FULL PRESCRIBING INFORMATION
WARNING: BIRTH DEFECTS
Bexarotene is a member of the retinoid class of drugs that is associated with birth defects in humans. Bexarotene also caused birth defects when administered orally to pregnant rats. Bexarotene must not be administered to a pregnant woman. (8.1)
1 INDICATIONS AND USAGE
Bexarotene capsules are indicated for the treatment of cutaneous manifestations of cutaneous T-cell lymphoma in patients who are refractory to at least one prior systemic therapy.
2 DOSAGE AND ADMINISTRATION
The recommended initial dose of bexarotene is 300 mg/m2/day (see Table 1). Bexarotene capsules should be taken as a single oral daily dose with a meal. For precautions to prevent pregnancy and birth defects in women of child-bearing potential [see Use in Specific Populations (8.1)].
| Initial Dose Level (300 mg/m2/day) | Number of 75 mg Bexarotene Capsules |
|
|---|---|---|
| Body Surface Area (m2) | Total Daily Dose (mg/day) |
|
| 0.88 – 1.12 | 300 | 4 |
| 1.13 – 1.37 | 375 | 5 |
| 1.38 – 1.62 | 450 | 6 |
| 1.63 – 1.87 | 525 | 7 |
| 1.88 – 2.12 | 600 | 8 |
| 2.13 – 2.37 | 675 | 9 |
| 2.38 – 2.62 | 750 | 10 |
Dose Modification Guidelines: The 300 mg/m2/day dose level of bexarotene may be adjusted to 200 mg/m2/day then to 100 mg/m2/day, or temporarily suspended, if necessitated by toxicity. When toxicity is controlled, doses may be carefully readjusted upward. If there is no tumor response after eight weeks of treatment and if the initial dose of 300 mg/m2/day is well tolerated, the dose may be escalated to 400 mg/m2/day with careful monitoring.
3 DOSAGE FORMS AND STRENGTHS
Capsules: 75 mg, off-white, oblong soft gelatin capsules, imprinted with black ink " US285".
4 CONTRAINDICATIONS
4.1 Pregnancy
Bexarotene can cause fetal harm when administered to a pregnant female. Bexarotene is a member of the retinoid class of drugs that is associated with birth defects in humans and is contraindicated in females who are pregnant. Bexarotene was also teratogenic and caused developmental mortality when administered orally to pregnant rats. If this drug is used during pregnancy or if the patient becomes pregnant while taking this drug, the patient should be advised of the potential risk to a fetus.
5 WARNINGS AND PRECAUTIONS
5.1 Hyperlipidemia
Bexarotene induces substantial elevations in lipids in most patients. About 70% of patients with CTCL who received an initial dose of ≥300 mg/m2/day of bexarotene had fasting triglyceride levels greater than 2.5 times the upper limit of normal. About 55% had values over 800 mg/dL with a median of about 1200 mg/dL in those patients. Cholesterol elevations above 300 mg/dL occurred in approximately 60% and 75% of patients with CTCL who received an initial dose of 300 mg/m2/day or greater than 300 mg/m2/day, respectively. Decreases in high density lipoprotein (HDL) cholesterol to less than 25 mg/dL were seen in about 55% and 90% of patients receiving an initial dose of 300 mg/m2/day or greater than 300 mg/m2/day, respectively, of bexarotene. Monitor lipid changes and treat abnormalities during therapy. The effects on triglycerides, HDL cholesterol, and total cholesterol were reversible with cessation of therapy, and could generally be mitigated by dose reduction and/or concomitant antilipemic therapy.
Perform fasting blood lipid determinations before bexarotene therapy is initiated and weekly until the lipid response to bexarotene is established, which usually occurs within two to four weeks, and monitor at eight week intervals thereafter. Fasting triglycerides should be normal or normalized with appropriate intervention prior to initiating bexarotene therapy. Maintain triglyceride levels below 400 mg/dL to reduce the risk of clinical sequelae [see Warnings and Precautions (5.2)]. If fasting triglycerides are elevated or become elevated during treatment, institute antilipemic therapy, and if necessary, reduce or interrupt the dose of bexarotene. In the 300 mg/m2/day initial dose group, 60% of patients were given lipid lowering drugs. Atorvastatin was used in 48% (73/152) of patients with CTCL. Because of a potential drug-drug interaction, avoid gemfibrozil use with bexarotene [see Drug Interactions (7)].
5.2 Pancreatitis
Acute pancreatitis, including a fatal case, has been reported in four patients with CTCL and in six patients with non-CTCL cancers treated with bexarotene; the cases were associated with marked elevations of fasting serum triglycerides, the lowest being 770 mg/dL in one patient. One patient with advanced non-CTCL cancer died of pancreatitis. Interrupt bexarotene and evaluate if pancreatitis is suspected. Patients with CTCL who have risk factors for pancreatitis (e.g., prior pancreatitis, uncontrolled hyperlipidemia, excessive alcohol consumption, uncontrolled diabetes mellitus, biliary tract disease, and medications known to increase triglyceride levels or to be associated with pancreatic toxicity) may be at greater risk for pancreatitis associated with bexarotene [see Warnings and Precautions (5.1)].
5.3 Hepatotoxicity, Cholestasis, and Hepatic Failure
Bexarotene caused elevations in liver chemistry tests (LFTs) in 5% (AST), 2% (ALT), and 0% (bilirubin) in patients with CTCL receiving an initial dose of 300 mg/m2/day. In contrast, with an initial dose greater than 300 mg/m2/day of bexarotene, the incidence of LFT elevations was higher at 7% (SGOT/AST), 9% (SGPT/ALT), and 6% (bilirubin). Two patients developed cholestasis, including one patient who died of liver failure. In clinical trials, elevated LFTs resolved within one month in 80% of patients following a decrease in dose or discontinuation of therapy. Obtain baseline LFTs and monitor LFTs after one, two and four weeks of treatment initiation, and if stable, at least every eight weeks thereafter during treatment. Interrupt or discontinue bexarotene if test results exceed three times the upper limit of normal values for AST, ALT, or bilirubin.
5.4 Hypothyroidism
Bexarotene induces hypothyroidism in about half of all patients treated by causing a reversible reduction in levels of thyroid hormone (total thyroxine [total T4]) and thyroid-stimulating hormone (TSH). The incidence of decreases in TSH and total T4 were about 60% and 45%, respectively, in patients with CTCL receiving an initial dose of 300 mg/m2/day. Hypothyroidism was reported as an adverse event in 29% of patients. Consider treatment with thyroid hormone supplementation in patients with hypothyroidism. In the 300 mg/m2/day initial dose group, 37% of patients were treated with thyroid hormone replacement. Obtain baseline thyroid function tests and monitor patients during treatment.
5.5 Neutropenia
Leukopenia in the range of 1000 to <3000 WBC × 106/L occurred in 18% of patients with CTCL receiving an initial dose of 300 mg/m2/day of bexarotene . Patients receiving an initial dose greater than 300 mg/m2/day of bexarotene had an incidence of leukopenia of 43%. No patient with CTCL treated with bexarotene developed leukopenia of less than 1000 WBC × 106/L. The usual time to onset of leukopenia was four to eight weeks after initiating bexarotene. The leukopenia observed in most patients was predominately neutropenia. In the 300 mg/m2/day initial dose group, the incidence of NCI Grade 3 and Grade 4 neutropenia, respectively, was 12% and 4%. The leukopenia and neutropenia experienced during bexarotene therapy resolved after dose reduction or discontinuation of treatment, on average within 30 days in 93% of the patients with CTCL and 82% of patients with non-CTCL cancers. Leukopenia and neutropenia were rarely associated with severe sequelae or serious adverse events. Obtain complete blood counts (CBC) at baseline and periodically during treatment.
5.6 Cataracts
Posterior subcapsular cataracts occurred in preclinical toxicity studies in rats and dogs administered bexarotene daily for 6 months. New cataracts or worsening of previous cataracts occurred in 15 of 79 patients who were monitored with serial slit lamp examinations. Because of the high prevalence and rate of cataract formation in older patient populations, the relationship of bexarotene and cataracts cannot be determined in the absence of an appropriate control group. Patients treated with bexarotene who experience visual difficulties should have an appropriate ophthalmologic evaluation.
5.7 Vitamin A Supplementation Hazard
In clinical studies, patients were advised to limit vitamin A intake to ≤15,000 IU/day. Because of the relationship of bexarotene to vitamin A, patients should be advised to limit vitamin A supplements to avoid potential additive toxic effects.
5.8 Hypoglycemia Risk in Patients with Diabetes Mellitus
In patients using insulin, agents enhancing insulin secretion (e.g., sulfonylureas), or insulin-sensitizers (e.g., thiazolidinedione class), based on the mechanism of action, bexarotene could enhance the action of these agents, resulting in hypoglycemia. Hypoglycemia has not been associated with the use of bexarotene as monotherapy.
5.9 Photosensitivity
Retinoids as a class have been associated with photosensitivity. In vitro assays indicate that bexarotene is a potential photosensitizing agent. Phototoxicity manifested as sunburn and skin sensitivity to sunlight occurred in patients who were exposed to direct sunlight while receiving bexarotene. Advise patients to minimize exposure to sunlight and artificial ultraviolet light while receiving bexarotene.
5.10 Laboratory Tests
Before initiating bexarotene therapy, obtain a CBC, fasting lipid profile, liver function tests, and a thyroid profile. Fasting triglycerides should be normal or normalized with appropriate intervention prior to therapy. Monitor lab tests during bexarotene therapy as described above.
Hyperlipidemia usually occurs within the initial two to four weeks. Therefore, weekly lipid determinations are recommended during this interval. Subsequently, in patients not hyperlipidemic, determinations can be performed less frequently [see Warnings and Precautions (5.1)].
A white blood cell count with differential should be obtained at baseline and periodically during treatment. Baseline liver function tests should be obtained and should be carefully monitored after one, two and four weeks of treatment initiation, and if stable, periodically thereafter during treatment. Baseline thyroid function tests should be obtained and then monitored during treatment as indicated [see Warnings and Precautions (5.3, 5.4, 5.5, 5.6)].
6 ADVERSE REACTIONS
The following serious adverse reactions are discussed in greater detail in other sections of the prescribing information:
- Hyperlipidemia [see Warnings and Precautions (5.1)]
- Pancreatitis [see Warnings and Precautions (5.2)]
- Hepatotoxicity, cholestasis, and hepatic failure [see Warnings and Precautions (5.3)]
- Hypothyroidism [see Warnings and Precautions (5.4)]
- Neutropenia [see Warnings and Precautions (5.5)]
- Cataracts [see Warnings and Precautions (5.6)]
- Vitamin A Supplementation Hazard [see Warnings and Precautions (5.7)]
- Hypoglycemia Risk in Patients with Diabetes Mellitus [see Warnings and Precautions (5.8)]
- Photosensitivity [see Warnings and Precautions (5.9)]
- Laboratory Tests [see Warnings and Precautions (5.10)]
- Drug/Laboratory Test Interactions [see Warnings and Precautions (5.11)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of bexarotene has been evaluated in two clinical trials of 152 patients with CTCL who received bexarotene for up to 97 weeks and in 352 patients in other trials. The mean duration of therapy for the 152 patients with CTCL was 166 days. The most common adverse events reported with an incidence of at least 10% in patients with CTCL treated at an initial dose of 300 mg/m2/day of bexarotene are shown in Table 2. The events at least possibly related to treatment are lipid abnormalities (elevated triglycerides, elevated total and LDL cholesterol and decreased HDL cholesterol), hypothyroidism, headache, asthenia, rash, leukopenia, anemia, nausea, infection, peripheral edema, abdominal pain, and dry skin. Most adverse events occurred at a greater incidence in patients treated at starting doses of greater than 300 mg/m2/day (see Table 2).
Adverse reactions leading to bexarotene dose reduction or discontinuation in at least two patients were hyperlipemia, neutropenia/leukopenia, diarrhea, fatigue/lethargy, hypothyroidism, headache, liver function test abnormalities, rash, pancreatitis, nausea, anemia, allergic reaction, muscle spasm, pneumonia, and confusion.
The NCI Grade 3 and NCI Grade 4 adverse reactions reported in two or more patients with CTCL treated at an initial dose of 300 mg/m2/day of bexarotene (see Table 3) were hypertriglyceridemia, pruritus, headache, peripheral edema, leukopenia, rash, and hypercholesteremia. Most of these moderately severe or severe adverse events occurred at a higher rate in patients treated at starting doses of greater than 300 mg/m2/day than in patients treated at a starting dose of 300 mg/m2/day.
In patients with CTCL receiving an initial dose of 300 mg/m2/day, the incidence of NCI Grade 3 or 4 elevations in triglycerides and total cholesterol was 28% and 25%, respectively (Table 4). In contrast, in patients with CTCL receiving greater than 300 mg/m2/day, the incidence of NCI Grade 3 or 4 elevated triglycerides and total cholesterol was 45% and 45%, respectively. Other Grade 3 and 4 laboratory abnormalities are shown in Table 3.
In addition to the 152 patients enrolled in the two CTCL trials, 352 patients received bexarotene as monotherapy for various advanced malignancies at doses from 5 mg/m2/day to 1000 mg/m2/day. The common adverse reactions (incidence greater than 10%) were similar to those seen in patients with CTCL.
In the 504 patients (CTCL and non-CTCL) who received bexarotene as monotherapy, drug-related serious adverse reactions that were fatal, in one patient each, were acute pancreatitis, subdural hematoma, and liver failure.
In the patients with CTCL receiving an initial dose of 300 mg/m2/day of bexarotene, adverse reactions reported at an incidence of less than 10% and not included in Tables 2 to 4 or discussed in other parts of labeling and possibly related to treatment were as follows:
Body as a Whole: chills, cellulitis, chest pain, breast pain, sepsis, and monilia infection.
Cardiovascular: hemorrhage, hypertension, angina pectoris, right heart failure, syncope, and tachycardia.
Digestive: constipation, dry mouth, flatulence, colitis, dyspepsia, cheilitis, gastroenteritis, gingivitis, liver failure, and melena.
Hemic and Lymphatic: eosinophilia, thrombocythemia, coagulation time increased, lymphocytosis, and thrombocytopenia.
Metabolic and Nutritional: LDH increased, creatinine increased, hypoproteinemia, hyperglycemia, weight decreased, weight increased, and amylase increased.
Musculoskeletal: arthralgia, myalgia, bone pain, myasthenia, and arthrosis.
Nervous: depression, agitation, ataxia, cerebrovascular accident, confusion, dizziness, hyperesthesia, hypesthesia, and neuropathy.
Respiratory: pharyngitis, rhinitis, dyspnea, pleural effusion, bronchitis, cough increased, lung edema, hemoptysis, and hypoxia.
Skin and Appendages: skin ulcer, acne, alopecia, skin nodule, macular papular rash, pustular rash, serous drainage, and vesicular bullous rash.
Special Senses: dry eyes, conjunctivitis, ear pain, blepharitis, corneal lesion, keratitis, otitis externa, and visual field defect.
Urogenital: albuminuria, hematuria, urinary incontinence, urinary tract infection, urinary urgency, dysuria, and kidney function abnormal.
| Initial Assigned Dose Group (mg/m2/day) |
|||||
|---|---|---|---|---|---|
| 300 | >300 | ||||
| Body System | N=84 | N=53 | |||
| Adverse Event*,† | N (%) | N (%) | |||
| Metabolic and Nutritional Disorders | |||||
| Hyperlipemia | 66 | (79) | 42 | (79) | |
| Hypercholesteremia | 27 | (32) | 33 | (62) | |
| Lactic dehydrogenase increased | 6 | (7) | 7 | (13) | |
| Body as a Whole | |||||
| Headache | 25 | (30) | 22 | (42) | |
| Asthenia | 17 | (20) | 24 | (45) | |
| Infection | 11 | (13) | 12 | (23) | |
| Abdominal pain | 9 | (11) | 2 | (4) | |
| Chills | 8 | (10) | 7 | (13) | |
| Fever | 4 | (5) | 9 | (17) | |
| Flu syndrome | 3 | (4) | 7 | (13) | |
| Back pain | 2 | (2) | 6 | (11) | |
| Infection bacterial | 1 | (1) | 7 | (13) | |
| Endocrine | |||||
| Hypothyroidism | 24 | (29) | 28 | (53) | |
| Skin and Appendages | |||||
| Rash | 14 | (17) | 12 | (23) | |
| Dry skin | 9 | (17) | 5 | (9) | |
| Exfoliative dermatitis | 8 | (10) | 15 | (28) | |
| Alopecia | 3 | (4) | 6 | (11) | |
| Hemic and Lymphatic System | |||||
| Leukopenia | 14 | (17) | 25 | (47) | |
| Anemia | 5 | (6) | 13 | (25) | |
| Hypochromic anemia | 3 | (4) | 7 | (13) | |
| Digestive System | |||||
| Nausea | 13 | (16) | 4 | (8) | |
| Diarrhea | 6 | (7) | 22 | (42) | |
| Vomiting | 3 | (4) | 7 | (13) | |
| Anorexia | 2 | (2) | 12 | (23) | |
| Cardiovascular System | |||||
| Peripheral edema | 11 | (13) | 6 | (11) | |
| Nervous System | |||||
| Insomnia | 4 | (5) | 6 | (11) | |
| Initial Assigned Dose Group (mg/m2/day) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 300 (N=84) | >300 (N=53) | ||||||||
| Mod Severe | Severe | Mod Severe | Severe | ||||||
| Body System | |||||||||
| Adverse Event*,† | N | (%) | N | (%) | N | (%) | N | (%) | |
| Body as a Whole | |||||||||
| Asthenia | 1 | (1) | 0 | (0) | 11 | (21) | 0 | (0) | |
| Headache | 3 | (4) | 0 | (0) | 5 | (9) | 1 | (2) | |
| Infection bacterial | 1 | (1) | 0 | (0) | 0 | (0) | 2 | (4) | |
| Cardiovascular System | |||||||||
| Peripheral edema | 2 | (2) | 1 | (1) | 0 | (0) | 0 | (0) | |
| Digestive System | |||||||||
| Anorexia | 0 | (0) | 0 | (0) | 3 | (6) | 0 | (0) | |
| Diarrhea | 1 | (1) | 1 | (1) | 2 | (4) | 1 | (2) | |
| Pancreatitis | 1 | (1) | 0 | (0) | 3 | (6) | 0 | (0) | |
| Vomiting | 0 | (0) | 0 | (0) | 2 | (4) | 0 | (0) | |
| Endocrine | |||||||||
| Hypothyroidism | 1 | (1) | 1 | (1) | 2 | (4) | 0 | (0) | |
| Hemic and Lymphatic System | |||||||||
| Leukopenia | 3 | (4) | 0 | (0) | 6 | (11) | 1 | (2) | |
| Metabolic and Nutritional Disorders | |||||||||
| Bilirubinemia | 0 | (0) | 1 | (1) | 2 | (4) | 0 | (0) | |
| Hypercholesteremia | 2 | (2) | 0 | (0) | 5 | (9) | 0 | (0) | |
| Hyperlipemia | 16 | (19) | 6 | (7) | 17 | (32) | 5 | (9) | |
| SGOT/AST increased | 0 | (0) | 0 | (0) | 2 | (4) | 0 | (0) | |
| SGPT/ALT increased | 0 | (0) | 0 | (0) | 2 | (4) | 0 | (0) | |
| Respiratory System | |||||||||
| Pneumonia | 0 | (0) | 0 | (0) | 2 | (4) | 2 | (4) | |
| Skin and Appendages | |||||||||
| Exfoliative dermatitis | 0 | (0) | 1 | (1) | 3 | (6) | 1 | (2) | |
| Rash | 1 | (1) | 2 | (2) | 1 | (2) | 0 | (0) | |
| Initial Assigned Dose (mg/m2/day) | |||||
|---|---|---|---|---|---|
| 300 | >300 | ||||
| N=83* | N=53* | ||||
| Grade 3† | Grade 4† | Grade 3 | Grade 4 | ||
| Analyte | (%) | (%) | (%) | (%) | |
|
|||||
| Triglycerides‡ | 21 | 7 | 32 | 14 | |
| Total Cholesterol‡ | 19 | 7 | 16 | 30 | |
| Alkaline Phosphatase | 1 | 0 | 0 | 2 | |
| Hyperglycemia | 1 | 0 | 6 | 0 | |
| Hypocalcemia | 1 | 0 | 0 | 0 | |
| Hyponatremia | 1 | 0 | 9 | 0 | |
| SGPT/ALT | 1 | 0 | 2 | 2 | |
| Hyperkalemia | 0 | 0 | 2 | 0 | |
| Hypernatremia | 0 | 1 | 0 | 0 | |
| SGOT/AST | 0 | 0 | 2 | 2 | |
| Total Bilirubin | 0 | 0 | 0 | 2 | |
| ANC decreased | 12 | 4 | 19 | 8 | |
| ALC decreased | 7 | 0 | 15 | 0 | |
| WBC decreased | 4 | 0 | 11 | 0 | |
| Hemoglobin decreased | 0 | 0 | 2 | 0 | |
7 DRUG INTERACTIONS
Effect of Bexarotene on Other Drugs
Bexarotene may be an inducer for the CYP3A4 enzymes, and may reduce plasma concentrations of other substrates metabolized by CYP3A4. Drug products which may be affected include oral or other systemic hormonal contraceptives. Thus, if treatment with bexarotene is intended for a female with reproductive potential, it is strongly recommended that a non-hormonal contraception be considered [see Use in Specific Populations (8.3), Clinical Pharmacology (12.3)].
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Bexarotene, a retinoid, can cause fetal harm based on findings from animal studies when administered to a pregnant female and is contraindicated during pregnancy. Bexarotene was teratogenic and caused developmental mortality in rats following oral administration during organogenesis [see Data]. Bexarotene must not be given to a pregnant female or a female who intends to become pregnant. If pregnancy does occur during treatment with bexarotene, immediately discontinue the drug and advise the pregnant female of the potential risk to a fetus.
The background risk of major birth defects and miscarriage for the indicated populations are unknown. However, the background risk in the U.S. general population of major birth defects is 2 to 4% and of miscarriage is 15 to 20% of clinically recognized pregnancies.
Data
Animal Data
Bexarotene caused malformations when administered orally to pregnant rats during days 7 to 17 of gestation. Developmental abnormalities included incomplete ossification at 4 mg/kg/day and cleft palate, depressed eye bulge/microphthalmia, and small ears at 16 mg/kg/day. The plasma AUC of bexarotene in rats at 4 mg/kg/day is approximately one third the AUC in humans at the recommended daily dose. At doses greater than 10 mg/kg/day, bexarotene caused developmental mortality. The no effect dose for fetal effects in rats was 1 mg/kg/day (producing an AUC approximately one sixth of the AUC at the recommended human daily dose).
8.2 Lactation
Risk Summary
There is no information regarding the presence of bexarotene in human milk, the effects on the breast fed infant, or the effects on milk production. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from bexarotene, discontinue breastfeeding during treatment with bexarotene.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Obtain a negative serum pregnancy test (e.g., serum beta-human chorionic gonadotropin [beta-HCG]) with a sensitivity of at least 50 mlU/L within one week prior to bexarotene therapy. Obtain another pregnancy test at monthly intervals while the patient remains on bexarotene.
Contraception
Females
Bexarotene can cause fetal harm when administered to a pregnant female [see Use in Specific Populations (8.1)]. Females of reproductive potential should be advised to avoid becoming pregnant when bexarotene is used. Effective contraception must be used for one month prior to the initiation of therapy, during therapy and for at least one month following discontinuation of therapy; it is recommended that two reliable forms of contraception be used simultaneously unless abstinence is the chosen method. Bexarotene can potentially induce metabolic enzymes and thereby theoretically reduce the plasma concentrations of oral or other systemic hormonal contraceptives [see Drug Interactions (7)].
Thus, if treatment with bexarotene is intended in a female with reproductive potential, it is strongly recommended that one of the two reliable forms of contraception should be non-hormonal.
Bexarotene therapy should be initiated on the second or third day of a normal menstrual period. No more than a one month supply of bexarotene should be given to the patient so that the results of pregnancy testing can be assessed and counseling regarding avoidance of pregnancy and birth defects can be reinforced.
8.4 Pediatric Use
Safety and effectiveness of bexarotene in pediatric patients have not been established.
8.5 Geriatric Use
Of the total patients with CTCL in clinical trials of bexarotene, 64% were 60 years or older, while 33% were 70 years or older. No overall differences in safety were observed between patients 70 years or older and younger patients, but greater sensitivity of some older individuals to bexarotene cannot be ruled out. Responses to bexarotene were observed across all age group decades, without preference for any individual age group decade.
8.6 Hepatic Impairment
No specific studies have been conducted with bexarotene in subjects with hepatic impairment. Hepatic impairment is expected to lead to decreased clearance [see Clinical Pharmacology (12.3)]. If bexarotene is used in patients with hepatic impairment, monitor for signs of toxicity that may be due to increased exposure.
10 OVERDOSAGE
Doses up to 1000 mg/m2/day of bexarotene have been administered in short-term trials in patients with advanced cancer without acute toxic effects. Single doses of 1500 mg/kg and 720 mg/kg were tolerated without significant toxicity in rats and dogs, respectively. These doses are approximately 30 and 50 times, respectively, the recommended human dose on a mg/m2 basis.
11 DESCRIPTION
Bexarotene capsules are a member of a subclass of retinoids that selectively activate retinoid X receptors (RXRs). These retinoid receptors have biologic activity distinct from that of retinoic acid receptors (RARs).
The chemical name of bexarotene is 4-[1-(5,6,7,8-tetrahydro-3,5,5,8,8-pentamethyl-2-naphthalenyl) ethenyl] benzoic acid, and the structural formula is as follows:
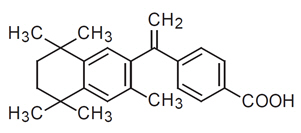
Bexarotene is an off-white to white powder with a molecular weight of 348.48 and a molecular formula of C24H28O2. It is insoluble in water and slightly soluble in vegetable oils and ethanol, USP.
Each bexarotene capsule contains 75 mg of bexarotene for oral administration. It also contains the following inactive ingredients: butylated hydroxyanisole, polyethylene glycol, polysorbate and povidone. The capsule shell contains gelatin, glycerin, sorbitol, titanium dioxide and water.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Bexarotene selectively binds and activates retinoid X receptor subtypes (RXRα, RXRβ, RXRγ). RXRs can form heterodimers with various receptor partners such as retinoic acid receptors (RARs), vitamin D receptor, thyroid receptor, and peroxisome proliferator activator receptors (PPARs). Once activated, these receptors function as transcription factors that regulate the expression of genes that control cellular differentiation and proliferation. Bexarotene inhibits the growth in vitro of some tumor cell lines of hematopoietic and squamous cell origin. It also induces tumor regression in vivo in some animal models. The exact mechanism of action of bexarotene in the treatment of cutaneous T-cell lymphoma (CTCL) is unknown.
12.3 Pharmacokinetics
Terminal half-life of bexarotene is approximately seven hours. Studies in patients with advanced malignancies show approximate single dose linearity within the therapeutic range.
Absorption
After oral administration of bexarotene capsules, bexarotene is absorbed with a Tmax of about two hours. Plasma bexarotene AUC and Cmax values resulting from a 75 to 300 mg dose were 35% and 48% higher, respectively, after a fat-containing meal than after a glucose solution.
Distribution
Bexarotene is highly bound (>99%) to plasma proteins. The plasma proteins to which bexarotene binds have not been elucidated, and the ability of bexarotene to displace drugs bound to plasma proteins and the ability of drugs to displace bexarotene binding have not been studied.
Elimination
Metabolism
Four bexarotene metabolites have been identified in plasma: 6- and 7-hydroxy-bexarotene and 6- and 7-oxo-bexarotene. In vitro studies suggest that cytochrome P450 3A4 is the major cytochrome P450 responsible for formation of the oxidative metabolites and that the oxidative metabolites may be glucuronidated. The oxidative metabolites are active in in vitro assays of retinoid receptor activation, but the relative contribution of the parent and any metabolites to the efficacy and safety of bexarotene is unknown.
Pharmacokinetics in Specific Populations
Age: Based on the population pharmacokinetic analysis of data for 232 patients aged ≥65 years and 343 patients aged <65 years, age has no statistically significant effect on bexarotene pharmacokinetics.
Body Weight and Gender: Based on the population pharmacokinetics analysis of data for 614 patients with a weight range of 26 to 145 kg, the bexarotene apparent clearance increases with increasing body weight. Gender has no statistically significant effect on bexarotene pharmacokinetics.
Race: Based on the population pharmacokinetic analysis of data for 540 Caucasian and 44 Black patients, bexarotene pharmacokinetics are similar in Blacks and Caucasians. There are insufficient data to evaluate potential differences in the pharmacokinetics of bexarotene for other races.
Renal Impairment: No formal studies have been conducted with bexarotene in patients with renal impairment. Urinary elimination of bexarotene and its known metabolites is a minor excretory pathway (<1% of administered dose), but because renal impairment can result in significant protein binding changes, pharmacokinetics may be altered in patients with renal impairment.
Hepatic Impairment: No specific studies have been conducted with bexarotene in patients with hepatic impairment. Because less than 1% of the dose is excreted in the urine unchanged and there is in vitro evidence of extensive hepatic contribution to bexarotene elimination, hepatic impairment would be expected to lead to greatly decreased clearance.
Drug Interactions
Effect of Other Drugs on Bexarotene
CYP3A4 Inhibitors/Inducers: In a clinical study, concomitant administration of multiple doses of ketoconazole with bexarotene did not alter bexarotene plasma concentrations. This suggests that bexarotene elimination is not dependent on CYP3A4 metabolism.
Paclitaxel plus Carboplatin: The coadministration of paclitaxel (200 mg/m2 IV dose over 3 hours) plus carboplatin (at a dose expected to achieve an AUC of 6 mg∙min/mL) with bexarotene (400 mg/m2 orally once daily) increased the exposure to bexarotene (AUC0–24 and Cmax) by 2-fold compared to bexarotene alone.
Effect of Bexarotene on Other Drugs
Bexarotene did not significantly inhibit the following enzymes in human liver microsomes: CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1 and CYP3A4. In vitro data suggested a potential for bexarotene to inhibit CYP2C8 and induce CYP3A4.
Atorvastatin: The exposure (AUC) to atorvastatin (a substrate for CYP3A4) decreased by half when atorvastatin was coadministered with bexarotene (400 mg/m2 orally once daily).
Tamoxifen: Based on interim data, concomitant administration of bexarotene and tamoxifen resulted in approximately a 35% decrease in plasma concentrations of tamoxifen, possibly through induction of CYP3A4 by bexarotene.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term studies in animals to assess the carcinogenic potential of bexarotene have not been conducted. Bexarotene is not mutagenic to bacteria (Ames assay) or mammalian cells (mouse lymphoma assay). Bexarotene was not clastogenic in vivo (micronucleus test in mice).
No formal fertility studies were conducted with bexarotene. Bexarotene caused testicular degeneration when oral doses of 1.5 mg/kg/day were given to dogs for 91 days (producing an AUC of approximately one fifth the AUC at the recommended human daily dose).
14 CLINICAL STUDIES
Bexarotene was evaluated in two clinical trials in 152 patients with advanced and early stage cutaneous T-cell lymphoma (CTCL) in two multicenter, open-label, historically-controlled clinical trials conducted in the U.S., Canada, Europe, and Australia.
The advanced disease patients had disease refractory to at least one prior systemic therapy (median of two, range one to six prior systemic therapies) and had been treated with a median of five (range 1 to 11) prior systemic, irradiation, and/or topical therapies. Early disease patients were intolerant to, had disease that was refractory to, or had reached a response plateau of six months on, at least two prior therapies. The patients entered had been treated with a median of 3.5 (range 2 to 12) therapies (systemic, irradiation, and/or topical).
The two clinical trials enrolled a total of 152 patients, 102 of whom had disease refractory to at least one prior systemic therapy, 90 with advanced disease and 12 with early disease. This is the patient population for whom bexarotene is indicated.
Patients were initially treated with a starting dose of 650 mg/m2/day with a subsequent reduction of starting dose to 500 mg/m2/day. Neither of these starting doses was tolerated, and the starting dose was then reduced to 300 mg/m2/day. If, however, a patient on 300 mg/m2/day of bexarotene showed no response after eight or more weeks of therapy, the dose could be increased to 400 mg/m2/day.
Tumor response was assessed in both trials by observation of up to five baseline-defined index lesions using a Composite Assessment of Index Lesion Disease Severity (CA). This endpoint was based on a summation of the grades, for all index lesions, of erythema, scaling, plaque elevation, hypopigmentation or hyperpigmentation, and area of involvement. Also considered in response assessment was the presence or absence of cutaneous tumors and extracutaneous disease manifestations.
All tumor responses required confirmation over at least two assessments separated by at least four weeks. A partial response was defined as an improvement of at least 50% in the index lesions without worsening, or development of new cutaneous tumors or non-cutaneous manifestations. A complete clinical response required complete disappearance of all manifestations of disease, but did not require confirmation by biopsy.
At the initial dose of 300 mg/m2/day, 1/62 (1.6%) of patients had a complete clinical tumor response and 19/62 (30%) of patients had a partial tumor response. The rate of relapse (25% increase in CA or worsening of other aspects of disease) in the 20 patients who had a tumor response was 6/20 (30%) over a median duration of observation of 21 weeks, and the median duration of tumor response had not been reached. Responses were seen as early as 4 weeks and new responses continued to be seen at later visits.
16 HOW SUPPLIED/STORAGE AND HANDLING
Bexarotene Capsules are supplied as 75 mg off-white, oblong soft gelatin capsules, imprinted "US285" in black ink. They are supplied as follows:
Bottles of 100 capsules: NDC 0832-0285-00
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Inform the patient or caregiver about the following:
Birth Defects
Advise patients that bexarotene is contraindicated in pregnancy [see Contraindications (4.1)]. Bexarotene is a member of the retinoid class of drugs that is associated with birth defects in humans [see Use in Specific Populations (8.1)].
- Advise females of reproductive potential that they must avoid pregnancy while taking bexarotene and for at least one month following discontinuation of therapy.
- Advise females of reproductive potential of the importance of monthly pregnancy testing while taking bexarotene.
- Advise females of reproductive potential to use effective contraception for one month prior to the initiation of therapy, during therapy, and for at least one month following discontinuation of therapy and that two reliable forms of contraception should be used simultaneously, one of which should be non-hormonal.
- Advise females of reproductive potential that bexarotene therapy should be initiated on the second or third day of a normal menstrual period.
- Instruct patient to immediately stop taking bexarotene if she becomes pregnant while taking this drug.
- Advise male patients with sexual partners who are pregnant, possibly pregnant, or who could become pregnant that they must use condoms during sexual intercourse while taking bexarotene and for at least one month after the last dose of the drug.
Pancreatitis
Advise patients of the risk of developing pancreatitis, which may be accompanied by nausea, vomiting, and abdominal or back pain and to immediately contact their healthcare provider if these symptoms occur [see Warnings and Precautions (5.2)].
Hepatotoxicity
Inform patients of the possibility of developing liver function abnormalities and serious hepatic toxicity. Advise patients to immediately contact their healthcare provider if signs of liver failure occur, including jaundice, anorexia, bleeding, or bruising [see Warnings and Precautions (5.3)].
Neutropenia
Advise patients of the possibility of developing neutropenia and to immediately contact their healthcare provider should they develop a fever, particularly in association with any suggestion of infection [see Warnings and Precautions (5.5)].
Cataracts
Advise patients of the possibility of developing new or worsening cataracts and to inform their healthcare provider about any changes in their vision during treatment with bexarotene [see Warnings and Precautions (5.6)].
Vitamin A Supplementation Hazard
Advise patients to limit vitamin A intake to ≤15,000 IU/day to avoid potential additive toxic effects.
Hypoglycemia and Diabetes Mellitus
Advise patients of the possibility of developing hypoglycemia when using insulin, agents enhancing insulin secretion, or insulin-sensitizers while on bexarotene therapy. Instruct patients on these medications to check their blood sugar frequently and to notify their physicians of any changes in blood sugar level [see Warnings and Precautions (5.8)].
Photosensitivity
Advise patients of potential increased skin sensitivity to sunlight while taking bexarotene and to minimize exposure to sunlight and artificial ultraviolet light [see Warnings and Precautions (5.9)].
Laboratory Tests
Advise patients of laboratory testing which will occur during therapy to monitor lipids, liver function, thyroid function, and white blood cell counts [see Warnings and Precautions (5.10)]. If applicable, advise patients of monthly pregnancy testing [see Use In Specific Populations (8.3)].
Administration Instructions
Advise patients to take bexarotene with a meal [see DOSAGE AND ADMINISTRATION].
Manufactured for
UPSHER-SMITH LABORATORIES, LLC
Maple Grove, MN 55369
Made in New Zealand
Revised 0617
| PATIENT INFORMATION |
Patient Information
Bexarotene (bek-sar´ah-tēn) Capsules
What is the most important information I should know about bexarotene?
Bexarotene can cause serious side effects, including major birth defects to an unborn baby, if taken during pregnancy.
For females who can become pregnant:
- You should avoid becoming pregnant during treatment with bexarotene.
- Do not take bexarotene if you are pregnant or plan to become pregnant.
- Your healthcare provider will do a pregnancy test, within one week before you start bexarotene, and each month during treatment with bexarotene to make sure that you are not pregnant.
- You must use two effective forms of birth control together starting one month before you begin treatment with bexarotene, during treatment, and for one month after stopping bexarotene. Birth control pills (oral contraceptives) and other hormonal forms of birth control may not be effective if used with bexarotene. At least one of the forms of birth control that you choose should not contain hormones. Talk to your healthcare provider about what forms of birth control may be right for you during treatment with bexarotene.
- You should start taking bexarotene on the second or third day of a normal menstrual period. Follow your healthcare provider's instructions about when to start bexarotene.
- Call your healthcare provider right away, if you become pregnant or think that you are pregnant during treatment with bexarotene, and for one month after you stop taking bexarotene.
For males:
- You must use a condom with female partners who are pregnant, might be pregnant, or who are able to become pregnant, during treatment with bexarotene and for at least one month after your last dose.
What is bexarotene?
Bexarotene is a prescription medicine used to treat the skin problems that happen with a type of cancer called cutaneous T-cell lymphoma (CTCL) after treatment with at least one other type of medicine by mouth or injection, did not work or has stopped working.
It is not known if bexarotene is safe and effective in children.
Who should not take bexarotene?
Do not take bexarotene:
- if you are pregnant or plan to become pregnant. See "What is the most important information I should know about bexarotene?"
- if you are allergic to any of the ingredients in bexarotene. See the end of this leaflet for a complete list of ingredients in bexarotene.
Before taking bexarotene, tell your healthcare provider about all of your medical conditions, including if you:
- have or have had problems with your pancreas, including pancreatitis
- have or have had gallbladder problems
- have or have had liver problems
- have thyroid problems
- have diabetes
- have high levels of fats (lipids) called cholesterol or triglycerides in your blood
- have cataracts or a history of cataracts
- drink alcohol
- are pregnant, plan to become pregnant, or think you may be pregnant. See "What is the most important information I should know about bexarotene?"
- are breastfeeding or plan to breastfeed. It is not known if bexarotene passes into your breast milk. You should not breastfeed during treatment with bexarotene. Talk to your healthcare provider about the best way to feed your baby during treatment with bexarotene.
Tell your healthcare provider about all of the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Using bexarotene with certain other medicines can affect each other.
How should I take bexarotene?
- Take bexarotene exactly as your healthcare provider tells you.
- Your healthcare provider will tell you how many bexarotene capsules to take each day. Your healthcare provider may change your daily dose of bexarotene as needed to treat your CTCL or if you get certain side effects. You should not change your dose unless your healthcare provider tells you to.
Take your dose of bexarotene 1 time a day with a meal. Your healthcare provider will do blood tests before you start bexarotene and during treatment to check for side effects.
What should I avoid while taking bexarotene?
- Limit your exposure to sunlight and artificial types of sunlight. Bexarotene can make your skin more sensitive to sunlight and you can get sunburn.
- Limit the amount of vitamin A that you take during treatment with bexarotene. Large doses of vitamin A may cause side effects that are similar to side effects that can happen in people who take bexarotene.
What are the possible side effects of bexarotene?
Bexarotene can cause serious side effects, including:
- See "What is the most important information I should know about bexarotene?"
- Increased levels of fats (lipids) called cholesterol or triglycerides in your blood are common with bexarotene, but can also be serious. Your healthcare provider may prescribe you medicines to treat high cholesterol or triglycerides levels, reduce your dose, temporarily stop treatment or completely stop treatment with bexarotene if you have this problem.
-
Inflammation of the pancreas (acute pancreatitis). Bexarotene can sometimes cause pancreatitis that comes on suddenly (acute), and that can lead to death. Your risk of developing acute pancreatitis may be greater if you:
- have had pancreatitis in the past
- high levels of fats in your blood that are not controlled
- drink large amounts of alcohol
- have gallbladder problems
- have diabetes that is not controlled
- take certain medicines that can increase the amount of triglycerides in your blood
- take medicines that can harm your pancreas
- nausea that will not go away
- vomiting
- stomach-area (abdominal) or back pain
- Liver problems, including liver failure. Bexarotene can cause increased liver function blood test results, or liver problems that can lead to death.
- Thyroid problems (hypothyroidism) are common with bexarotene. Your healthcare provider may prescribe thyroid hormone treatment for you if needed.
- Low white blood cell count is common with bexarotene, but may sometimes be severe.
- New or worse cataracts. Tell your healthcare provider about any changes in your vision during treatment with bexarotene. You may need to have an eye examination.
- Risk of low blood sugar in people who have diabetes. Bexarotene can interact with certain medicines used to treat diabetes, such as insulin, sulfonylurea medicines, and thiazolinedione medicines. If you have diabetes, talk to your healthcare provider about your diabetes medicines and your risk for low blood sugar if you take bexarotene.
The most common side effects of bexarotene include:
- headache
- asthenia
- rash
- nausea
- infection
- stomach-area (abdomen) pain
- swelling of your hands, arms, feet or legs
- dry skin
These are not all the possible side effects of bexarotene. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store bexarotene?
- Store bexarotene between 2° to 25°C (36° to 77°F).
- Store the bexarotene bottle away from light, heat, and humidity.
- The capsules should not be taken after the expiration date printed on the bottle.
Keep bexarotene and all medicines out of the reach of children.
General information about the safe and effective use of bexarotene
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use bexarotene for a condition for which it was not prescribed. Do not give bexarotene to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about bexarotene that is written for health professionals
What are the ingredients in bexarotene?
Active ingredient: bexarotene
Inactive ingredients: butylated hydroxyanisole, polyethylene glycol, polysorbate and povidone. The capsule shell contains gelatin, glycerin, sorbitol, titanium dioxide and water.
For more information, visit www.upsher-smith.com or call 1-888-650-3789.
This Patient Information has been approved by the U. S. Food and Drug Administration.
Manufactured for
UPSHER-SMITH LABORATORIES, LLC
Maple Grove, MN 55369
Made in New Zealand
Revised 0617
