DESCRIPTION
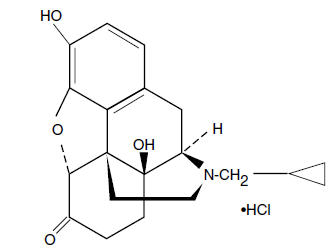
Naltrexone hydrochloride, an opioid antagonist, is a synthetic congener of oxymorphone with no opioid agonist properties. Naltrexone hydrochloride differs in structure from oxymorphone in that the methyl group on the nitrogen atom is replaced by a cyclopropylmethyl group. Naltrexone hydrochloride is also related to the potent opioid antagonist, naloxone, or n-allylnoroxymorphone. The chemical name for naltrexone hydrochloride is Morphinan-6-one, 17-(cyclopropylmethyl)-4,5-epoxy-3,14-dihydroxy-, hydrochloride, (5a)-. The structural formula is as follows:
| C20H23NO4∙HCl | Molecular Weight: 377.87 |
Naltrexone hydrochloride is a white, crystalline compound. The hydrochloride salt is soluble in water to the extent of about 100 mg/ml. Each film-coated tablet, for oral administration, contains 50 mg of naltrexone hydrochloride. In addition each film-coated tablet contains the following inactive ingredients: carnauba wax powder, colloidal silicon dioxide, croscarmellose sodium, hypromellose, hydroxypropyl cellulose, lactose anhydrous, magnesium stearate, microcrystalline cellulose, polyethylene glycol, titanium dioxide and yellow iron oxide.
CLINICAL PHARMACOLOGY
Pharmacodynamic Actions
Naltrexone hydrochloride is a pure opioid antagonist. It markedly attenuates or completely blocks, reversibly, the subjective effects of intravenously administered opioids.
When co-administered with morphine, on a chronic basis, naltrexone blocks the physical dependence to morphine, heroin and other opioids.
Naltrexone has few, if any, intrinsic actions besides its opioid blocking properties. However, it does produce some pupillary constriction, by an unknown mechanism.
The administration of naltrexone is not associated with the development of tolerance or dependence. In subjects physically dependent on opioids, naltrexone will precipitate withdrawal symptomatology.
Clinical studies indicate that 50 mg of naltrexone hydrochloride will block the pharmacologic effects of 25 mg of intravenously administered heroin for periods as long as 24 hours. Other data suggest that doubling the dose of naltrexone hydrochloride provides blockade for 48 hours, and tripling the dose of naltrexone hydrochloride provides blockade for about 72 hours.
Naltrexone blocks the effects of opioids by competitive binding (i.e., analogous to competitive inhibition of enzymes) at opioid receptors. This makes the blockade produced potentially surmountable, but overcoming full naltrexone blockade by administration of very high doses of opiates has resulted in excessive symptoms of histamine release in experimental subjects. The mechanism of action of naltrexone in alcoholism is not understood; however, involvement of the endogenous opioid system is suggested by preclinical data. Naltrexone, an opioid receptor antagonist, competitively binds to such receptors and may block the effects of endogenous opioids. Opioid antagonists have been shown to reduce alcohol consumption by animals, and naltrexone has been shown to reduce alcohol consumption in clinical studies.
Naltrexone is not aversive therapy and does not cause a disulfiram-like reaction either as a result of opiate use or ethanol ingestion.
Pharmacokinetics
Naltrexone is a pure opioid receptor antagonist. Although well absorbed orally, naltrexone is subject to significant first pass metabolism with oral bioavailability estimates ranging from 5 to 40%. The activity of naltrexone is believed to be due to both parent and the 6-β-naltrexol metabolite. Both parent drug and metabolites are excreted primarily by the kidney (53% to 79% of the dose), however, urinary excretion of unchanged naltrexone accounts for less than 2% of an oral dose and fecal excretion is a minor elimination pathway. The mean elimination half-life (T-1/2) values for naltrexone and 6-β-naltrexol are 4 hours and 13 hours, respectively. Naltrexone and 6-β-naltrexol are dose proportional in terms of AUC and Cmax over the range of 50 to 200 mg and do not accumulate after 100 mg daily doses.
Absorption
Following oral administration, naltrexone undergoes rapid and nearly complete absorption with approximately 96% of the dose absorbed from the gastrointestinal tract. Peak plasma levels of both naltrexone and 6-β-naltrexol occur within one hour of dosing.
Distribution
The volume of distribution for naltrexone following intravenous administration is estimated to be 1350 liters. In vitro test with human plasma show naltrexone to be 21% bound to plasma proteins over the therapeutic dose range.
Metabolism
The systemic clearance (after intravenous administration) of naltrexone is ~3.5 L/min, which exceeds liver blood flow (~1.2 L/min). This suggests both that naltrexone is a highly extracted drug (>98% metabolized) and that extra hepatic sites of drug metabolism exist. The major metabolite of naltrexone is 6-β-naltrexol. Two other minor metabolites are 2- hydroxy-3-methoxy-6-β-naltrexol and 2-hydroxy-3-methyl-naltrexone. Naltrexone and its metabolites are also conjugated to form additional metabolic products.
Elimination
The renal clearance for naltrexone ranges from 30 to 127 mL/min and suggests that renal elimination is primarily by glomerular filtration. In comparison, the renal clearance for 6-β-naltrexol ranges from 230 to 369 mL/min, suggesting an additional renal tubular secretory mechanism. The urinary excretion of unchanged naltrexone accounts for less than 2% of an oral dose; urinary excretion of unchanged and conjugated 6-β-naltrexol accounts for 43% of an oral dose. The pharmacokinetic profile of naltrexone suggest that naltrexone and its metabolites may undergo enterohepatic recycling.
Hepatic and Renal Impairment
Naltrexone appears to have extra-hepatic sites of drug metabolism and its major metabolite undergoes active tubular secretion (see Metabolism above). Adequate studies of naltrexone in patients with severe hepatic or renal impairment have not been conducted (see PRECAUTIONS, Special Risk Patients).
Clinical Trials
Alcoholism
The efficacy of naltrexone as an aid to the treatment of alcoholism was tested in placebocontrolled, outpatient, double blind trials. These studies used a dose of naltrexone hydrochloride 50 mg once daily for 12 weeks as an adjunct to social and psychotherapeutic methods when given under conditions that enhanced patient compliance. Patients with psychosis, dementia, and secondary psychiatric diagnoses were excluded from these studies.
In one of these studies, 104 alcohol-dependent patients were randomized to receive either naltrexone hydrochloride 50 mg once daily or placebo. In this study, naltrexone proved superior to placebo in measures of drinking including abstention rates (51% vs. 23%), number of drinking days, and relapse (31% vs. 60%). In a second study with 82 alcoholdependent patients, the group of patients receiving naltrexone were shown to have lower relapse rates (21% vs. 41%), less alcohol craving, and fewer drinking days compared with patients who received placebo, but these results depended on the specific analysis used.
The clinical use of naltrexone as adjunctive pharmacotherapy for the treatment of alcoholism was also evaluated in a multicenter safety study. This study of 865 individuals with alcoholism included patients with the comorbid psychiatric conditions, concomitant medications, polysubstance abuse and HIV disease. Results of this study demonstrated that the side effect profile of naltrexone appears to be similar in both alcoholic and opioid dependent populations, and that serious side effects are uncommon.
In the clinical studies, treatment with naltrexone supported abstinence, prevented relapse and decreased alcohol consumption. In the uncontrolled study, the patterns of abstinence and relapse were similar to those observed in the controlled studies. Naltrexone was not uniformly helpful to all patients, and the expected effect of the drug is a modest improvement in the outcome of conventional treatment.
Treatment of Opioid Addiction
Naltrexone has been shown to produce complete blockade of the euphoric effects of opioids in both volunteer and addict populations. When administered by means that enforce compliance, it will produce an effective opioid blockade, but has not been shown to affect the use of cocaine or other non-opioid drugs of abuse.
There are no data that demonstrate an unequivocally beneficial effect of naltrexone on rates of recidivism among detoxified, formerly opioid-dependent individuals who self-administer the drug. The failure of the drug in this setting appears to be due to poor medication compliance.
The drug is reported to be of greatest use in good prognosis opioid addicts who take the drug as part of a comprehensive occupational rehabilitative program, behavioral contract, or other compliance-enhancing protocol. Naltrexone, unlike methadone or LAAM (levo- alpha-acetyl-methadol), does not reinforce medication compliance and is expected to have a therapeutic effect only when given under external conditions that support continued use of the medication.
Individualization of Dosage
DO NOT ATTEMPT TREATMENT WITH NALTREXONE UNLESS, IN THE MEDICAL JUDGEMENT OF THE PRESCRIBING PHYSICIAN, THERE IS NO REASONABLE POSSIBILITY OF OPIOID USE WITHIN THE PAST 7 TO 10 DAYS. IF THERE IS ANY QUESTION OF OCCULT OPIOID DEPENDENCE, PERFORM A NALOXONE CHALLENGE TEST.
Treatment of Alcoholism
The placebo-controlled studies that demonstrated the efficacy of naltrexone as an adjunctive treatment of alcoholism used a dose regimen of naltrexone hydrochloride 50 mg once daily for up to 12 weeks. Other dose regimens or durations of therapy were not studied in these trials.
Physicians are advised that 5 to 15% of patients taking naltrexone for alcoholism will complain of non-specific side effects, chiefly gastrointestinal upset. Prescribing physicians have tried using an initial 25 mg dose, splitting the daily dose, and adjusting the time of dosing with limited success. No dose or pattern of dosing has been shown to be more effective than any other in reducing these complaints for all patients.
Treatment of Opioid Dependence
Once the patient has been started on naltrexone hydrochloride, 50 mg once a day will produce adequate clinical blockade of the actions of parenterally administered opioids. As with many non-agonist treatments for addiction, naltrexone is of proven value only when given as part of a comprehensive plan of management that includes some measure to ensure the patient takes the medication.
A flexible approach to a dosing regimen may be employed to enhance compliance. Thus, patients may receive 50 mg of naltrexone hydrochloride every weekday with a 100 mg dose on Saturday or patients may receive 100 mg every other day, or 150 mg every third day. Several of the clinical studies reported in the literature have employed the following dosing regimen: 100 mg on Monday, 100 mg on Wednesday, and 150 mg on Friday. This dosing schedule appeared to be acceptable to many naltrexone patients successfully maintaining their opiod- free state.
Experience with the supervised administration of a number of potentially hepatotoxic agents suggests that supervised administration and single doses of naltrexone hydrochloride higher than 50 mg may have an associated increased risk of hepatocellular injury, even though three-times a week dosing has been well-tolerated in the addict population and in initial clinical trials in alcoholism. Clinics using this approach should balance the possible risks against the probable benefits and may wish to maintain a higher index of suspicion for drug-associated hepatitis and ensure patients are advised of the need to report non-specific abdominal complaints (see PRECAUTIONS, Information for Patients).
INDICATIONS AND USAGE
Naltrexone hydrochloride tablets are indicated in the treatment of alcohol dependence and for the blockade of the effects of exogenously administered opioids.
Naltrexone has not been shown to provide any therapeutic benefit except as part of an appropriate plan of management for the addictions.
CONTRAINDICATIONS
Naltrexone is contraindicated in:
- Patients receiving opioid analgesics.
- Patients currently dependent on opioids, including those currently maintained on opiate agonists [e.g., methadone or LAAM (levo- alpha-acetyl-methadol)].
- Patients in acute opioid withdrawal (see WARNINGS).
- Any individual who has failed the naloxone challenge test or who has a positive urine screen for opioids.
- Any individual with a history of sensitivity to naltrexone or any other components of this product. It is not known if there is any cross-sensitivity with naloxone or the phenanthrene containing opioids.
- Any individual with acute hepatitis or liver failure.
WARNINGS
Hepatotoxicity
| Naltrexone has the capacity to cause hepatocellular injury when given in excessive doses. |
| Naltrexone is contraindicated in acute hepatitis or liver failure, and its use in patients with active liver disease must be carefully considered in light of its hepatotoxic effects. |
| The margin of separation between the apparently safe dose of naltrexone and the dose causing hepatic injury appears to be only five-fold or less. Naltrexone does not appear to be a hepatotoxin at the recommended doses. |
| Patients should be warned of the risk of hepatic injury and advised to stop the use of naltrexone and seek medical attention if they experience symptoms of acute hepatitis. |
Evidence of the hepatotoxic potential of naltrexone is derived primarily from a placebo controlled study in which naltrexone hydrochloride was administered to obese subjects at a dose approximately five-fold that recommended for the blockade of opiate receptors (300 mg per day). In that study, 5 of 26 naltrexone recipients developed elevations of serum transaminases (i.e., peak ALT values ranging from a low of 121 to a high of 532; or 3 to 19 times their baseline values) after three to eight weeks of treatment. Although the patients involved were generally clinically asymptomatic and the transaminase levels of all patients on whom follow-up was obtained returned to (or toward) baseline values in a matter of weeks, the lack of any transaminase elevations of similar magnitude in any of the 24 placebo patients in the same study is persuasive evidence that naltrexone is a direct (i.e., not idiosyncratic) hepatotoxin.
This conclusion is also supported by evidence from other placebo controlled studies in which exposure to naltrexone hydrochloride at doses above the amount recommended for the treatment of alcoholism or opiate blockade (50 mg/day) consistently produced more numerous and more significant elevations of serum transaminases than did placebo. Transaminase elevations in 3 of 9 patients with Alzheimer's Disease who received naltrexone hydrochloride (at doses up to 300 mg/day) for 5 to 8 weeks in an open clinical trial have been reported.
Although no cases of hepatic failure due to naltrexone administration have ever been reported, physicians are advised to consider this as a possible risk of treatment and to use the same care in prescribing naltrexone as they would other drugs with the potential for causing hepatic injury.
Unintended Precipitation of Abstinence
To prevent occurrence of an acute abstinence syndrome, or exacerbation of a preexisting subclinical abstinence syndrome, patients must be opioid-free for a minimum of 7-10 days before starting naltrexone. Since the absence of an opioid drug in the urine is often not sufficient proof that a patient is opioid-free, a naloxone challenge should be employed if the prescribing physician feels there is a risk of precipitating a withdrawal reaction following administration of naltrexone. The naloxone challenge test is described in the DOSAGE AND ADMINISTRATION section.
Attempt to Overcome Blockade
While naltrexone is a potent antagonist with a prolonged pharmacologic effect (24 to 72 hours), the blockade produced by naltrexone is surmountable. This is useful in patients who may require analgesia, but poses a potential risk to individuals who attempt, on their own, to overcome the blockade by administering large amounts of exogenous opioids. Indeed, any attempt by a patient to overcome the antagonism by taking opioids is very dangerous and may lead to fatal overdose. Injury may arise because the plasma concentration of exogenous opioids attained immediately following their acute administration may be sufficient to overcome the competitive receptor blockade. As a consequence, the patient may be in immediate danger of suffering life endangering opioid intoxication (e.g., respiratory arrest, circulatory collapse). Patients should be told of the serious consequences of trying to overcome the opiate blockade (see PRECAUTIONS, Information for Patients).
There is also the possibility that a patient who has been treated with naltrexone will respond to lower doses of opioids than previously used, particularly if taken in such a manner that high plasma concentrations remain in the body beyond the time that naltrexone exerts its therapeutic effects. This could result in potentially life-threatening opioid intoxication (respiratory compromise or arrest, circulatory collapse, etc.). Patients should be aware that they may be more sensitive to lower doses of opioids after naltrexone treatment is discontinued.
Ultra Rapid Opioid Withdrawal
Safe use of naltrexone in ultra rapid opiate detoxification programs has not been established (see ADVERSE REACTIONS).
PRECAUTIONS
General
When Reversal of Naltrexone Blockade is Required
In an emergency situation in patients receiving fully blocking doses of naltrexone, a suggested plan of management is regional analgesia, conscious sedation with a benzodiazepine, use of non-opioid analgesics or general anesthesia.
In a situation requiring opioid analgesia, the amount of opioid required may be greater than usual, and the resulting respiratory depression may be deeper and more prolonged.
A rapidly acting opioid analgesic which minimizes the duration of respiratory depression is preferred. The amount of analgesic administered should be titrated to the needs of the patient. Non-receptor mediated actions may occur and should be expected (e.g., facial swelling, itching, generalized erythema, or bronchoconstriction) presumably due to histamine release.
Irrespective of the drug chosen to reverse naltrexone blockade, the patient should be monitored closely by appropriately trained personnel in a setting equipped and staffed for cardiopulmonary resuscitation.
Accidentally Precipitated Withdrawal
Severe opioid withdrawal syndromes precipitated by the accidental ingestion of naltrexone have been reported in opioid-dependent individuals. Symptoms of withdrawal have usually appeared within five minutes of ingestion of naltrexone and have lasted for up to 48 hours. Mental status changes including confusion, somnolence and visual hallucinations have occurred. Significant fluid losses from vomiting and diarrhea have required intravenous fluid administration. In all cases patients were closely monitored and therapy with nonopioid medications was tailored to meet individual requirements.
Use of naltrexone does not eliminate or diminish withdrawal symptoms. If naltrexone is initiated early in the abstinence process, it will not preclude the patient's experience of the full range of signs and symptoms that would be experienced if naltrexone had not been started. Numerous adverse events are known to be associated with withdrawal.
Special Risk Patients
Renal Impairment
Naltrexone and its primary metabolite are excreted primarily in the urine, and caution is recommended in administering the drug to patients with renal impairment.
Hepatic Impairment
Caution should be exercised when naltrexone hydrochloride is administered to patients with liver disease. An increase in naltrexone AUC of approximately 5- and 10-fold in patients with compensated and decompensated liver cirrhosis, respectively, compared with subjects with normal liver function has been reported. These data also suggest that alterations in naltrexone bioavailability are related to liver disease severity.
Suicide
The risk of suicide is known to be increased in patients with substance abuse with or without concomitant depression. This risk is not abated by treatment with naltrexone (see ADVERSE REACTIONS).
Information for Patients
It is recommended that the prescribing physician relate the following information to patients being treated with naltrexone:
You have been prescribed naltrexone hydrochloride tablets as part of the comprehensive treatment for your alcoholism or drug dependence. You should carry identification to alert medical personnel to the fact that you are taking naltrexone hydrochloride. A naltrexone medication card may be obtained from your physician and can be used for this purpose. Carrying the identification card should help to ensure that you can obtain adequate treatment in an emergency. If you require medical treatment, be sure to tell the treating physician that you are receiving naltrexone therapy.
You should take naltrexone as directed by your physician. If you attempt to self-administer heroin or any other opiate drug, in small doses while on naltrexone, you will not perceive any effect. Most important, however, if you attempt to self-administer large doses of heroin or any other opioid (including methadone or LAAM) while on naltrexone, you may die or sustain serious injury, including coma.
Naltrexone is well-tolerated in the recommended doses, but may cause liver injury when taken in excess or in people who develop liver disease from other causes. If you develop abdominal pain lasting more than a few days, white bowel movements, dark urine, or yellowing of your eyes, you should stop taking naltrexone immediately and see your doctor as soon as possible.
Laboratory Tests
A high index of suspicion for drug-related hepatic injury is critical if the occurrence of liver damage induced by naltrexone is to be detected at the earliest possible time. Evaluations, using appropriate batteries of tests to detect liver injury are recommended at a frequency appropriate to the clinical situation and the dose of naltrexone.
Naltrexone does not interfere with thin-layer, gas-liquid, and high pressure liquid chromatographic methods which may be used for the separation and detection of morphine, methadone or quinine in the urine. Naltrexone may or may not interfere with enzymatic methods for the detection of opioids depending on the specificity of the test. Please consult the test manufacturer for specific details.
Drug Interactions
Studies to evaluate possible interactions between naltrexone and drugs other than opiates have not been performed. Consequently, caution is advised if the concomitant administration of naltrexone and other drugs is required.
The safety and efficacy of concomitant use of naltrexone and disulfiram is unknown, and the concomitant use of two potentially hepatotoxic medications is not ordinarily recommended unless the probable benefits outweigh the known risks.
Lethargy and somnolence have been reported following doses of naltrexone and thioridazine.
Patients taking naltrexone may not benefit from opioid containing medicines, such as cough and cold preparations, antidiarrheal preparations, and opioid analgesics. In an emergency situation when opioid analgesia must be administered to a patient receiving naltrexone, the amount of opioid required may be greater than usual, and the resulting respiratory depression may be deeper and more prolonged (see PRECAUTIONS).
Carcinogenesis, Mutagenesis and Impairment of Fertility
The following statements are based on the results of experiments in mice and rats. The potential carcinogenic, mutagenic and fertility effects of the metabolite 6-β-naltrexol are unknown.
In a two-year carcinogenicity study in rats, there were small increases in the numbers of testicular mesotheliomas in males and tumors of vascular origin in males and females. The incidence of mesothelioma in males given naltrexone at a dietary dose of 100 mg/kg/day (600 mg/m2/day; 16 times the recommended therapeutic dose, based on body surface area) was 6%, compared with a maximum historical incidence of 4%. The incidence of vascular tumors in males and females given dietary doses of 100 mg/kg/day (600 mg/m2/day) was 4% but only the incidence in females was increased compared with a maximum historical control incidence of 2%. There was no evidence of carcinogenicity in a two-year dietary study with naltrexone in male and female mice.
There was limited evidence of a weak genotoxic effect of naltrexone in one gene mutation assay in a mammalian cell line, in the Drosophila recessive lethal assay, and in non-specific DNA repair tests with E.coli. However, no evidence of genotoxic potential was observed in a range of other in vitro tests, including assays for gene mutation in bacteria, yeast, or in a second mammalian cell line, a chromosomal aberration assay, and an assay for DNA damage in human cells. Naltrexone did not exhibit clastogenicity in an in vivo mouse micronucleus assay.
Naltrexone (100 mg/kg/day [600 mg/m2/day] PO; 16 times the recommended therapeutic dose, based on body surface area) caused a significant increase in pseudopregnancy in the rat. A decrease in the pregnancy rate of mated female rats also occurred. There was no effect on male fertility at this dose level. The relevance of these observations to human fertility is not known.
Pregnancy
Teratogenic Effects
Pregnancy Category C
Naltrexone has been shown to increase the incidence of early fetal loss when given to rats at doses ≥30 mg/kg/day (180 mg/m2/day; 5 times the recommended therapeutic dose, based on body surface area) and to rabbits at oral doses ≥60 mg/kg/day (720 mg/m2/day; 18 times the recommended therapeutic dose, based on body surface area). There was no evidence of teratogenicity when naltrexone was administered orally to rats and rabbits during the period of major organogenesis at doses up to 200 mg/kg/day (32 and 65 times the recommended therapeutic dose, respectively, based on body surface area).
Rats do not form appreciable quantities of the major human metabolite, 6-β-naltrexol; therefore, the potential reproductive toxicity of the metabolites in rats is not known.
There are no adequate and well-controlled studies in pregnant women. Naltrexone should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Nursing Mothers
In animal studies, naltrexone and 6-β-naltrexol were excreted in the milk of lactating rats dosed orally with naltrexone. Whether or not naltrexone is excreted in human milk is unknown. Because many drugs are excreted in human milk, caution should be exercised when naltrexone is administered to a nursing woman.
ADVERSE REACTIONS
During two randomized, double-blind placebo-controlled 12-week trails to evaluate the efficacy of naltrexone as an adjunctive treatment of alcohol dependence, most patients tolerated naltrexone well. In these studies, a total of 93 patients received naltrexone hydrochloride at a dose of 50 mg once daily. Five of these patients discontinued naltrexone because of nausea. No serious adverse events were reported during these two trials.
While extensive clinical studies evaluating the use of naltrexone in detoxified, formerly opioid-dependent individuals failed to identify any single, serious untoward risk of naltrexone use, placebo-controlled studies employing up to fivefold higher doses of naltrexone hydrochloride (up to 300 mg per day) than that recommended for use in opiate receptor blockade have shown that naltrexone causes hepatocellular injury in a substantial proportion of patients exposed at higher doses (see WARNINGS and PRECAUTIONS, Laboratory Tests).
Aside from this finding, and the risk of precipitated opioid withdrawal, available evidence does not incriminate naltrexone, used at any dose, as a cause of any other serious adverse reaction for the patient who is "opioid-free". It is critical to recognize that naltrexone hydrochloride can precipitate or exacerbate abstinence signs and symptoms in any individual who is not completely free of exogenous opioids.
Patients with addictive disorders, especially opioid addiction, are at risk for multiple numerous adverse events and abnormal laboratory findings, including liver function abnormalities. Data from both controlled and observational studies suggest that these abnormalities, other than the dose- related hepatotoxicity described above, are not related to the use of naltrexone.
Among opioid-free individuals, naltrexone administration at the recommended dose has not been associated with a predictable profile of serious adverse or untoward events. However, as mentioned above, among individuals using opioids, naltrexone may cause serious withdrawal reactions (see CONTRAINDICATIONS, WARNINGS, DOSAGE AND ADMINISTRATION).
Reported Adverse Events
Naltrexone has not been shown to cause significant increases in complaints in placebocontrolled trials in patients known to be free of opioids for more than 7 to 10 days. Studies in alcoholic populations and in volunteers in clinical pharmacology studies have suggested that a small fraction of patients may experience an opioid withdrawal-like symptom complex consisting of tearfulness, mild nausea, abdominal cramps, restlessness, bone or joint pain, myalgia, and nasal symptoms. This may represent the unmasking of occult opioid use, or it may represent symptoms attributable to naltrexone. A number of alternative dosing patterns have been recommended to try to reduce the frequency of these complaints (see CLINICAL PHARMACOLOGY, Clinical Trials, Individualization of Dosage).
Alcoholism
In an open label safety study with approximately 570 individuals with alcoholism receiving naltrexone, the following new-onset adverse reactions occurred in 2% or more of the patients: nausea (10%), headache (7%), dizziness (4%), nervousness (4%), fatigue (4%), insomnia (3%), vomiting (3%), anxiety (2%) and somnolence (2%).
Depression, suicidal ideation, and suicidal attempts have been reported in all groups when comparing naltrexone, placebo, or controls undergoing treatment for alcoholism.
| RATE RANGES OF NEW ONSET EVENTS | ||
|---|---|---|
| Naltrexone | Placebo | |
| Depression | 0 - 15% | 0 - 17% |
| Suicide Attempt/ldeation | 0 - 1% | 0 - 3% |
Although no causal relationship with naltrexone is suspected, physicians should be aware that treatment with naltrexone does not reduce the risk of suicide in these patients (see PRECAUTIONS).
Opioid Addiction
The following adverse reactions have been reported both at baseline and during the naltrexone clinical trials in opioid addiction at an incidence rate of more than 10%:
Difficulty sleeping, anxiety, nervousness, abdominal pain/cramps, nausea and/or vomiting, low energy, joint and muscle pain, and headache.
The incidence was less than 10% for:
Loss of appetite, diarrhea, constipation, increased thirst, increased energy, feeling down, irritability, dizziness, skin rash, delayed ejaculation, decreased potency, and chills.
The following events occurred in less than 1% of subjects:
Respiratory:
nasal congestion, itching, rhinorrhea, sneezing, sore throat, excess mucus or phlegm, sinus trouble, heavy breathing, hoarseness, cough, shortness of breath.
Cardiovascular:
nose bleeds, phlebitis, edema, increased blood pressure, non-specific ECG changes, palpitations, tachycardia.
Gastrointestinal: excessive gas, hemorrhoids, diarrhea, ulcer.
Musculoskeletal:
painful shoulders, legs or knees; tremors, twitching.
Genitourinary:
increased frequency of, or discomfort during, urination; increased or decreased sexual interest.
Dermatologic:
oily skin, pruritus, acne, athlete's foot, cold sores, alopecia.
Psychiatric:
depression, paranoia, fatigue, restlessness, confusion, disorientation, hallucinations, nightmares, bad dreams.
Special senses:
eyes-blurred, burning, light sensitive, swollen, aching, strained; ears-"clogged," aching, tinnitus.
General:
increased appetite, weight loss, weight gain, yawning, somnolence, fever, dry mouth, head "pounding," inguinal pain, swollen glands, "side" pains, cold feet, "hot spells."
Post-marketing Experience
Data collected from post-marketing use of naltrexone show that most events usually occur early in the course of drug therapy and are transient. It is not always possible to distinguish these occurrences from those signs and symptoms that may result from a withdrawal syndrome. Events that have been reported include anorexia, asthenia, chest pain, fatigue, headache, hot flushes, malaise, changes in blood pressure, agitation, dizziness, hyperkinesia, nausea, vomiting, tremor, abdominal pain, diarrhea, elevations in liver enzymes or bilirubin, hepatic function abnormalities or hepatitis, palpitations, myalgia, anxiety, confusion, euphoria, hallucinations, insomnia, nervousness, somnolence, abnormal thinking, dyspnea, rash, increased sweating, and vision abnormalities.
Depression, suicide, attempted suicide and suicidal ideation have been reported in the postmarketing experience with naltrexone used in the treatment of opioid dependence. No causal relationship has been demonstrated. In the literature, endogenous opioids have been theorized to contribute to a variety of conditions. In some individuals the use of opioid antagonists has been associated with a change in baseline levels of some hypothalamic, pituitary, adrenal, or gonadal hormones. The clinical significance of such changes is not fully understood.
Adverse events, including withdrawal symptoms and death, have been reported with the use of naltrexone hydrochloride in ultra rapid opiate detoxification programs. The cause of death in these cases is not known (see WARNINGS).
Laboratory Tests
With the exception of liver test abnormalities (see WARNINGS and PRECAUTIONS), results of laboratory tests, like adverse reaction reports, have not shown consistent patterns of abnormalities that can be attributed to treatment with naltrexone.
Idiopathic thrombocytopenic purpura was reported in one patient who may have been sensitized to naltrexone in a previous course of treatment with naltrexone. The condition cleared without sequelae after discontinuation of naltrexone and corticosteroid treatment.
DRUG ABUSE AND DEPENDENCE
Naltrexone is a pure opioid antagonist. It does not lead to physical or psychological dependence. Tolerance to the opioid antagonist effect is not known to occur.
OVERDOSAGE
There is limited clinical experience with naltrexone overdosage in humans. In one study, subjects who received 800 mg daily of naltrexone hydrochloride for up to one week showed no evidence of toxicity.
In the mouse, rat and guinea pig, the oral LD50s were 1,100 to 1,550 mg/kg; 1,450 mg/kg; and 1,490 mg/kg; respectively. High doses of naltrexone (generally ≥ 1,000 mg/kg) produced salivation, depression/reduced activity, tremors, and convulsions. Mortalities in animals due to high-dose naltrexone administration usually were due to clonic-tonic convulsions and/or respiratory failure.
DOSAGE AND ADMINISTRATION
IF THERE IS ANY QUESTION OF OCCULT OPIOID DEPENDENCE, PERFORM A NALOXONE CHALLENGE TEST AND DO NOT INITIATE NALTREXONE THERAPY UNTIL THE NALOXONE CHALLENGE IS NEGATIVE.
Treatment of Alcoholism
A dose of 50 mg once daily is recommended for most patients (see CLINICAL PHARMACOLOGY, Clinical Trials, Individualization of Dosage). The placebo-controlled studies that demonstrated the efficacy of naltrexone as an adjunctive treatment of alcoholism used a dose regimen of naltrexone 50 mg once daily for up to 12 weeks. Other dose regimens or durations of therapy were not evaluated in these trials.
A patient is a candidate for treatment with naltrexone if:
- the patient is willing to take a medicine to help with alcohol dependence
- the patient is opioid-free for 7 to 10 days
- the patient does not have severe or active liver or kidney problems (Typical guidelines suggest liver function tests no greater than 3 times the upper limits of normal, and bilirubin normal.)
- the patient is not allergic to naltrexone, and no other contraindications are present
Refer to CONTRAINDICATIONS, WARNINGS, and PRECAUTIONS sections for additional information.
Naltrexone should be considered as only one of many factors determining the success of treatment of alcoholism. Factors associated with a good outcome in the clinical trials with naltrexone were the type, intensity, and duration of treatment; appropriate management of comorbid conditions; use of community-based support groups; and good medication compliance. To achieve the best possible treatment outcome, appropriate complianceenhancing techniques should be implemented for all components of the treatment program, especially medication compliance.
Treatment of Opioid Dependence
Initiate treatment with Naltrexone using the following guidelines
- Treatment should not be attempted unless the patient has remained opioid-free for at least 7 to 10 days. Self-reporting of abstinence from opioids in opioid addicts should be verified by analysis of patient's urine for absence of opioids. The patient should not be manifesting withdrawal signs or reporting withdrawal symptoms.
- If there is any questions of occult opioid dependence, perform a naloxone challenge test. If signs of opioid withdrawal are still observed following naloxone challenge, treatment with naltrexone should not be attempted. The naloxone challenge can be repeated in 24 hours.
- Treatment should be initiated carefully, with an initial dose of 25 mg of naltrexone hydrochloride. If no withdrawal signs occur, the patient may be started on 50 mg a day thereafter.
Naloxone Challenge Test
The naloxone challenge test should not be performed in a patient showing clinical signs or symptoms of opioid withdrawal, or in a patient whose urine contains opioids. The naloxone challenge test may be administered by either the intravenous or subcutaneous routes.
Interpretation of the Challenge
Monitor vital signs and observe the patient for signs and symptoms of opioid withdrawal. These may include, but are not limited to: nausea, vomiting, dysphoria, yawning, sweating, tearing, rhinorrhea, stuffy nose, craving for opioids, poor appetite, abdominal cramps, sense of fear, skin erythema, disrupted sleep patterns, fidgeting, uneasiness, poor ability to focus, mental lapses, muscle aches or cramps, pupillary dilation, piloerection, fever, changes in blood pressure, pulse or temperature, anxiety, depression, irritability, backache, bone or joint pains, tremors, sensations of skin crawling or fasciculations. If signs or symptoms of withdrawal appear, the test is positive and no additional naloxone should be administered.
Warning: If the test is positive, do NOT initiate naltrexone therapy. Repeat the challenge in 24 hours. If the test is negative, naltrexone therapy may be started if no other contraindications are present. If there is any doubt about the result of the test, hold naltrexone and repeat the challenge in 24 hours.
Alternative Dosing Schedules
Once the patient has been started on naltrexone hydrochloride, 50 mg every 24 hours will produce adequate clinical blockade of the actions of parenterally administered opioids (i.e., this dose will block the effects of a 25 mg intravenous heroin challenge). A flexible approach to a dosing regimen may need to be employed in cases of supervised administration. Thus, patients may receive 50 mg of naltrexone hydrochloride every weekday with a 100 mg dose on Saturday, 100 mg every other day, or 150 mg every third day. The degree of blockade produced by naltrexone may be reduced by these extended dosing intervals.
There may be a higher risk of hepatocellular injury with single doses above 50 mg, and use of higher doses and extended dosing intervals should balance the possible risks against the probable benefits (see WARNINGS and CLINICAL PHARMACOLOGY, Clinical Trials, Individualization of Dosage).
Patient Compliance
Naltrexone should be considered as only one of many factors determining the success of treatment. To achieve the best possible treatment outcome, appropriate complianceenhancing techniques should be implemented for all components of the treatment program, including medication compliance.
HOW SUPPLIED
Naltrexone Hydrochloride Tablets, USP 50 mg are yellow, round film-coated tablets, bisected on one side, debossed with "EL" on one side of the bisect and "15" on the other side of the bisect.
NDC 68094-853-62
Thirty (30) unit dose 50 mg tablets per carton
Manufactured by:
Elite Laboratories, Inc.
Northvale, NJ 07647
Distributed by:
Precision Dose, Inc.
South Beloit, IL 61080
LI966 Rev. 06/14
PRINCIPAL DISPLAY PANEL - 50 mg Tablet Carton Label
PrecisionDose™
NDC 68094-853-62
Unit Dose
Naltrexone
Hydrochloride
Tablets, USP
50 mg
30 TABLETS
(3x10)
EACH FILM-COATED TABLET CONTAINS:
Naltrexone Hydrochloride, USP
50 mg
USUAL DOSE: SEE ENCLOSED INSERT.
Store at 20° to 25°C (68° to 77°F).
[see USP Controlled Room Temperature].
Keep this and all medication out of reach of children.
Rx Only
LC965 R1
Distributed by:
Precision Dose, Inc.
South Beloit, IL 61080
