FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
TNKase® is indicated to reduce the risk of death associated with acute ST elevation myocardial infarction (STEMI).
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
Initiate treatment as soon as possible after the onset of STEMI symptoms.
TNKase is for intravenous (IV) administration only, administered as a single bolus over 5 seconds. Individualize dosage based on the patient's weight (see Table 1).
| Patient Weight (kg) | TNKase (mg) | Volume TNKase* to be administered (mL) |
|---|---|---|
|
||
| < 60 | 30 | 6 |
| ≥ 60 to < 70 | 35 | 7 |
| ≥ 70 to < 80 | 40 | 8 |
| ≥ 80 to < 90 | 45 | 9 |
| ≥ 90 | 50 | 10 |
2.2 Preparation
Follow the below steps to prepare TNKase for administration:
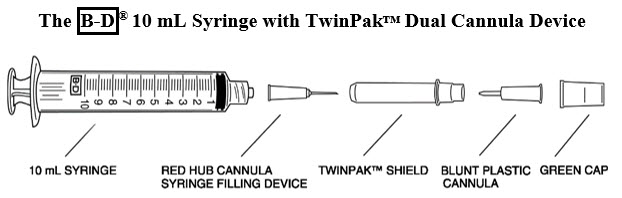
Remove the shield assembly from the supplied B-D® 10 mL syringe with TwinPak™ Dual Cannula Device (see Figure 1) and aseptically withdraw 10 mL of Sterile Water for Injection, USP, from the supplied diluent vial using the red hub cannula syringe filling device. Only use the supplied Sterile Water for Injection, USP for reconstitution.
- Note: Do not discard the shield assembly.
- Aseptically reconstitute the vial with 10 mL Sterile Water for Injection, USP by directing the stream into the lyophilized powder to obtain a final concentration of 5 mg/mL. Slight foaming upon reconstitution is not unusual; any large bubbles will dissipate if the product is allowed to stand undisturbed for several minutes.
- Gently swirl until contents are completely dissolved. DO NOT SHAKE. The reconstituted preparation results in a colorless to pale yellow transparent solution.
- Determine the appropriate dose of TNKase [see Dosage and Administration (2.1)] and withdraw this volume (in milliliters) from the reconstituted vial with the syringe. Discard any unused solution.
- Stand the shield vertically on a flat surface (with green side down) and passively recap the red hub cannula.
- Remove the entire shield assembly, including the red hub cannula, by twisting counterclockwise. Note: The shield assembly also contains the clear-ended blunt plastic cannula; retain for split septum intravenous access.
2.3 Administration
Follow the below steps for administration of TNKase;
- Inspect the product prior to administration for particulate matter and discoloration. Administer TNKase as reconstituted at 5 mg/mL.
- Precipitation may occur when TNKase is administered in an intravenous line containing dextrose. Flush dextrose-containing lines with a saline-containing solution prior to and following single bolus administration of TNKase.
- Administer reconstituted TNKase as a single intravenous bolus over 5 seconds.
- Because TNKase contains no antibacterial preservatives, reconstitute immediately before use. If the reconstituted TNKase is not used immediately, refrigerate the TNKase vial at 2°C to 8°C (36°F to 46°F) and use within 8 hours.
- Although the supplied syringe is compatible with a conventional needle, this syringe is designed to be used with needleless intravenous systems. From the information below, follow the instructions applicable to the intravenous system in use.
Split septum intravenous system: - Remove the green cap.
- Attach the clear-ended blunt plastic cannula to the syringe.
- Remove the shield and use the blunt plastic cannula to access the split septum injection port.
- Because the blunt plastic cannula has two side ports, air or fluid expelled through the cannula will exit in two sideways directions; direct away from face or mucous membranes.
Luer-Lok® system: Connect syringe directly to intravenous port. Conventional needle
(not supplied in this kit):Attach a large bore needle, e.g., 18 gauge, to the syringe's universal Luer-Lok®. - Dispose of the syringe, cannula and shield per established procedures.
3 DOSAGE FORMS AND STRENGTHS
For injection: 50 mg as a white to pale yellow lyophilized powder in a single-dose vial for reconstitution with the co-packaged 10 mL single-dose vial of Sterile Water for Injection, USP (diluent).
4 CONTRAINDICATIONS
TNKase is contraindicated in patients with [see Warnings and Precautions (5.1)]:
- Active internal bleeding
- History of cerebrovascular accident
- Intracranial or intraspinal surgery or trauma within 2 months
- Intracranial neoplasm, arteriovenous malformation, or aneurysm
- Known bleeding diathesis
- Severe uncontrolled hypertension
5 WARNINGS AND PRECAUTIONS
5.1 Bleeding
TNKase can cause bleeding, including intracranial hemorrhage and fatal bleeding. Concomitant use of other drugs that impair hemostasis increases the risk of bleeding.
Should serious bleeding that is not controlled by local pressure occur, discontinue any concomitant heparin or antiplatelet agents immediately and treat appropriately.
Avoid intramuscular injections and nonessential handling of the patient for the first few hours following treatment with TNKase. Perform arterial and venous punctures carefully and only as required. To minimize bleeding from noncompressible sites, avoid internal jugular and subclavian venous punctures. If an arterial puncture is necessary during TNKase infusion, use an upper extremity vessel that is accessible to manual compression. Apply pressure for at least 30 minutes.
5.2 Thromboembolism
The use of thrombolytics can increase the risk of thrombo-embolic events in patients with high likelihood of left heart thrombus, such as patients with mitral stenosis or atrial fibrillation.
5.3 Cholesterol Embolization
Cholesterol embolism has been reported in patients treated with thrombolytic agents. Investigate cause of any new embolic event and treat appropriately.
5.4 Arrhythmias
Coronary thrombolysis may result in arrhythmias associated with reperfusion. These arrhythmias (such as sinus bradycardia, accelerated idioventricular rhythm, ventricular premature depolarizations, ventricular tachycardia) may be managed with standard anti-arrhythmic measures. It is recommended that anti-arrhythmic therapy for bradycardia and/or ventricular irritability be available when TNKase is administered.
5.5 Increased Risk of Heart Failure and Recurrent Ischemia when used with Planned Percutaneous Coronary Intervention (PCI) in STEMI.
In a trial of patients with STEMI, there were trends toward worse outcomes in the individual components of the primary endpoint between TNKase plus PCI versus PCI alone (mortality 6.7% vs. 4.9%, respectively; cardiogenic shock 6.3% vs. 4.8%, respectively; and CHF 12% vs. 9.2%, respectively). In addition, there were trends towards worse outcomes in recurrent MI (6.1% vs. 3.7%, respectively; p = 0.03) and repeat target vessel revascularization (6.6% vs. 3.4%, respectively; p = 0.0045) in patients receiving TNKase plus PCI versus PCI alone [see Clinical Studies (14.1)]. In patients with large ST segment elevation myocardial infarction, physicians should choose either thrombolysis or PCI as the primary treatment strategy for reperfusion. Rescue PCI or subsequent elective PCI may be performed after administration of thrombolytic therapies if medically appropriate; however, the optimal use of adjunctive antithrombotic and antiplatelet therapies in this setting is unknown.
5.6 Hypersensitivity
Hypersensitivity, including urticarial / anaphylactic reactions, have been reported after administration of TNKase (e.g., anaphylaxis, angioedema, laryngeal edema, rash, and urticaria). Monitor patients treated with TNKase during and for several hours after infusion. If symptoms of hypersensitivity occur, initiate appropriate therapy (e.g., antihistamines, corticosteroids).
6 ADVERSE REACTIONS
The following adverse reactions are discussed in other sections of the label:
- Bleeding [see Contraindications (4), Warnings and Precautions (5.1)]
- Hypersensitivity [see Warnings and Precautions (5.6)]
6.1 Immunogenicity
Four of 625 (0.64%) patients tested for antibody formation to TNKase had a positive antibody titer at 30 days in studies with TNKase. The observed incidence of antibody positivity in an assay may be influenced by several factors including sample handling, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to TNKase with the incidence of antibodies to other products may be misleading.
7 DRUG INTERACTIONS
7.1 Drug/Laboratory Test Interactions
During TNKase therapy, results of coagulation tests and/or measures of fibrinolytic activity may be unreliable unless specific precautions are taken to prevent in vitro artifacts. Tenecteplase is an enzyme that, when present in blood in pharmacologic concentrations, remains active under in vitro conditions. This can lead to degradation of fibrinogen in blood samples removed for analysis.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are risks to the mother and fetus from acute ST elevation myocardial infarction, which is a medical emergency in pregnancy and can be fatal if left untreated (see Clinical Considerations). Published data consisting of a small number of case reports involving the use of related thrombolytic agents in pregnant women have not identified an increased risk of major birth defects. There are no data on the use of tenecteplase during pregnancy to evaluate for a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes.
TNKase does not elicit maternal and direct embryo toxicity in rabbits following a single IV administration. In developmental toxicity studies conducted in rabbits, the no observable effect level (NOEL) of a single IV administration of TNKase on maternal or developmental toxicity (5 mg/kg) was approximately 7 times human exposure (based on AUC) at the dose for STEMI.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
8.2 Lactation
Risk Summary
There are no data on the presence of tenecteplase in either human or animal milk, the effects on the breastfed infant, or the effect on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for TNKase and any potential adverse effects on the breastfed infant from the TNKase or from the underlying maternal condition.
8.4 Pediatric Use
The safety and effectiveness of TNKase in pediatric patients have not been established.
8.5 Geriatric Use
In the ASSENT-2 study, 41% (3500/8458) of patients who were treated with TNKase were aged 65 years or older. In this population, rates of 30-day mortality, stroke, intracranial hemorrhage and major bleeds requiring blood transfusion or leading to hemodynamic complications were higher than in those aged less than 65 years.
11 DESCRIPTION
Tenecteplase is a tissue plasminogen activator (tPA) produced by recombinant DNA technology using a mammalian cell line (Chinese Hamster Ovary cells). Tenecteplase is a 527-amino acid glycoprotein developed by introducing the following modifications to the complementary DNA (cDNA) for natural human tPA: a substitution of threonine 103 with asparagine, and a substitution of asparagine 117 with glutamine, both within the kringle 1 domain, and a tetra-alanine substitution at amino acids 296–299 in the protease domain. It has a molecular weight of 58,742 daltons. Biological potency is determined by an in vitro clot lysis assay and is expressed in tenecteplase specific units. The specific activity of tenecteplase has been defined as 200 units/mg.
TNKase (tenecteplase) for injection is a sterile, white to pale yellow, lyophilized powder for intravenous bolus administration after reconstitution with Sterile Water for Injection, USP. Each single-dose vial of TNKase nominally contains 50 mg of tenecteplase, arginine (522 mg), phosphoric acid (approximately 160 mg), and polysorbate 20 (4.0 mg). Following reconstitution with the supplied 10 mL single-dose vial of Sterile Water for Injection, USP, the final concentration is 5 mg/mL with a pH of approximately 7.3.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Tenecteplase is a modified form of human tissue plasminogen activator (tPA) that binds to fibrin and converts plasminogen to plasmin. In the presence of fibrin, in vitro studies demonstrate that tenecteplase-mediated conversion of plasminogen to plasmin is increased relative to its conversion in the absence of fibrin. This fibrin specificity decreases systemic activation of plasminogen and the resulting degradation of circulating fibrinogen as compared to a molecule lacking this property. The clinical significance of fibrin-specificity on safety (e.g., bleeding) or efficacy has not been established.
12.2 Pharmacodynamics
Following administration of 30, 40, or 50 mg of TNKase, there are decreases in circulating fibrinogen (4%–15%) and plasminogen (11%–24%).
12.3 Pharmacokinetics
Distribution
In patients with STEMI, TNKase administered as a single IV bolus exhibits a biphasic disposition from the plasma.
Volume of distribution at central compartment ranges from 4.22 to 5.43 L, approximating plasma volume. Steady-state volume of distribution was approximately 50% greater (6.12 to 8.01 L), suggestive of some extravascular distribution.
Elimination
After IV bolus administration, the terminal phase half-life of tenecteplase was 90 to 130 minutes. In 99 of 104 patients treated with TNKase, TNKase has linear PK with mean maximum concentrations increased in a dose-proportional manner and mean plasma clearance was similar for the 30, 40, and 50 mg doses ranging from 99 to 119 mL/min.
14 CLINICAL STUDIES
14.1 Acute Myocardial Infarction
ASSENT-2
The Assessment of the Safety and Efficacy of a New Thrombolytic (ASSENT-2) study was an international, randomized, double-blind trial that compared 30-day mortality rates in 16,949 patients assigned to receive an IV bolus dose of TNKase or an accelerated infusion of Activase® (alteplase). Eligibility criteria included onset of chest pain within 6 hours of randomization and ST-segment elevation or left bundle branch block on electrocardiogram (ECG). Patients were to be excluded from the trial if they received GP IIb/IIIa inhibitors within the previous 12 hours. TNKase was dosed using actual or estimated weight in a weight-tiered fashion as described in Dosage and Administration (2.1). All patients were to receive 150–325 mg of aspirin administered as soon as possible, followed by 150–325 mg daily. Intravenous heparin was to be administered as soon as possible: for patients weighing ≤ 67 kg, heparin was administered as a 4000-unit IV bolus followed by infusion at 800 U/hr; for patients weighing > 67 kg, heparin was administered as a 5000-unit IV bolus followed by infusion at 1000 U/hr. Heparin was continued for 48 to 72 hours with infusion adjusted to maintain aPTT at 50–75 seconds. The use of GP IIb/IIIa inhibitors was discouraged for the first 24 hours following randomization. The results of the primary endpoint (30-day mortality rates with non-parametric adjustment for the covariates of age, Killip class, heart rate, systolic blood pressure and infarct location) along with selected other 30-day endpoints are shown in Table 2.
| 30-Day Events | TNKase (n = 8461) | Accelerated Activase (n = 8488) | Relative Risk TNKase/Activase (95% CI) |
|---|---|---|---|
| Mortality | 6.2% | 6.2% | 1.00 (0.89, 1.12) |
| Intracranial Hemorrhage (ICH) | 0.9% | 0.9% | 0.99 (0.73, 1.35) |
| Any Stroke | 1.8% | 1.7% | 1.07 (0.86, 1.35) |
| Death or Nonfatal Stroke | 7.1% | 7.0% | 1.01 (0.91, 1.13) |
Rates of mortality and the combined endpoint of death or stroke among pre-specified subgroups, including age, gender, time to treatment, infarct location, and history of previous myocardial infarction, demonstrate consistent relative risks across these subgroups. There was insufficient enrollment of non-Caucasian patients to draw any conclusions regarding relative efficacy in racial subsets.
Rates of in-hospital procedures, including percutaneous transluminal coronary angioplasty (PTCA), stent placement, intra-aortic balloon pump (IABP) use, and coronary artery bypass graft (CABG) surgery, were similar between the TNKase and Activase (alteplase) groups.
TIMI-10B
TIMI 10B was an open-label, controlled, randomized, dose-ranging, angiography study which utilized a blinded core laboratory for review of coronary arteriograms. Patients (n = 837) presenting within 12 hours of symptom onset were treated with fixed doses of 30, 40, or 50 mg of TNKase or the accelerated infusion of Activase and underwent coronary arteriography at 90 minutes. The primary endpoint was the rate of TIMI Grade 3 flow at 90 minutes. The results showed that the 40 mg and 50 mg doses were similar to accelerated infusion of Activase in restoring patency. TIMI Grade 3 flow and TIMI Grade 2/3 flow at 90 minutes are shown in Table 3. The exact relationship between coronary artery patency and clinical activity has not been established.
| Activase ≤100 mg (n=311) | TNKase 30 mg (n=302) | TNKase 40 mg (n=148) | TNKase 50 mg (n=76) |
|
|---|---|---|---|---|
| TIMI Grade 3 Flow | 62.7% | 54.3% | 62.8% | 65.8% |
| (95% CI) | (57.1%, 68.1%) | (48.5%, 60.0%) | (54.5%, 70.6%) | (54.0%, 76.3%) |
| TIMI Grade 2/3 Flow | 81.7% | 76.8% | 79.1% | 88.2% |
| (95% CI) | (76.9%, 85.8%) | (71.6%, 81.5%) | (71.6%, 85.3%) | (78.7%, 94.4%) |
The angiographic results from TIMI 10B and the safety data from ASSENT-1, an additional uncontrolled safety study of 3,235 TNKase-treated patients, provided the framework to develop a weight-tiered TNKase dose regimen. Exploratory analyses suggested that a weight-adjusted dose of 0.5 to 0.6 mg/kg of TNKase resulted in a better patency to bleeding relationship than fixed doses of TNKase across a broad range of patient weights.
ASSENT 4 PCI
The Assessment of the Safety and Efficacy of a New Treatment Strategy with Percutaneous Coronary Intervention (ASSENT 4 PCI) was a phase IIIb/IV study designed to assess the safety and effectiveness of a strategy of administering full dose TNKase with a single bolus of 4000 U of unfractionated heparin in patients with STEMI, in whom primary percutaneous coronary intervention (PCI) was planned, but in whom a delay of 1-3 hours was anticipated before PCI. The trial was prematurely terminated with 1667 randomized patients (75 of whom were in the United States) due to a numerically higher mortality in the patients receiving TNKase prior to primary PCI versus PCI without TNKase (median time from randomization to balloon was 115 minutes in patients who were treated with TNKase plus PCI versus 107 minutes in patients who were treated with PCI alone). The incidence of the 90-day primary endpoint, a composite of death or cardiogenic shock or congestive heart failure (CHF) within 90 days, was 18.6% in patients treated with TNKase plus PCI versus 13.4% in those treated with PCI alone (p = 0.0045; RR 1.39 (95% CI 1.11–1.74)).
There were trends toward worse outcomes in the individual components of the primary endpoint between TNKase plus PCI versus PCI alone (mortality 6.7% vs. 4.9%, respectively; cardiogenic shock 6.3% vs. 4.8%, respectively; and CHF 12.0% vs. 9.2%, respectively). In addition, there were trends towards worse outcomes in recurrent MI (6.1% vs. 3.7%, respectively; p = 0.03) and repeat target vessel revascularization (6.6% vs. 3.4%, respectively; p = 0.004) in patients receiving TNKase plus PCI versus PCI alone [see Warnings and Precautions (5.5)].
There was no difference in in-hospital major bleeding between the two groups (5.6% vs. 4.4% for TNKase plus PCI vs. PCI alone, respectively). For patients treated with TNKase plus PCI, in-hospital rates of intracranial hemorrhage and total stroke were similar to those observed in previous trials (0.97% and 1.8%, respectively); however, none of the patients treated with PCI alone experienced a stroke (ischemic, hemorrhagic or other).
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
TNKase (tenecteplase) for injection is supplied as a sterile, white to pale yellow lyophilized powder in a 50 mg single-dose vial under partial vacuum.
Each 50 mg single-dose vial of TNKase is packaged with one 10 mL single-dose vial of Sterile Water for Injection, USP for reconstitution, and the B-D® 10 mL syringe with TwinPak™ Dual Cannula Device: NDC 50242-120-47.
TNKase® (tenecteplase)
Manufactured by:
Genentech, Inc.
A Member of the Roche Group
1 DNA Way
South San Francisco, CA
94080-4990
U.S. License No. 1048
TNKase® is a registered trademark of Genentech, Inc.
©2023 Genentech, Inc.
PRINCIPAL DISPLAY PANEL - Kit Carton
NDC 50242-120-47
Tenecteplase
TNKase®
50 mg
For use in myocardial infarction
Kit Contents: Each kit contains one 50 mg vial of TNKase, one 10 mL vial of preservative-free
Sterile Water for Injection, USP, one BD® 10 mL syringe with TwinPak™ Dual Cannula Device,
and package insert containing full prescribing information.
Vial Contents: The preservative-free single-use vial of TNKase contains 52.5 mg
Tenecteplase, 0.55 g L-arginine, 0.17 g phosphoric acid, and 4.3 mg polysorbate 20,
under partial vacuum. No U.S. standard of potency.
Rx only
US License No.: 1048
Genentech
10200128
