEN were administered, 272 in the 3-week schedule and 766 in the 4-week schedule.
Routine premedication was not allowed. However, subjects who experienced two consecutive infusion-related ARs that were likely to be prevented by premedication were permitted to receive antipyretics, antihistamines, NSAIDs, or antiemetic agents. During the study, 8 (10%) subjects received premedication prior to 51 (4.9%) of the 1038 infusions administered.
Table 2 summarizes the most frequent ARs that occurred in >5% of subjects.
| AR* | Number (%) of Subjects [n=80] | Number (Rate) of Infusions with AR [n=1038] |
|---|---|---|
|
||
| Headache | 36 (45.0) | 100 (0.096) |
| Fatigue | 13 (16.3) | 29 (0.028) |
| Nausea | 11 (13.8) | 23 (0.022) |
| Chills | 9 (11.3) | 15 (0.014) |
| Vomiting | 9 (11.3) | 15 (0.014) |
| Back pain | 8 (10.0) | 15 (0.014) |
| Pain | 7 (8.8) | 14 (0.013) |
| Elevated body temperature | 7 (8.8) | 12 (0.012) |
| Diarrhea | 6 (7.5) | 6 (0.006) |
| Cough | 5 (6.3) | 5 (0.005) |
| Stomach discomfort | 5 (6.3) | 5 (0.005) |
Of the 192 ARs reported (including 5 serious, severe ARs described below) 91 were mild (awareness of sign, symptom or event, but easily tolerated), 81 were moderate (discomfort enough to cause interference with usual activity and may have warranted intervention), 19 were severe (incapacitating with inability to do usual activities or significantly affected clinical status, and warranted intervention), and 1 was of unknown severity.
The five serious ARs (hypersensitivity, chills, fatigue, dizziness, and increased body temperature, all severe), occurred in one subject, and resulted in the subject's withdrawal from the study. Two other subjects withdrew from the study due to ARs (chills and headache in one subject; vomiting in the other).
Seventy-seven of the 80 subjects enrolled in this study had a negative DAT at baseline. Of these 77 subjects, 36 (46.8%) developed a positive DAT at some time during the study. However, no subjects showed evidence of hemolytic anemia.
During this study, no subjects tested positive for infection due to human immunodeficiency virus (HIV), hepatitis B virus (HBV), hepatitis C virus (HCV), or B19 virus (B19V).
An extension of the study described above was conducted in 55 adult and pediatric subjects with PI to collect additional efficacy, safety, and tolerability data. This study included 45 subjects from the pivotal study who were receiving PRIVIGEN and 10 new subjects who were receiving another IGIV product prior to enrolling in the extension study. Subjects ranged in age from 4 to 81 years; 26 (47.3%) were male and 29 (52.7%) were female.
Subjects were treated with PRIVIGEN at median doses ranging from 286 to 832 mg/kg per infusion over a treatment period ranging from 1 to 27 months. Twelve (21.8%) subjects were on a 3-week treatment schedule with the number of infusions per subject ranging from 4 to 38 (median: 8 infusions); 43 (78%) subjects were on a 4-week schedule with the number of infusions ranging from 1 to 31 (median: 15 infusions). A total of 771 infusions were administered in this study.
In this study, subjects who continued from the pivotal study were permitted to receive infusions of PRIVIGEN at a rate up to 12 mg/kg/min (as opposed to the maximum of 8 mg/kg/min allowed in the pivotal study) at the discretion of the investigator based on individual tolerability. Twenty-three (51%) of the 45 subjects from the pivotal study (42% of the 55 subjects in the extension study) received 265 (38%) infusions at a maximum rate greater than the recommended rate of 8 mg/kg/min [see Dosage and Administration (2.5)]. The median of the maximum infusion rate in this subset was 12 mg/kg/min. However, because the study was not designed to compare infusion rates, no definitive conclusions regarding tolerability could be drawn for infusion rates higher than the recommended rate of 8 mg/kg/min.
Table 3 summarizes the ARs that occurred in >5% of subjects.
| AR* | Number (%) of Subjects [n=55] | Number (Rate) of Infusions with AR [n=771] |
|---|---|---|
| Note: The AR rates in this study cannot be compared directly to the rates in other IGIV studies, including the original pivotal study described earlier in this section, because (1) the extension study used an enriched population and (2) the selective use of higher infusion rates at the investigators' discretion in a subset of subjects may have introduced bias. | ||
| Headache | 18 (32.7) | 76 (0.099) |
| Nausea | 6 (10.9) | 10 (0.013) |
| Elevated body temperature | 4 (7.3) | 12 (0.016) |
| Abdominal pain† | 4 (7.3) | 7 (0.009) |
| Chest pain | 3 (5.5) | 4 (0.005) |
| Chills | 3 (5.5) | 7 (0.009) |
| Joint swelling/effusion | 3 (5.5) | 7 (0.009) |
| Pain | 3 (5.5) | 6 (0.008) |
| Fatigue | 3 (5.5) | 5 (0.006) |
| Influenza-like illness | 3 (5.5) | 5 (0.006) |
| Pharyngolaryngeal pain | 3 (5.5) | 4 (0.005) |
| Urticaria | 3 (5.5) | 4 (0.005) |
| Dizziness | 3 (5.5) | 3 (0.004) |
Of the 125 reported ARs, 76 were mild (did not interfere with routine activities), 40 were moderate (interfered somewhat with routine activities), and 9 were severe (impossible to perform routine activities).
Three subjects experienced ARs: dyspnea and pancytopenia in one subject, a transient ischemic attack 16 days after the infusion in one subject, and mild urticaria in one subject, resulting in the subject's withdrawal from the study.
Treatment of Chronic Immune Thrombocytopenic Purpura
In a prospective, open-label, single-arm, multicenter premarketing clinical study, 57 subjects with chronic ITP and a platelet count of 20 × 109/L or less received a total of 2 g/kg dose of PRIVIGEN administered as 1 g/kg infusions daily for 2 consecutive days [see Clinical Studies (14.2)]. Subjects ranged in age from 15 to 69; 23 (40%) were male and 34 (60%) were female.
Concomitant medications affecting platelets or other treatments for chronic ITP were not allowed. Thirty-two (56%) subjects received premedication with acetaminophen and/or an antihistamine.
Table 4 summarizes the most frequent ARs that occurred in >5% of subjects with chronic ITP.
| AR* | Number (%) of Subjects [n=57] | Number (Rate) of Infusions with AR [n=114] |
|---|---|---|
|
||
| Headache | 37 (64.9) | 52 (0.456) |
| Elevated body temperature | 21 (36.8) | 23 (0.202) |
| Positive DAT | 7 (12.3) | 8 (0.070) |
| Anemia | 6 (10.5) | 6 (0.053) |
| Nausea | 6 (10.5) | 8 (0.070) |
| Epistaxis | 6 (10.5) | 8 (0.070) |
| Vomiting | 6 (10.5) | 7 (0.061) |
| Blood bilirubin unconjugated increased | 6 (10.5) | 6 (0.053) |
| Blood bilirubin conjugated increased | 5 (8.8) | 5 (0.044) |
| Blood total bilirubin increased | 3 (5.3) | 3 (0.026) |
| Hematocrit decreased | 3 (5.3) | 3 (0.026) |
| Blood lactate dehydrogenase increased | 3 (5.3) | 3 (0.026) |
Of the 149 non-serious ARs, 103 were mild (awareness of sign, symptom or event, but easily tolerated), 37 were moderate (discomfort enough to cause interference with usual activity and may have warranted intervention), and 9 were severe (incapacitating with inability to do usual activities or significantly affected clinical status, and warranted intervention).
One subject experienced a serious AR (aseptic meningitis).
Eight subjects, all of whom had a positive DAT, experienced transient drug-related hemolytic reactions, which were associated with elevated bilirubin, elevated lactate dehydrogenase, and a decrease in hemoglobin level within two days after the infusion of PRIVIGEN. Two of the eight subjects were clinically anemic but did not require clinical intervention; these cases resolved uneventfully.
Four other subjects with active bleeding were reported to have developed anemia without evidence of hemolysis.
In this study, there was a decrease in hemoglobin after the first PRIVIGEN infusion (median decrease of 1.2 g/dL by Day 8) followed by a return to near baseline by Day 29.
Fifty-six of the 57 subjects in this study had a negative DAT at baseline. Of these 56 subjects, 12 (21%) developed a positive DAT during the 29-day study period.
Postmarketing Commitment Study in Chronic Immune Thrombocytopenic Purpura
In a prospective, open-label, single-arm, multicenter postmarketing clinical study whose primary objective was to evaluate mechanisms of hemolysis, 57 subjects with chronic ITP and a platelet count of <30 × 109/L at screening were studied following treatment with PRIVIGEN. Twenty-one (21) subjects (37%) received 1 infusion of 1 g/kg on Day 1 and 36 subjects (63%) received 2 infusions each of 1 g/kg (Day 1 and Day 3). Concomitant medications affecting platelets or other treatments for chronic ITP were not allowed. Subjects received premedication with acetaminophen and/or an antihistamine [see Clinical Studies (14.3)].
The most frequent ARs (adverse events at least possibly related or events occurring during or within 72 hours after the end of treatment) that occurred in >5% of subjects with chronic ITP were headache (16 subjects [28%]) and pyrexia (3 subjects [5%]).
No subject experienced a serious adverse reaction.
Of the 23 non-serious ARs, 22 were mild (does not interfere with routine activities), 1 was moderate (interferes somewhat with routine activities), and none were severe (impossible to perform routine activities).
All 57 subjects had a negative DAT at baseline. Twenty-two (38%) developed a positive DAT by Day 4, 19 of these subjects were from blood group A.
Fifteen subjects were adjudicated by an independent expert committee, for presumptive/possible hemolysis, all of whom received 2 g/kg IGIV during the study [see Clinical Studies (14.3)]. Twelve subjects (21%) were judged to have mild hemolysis. In these 12 subjects there was a median hemoglobin drop from baseline at Day 9 (nadir) of -3.0 g/dL (range -0.9 to -5.8 g/dL) with Day 9 hemoglobin values ranging from 9.9 to 13.2 g/dL and a median drop from baseline in hemogloblin of -1.2 g/dL (range -0.1 to -2.7 g/dL) at Day 29 (end of study) with hemoglobin values ranging from 11.8 to 15.8 g/dL. Ten subjects were blood group A and 2 subjects were blood group B. These hemoglobin drops were transient and were followed by recovery or partial recovery by Day 29. One subject experienced mild dyspnea between Day 9 and Day 16; 1 subject experienced mild dizziness on Day 4. No subject was judged as having experienced clinically significant intravascular hemolysis. Three of the 15 adjudicated subjects were judged not to have experienced hemolysis.
Treatment of Chronic Inflammatory Demyelinating Polyneuropathy (CIDP)
In a prospective, open-label, single-arm, multicenter clinical study (PRIVIGEN Impact on Mobility and Autonomy [PRIMA]), 28 subjects with CIDP received a PRIVIGEN loading dose of 2 g/kg followed by PRIVIGEN maintenance doses of 1 g/kg every 3 weeks for up to 21 weeks with 3 week follow up [see Clinical Studies (14.4)]. Administration of the loading dose occurred over 2 days and the maintenance dose over 1 day in the majority of cases.
Table 5 summarizes the most frequent ARs that occurred in ≥5% of subjects with CIDP.
| AR* | Number (%) of Subjects [n=28] | Number (Rate) of Infusions with AR [n=259] |
|---|---|---|
|
||
| Headache | 8 (28.6) | 19 (0.073) |
| Asthenia | 4 (14.3) | 4 (0.015) |
| Hypertension | 4 (14.3) | 6 (0.023) |
| Nausea | 3 (10.7) | 3 (0.012) |
| Pain in extremity | 3 (10.7) | 3 (0.012) |
| Hemolysis | 2 (7.1) | 2 (0.008) |
| Influenza like illness | 2 (7.1) | 2 (0.008) |
| Leukopenia | 2 (7.1) | 2 (0.008) |
| Rash | 2 (7.1) | 2 (0.008) |
Two hemolysis serious adverse reactions occurred after the start of the PRIVIGEN induction dose in subjects with non-O blood groups (A and AB). The reactions resolved after discontinuation without the need for transfusion.
Four subjects, three of whom had a history of hypertension, had reversible increases in systolic blood pressure to ≥180 mm Hg during or within 1 to 4 hours following PRIVIGEN infusion. One of these subjects who had a history of untreated hypertension had a reversible increase in diastolic blood pressure from 84 mm Hg pre-infusion to 135 mm Hg at 1 hour after the end of the infusion. All were resolved or significantly improved within 1 to 6 hours with either observation alone or changes in oral anti-hypertensive therapy.
A total of 71 ARs were reported: 46 were mild (does not interfere with routine activities), 23 were moderate (interferes somewhat with routine activities), and 2 were severe (impossible to perform routine activities) in intensity.
In a second prospective, open-label PRIVIGEN pre-randomization phase of a multicenter, randomized, double-blind, placebo-controlled clinical study (Polyneuropathy and Treatment with Hizentra [PATH]), 207 IGIV-pretreated subjects with CIDP received a PRIVIGEN loading dose of 2 g/kg followed by up to 4 PRIVIGEN maintenance doses of 1 g/kg every three weeks for up to 13 weeks. Additionally, 60 of these subjects received PRIVIGEN rescue treatment by the same dosing regimen following CIDP relapse during the double-blind post-randomization phase [see Clinical Studies (14.4)].
Eight subjects experienced a serious adverse reaction (acute rash cutaneous, blood pressure diastolic increased, exacerbation of CIDP [2], hypersensitivity, pulmonary embolism, respiratory failure, and migraine. The serious adverse reactions of pulmonary embolism and respiratory failure occurred in subjects with preexisting risk factors. All serious adverse reactions resolved without sequelae.
Adverse reactions that occurred in >5% of subjects with CIDP were headache (33 subjects, 15.9% [rate per infusion 56/1894, 0.030]).
A total of 225 ARs were reported: 160 were mild (is transient, does not usually interfere with routine activities but minimal treatment or therapeutic intervention may be required), 59 were moderate (interferes somewhat with routine activities and usually alleviated with specific intervention but poses no significant or permanent risk of harm), and 6 were severe (interrupts usual activities of daily living, significantly affects clinical status, or may require intensive therapeutic intervention) in intensity.
6.2 Postmarketing Experience
Because adverse reactions are reported voluntarily post-approval from a population of uncertain size, it is not always possible to reliably estimate the frequency of these reactions or establish a causal relationship to product exposure.
PRIVIGEN
The following adverse reactions have been identified during postmarketing use of PRIVIGEN. This list does not include reactions already reported in clinical studies with PRIVIGEN [see Adverse Reactions (6.1)].
- Blood and lymphatic system disorders: Decreased neutrophil count
- Infusion reactions: Changes in blood pressure, dyspnea, tachycardia, flushing
- Hematologic: hemoglobinuria/hematuria/chromaturia, renal failure
- Neurological: photophobia
- Integumentary: pruritus
General
In addition, the following adverse reactions have been identified and reported during the post-approval use of immune globulin products.14
- Infusion Reactions: Tachycardia, malaise, flushing, rigors
- Renal: Acute renal dysfunction/failure, osmotic nephropathy
- Respiratory: Apnea, Acute Respiratory Distress Syndrome (ARDS), TRALI, cyanosis, hypoxemia, pulmonary edema, bronchospasm
- Cardiovascular: Cardiac arrest, thromboembolism, vascular collapse, hypotension
- Neurological: Coma, loss of consciousness, seizures, tremor
- Integumentary: Stevens-Johnson syndrome, epidermolysis, erythema multiforme, bullous dermatitis
- Hematologic: Pancytopenia, leukopenia
- Gastrointestinal: Hepatic dysfunction
7 DRUG INTERACTIONS
7.1 Live Virus Vaccines
The passive transfer of antibodies with immunoglobulin administration may interfere with the response to live virus vaccines such as measles, mumps, rubella, and varicella [see Patient Counseling Information (17)].15
Inform the immunizing physician of recent therapy with PRIVIGEN so that appropriate measures can be taken.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
No human data are available to indicate the presence or absence of drug-associated risk. Animal reproduction studies have not been conducted with PRIVIGEN. It is not known whether PRIVIGEN can cause fetal harm when administered to a pregnant woman or can affect reproduction capacity. Immune globulins cross the placenta from maternal circulation increasingly after 30 weeks of gestation.16,17 PRIVIGEN should be given to pregnant women only if clearly needed. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
8.2 Lactation
Risk Summary
No human data are available to indicate the presence or absence of drug-associated risk. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for PRIVIGEN and any potential adverse effects on the breastfed infant from PRIVIGEN or from the underlying maternal condition.
8.4 Pediatric Use
Treatment of Primary Humoral Immunodeficiency
PRIVIGEN was evaluated in 31 pediatric subjects (19 children and 12 adolescents) with PI (prospective, open label, single arm, multicenter clinical study). There were no apparent differences in the safety and efficacy profiles as compared to those in adult subjects. No pediatric-specific dose requirements were necessary to achieve the desired serum IgG levels. The safety and effectiveness of PRIVIGEN have not been studied in clinical trials in pediatric patients with PI who are under the age of 3.
8.5 Geriatric Use
Clinical studies of PRIVIGEN in PID and ITP did not include sufficient numbers of subjects age 65 and over to determine whether they respond differently from younger subjects.
The safety and effectiveness of PRIVIGEN in CIDP subjects age 65 and over was similar to those under age 65.
Use caution when administering PRIVIGEN to patients age 65 and over who are judged to be at increased risk of developing acute renal insufficiency and thrombosis [see Boxed Warning, Warnings and Precautions (5.2, 5.3)]. Do not exceed recommended doses, and administer PRIVIGEN at the minimum dose and infusion rate practicable.
10 OVERDOSAGE
Overdose may lead to fluid overload and hyperviscosity, particularly in the elderly and in patients with impaired renal function.
11 DESCRIPTION
PRIVIGEN is a ready-to-use, sterile, 10% protein liquid preparation of polyvalent human immunoglobulin G (IgG) for intravenous administration. PRIVIGEN has a purity of at least 98% IgG, consisting primarily of monomers. The balance consists of IgG dimers (≤12%), small amounts of fragments and polymers, and albumin. PRIVIGEN contains ≤25 mcg/mL IgA. The IgG subclass distribution is similar to that of normal human plasma. PRIVIGEN has an osmolality of approximately 320 mOsmol/kg (range: 240 to 440) and a pH of 4.8 (range: 4.6 to 5.0).
PRIVIGEN contains approximately 250 mmol/L (range: 210 to 290) of L-proline (a nonessential amino acid) as a stabilizer and trace amounts of sodium. PRIVIGEN contains no carbohydrate stabilizers (e.g., sucrose, maltose) and no preservative.
PRIVIGEN is prepared from large pools of human plasma by a combination of cold ethanol fractionation, octanoic acid fractionation, and anion exchange chromatography. The IgG proteins are not subjected to heating or to chemical or enzymatic modification. The Fc and Fab functions of the IgG molecule are retained. Fab functions tested include antigen binding capacities, and Fc functions tested include complement activation and Fc-receptor-mediated leukocyte activation (determined with complexed IgG). PRIVIGEN does not activate the complement system or prekallikrein in an unspecific manner.
To specifically reduce blood group A and B antibodies (isoagglutinins A and B) the manufacturing process for PRIVIGEN includes an immunoaffinity chromatography step.
All plasma units used in the manufacture of PRIVIGEN have been tested and approved for manufacture using FDA-licensed serological assays for hepatitis B surface antigen and antibodies to HCV and HIV-1/2 as well as FDA-licensed Nucleic Acid Testing (NAT) for HBV, HCV and HIV-1 and found to be nonreactive (negative). In addition, the plasma has been tested for B19 virus (B19V) DNA by NAT. Only plasma that passed virus screening is used for production, and the limit for B19V in the fractionation pool is set not to exceed 104 IU of B19V DNA per mL.
The manufacturing process for PRIVIGEN includes three steps to reduce the risk of virus transmission. Two of these are dedicated virus clearance steps: pH 4 incubation to inactivate enveloped viruses and virus filtration to remove, by size exclusion, both enveloped and non-enveloped viruses as small as approximately 20 nanometers. In addition, a depth filtration step contributes to the virus reduction capacity.
These steps have been independently validated in a series of in vitro experiments for their capacity to inactivate and/or remove both enveloped and non-enveloped viruses.
Table 6 shows the virus clearance during the manufacturing process for PRIVIGEN, expressed as the mean log10 reduction factor (LRF).
| HIV-1 | PRV | BVDV | WNV | EMCV | MVM | |
|---|---|---|---|---|---|---|
| HIV-1, human immunodeficiency virus type 1, a model for HIV-1 and HIV-2; PRV, pseudorabies virus, a nonspecific model for large enveloped DNA viruses (eg, herpes virus); BVDV, bovine viral diarrhea virus, a model for hepatitis C virus; WNV, West Nile virus; EMCV, encephalomyocarditis virus, a model for hepatitis A virus; MVM, minute virus of mice, a model for a small highly resistant non-enveloped DNA virus (eg, parvovirus); LRF, log10 reduction factor; nt, not tested. | ||||||
|
||||||
| Virus property | ||||||
| Genome | RNA | DNA | RNA | RNA | RNA | DNA |
| Envelope | Yes | Yes | Yes | Yes | No | No |
| Size (nm) | 80-100 | 120-200 | 50-70 | 50-70 | 25-30 | 18-24 |
| Manufacturing step | Mean LRF | |||||
| pH 4 incubation | ≥5.6 | ≥6.1 | 4.6 | ≥7.8 | nt | nt |
| Depth filtration | ≥6.7 | ≥5.7 | 3.5±0.2 | 3.0±0.4 | 5.7±0.2 | 3.7±0.3 |
| Virus filtration | ≥4.7 | ≥5.8 | ≥4.6 | ≥6.8 | ≥6.3 | ≥6.5 |
| Overall reduction (log10 units) | ≥17.0 | ≥17.6 | ≥12.7 | ≥17.6 | ≥12.0 | ≥10.2 |
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
PRIVIGEN supplies a broad spectrum of opsonizing and neutralizing IgG antibodies against a wide variety of bacterial and viral agents. The mechanism of action has not been fully elucidated, but may include immunomodulatory effects.
12.3 Pharmacokinetics
Treatment of Primary Humoral Immunodeficiency
In the clinical study assessing the efficacy and safety of PRIVIGEN in 80 subjects with PI [see Clinical Studies (14.1)], serum concentrations of total IgG and IgG subclasses were measured in 25 subjects (ages 13 to 69) following the 7th infusion for the 3 subjects on the 3-week dosing interval and following the 5th infusion for the 22 subjects on the 4-week dosing interval. The dose of PRIVIGEN used in these subjects ranged from 200 mg/kg to 714 mg/kg. After the infusion, blood samples were taken until Day 21 and Day 28 for the 3-week and 4-week dosing intervals, respectively.
Table 7 summarizes the pharmacokinetic parameters of PRIVIGEN, based on serum concentrations of total IgG.
| Parameter | 3-Week Dosing Interval (n=3) | 4-Week Dosing Interval (n=22) |
||
|---|---|---|---|---|
| Mean (SD) | Median (Range) | Mean (SD) | Median (Range) |
|
| Cmax, maximum serum concentration; Cmin, trough (minimum level) serum concentration; t½, elimination half-life; AUC0-t, area under the curve from 0 hour to last sampling time; AUC0-∞, area under the curve from 0 hour to infinite time. | ||||
|
||||
| Cmax (peak, mg/dL) | 2,550 (400) | 2,340 (2,290-3,010) | 2,260 (530) | 2,340 (1,040-3,460) |
| Cmin (trough, mg/dL) | 1,230 (230) | 1,200 (1,020-1,470) | 1,000 (200) | 1,000 (580-1,360) |
| t½ (days) | 27.6 (5.9) | 27.8 (21.6-33.4) | 45.4 (18.5) | 37.3 (20.6-96.6) |
| AUC0-t
(day × mg/dL)* | 32,820 (6,260) | 29,860 (28,580-40,010) | 36,390 (5,950) | 36,670 (19,680-44,340) |
| AUC0-∞
(day × mg/dL)* | 79,315 (20,170) | 78,748 (59,435-99,762) | 104,627 (33,581) | 98,521 (64,803-178,600) |
| Clearance (mL/day/kg)* | 1.3 (0.1) | 1.3 (1.1-1.4) | 1.3 (0.3) | 1.3 (0.9-2.1) |
| Mean residence time (days)* | 38.6 (8.1) | 39.5 (30.1-46.2) | 65.2 (24.7) | 59.0 (33.2-129.6) |
| Volume of distribution at steady state (mL/kg)* | 50 (13) | 44 (40-65) | 84 (35) | 87 (40-207) |
Although no systematic study was conducted to evaluate the effect of gender and age on the pharmacokinetics of PRIVIGEN, based on the small sample size (11 males and 14 females), it appears that clearance of PRIVIGEN is comparable in males (1.27 ± 0.35 mL/day/kg) and females (1.34 ± 0.22 mL/day/kg). In six subjects between 13 and 15 years of age, the clearance of PRIVIGEN (1.35 ± 0.44 mL/day/kg) is comparable to that observed in 19 adult subjects 19 years of age or older (1.29 ± 0.22 mL/day/kg). The IgG subclass levels observed in the pharmacokinetic study were consistent with a physiologic distribution pattern.
Treatment of Chronic Immune Thrombocytopenic Purpura
Pharmacokinetic studies with PRIVIGEN were not performed in subjects with chronic ITP.
Treatment of Chronic Inflammatory Demyelinating Polyneuropathy
Trough concentrations:
In both the PRIMA and PATH studies, on Day 1, subjects received an induction dose (2 g/kg) given over 2 to 5 days, followed by maintenance doses of 1 g/kg every 3 weeks.
In the PRIMA study, from Day 1 (reference) to Day 2, the mean serum IgG trough concentration increased from 12.6 ± 3.8 g/L to 24.4 ± 7.0 g/L. At Week 7, before the second maintenance treatment of (1 g/kg) given over 1 or 2 days every 3 weeks, the mean IgG trough concentration was 17.5 ± 3.1 g/L and remained stable from Week 7 to Week 19.
In the PATH study, from Day 1 (reference) to Day 5, the mean serum IgG trough concentration increased from 12.7 ± 3.2 g/L to 33.2 ± 6.9 g/L. At Week 7, before the second maintenance treatment of (1 g/kg) given over 1 or 2 days every 3 weeks, the mean IgG trough concentration was 17.7 ± 4.0 g/L and remained stable from Week 7 to Week 13.
Post-infusion concentrations:
In the PRIMA study, from Day 1 to Day 2, the post-infusion serum IgG concentration increased from 28.6 ± 8.5 g/L to 40.0 ± 11.5 g/L. At Week 7 (after the second maintenance treatment), the post-infusion IgG concentration was 32.3 ± 8.0 g/L and remained stable from Week 7 to Week 19.
14 CLINICAL STUDIES
14.1 Treatment of Primary Humoral Immunodeficiency
A prospective, open-label, single-arm, multicenter study assessed the efficacy, safety, and pharmacokinetics of PRIVIGEN in adult and pediatric subjects with PI, who were treated for 12 months at a 3-week or 4-week dosing interval. Subjects ranged in age from 3 to 69; 46 (57.5%) were male and 34 (42.5%) were female; 77.5% were Caucasian, 15% were Hispanic, and 7.5% were African-American. All subjects had been on regular IGIV replacement therapy for at least 6 months prior to participating in the study.
The efficacy analysis included 80 subjects, 16 (20%) on the 3-week dosing interval and 64 (80%) on the 4-week dosing interval. Doses ranged from 200 mg/kg to 888 mg/kg per infusion. The median dose for the 3-week interval was 428.3 mg/kg per infusion; the median dose for the 4-week interval was 440.6 mg/kg per infusion. Subjects received a total of 1038 infusions of PRIVIGEN, 272 for the 3-week dosing regimen and 766 for the 4-week dosing regimen. The maximum infusion rate allowed during this study was 8 mg/kg/min with 715 (69%) of the infusions administered at a rate of 7 mg/kg/min or greater.
The primary analysis for efficacy was based on the annual rate of acute serious bacterial infections (aSBIs), defined as pneumonia, bacteremia/septicemia, osteomyelitis/septic arthritis, bacterial meningitis, and visceral abscess, per subject per year. Secondary analyses were based on the annual rate of other infections, antibiotic use, days out of work/school/day care or unable to perform normal activities due to illness, and days of hospitalization.
During the 12-month study period, the aSBI rate was 0.08 (with an upper 1-sided 99% confidence interval of 0.203), which met the predefined success rate of less than one aSBI per subject per year. Six subjects experienced an aSBI, including three cases of pneumonia and one case each of septic arthritis, osteomyelitis, and visceral abscess. All six subjects completed the study.
The rate of other infections was 3.55 infections per subject per year. The infections that occurred most frequently were sinusitis (31.3%), nasopharyngitis (22.5%), upper respiratory tract infection (18.8%), bronchitis (13.8%), and rhinitis (13.8%). Among the 255 infections, 16 (6.3%) occurring in 10 subjects were considered severe.
Table 8 summarizes the efficacy results for all 80 subjects.
| Number of Subjects | 80 |
| Results from Case Report Forms | |
| Total Number of Subject Days | 26,198 |
| Infections | |
| Annual rate of confirmed aSBIs* | 0.08 aSBIs/subject year† |
| Annual rate of other infections | 3.55 infections/subject year |
| Antibiotic use | |
| Number of subjects (%) | 64 (80%) |
| Annual rate | 87.4 days/subject year |
| Results from Subject Diaries | |
| Total Number of Diary Days | 24,059 |
| Out of work/school/day care or unable to perform normal activities due to illness | |
| Number of days (%) | 570 (2.37%) |
| Annual rate | 8.65 days/subject year |
| Hospitalization | |
| Number of days (%) | 166 (0.69%) |
| Annual rate | 2.52 days/subject year |
14.2 Treatment of Chronic Immune Thrombocytopenic Purpura
A prospective, open-label, single-arm, multicenter study assessed the efficacy, safety, and tolerability of PRIVIGEN in 57 subjects with chronic ITP and a platelet count of 20 × 109/L or less. Subjects ranged in age from 15 to 69; 23 (40.4%) were male and 34 (59.6%) were female; all were Caucasian.
Subjects received a 2 g/kg dosage of PRIVIGEN administered as 1 g/kg (10 mL/kg) intravenous infusion daily for 2 consecutive days, and were observed for 29 days. Fifty-three (93%) subjects received PRIVIGEN at the maximum infusion rate allowed (4 mg/kg/min [0.04 mL/kg/min]).
The primary analysis was based on the response rate defined as the percentage of subjects with an increase in platelet counts to at least 50 × 109/L within 7 days after the first infusion (responders). Secondary analyses were based on the increase in platelet counts and the time to reach a platelet count of at least 50 × 109/L at any point within the study period, the duration of that response, and the regression (decrease in the severity) of hemorrhage in subjects who had bleeding at baseline. Platelet counts were measured on Days 1, 2, 4, 6, 8, 15, 22, and 29. Additional measurements on Days 57 and 85 occurred in subjects with a platelet count of at least 50 × 109/L at the previous visit.
Of the 57 subjects in the efficacy analysis, 46 (80.7%) responded to PRIVIGEN with a rise in platelet counts to at least 50 × 109/L within 7 days after the first infusion. The lower bound of the 95% confidence interval for the response rate (69.2%) is above the predefined response rate of 50%.
The highest median increase in platelet counts was seen 7 days after the first infusion (123 × 109/L). The median maximum platelet count achieved was 154 × 109/L. The median time to reach a platelet response of more than 50 × 109/L was 2.5 days after the first infusion. Twenty-five (43%) of the 57 subjects reached this response by Day 2 prior to the second infusion and 43 (75%) subjects reached this response by Day 6.
The duration of platelet response was analyzed for the 48 subjects who achieved a response any time after the first infusion. The median duration of platelet response in these subjects was 15.4 days (range: 1 to >82 days). Thirty-six (75%) of the 48 subjects maintained the response for at least 8.8 days and 12 (25%) of them for at least 21.9 days. Five (9%) subjects maintained a response up to Day 29 and two (4%) up to Day 85.
A decrease in the severity of hemorrhage from baseline was observed in the following bleeding locations: skin (31 of 36 subjects), oral cavity (11 of 11 subjects), and genitourinary tract (7 of 9 subjects). This decrease was not sustained in all subjects up to the end of the 29-day study period.
14.3 Postmarketing Commitment Study in Chronic Immune Thrombocytopenic Purpura
A prospective, open-label, single-arm, multicenter study assessed efficacy and safety parameters in 57 IGIV-treated subjects with chronic ITP with a platelet count of <30 × 109/L at screening. Fifty-three subjects had a history of chronic ITP with a duration of greater than 6 months and 4 subjects, all of whom had received prior treatment for ITP with subsequent elevation followed by falls in platelet counts, had a duration of ITP less than 6 months. The study examined the incidence of subjects who met laboratory and clinical criteria for hemolysis and was intended to identify antibodies most frequently bound to erythrocytes in subjects who experienced clinically significant intravascular hemolysis. Subjects ranged in age from 18 to 65; 20 (35.1%) were male and 37 (64.9%) were female; all were Caucasian.
Twenty-one (21) subjects (37%) received 1 infusion of 1 g/kg on Day 1 and 36 subjects (63%) received 2 infusions of 1 g/kg (Day 1 and Day 3). The second infusion was administered based on the subject's platelet response to the Day 1 dose (<50 × 109/L) and investigator's discretion.
The efficacy endpoint platelet response (increase in platelet count at least once to at least 50 × 109/L within 6 days after the first infusion) was achieved in 42 subjects (74%; 95% confidence interval [CI]: 61% to 83%).
Fifteen subjects with a suspicion of hemolysis based on laboratory data were referred for independent expert adjudication during the study. The adjudication committee selected from 3 options for their determination: no hemolysis, hemolysis, or clinically significant intravascular hemolysis. The set of antibodies most frequently bound to erythrocytes in subjects with clinically significant intravascular hemolysis could not be analyzed, because no subject experienced clinically significant intravascular hemolysis. No irregular antibodies were detected in any subject; therefore, no association between such antibodies and hemolytic laboratory changes could be established. Hemolytic laboratory changes were most often found in non-O blood group (especially the A blood group) subjects and those receiving 2 infusions. These laboratory parameters improved or normalized by the end of the study in the majority of subjects. Seven subjects (12% of the study population) with a normal hemoglobin at baseline had an abnormal hemoglobin at Day 29 (end of study) with a hemoglobin range from 11.2 to 13.6 g/dL.
Post-hoc analyses were performed using a set of defined criteria for hemolysis. The hemolysis group (18 subjects, 32%) met the criterion for greater than 1 g/dL drop in hemoglobin within a 21-day interval since the last IGIV administration not explained by blood loss or repeated phlebotomy, were treatment-emergent DAT positive, and met at least one other minor criterion (eg, fall in serum haptoglobin level to below the lower limit of normal, rise in lactate dehydrogenase level above the upper limit of normal, rise in indirect or total bilirubin to above the upper limit of normal, or rise in plasma-free hemoglobin above the upper limit of normal). Fourteen of 15 previously adjudicated presumptive hemolysis cases during the study were included in this post-hoc hemolysis group.
14.4 Treatment of Chronic Inflammatory Demyelinating Polyneuropathy
In a prospective, open-label, single-arm, multicenter clinical study (PRIVIGEN Impact on Mobility and Autonomy [PRIMA]), 28 subjects with CIDP (13 IGIV-pretreated and 15 IGIV-untreated) received a PRIVIGEN loading dose of 2 g/kg followed by PRIVIGEN maintenance doses of 1 g/kg for up to 21 weeks with a 3 week follow up.
Efficacy in the PRIMA study was based on the responder rate of PRIVIGEN in comparison to an historical control in the adjusted 10-point Inflammatory Neuropathy Cause and Treatment (INCAT) score.19 The responder rate was defined as the proportion of subjects who demonstrated clinically meaningful improvement (at least 1 point decrease on adjusted Inflammatory Neuropathy Cause and Treatment [INCAT] score) between baseline and Week 25, with a pre-specified threshold of 35% in the lower limit of the 2-sided 95% Wilson-Score confidence interval (CI). The overall percentage of responders in PRIMA was 61% (95% CI: 42.4% to 76.4%). Response rates were 47% in IGIV-untreated and 77% in IGIV-pretreated subject subgroups. In a post-hoc analysis, the overall percentage of subjects in PRIMA who responded by week 10 and maintained the response through week 25 and lacked confounding changes in glucocorticoid/immunosuppressant dosage was 53.6% (95% CI: 35.8% to 70.5%).
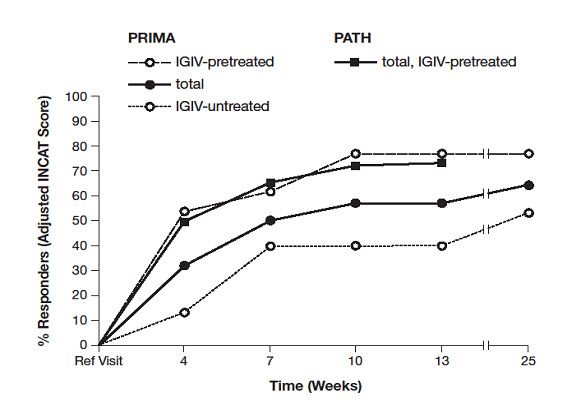
In a second study (PATH) with the same PRIVIGEN dosing regimen, all 207 subjects were IGIV-pretreated and had relapsed following withdrawal of IGIV prior to being administered PRIVIGEN [see Dosage and Administration (2.3)]. The response rate was 73% (see Figure 1). Among the subset of 151 subjects in the PATH study who had deteriorated by one or more points in adjusted INCAT score following withdrawal of IGIV, 137 subjects (90.7%) responded during the PRIVIGEN "restabilization" period with an increase of one or more adjusted INCAT score points.
The overall median time to first adjusted INCAT response in PRIMA was 7.5 weeks (18 weeks in IGIV-untreated and 3 weeks in IGIV-pretreated). The median time to first adjusted INCAT response in PATH (all IGIV-pretreated) was 3.7 weeks (95% CI: 3.4 to 5.9 weeks). Mean INCAT score in PRIMA showed a clinically meaningful improvement by 1.4 points (1.1 points for IGIV-untreated, and 1.8 points for IGIV-pretreated [1.2 points in PATH]).
Figure 1. Percentage of Responders (Adjusted INCAT Score)
Medical Research Council (MRC) sum score in PRIMA improved by a mean of 6.9 points (7.7 points for IGIV-untreated and 6.1 points for IGIV-pretreated). MRC sum score in PATH improved by a mean of 3.6 points.
Grip strength of the dominant hand improved in PRIMA by a mean of 14.1 kPa (17.0 kPa for IGIV-untreated and 10.8 kPa for IGIV-pretreated subgroups). Grip strength of the dominant hand improved in PATH by a mean of 12.2 kPa. Similar results were observed for the non-dominant hand in both studies.
15 REFERENCES
- Dalakas MC. High-dose intravenous immunoglobulin and serum viscosity: risk of precipitating thromboembolic events. Neurology 1994;44:223-226.
- Woodruff RK, Grigg AP, Firkin FC, Smith IL. Fatal thrombotic events during treatment of autoimmune thrombocytopenia with intravenous immunoglobulin in elderly patients. Lancet 1986;2:217-218.
- Wolberg AS, Kon RH, Monroe DM, Hoffman M. Coagulation factor XI is a contaminant in intravenous immunoglobulin preparations. Am J Hematol 2000;65:30-34.
- Cayco AV, Perazella MA, Hayslett JP. Renal insufficiency after intravenous immune globulin therapy: a report of two cases and an analysis of the literature. J Am Soc Nephrol 1997;8:1788-1793.
- Steinberger BA, Ford SM, Coleman TA. Intravenous immunoglobulin therapy results in post-infusional hyperproteinemia, increased serum viscosity, and pseudohyponatremia. Am J Hematol 2003;73:97-100.
- Gabor EP. Meningitis and skin reaction after intravenous immune globulin therapy. Ann Intern Med 1997;127:1130.
- Copelan EA, Strohm PL, Kennedy MS, Tutschka PJ. Hemolysis following intravenous immune globulin therapy. Transfusion 1986;26:410-412.
- Thomas MJ, Misbah SA, Chapel HM, Jones M, Elrington G, Newsom-Davis J. Hemolysis after high-dose intravenous Ig. Blood 1993;15:3789.
- Wilson JR, Bhoopalam N, Fisher M. Hemolytic anemia associated with intravenous immunoglobulin. Muscle Nerve 1997;20:1142-1145.
- Kessary-Shoham H, Levy Y, Shoenfeld Y, Lorber M, Gershon H. In vivo administration of intravenous immunoglobulin (IVIg) can lead to enhanced erythrocyte sequestration. J Autoimmun 1999;13:129-135.
- Kahwaji J, Barker E, Pepkowitz S, et al. Acute Hemolysis After High-Dose Intravenous Immunoglobulin Therapy in Highly HLA Sensitized Patients. Clin J Am Soc Nephrol 2009;4:1993-1997.
- Daw Z, Padmore R, Neurath D, et al. Hemolytic transfusion reactions after administration of intravenous immune (gamma) globulin: A case series analysis. Transfusion 2008;48:1598-1601.
- Rizk A, Gorson KC, Kenney L, Weinstein R. Transfusion-related acute lung injury after the infusion of IVIG. Transfusion 2001;41:264-268.
- Pierce LR, Jain N. Risks associated with the use of intravenous immunoglobulin. Trans Med Rev 2003;17:241-251.
- Siber GA, Werner BG, Halsey NA, et al. Interference of immune globulin with measles and rubella immunization. J Pediatr 1993;122:204-211.
- Hammarström L, Smith CIE. Placental transfer of intravenous immunoglobulin. Lancet 1986;1:681.
- Sidiropoulos D, Herrmann U, Morell A, von Muralt G, Barandun S. Transplacental passage of intravenous immunoglobulin in the last trimester of pregnancy. J Pediatr 1986;109:505-508.
- Gregori L, Maring J-A, MacAuley C, Stühler A, Löwer J, Blümel J. Partitioning of TSE infectivity during ethanol fractionation of human plasma. Biologicals 2004;32:1-10.
- Hughes RAC, Donofrio P, Bril V, et al. Intravenous immune globulin (10% caprylate/chromatography purified) for the treatment of chronic inflammatory demyelinating polyradiculoneuropathy (ICE study): a randomised placebo-controlled trial. Lancet Neurol 2008;7:136-44.
16 HOW SUPPLIED/STORAGE AND HANDLING
PRIVIGEN is supplied in a single-use, tamper-evident vial containing the labeled amount of functionally active IgG. The PRIVIGEN packaging components are not made with natural rubber latex.
Each product presentation includes a package insert and the following components:
| Presentation | Carton NDC Number | Components |
|---|---|---|
| 50 mL | 44206-436-05 | Vial containing 5 grams of protein (NDC 44206-436-90) |
| 100 mL | 44206-437-10 | Vial containing 10 grams of protein (NDC 44206-437-91) |
| 200 mL | 44206-438-20 | Vial containing 20 grams of protein (NDC 44206-438-92) |
| 400 mL | 44206-439-40 | Vial containing 40 grams of protein (NDC 44206-439-93) |
Storage and Handling
- Keep PRIVIGEN in its original carton to protect it from light.
- Each vial has an integral suspension band and a label with two peel-off strips showing the product name, lot number, and expiration date.
- When stored at room temperature (up to 25ºC [77ºF]), PRIVIGEN is stable for up to 36 months, as indicated by the expiration date printed on the outer carton and vial label.
- Do not freeze.
17 PATIENT COUNSELING INFORMATION
Inform patients of the early signs of hypersensitivity reactions to PRIVIGEN (including hives, generalized urticaria, tightness of the chest, wheezing, hypotension, and anaphylaxis), and advise them to notify their physician if they experience any of these symptoms [see Warnings and Precautions (5.1)].
Inform patients to immediately report the following signs and symptoms to their physician:
- Decreased urine output, sudden weight gain, fluid retention/edema, and/or shortness of breath, which may suggest kidney problems [see Warnings and Precautions (5.2)].
- Instruct patients to immediately report symptoms of thrombosis. These symptoms may include: pain and/or swelling of an arm or leg with warmth over the affected area, discoloration of an arm or leg, unexplained shortness of breath, chest pain or discomfort that worsens on deep breathing, unexplained rapid pulse, numbness or weakness on one side of the body [see Warnings and Precautions (5.3)].
- Severe headache, neck stiffness, drowsiness, fever, sensitivity to light, painful eye movements, nausea, and vomiting, which may suggest aseptic meningitis syndrome [see Warnings and Precautions (5.5)].
- Fatigue, increased heart rate, yellowing of skin or eyes, and dark-colored urine, which may suggest hemolysis [see Warnings and Precautions (5.6)].
- Severe breathing problems, lightheadedness, drops in blood pressure, and fever, which may suggest TRALI (a condition typically occurring within 1 to 6 hours following transfusion) [see Warnings and Precautions (5.8)].
Inform patients that PRIVIGEN is made from human blood and may contain infectious agents that can cause disease (eg, viruses, the variant Creutzfeldt-Jakob disease [vCJD] agent and, theoretically the CJD agent). Explain that the risk that PRIVIGEN may transmit an infectious agent has been reduced by screening the plasma donors, by testing donated plasma for certain virus infections, and by inactivating or removing certain viruses during manufacturing, and counsel patients to report any symptoms that concern them [see Warnings and Precautions (5.10)].
Inform patients that administration of IgG may interfere with the response to live virus vaccines (eg, measles, mumps, rubella, and varicella), and instruct them to notify their immunizing physician of recent therapy with PRIVIGEN [see Warnings and Precautions (5.11)].
Manufactured by:
CSL Behring AG
Bern, Switzerland
US License No. 1766
Distributed by:
CSL Behring LLC
Kankakee, IL 60901 USA
PRINCIPAL DISPLAY PANEL - 50 mL Vial Carton
NDC 44206-436-05
5 g
Immune Globulin
Intravenous (Human),
10% Liquid
50 mL
privigen®
10% Liquid Preparation
Single-dose vial
For Intravenous Administration
only
Rx only
CSL Behring
PRINCIPAL DISPLAY PANEL - 100 mL Vial Carton
NDC 44206-437-10
10 g
Immune Globulin
Intravenous (Human),
10% Liquid
100 mL
privigen®
10% Liquid Preparation
Single-dose vial
For Intravenous Administration
only
Rx only
CSL Behring